

Background: Soluble Aβ42 oligomers, rather than insoluble amyloid fibrils, are toxic species in Alzheimer's disease.
Results: We obtained structural restraints at all 42 residue positions in Aβ42 oligomers and performed structural modeling.
Conclusion: In oligomers, each Aβ42 protein forms a single β-sheet with three antiparallel β-strands.
Significance: Our novel structural model provides new structural framework for understanding oligomer-fibril interconversion and designing oligomer-targeted therapeutics.
Keywords: Alzheimer Disease, Amyloid, Electron Paramagnetic Resonance (EPR), Protein Aggregation, Protein Structure, Spin Labeling
Abstract
Aβ42 oligomers play key roles in the pathogenesis of Alzheimer disease, but their structures remain elusive partly due to their transient nature. Here, we show that Aβ42 in a fusion construct can be trapped in a stable oligomer state, which recapitulates characteristics of prefibrillar Aβ42 oligomers and enables us to establish their detailed structures. Site-directed spin labeling and electron paramagnetic resonance studies provide structural restraints in terms of side chain mobility and intermolecular distances at all 42 residue positions. Using these restraints and other biophysical data, we present a novel atomic-level oligomer model. In our model, each Aβ42 protein forms a single β-sheet with three β-strands in an antiparallel arrangement. Each β-sheet consists of four Aβ42 molecules in a head-to-tail arrangement. Four β-sheets are packed together in a face-to-back fashion. The stacking of identical segments between different β-sheets within an oligomer suggests that prefibrillar oligomers may interconvert with fibrils via strand rotation, wherein β-strands undergo an ∼90° rotation along the strand direction. This work provides insights into rational design of therapeutics targeting the process of interconversion between toxic oligomers and non-toxic fibrils.
Introduction
Alzheimer disease is a fatal neurodegenerative disorder and the most common form of dementia (1). Deposition of amyloid β (Aβ)3 peptide in the form of amyloid plaques is a hallmark of Alzheimer pathology. Amyloids refer to fibrillar protein aggregates with common tinctorial and structural characteristics and are involved in a range of neurodegenerative, localized, and systemic disorders including Parkinson and Huntington diseases, and type II diabetes (2). Soluble Aβ oligomers have been increasingly recognized as primary neurotoxins in Alzheimer disease (3–5). Several Aβ oligomers have been identified in vivo (6, 7), including dimers, trimers, and Aβ*56. Different in vitro protocols have been used to prepare oligomers such as Aβ-derived diffusible ligands (8), globulomers (9), prefibrillar oligomers (10), and amylospheroids (11). Because the molecular structures of these oligomers are unknown, it is impossible to know how many unique structures exist in these Aβ oligomers. Currently, structural classification of these oligomers is largely restricted to the use of conformation-specific antibodies (12). Based on immunoreactivity to the oligomer-specific polyclonal antibody A11, Aβ oligomers can be classified into A11-positive prefibrillar oligomers and A11-negative fibrillar oligomers (12).
One challenge in the structural studies of Aβ oligomers is related to their transient and heterogeneous nature. Aβ oligomers represent a series of intermediate assemblies on or off the pathway to fibril formation. Oligomers prepared using different protocols have been shown to be structurally diverse (13). Some Aβ oligomers have been shown to have similar parallel in-register β structures as amyloid fibrils (14), and other oligomers adopt distinct structures (15–19). Heterogeneity can also occur within the same oligomer sample (20, 21). Structural heterogeneity has been a major obstacle in obtaining high-resolution structural data.
Site-directed spin labeling (SDSL) in combination with electron paramagnetic resonance (EPR) spectroscopy has emerged as a powerful approach to characterize the structures of amyloid fibrils (22). The general strategy of SDSL includes substitution of a selected residue with cysteine and subsequent modification of the cysteine residue to produce a spin label side chain. The EPR sample can be in solutions, aggregates, or membrane environments, and of any size. As shown previously in the studies of Aβ and yeast prion protein Ure2p, EPR can resolve structural heterogeneity and separate different structural states (23–26). Distance measurements with continuous-wave and pulsed EPR can cover a wide range of distances from 5 to 70 Å (27, 28). These advantages make SDSL EPR a promising technique to obtain detailed structural information of the inherently heterogeneous Aβ oligomers.
In this work, we performed a comprehensive structural study on Aβ42 oligomers prepared using a fusion protein, GroES-ubiquitin-Aβ42 (GU-Aβ42). This fusion protein construct forms highly ordered oligomers without further assembling into fibrils, and enables us to obtain detailed structural information of these Aβ42 oligomers. The fusion protein system is similar to yeast prion proteins such as Sup35p and Ure2p, which contain both a prion domain and a globular domain, and the globular domain does not participate in the amyloid formation of these yeast prion proteins (29). The fusion protein approach also provides for some other unique applications. For example, a split luciferase-Aβ system allows high sensitivity detection of oligomer formation in mammalian cells (30). Fusion protein approaches also enable studies of mutational effects at specific residue positions in yeast (31) and Escherichia coli (32) cells and in vivo high throughput screening of small molecule inhibitor libraries (33). Fusion proteins also facilitate structural characterization of Aβ fragments using x-ray crystallography (34).
These GU-Aβ42 oligomers recapitulate the characteristics of prefibrillar oligomers, such as immunoreactivity to oligomer-specific antibody A11 (12). For structural studies with EPR, spin labels are introduced, one at a time, at all 42 residue positions of Aβ42 sequence. Residue-specific mobility analysis using EPR reveals three ordered segments at residues 1–10, 13–23, and 28–42. Distance measurements show two major intermolecular distance distributions at each of the 42 residue positions: 9–10 Å and 15–17 Å. These results allow us to suggest a triple-strand antiparallel model for the Aβ42 prefibrillar oligomers. Our model for prefibrillar oligomers points to a mechanism of oligomer-fibril interconversion wherein rotation of β-strands reorganizes the β-sheets of the oligomers into new fibril β-sheets that run (i.e. have β-hydrogen bonding) approximately perpendicular to the original β-sheets of the oligomers. We term this mechanism of nucleated conformational conversion (35) (conversion of an oligomer from one conformation to another without adding or losing material) to be “strand rotation.”
EXPERIMENTAL PROCEDURES
Preparation of Aβ42 Fusion Proteins and Full-length Aβ42
The DNA construct of GroES-ubiquitin-Aβ42 (36) and the deubiquitylating enzyme Usp2cc (37) were kindly provided by Dr. Rohan T. Baker at Australian National University (Australia) and Dr. Il-Seon Park at Chosun University (South Korea). Single cysteine mutations at various sites were introduced into Aβ42 sequence using QuikChange kit (Agilent Technologies). Mutations were confirmed with DNA sequencing. Expression of GroES-ubiquitin-Aβ42 in E. coli and their purification were performed as previously described in Ngo and Guo (38). Expression of Usp2cc and removal of fusion protein GU to prepare full-length Aβ42 was performed as previously described in Gu et al. (39). The purity of Aβ42 fusion proteins was checked with SDS-PAGE. The fusion protein GU without Aβ42 was prepared using the same protocol as GU-Aβ42.
Spin Labeling of Aβ42 Fusion Proteins
Dithiothreitol was added to purified Aβ42 fusion proteins to a final concentration of 10 mm and was allowed to incubate for 20 min at room temperature to break any disulfide bonds. Then Aβ42 fusion proteins were buffer exchanged into the labeling buffer (7 m guanidine hydrochloride, 50 mm NaCl, 20 mm MOPS, pH 6.8) using a 5-ml HiTrap desalting column (GE Healthcare). The spin labeling reagent MTSSL (1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl methanethiosulfonate, Enzo Life Sciences) was at 10-times molar excess and then incubated for 1 h at room temperature. Spin-labeled Aβ42 fusion proteins were then precipitated with methanol, air dried, and then stored at −80 °C.
Preparation of Aβ42 Fusion Protein Oligomers
Methanol precipitated Aβ42 fusion proteins were resuspended in PU buffer (50 mm phosphate, 8 m urea, pH 10.0) to a final concentration of 1 mm. Then Aβ42 fusion proteins were diluted 20-fold in PBS buffer (50 mm phosphate, 140 mm NaCl, pH 7.4) to 50 μm. After incubation at room temperature for 24 h, oligomers form loosely associated precipitates. As a control, the fusion protein GU without Aβ42 was also precipitated with methanol, resuspended in PU buffer, and followed by 20-fold dilution to PBS buffer and incubation at room temperature for 24 h.
Transmission Electron Microscopy
GU-Aβ42 oligomer samples were diluted with water to 5 μm and then applied onto glow discharged copper grids covered with 400 mesh formvar/carbon film (Ted Pella) and negatively stained with 1% uranyl acetate. Samples were examined under a JEOL JEM-1200EX transmission electron microscope with an accelerating voltage of 80 kV.
X-ray Powder Diffraction
GU-Aβ42 oligomers were collected by centrifugation at 20,000 × g for 20 min, and washed with water. The GU sample was buffer exchanged to water using a HiTrap desalting column (GE Healthcare). The lyophilized powders for both GU and GU-Aβ42 were mounted on the tip of a broken glass rod. Then, the specimen was placed on the goniometer of an in-house x-ray machine and shot using a Rigaku-FR-D x-ray generator equipped with a Rigaku HTC imaging plate detector.
Thioflavin T Fluorescence Assay
Thioflavin T (Sigma) was dissolved in PBS and filtered with a 0.22-μm filter. GU and GU-Aβ42 samples were diluted to a final concentration of 5 μm in PBS containing 50 μm ThT. Fluorescence was measured immediately on a Jasco FP-6200 spectrofluorometer. Excitation was at 440 nm (5-nm slit width), and emission was at 485 nm (5-nm slit width).
Dot Blot Assay with A11 Antibody
GU sample and GU-Aβ42 oligomers were diluted with PBS to 25 μm. Then 5 μl of GU and GU-Aβ42 samples were spotted on a nitrocellulose membrane (Bio-Rad). The membrane was blocked in 10% fat free milk in Tris-buffered saline (TBST) (50 mm Tris, 150 mm NaCl, 0.05% Tween20) at room temperature for 1 h, followed by incubation with the rabbit polyclonal A11 antibody at 2.4 μg/ml in 5% fat free milk, TBST at 4 °C overnight. TBST was used to wash the membranes for three times. Then, the membrane was incubated with anti-rabbit HRP-conjugated secondary antibodies (1:5000 in 5% fat free milk, TBST) (Jackson ImmunoResearch) for 1 h at room temperature, followed by further washing in TBST buffer for three times. The blots were developed using the Super Signal West Pico Chemiluminescent Substrate kit (Pierce).
SDS-PAGE
SDS-PAGE was performed on the 24-h samples of GU and GU-Aβ42 using Mini-PROTEAN tetra system (Bio-Rad). 4–20% gradient Tris-glycine gels (Bio-Rad) were used. Samples were mixed at 1:1 volume ratio with SDS loading buffer (4% SDS, 0.5 m β-mercaptoethanol, 125 mm Tris, 20% glycerol (v/v), 0.2 mg/ml bromphenol blue, pH 6.8) without boiling (unless specified otherwise).
Circular Dichroism Spectroscopy
Secondary structures of GU sample were analyzed by CD spectroscopy. The GU sample (200 μl), 24 h after dilution from PU to PBS, was placed in a 0.1-cm path length quartz cell (Starna). A Jasco J-715 CD spectrometer was employed. The measurement were carried out in a wavelength range of 190–260 nm at a rate of 20 nm min-1 with a step resolution of 0.5 nm, a time constant of 4 s and a bandwidth of 1 nm. The CD spectra were obtained by averaging 6 scans. The temperature was set at 25 °C. The spectra were corrected by subtracting the buffer background. The HT voltages were above 800 for the wavelength range of 190–203.5 nm, so only the CD data in the wavelength range of 204–260 nm were reported. Because of the formation of insoluble oligomers, the GU-Aβ42 samples were not studied with CD spectroscopy. The CD results are reported as mean residue ellipticity in Fig. 1F.
FIGURE 1.

Characterization of GU-Aβ42 oligomers. A, transmission electron microscopy image of GU-Aβ42 oligomers shows globular structures with diameters of 10–12 nm. Arrows point to several of the oligomers. B, dot blot analysis with A11 antibody shows that GU-Aβ42 oligomers bind strongly to A11, while GU alone shows very weak reactivity. C, GU-Aβ42 oligomers show a powder x-ray diffraction pattern consistent with β-sheet structure, and GU alone shows very weak diffraction. D, GU-Aβ42 oligomers have weak binding to ThT. A.U., arbitrary unit. Error bars are standard deviations of three independent measurements. E, SDS-PAGE shows that, in addition to monomer, GU-Aβ42 oligomers contain SDS-resistant tetramer and hexamer. F, circular dichroism spectrum of the GU sample.
Aggregation Kinetics of Aβ42
Purified full-length Aβ42, without fusion protein partner, was buffer exchanged to 30 mm ammonium acetate, pH 10.0, then lyophilized and stored at −80 °C. For aggregation experiments, lyophilized Aβ42 powder was dissolved in CG buffer (20 mm CAPS, 7 m guanidine hydrochloride, pH 11), filtered through 0.2-μm syringe filter (Corning 431212), and then buffer exchanged to PBS using a 5-ml HiTrap desalting column (GE Healthcare). The sample was then filtered through a 0.2-μm syringe filter (Corning 431212), and concentration was determined using an extinction coefficient of 1.28 mm−1 at 280 nm. The Aβ42 was diluted to 50 μm with PBS, supplemented with 20 μm ThT, either in the presence or absence of 2.5 μm GU or GU-Aβ42 samples. After mixing all components, 50 μl of each mixture was transferred to the 384 well black polystyrene plate with clear bottom and PEG coating (Corning 3655). The plate was then sealed with a plastic film (Corning 3095). All these steps were performed on ice if possible. The aggregation was initiated by placing the plate in a Victor 3V plate reader (Perkin Elmer). The plate is incubated at 37 °C with orbital shaking (1 mm shaking diameter, normal shaking speed). The thioflavin T fluorescence was measured through the bottom of the plate at every 5 min (with excitation filter of 450 nm and emission filter of 490 nm). Each sample was prepared in duplicates.
EPR Spectroscopy
EPR measurements were performed at X-band frequency on a Bruker EMX spectrometer equipped with the ER4102ST cavity at room temperature using a microwave power of 20 milliwatt. A modulation frequency of 100 kHz was used. Modulation amplitude was optimized to each individual spectrum. Scan width is 200 G. For each sample, 20 μl of oligomer sample was loaded into glass capillaries (VitroCom) sealed at one end. EPR spectra in each figure panel were normalized to the same number of spins.
EPR Distance Analysis
Distance analysis was performed using the program ShortDistances, developed by Dr. Christian Altenbach at UCLA. The detailed fitting procedure to obtain distances has been previously described (40). The 20% labeled spectra were used as the spectra without dipolar interactions. To avoid over fitting of the experimental data, we emphasized on using minimum number of variable parameters. The width of the distance distribution was fixed at 2 Å. The distance, percentage of the spin labels at the fitted distance, and the percentage of non-interacting spin labels were allowed to vary. The fitted spectra, distances, and their relative populations are plotted in Fig. 8.
FIGURE 8.

Intermolecular distances between spin labels in GU-Aβ42 oligomers. A, plot of intermolecular distances. B, plot of populations of spin labels at measured distances.
Modeling of Aβ42 Oligomers
EPR distance information was used to create harmonic distance constraints for backbone hydrogen bonding for Aβ42 plus four residues of ubiquitin (Lys-Arg-Gly-Gly) at the N terminus of Aβ42. Models were varied in number of oligomers in the directions of β-hydrogen bonding (direction of sheets), and in number of sheets. Models were also varied in β-twist, which changes the overall twist along the β-sheet direction. Models were energy-minimized by molecular dynamics with harmonic distance constraints using the CNS program (41). The best fit model satisfied the distance constraints from EPR and also fit well to the x-ray powder diffraction, using the Ro goodness of fit metric as previously described (18).
RESULTS
Aβ42 Fusion Protein Forms A11-positive Prefibrillar Oligomers
We studied the oligomer formation of Aβ42 using a fusion protein containing GroES-ubiquitin at the N terminus of Aβ42 sequence (36). Here this construct is termed GU-Aβ42. Oligomers are formed by a 20-fold dilution from a denaturing buffer containing 8 m urea to phosphate-buffered saline (PBS) and incubated at room temperature for 24 h without agitation. The final GU-Aβ42 concentration is 50 μm. Shortly after dilution from urea to PBS, GU-Aβ42 forms visible precipitates. Twenty-four hours after aggregation began, most of the GU-Aβ42 was in precipitate, and soluble GU-Aβ42 was below the detection limit by absorbance at 280 nm. We attribute the ability to form visible aggregates to the presence of GroES as a fusion protein partner. GroES is known to form oligomers (42, 43). When we performed the same aggregation assay using full-length Aβ42 without the fusion protein, we did not observe any visible aggregates. At the same time, Aβ42 quickly aggregates into amyloid fibrils in the absence of fusion protein partners. We conclude that the GroES fusion partner promotes the trapping of GU-Aβ42 into oligomers and prevents fibril formation, allowing us to further characterize its structure below. One disadvantage, however, is that formation of precipitates prohibited us from characterizing the properties of GU-Aβ42 oligomers using solution-based methods such as size exclusion chromatography, light scattering, and sedimentation.
Transmission electron microscopy (TEM) shows that the GU-Aβ42 sample consists of globular oligomers (Fig. 1A). Most of the GU-Aβ42 oligomers have a diameter of 10–12 nm. No fibrils or any elongated protofibrils were observed by electron microscopy, even after more than 2 weeks of incubation at room temperature.
To establish the relationship between the GU-Aβ42 oligomers and other Aβ42 oligomers, we performed dot blot analysis using the oligomer-specific A11 antibody (10). Fig. 1B shows that the GU-Aβ42 oligomers stained strongly with A11 antibody. This result suggests that the GU-Aβ42 oligomers should be classified as “prefibrillar” oligomers (12), which are distinct from fibrillar oligomers that share the same epitope as amyloid fibrils.
X-ray powder diffraction of GU-Aβ42 oligomers shows two sharp reflections at 4.6 and 10.0 Å (Fig. 1C), which have been previously suggested as characteristics of cross-β structure (44). The 4.6-Å reflection corresponds to the interstrand spacing within the same β-sheet, and the 10.0-Å reflection corresponds to the sheet to sheet spacing in the oligomers. The sharp nature of reflections at both 4.6 and 10.0 Å suggests that GU-Aβ42 oligomers contain highly ordered β-sheet structure.
Thioflavin T (ThT) binding assay shows that GU-Aβ42 has weak ThT binding (∼2.5-fold change in fluorescence intensity compared with ThT alone), and GU alone has only a marginally greater ThT signal (Fig. 1D). This is consistent with previous studies on full-length Aβ42 oligomers, which also show much weaker binding to ThT than mature fibrils (45).
On SDS gel, the GU-Aβ42 sample contains two major species of SDS-resistant oligomers (Fig. 1E). The apparent sizes for these two oligomers are 86 and 128 kDa according to the calibration with molecular weight standards. The molecular mass for GU-Aβ42 monomer is 25 kDa. The commercial molecular weight standards consist of denatured single polypeptide chains and thus adopt extended structures. SDS-resistant GU-Aβ42 oligomers, however, must adopt some compact structures to stay oligomeric. Therefore, GU-Aβ42 oligomers would migrate faster on the gel than the molecular weight standards of comparable size. For these reasons, we conclude that the SDS-resistant GU-Aβ42 oligomers are tetramers and hexamers. This finding is similar to our previous study showing that GU-Aβ42 oligomers also form tetramers and hexamers in the presence of 8 m urea (38). Here the oligomer sample contains only 0.4 m urea. Even with such low urea concentration, no other oligomeric species were observed within the detection limit of Coomassie staining on the gel. Tetramers and hexamers are also the major oligomer forms for Aβ42 without fusion protein partners, as revealed by ion mobility mass spectrometry (46), suggesting GU-Aβ42 oligomerizes similarly as Aβ42.
We also checked if the fusion protein GU alone is refolded under the condition of our oligomer preparation. As shown in Fig. 1F, the CD spectrum of the GU sample is qualitatively similar to previously published CD spectra of GroES (47) and ubiquitin (48), suggesting that the GU protein is refolded to the native structure under our oligomerization condition.
These results show that GU-Aβ42 forms globular oligomers. Lack of fibril formation suggests fusion protein partners effectively trap GU-Aβ42 in a stable oligomeric state. These GU-Aβ42 oligomers recapitulate the characteristics of full-length Aβ42 prefibrillar oligomers including A11 reactivity, weak ThT binding, and presence of SDS-resistant oligomers.
Residue-specific Side Chain Mobility in Aβ42 Oligomers
To study the structure of GU-Aβ42 oligomers with EPR, we introduced spin labels, one at a time, at all 42 residue positions of Aβ42 sequence. A commonly used spin labeling reagent (see “Experimental Procedures”) was used to generate the spin label side chain named R1 (Fig. 2A).
FIGURE 2.

Characterization of the spin-labeled GU-Aβ42 oligomers. A, a stick model of spin label R1 (PDB ID: 2Q9E). B, transmission electron microscopy images of representative spin-labeled GU-Aβ42 oligomers show globular structures with similar diameters as the wild-type oligomers. Arrows point to several of the oligomers. C, SDS-PAGE analysis of spin-labeled GU-Aβ42 oligomers. Note that overall spin labeling did not disrupt the formation of tetramers and hexamers.
TEM studies show that the spin-labeled GU-Aβ42 proteins form globular oligomers (Fig. 2B). The morphology of these oligomers is indistinguishable from the oligomers of wild-type GU-Aβ42, suggesting that spin labeling has little effect on the formation of oligomers.
Previously, we found that cysteine substitutions of hydrophobic residues (Ile-31, Ile-32, Leu-34, Val-39, Val-40, Ile-41) at the C-terminal region disrupted the formation of tetramers and hexamers (38). To check if spin labeling at these residue positions also disrupts oligomer formation, we performed SDS-PAGE analysis for the GU-Aβ42 oligomer samples labeled at C-terminal residues (Fig. 2C). We found that tetramers and hexamers are largely unaffected by spin labeling. We propose that the hydrophobic nature of the nitroxide ring makes it very tolerable in Aβ oligomers. These results also suggest that hydrophobicity, rather than size, of the amino acid side chain is critical for Aβ42 oligomerization.
To study the side chain mobility using SDSL EPR, we prepared GU-Aβ42 oligomers using a mixture of spin-labeled protein and wild-type protein at 1:4 molar ratio. This sample is referred to as “20% labeled.” The EPR spectra of 20% labeled samples are shown in Fig. 3. In the 20% labeled sample, intermolecular spin-spin interactions are minimized, so the EPR spectral lineshape is mainly determined by the mobility of the spin label, which reflects local structure at the labeling site. The side chain mobility is estimated using inverse center line width of the EPR spectra (49). Based on the plot of residue-specific side chain mobility (Fig. 4), Aβ42 consists of four structural segments. Segment 2 (residues 13–23) and segment 4 (residues 28–42) are the most ordered segments in the oligomers. The relatively more flexible segment 3 (residues 24–27) separates segments 2 and 4. X-ray powder diffraction studies suggest the presence of ordered β-sheet structures in GU-Aβ42 oligomers (Fig. 1C). Therefore, we conclude that both segments 2 and 4 adopt β structures, and segment 3 forms a turn connecting the two β strands.
FIGURE 3.

EPR spectra of spin-labeled GU-Aβ42 oligomers. The oligomers were prepared with spin-labeled and WT proteins at 1:4 molar ratio to minimize the effect of intermolecular spin-spin interactions on EPR lineshape. The sample preparation is referred to as “20% labeled” in the text. Scan width is 200 G.
FIGURE 4.

Residue-specific side chain mobility in GU-Aβ42 oligomers. The inverse center line width (δ−1) is determined using the EPR spectra of the 20% labeled GU-Aβ42 oligomers, in which spin-labeled and wild-type GU-Aβ42 are mixed at 1:4 molar ratio. The inset shows how the center line width was measured from the EPR spectrum.
Segment 1 (residues 1–12) has higher mobility than other segments (Fig. 4). However, the presence of two peaks at low-field resonance line (labeled M and N in Fig. 5A) suggests that the EPR spectra in this segment are composed of two components, which we termed M and N. The M component has similar mobility as the well ordered positions (Fig. 5B). Using spectral subtractions, we were able to reveal the lineshape of component N, whose relatively narrow lines indicate high mobility (Fig. 5). Therefore, even though center line width measurement, which has contributions from both components M and N, shows that residues 1–12 have higher mobility than other residues, spectral subtractions show that these residues consist of two structural states: a structured state and a locally disordered state. The structured state has mobility similar to segments 2 and 4 in Fig. 4.
FIGURE 5.
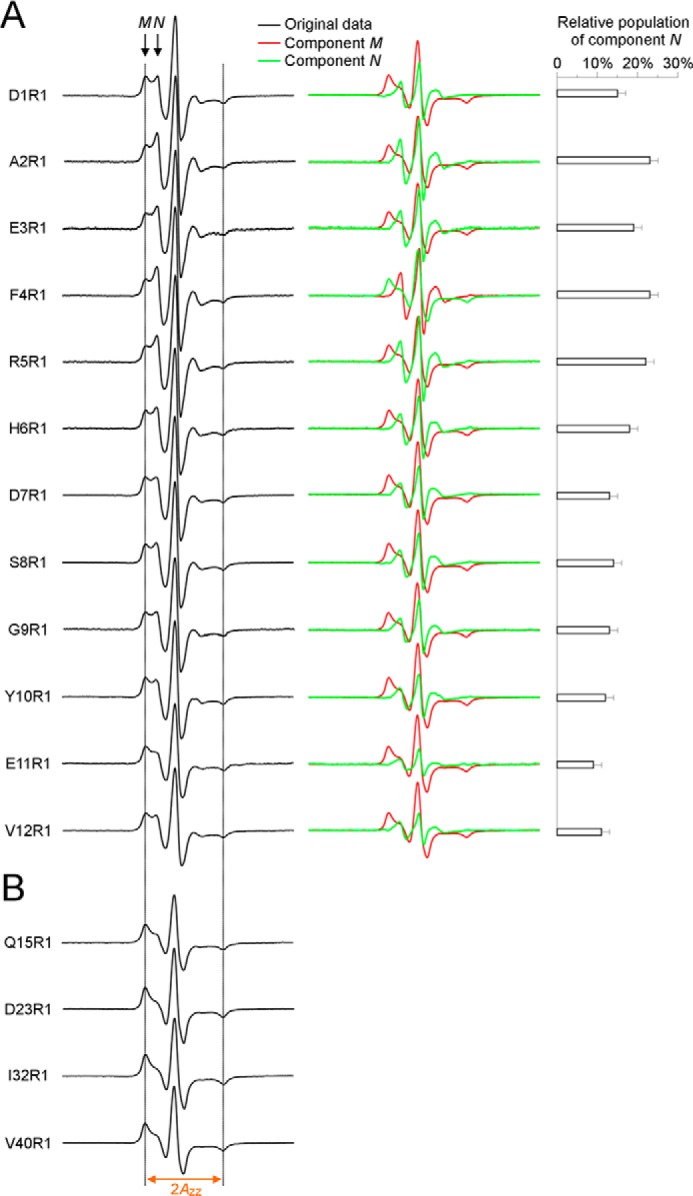
Spectral subtraction reveals a partially ordered structure for the N-terminal residues 1–12 in oligomers. A, EPR spectra at labeling sites 1–12 contain two spectral components M and N (arrows). Using the average EPR spectrum of Q15R1, D23R1, I32R1, and V40R1 as an approximation of component M, we obtained the lineshape of component N by subtracting component M from the experimental spectra. The component N accounts for 9–23% of total population, depending on the labeling sites. Dotted lines are drawn as visual aid to compare spectra. B, spectral lineshape is similar for the ordered residues 15, 23, 32, and 40, with similar 2Azz values. The same 2Azz is observed for the component M of spectra at residues 1–12, suggesting that the lineshape of component M may be similar as these ordered residues. Because of the similarity of these ordered spectra, an average spectrum was used as the approximation for the component M.
GU-Aβ42 Oligomers Adopt a Distinct Structure from Fibrils
Most amyloid fibrils studied to date adopt parallel in-register β-sheets (50), in which the side chains at the same residue position stack on top of each other. When spin label is introduced at a β-strand site in fibrils, the stacking of spin labels leads to strong spin exchange interactions (23, 26). As demonstrated in Fig. 6A, strong spin exchange interactions (i.e. spin exchange frequency >100 MHz) manifest as single-line EPR spectra due to the collapse of the low-field and high-field resonance lines to the center-field line. The EPR spectra of 100% labeled GU-Aβ42 oligomers show significant broadening as indicated by lower spectral amplitude compared with 20% labeled oligomers (Fig. 7). However, the EPR spectral lineshape of GU-Aβ42 oligomers is distinct from the single-line spectrum of Aβ42 fibrils (Fig. 6B). Therefore, the EPR data suggest that the structure of GU-Aβ42 oligomers is distinct from the parallel in-register β-sheet structure of Aβ42 fibrils.
FIGURE 6.
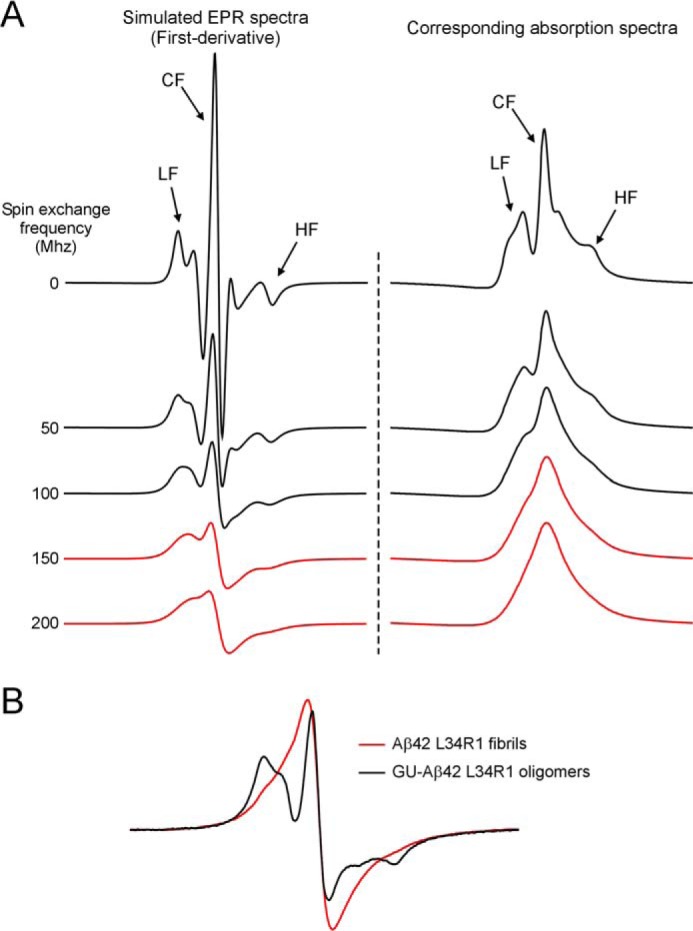
GU-Aβ42 oligomers adopt a distinct structure from Aβ42 fibrils. A, single-line EPR spectra indicate strong spin exchange interactions. Experimentally, EPR spectra are measured as the first-derivative of the absorption spectra. With increasing spin exchange interactions, the low-field (LF) and high-field (HF) resonance lines collapse with the center-field (CF) resonance line, giving rise to the so-called single-line spectra (red). Single-line spectra are characteristic of the parallel in-register β-sheet structure in amyloid fibrils. B, spectrum of Aβ42 fibrils, spin-labeled at Leu-34, is characterized by a single-line feature, suggesting strong spin exchange interactions in parallel in-register β-sheet structures. In contrast, the spectrum of GU-Aβ42 oligomers shows three spectral lines, suggesting that GU-Aβ42 oligomers adopt a distinct structure from fibrils. Scan width is 200 G.
FIGURE 7.

Intermolecular distance analysis for spin-labeled GU-Aβ42 oligomers. The 20% labeled EPR spectra are reproduced from Fig. 3 to show the effect of spin-spin interactions in the 100% labeled spectra. Intermolecular distances were obtained by simulating the 100% labeled spectra. The residual is the difference between simulated spectra and 100% labeled spectra.
Distance Analysis and Structural Modeling of GU-Aβ42 Oligomers
To gain detailed structural information, we analyzed the intermolecular distances between spin labels in the GU-Aβ42 oligomers by spectral fitting (Figs. 7 and 8). Distances were obtained for every labeling position. All labeling positions show two intermolecular distances at ∼9–10 and 15–17 Å (Fig. 8A). For the 9–10 Å distance, there are two possible structural origins. First, in a β-sheet structure, the interstrand distance is 4.75 Å, thus the distance between alternate β-strands is 9.5 Å. Second, the distance between adjacent β-sheets is ∼10 Å (Fig. 1C). A face-to-back packing between β-sheets would also give rise to inter-residue distance of ∼10 Å for all residue positions. For the 15–17 Å distance, a likely origin is the spacing between every third β-strands within the same β-sheet. A structural model consistent with both distances is presented below.
Distance analysis also shows the populations for spin labels at each measured distance. The population of spin labels at 9–10 Å increases from ∼20% for N-terminal regions to ∼40% for C-terminal regions (Fig. 8B), suggesting that the repeat giving rise to the 9–10 Å distances has differing structural orders between N- and C-terminal regions. The population of spin labels at 15–17 Å account for ∼30–40% of total spins, and it remains relatively unchanged from N terminus to C terminus (Fig. 8B). There is also a significant population of spin labels that are further apart (>20 Å) and do not contribute to the broadening of the continuous-wave EPR spectra. The different populations of spin labels reveal structural heterogeneity in GU-Aβ42 oligomers. The N-terminal region is least ordered, and ∼50% of spin labels at each labeling site are >20 Å apart. For C-terminal regions, only 20–30% of spin labels are >20 Å apart. A less ordered N-terminal region and more ordered C-terminal region are consistent with the two-component analysis for the N-terminal residues in Fig. 5. In structural studies of Aβ42 fibrils (51, 52), the N-terminal region is also less ordered than C-terminal region, suggesting that some common mechanisms may underlie the assembly of both oligomers and fibrils.
The x-ray powder diffraction and near atomic-scale EPR distance information provide several structural restraints for prefibrillar Aβ42 oligomers: (i) β-rich structure; (ii) sheet repeats of three strands; (iii) head-to-tail packing of Aβ42 subunits within the same β-sheet, (iv) intramolecular antiparallel β-sheets; and (v) face-to-back packing of sheets. From these restraints, we developed an atomic model of prefibrillar Aβ42 oligomers (Fig. 9). This model fits the powder diffraction data well, with a goodness of fit of 0.220 using the previously described Ro metric (18), which has a scale of 0 (perfect fit) to 1 (worst possible fit). The best-fit model of prefibrillar Aβ42 oligomers is composed of 16 Aβ42 molecules arranged into four face-to-back packed β-sheets (Fig. 9A). Although the architecture of these oligomers is fibril-like, their size (∼60 × 60 × 35 Å in the absence of fusion protein partners) suggests that they are expected to have a globular appearance by transmission electron microscopy (Fig. 1A). A schematic of the model of prefibrillar Aβ42 oligomers is shown in Fig. 9B to illustrate the measured distances from EPR. The model predicts another spin-spin distance at ∼20 Å, which was not observed from the distance measurements. This is likely due to the fact that the upper limit of distance measurements with continuous-wave EPR is ∼20 Å. Our analysis already revealed two interspin distances at 9–10 Å and 15–17 Å. Spin-spin interactions at ∼20 Å produce very weak broadening in the EPR spectral lineshape. We attempted to include a third distance, which did not significantly improve the fitting. Therefore, we limited our analysis to two interspin distances. This structural model is also capable of accommodating the fusion protein partners, as illustrated in Fig. 9C. The fibril-like β-sheets in our oligomer model suggest that oligomers may be able to convert to fibrils through a mechanism of β-stand rotation (Fig. 9D).
FIGURE 9.
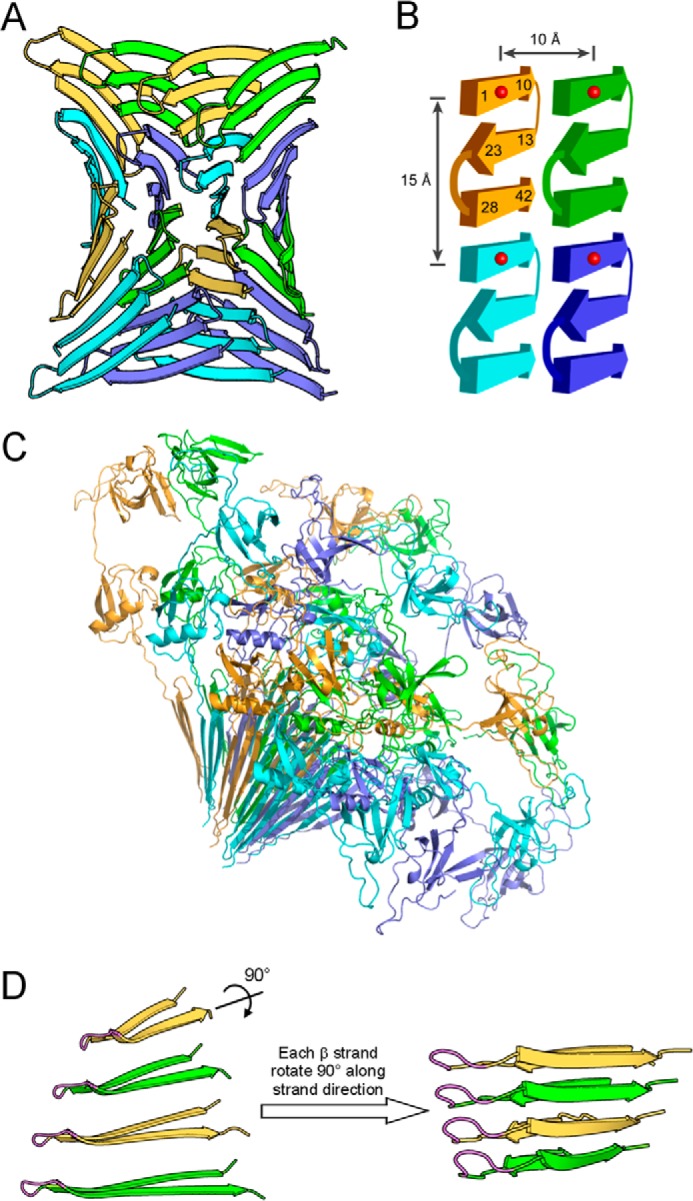
Atomic model of prefibrillar Aβ42 oligomers suggests a mechanism for oligomer-fibril interconversion. A, prefibrillar Aβ42 oligomers have a wrapped architecture wherein protofilament axes are correlated in phase with a central axis of 315 Å pitch. For clarity, Aβ subunits in oligomers are shown in four colors: green, yellow, slate blue, and cyan. B, each Aβ42 subunit consists of three β-strands that interact through backbone hydrogen bonds. The atomic model of panel A is schematized to illustrate measured distances at ∼10 and 15 Å. Numbers represent residue positions, and red balls represent spin labels. C, ribbon diagram of GU-Aβ42 oligomers with GroES and ubiquitin. D, strand rotation mechanism for oligomer-fibril interconversion. On the left, four Aβ42 monomers that interact through intersheet interaction are shown. These monomers are depicted as if looking down the β-sheet direction of the model in panel A. The N-terminal strand is omitted for clarity. The flexible turn identified by analysis of the inverse center line width (Fig. 4) is shown in magenta. Shown on the right is a fibril model of Aβ40 proposed previously (76). Prefibrillar oligomers may interconvert with fibril seeds by an ∼90° rotation around the long axes of the β-strands. We term this conversion mechanism as strand rotation.
GU-Aβ42 Oligomers Promote the Fibrillization of Aβ42
To further explore the relationship between the GU-Aβ42 oligomers and full-length Aβ42 oligomers (without the fusion protein partner), we studied the aggregation kinetics of Aβ42 in the presence and absence of GU-Aβ42 oligomers. As shown in Fig. 10, GU-Aβ42 oligomers shortened the lag phase of the Aβ42 aggregation. This suggests that GU-Aβ42 oligomers likely adopt a structure that is on pathway to amyloid formation and support the possibility of oligomer-to-fibril conversion as proposed in Fig. 9D. In contrast, GU alone dramatically lengthened the lag phase of Aβ42 fibrillization. This is likely due to the chaperone activity of GroES, consistent with reports in the literature that molecular chaperones generally have anti-amyloid activities (53).
FIGURE 10.

Aggregation kinetics of Aβ42 in the absence and presence of GU-Aβ42 oligomers. Aggregation of Aβ42 was followed with thioflavin T fluorescence. Aβ42 concentration is 50 μm in all aggregation reactions. Solid and dashed lines represent duplicates of aggregation experiments. A.U., arbitrary unit.
DISCUSSION
Oligomeric assemblies of Aβ, particularly Aβ42, have been widely hypothesized as the primary neurotoxins that cause the pathology in Alzheimer's disease. Structural knowledge of Aβ oligomers is critical for understanding the structural basis of toxicity and the mechanism of Alzheimer pathogenesis. An accumulating body of evidence supports the notion that Aβ oligomers adopt a different structure from the parallel in-register β-sheet structure of Aβ fibrils (15–19). Particularly, Fourier transform infrared spectroscopy (FTIR) studies on Aβ oligomers prepared using various protocols (15–19) show a common 1695-cm−1 peak, which is absent in fibril samples, suggesting that antiparallel β structure may be a common feature for Aβ oligomers (54). However, concerns have been raised regarding whether the 1695-cm−1 peak can distinguish parallel from antiparallel β-sheets (55, 56). For example, Khurana and Fink (57) show that β-helix proteins give rise to an FTIR peak at ∼1690 cm−1. Ahmed et al. (16) interpreted the high frequency FTIR peak at 1675–1695 cm−1 as side chain vibrations of arginine, asparagine, and glutamine in their Aβ42 oligomers. Detailed structural information such as inter-residue distances is very limited (16, 58). Currently there are no structural models of oligomers based on extensive experimental restraints. Computational modeling studies have provided a number of structural models (59, 60), but these models still await experimental validation.
In this work, we employed site-directed spin labeling to probe the side chain mobility (Fig. 4) and measure intermolecular distances (Fig. 8) at all 42 residue positions of Aβ42 sequence in a prefibrillar oligomer preparation. Together with x-ray powder diffraction (Fig. 1C), the spin label mobility analysis reveals a turn at residues 24–27, which connects two β-strands at residues 13–23 and 28–42. This β-turn-β motif has also been observed in other Aβ oligomer preparations (61, 62) and has been proposed in Aβ42 fibril structural models (51, 52). Mobility analysis also suggests that N-terminal residues consist of both a disordered state and a structured state (Fig. 5), consistent with recent hydrogen exchange and solid-state NMR studies (61, 62), which reveal a structured N-terminal segment. Distance measurements show two intermolecular distances at 9–10 Å and 15–17 Å for every residue position, providing unambiguous support for the lack of parallel in-register β structure in Aβ42 oligomers. This work echoes a recent finding that Aβ42 globulomers adopt structures distinct from fibrils (39).
Based on the EPR distance data, we developed a novel atomic model for Aβ42 prefibrillar oligomers (Fig. 9A). This model is distinct from all existing Aβ oligomer models in terms of both the Aβ42 tertiary structure and quaternary subunit packing. In our model, each Aβ42 subunit forms a single β-sheet with three β-strands in an antiparallel arrangement. Each β-sheet consists of four Aβ42 molecules in a head-to-tail arrangement. Four β-sheets are packed together in a face-to-back fashion. The β-sheets have internal helical axes correlated in phase with a central axis, and this feature resembles that of toxic Aβ42 fibrillar oligomers (named TABFOs) (18).
The size of our oligomer model is in general agreement with the reported size of in vitro Aβ42 oligomers. For example, size exclusion chromatography studies show that Aβ-derived diffusible ligands are ∼65–80 kDa, corresponding to 14–18 subunits (63). Globulomers are shown to consist of 12 subunits (9). Prefibrillar oligomers are eluted as a 90-kDa peak using size exclusion chromatography, corresponding to 20 subunits (64). The essence of our structural model is the antiparallel triple-strand architecture. Because of the head-to-tail arrangement between Aβ subunits within the same β-sheet, and the face-to-back packing between β-sheets, our oligomer model is open-ended in both the backbone hydrogen bonding direction and the side chain direction, allowing the growth of the oligomer in size.
The fibril-like appearance of β-sheets in prefibrillar oligomers suggests that prefibrillar oligomers may interconvert with fibrils via a transition from a β-hairpin to a heterosteric zipper (Fig. 9D). The EPR distance restraints determined herein (Fig. 8) dictate that intersheet interactions are mediated by identical segments in oligomers (Fig. 9). Oligomers may convert to fibril seeds by strand rotation, wherein these identical segments rotate about 90° around their β-strand axes to make β-sheets of identical segments. The strand rotation model is illustrated in Fig. 9D. Such strand rotations would be facilitated by flexibility in the turn connecting the two β-strands. Indeed, our EPR data unambiguously reveal that this turn is flexible (Fig. 4). Furthermore, the possibility of oligomer-to-fibril conversion is supported by the result that GU-Aβ42 oligomers reduced the lag phase of Aβ42 aggregation (Fig. 10).
Nussinov et al. previously modeled various triple β-sheet structures for Aβ42 amyloid (65). In their model, each Aβ42 molecule consists of three β-strands with two turn regions: one at residues 25–29, and the other one at residues 9–14. Each β-strand participates in a different β-sheet in the triple β-sheet structure, which has a better correlation with the hydrogen exchange data than the double β-sheet structure (65). The triple β-sheet structure has been previously reported for Aβ40 fibrils based on solid-state NMR data (66). Hydrogen exchange studies of Aβ42 fibrils also found that N-terminal region is protected in approximately half of the population (52). These studies highlighted the structural importance of N-terminal region, which is likely structured in the amyloid fibrils of both Aβ40 and Aβ42. In the structural model of Aβ42 oligomers, we also show that each Aβ42 molecule consists of three β-strands at similar residue positions as previously found in Aβ42 fibrils (52, 65). At the tertiary structure level, however, three β-strands from the same Aβ42 molecule participate in the same β-sheet in an antiparallel fashion. Therefore, the Aβ42 structure in the oligomers can be characterized as triple-strand, single β-sheet, which distinguishes it from the Aβ42 structure in fibrils.
Structural heterogeneity or polymorphism has been observed for Aβ fibrils (67, 68). For Aβ oligomers, different oligomer preparation protocols have been reported, and these oligomers are classified largely by morphology, size, and immunoreactivity to mono- and polyclonal antibodies (12). The findings in this work allow us to assess the heterogeneity of the underlying molecular structure in Aβ42 oligomers. The EPR spectral lineshape (Fig. 3) and spin label mobility profile (Fig. 4) suggest that residues 13–42 adopt a single β-turn-β conformation at secondary structure level. This β-turn-β secondary structure is very similar to those observed in Aβ42 fibrils (51, 52). This similarity can be rationalized in the general framework of hierarchical protein folding (69). Assuming the same force is driving both fibril and oligomer formation, it is not surprising to see structural similarity between oligomers and fibrils at secondary structure level. In contrast to the rest of Aβ42 molecule, the N-terminal residues 1–12 show significant amount of heterogeneity. In the N-terminal region, a locally disordered conformation co-exists with a well-ordered conformation. The population of the disordered conformation is ∼10–20% of the total population (Fig. 5). In Aβ42 fibrils, a previous hydrogen exchange study shows that ∼50% of the total population for the N-terminal region is structured (52). The higher percentage of the ordered population in oligomers is likely due to the presence of the fusion protein partner, which traps and stabilizes the oligomeric state. Intermolecular distance measurements reveal another layer of structural heterogeneity at quaternary structure level. We observed two major distance distributions at 9–10 Å and 15–17 Å, but the spin label population for these two distances combined is ∼50–70%. The rest of the spin labels, at ∼30–50% of total population, give rise to distances over the detection limit of continuous-wave EPR, which is ∼20 Å (Fig. 8B). The spin label population with >20 Å distances may represent other structures that are not currently modeled. The spin label mobility studies (Figs. 3 and 4) shows that these other structures are also highly ordered, at least at secondary structure level.
Our modeling explains both the distances at 9–10 Å and 15–17 Å with a unifying structure (Fig. 9). It is likely that other structures satisfying only one set of distances also exist. In Fig. 11, we modeled two such alternative structures in which only the 9–10 Å distance is accounted for. In alternative model A (Fig. 11), each Aβ42 molecule adopts the same three β-strands, but each β-strand participates in a different β-sheet in an antiparallel fashion. Except for the N-terminal β-strand, the rest of the residues adopt a structure that is very similar to the antiparallel structure of Aβ40 D23N fibrils (70). Although conceptually similar to the triple β-sheet models of Nussinov et al. (65), this alternative model distinguishes itself in the N-terminal region, which folds back to the double β-sheet of the C-terminal region. In alternative model B (Fig. 11), the N-terminal region adopts a locally disordered conformation. This may partly explain the structural heterogeneity observed for this region in our spin label mobility analysis (Figs. 4 and 5) and distance measurements (Fig. 8).
FIGURE 11.

Alternative models for prefibrillar Aβ42 oligomers. In alternative model A, the Aβ42 oligomer adopts a triple β-sheet structure. Alternative model B is very similar to model A except for the disordered N-terminal region. Balls represent spin labels to demonstrate the 10 Å spacing in model A. Only 6 Aβ42 molecules are modeled in each structure to show basic architecture, without consideration of the size of the oligomer. Note that the 15 Å distance is not accounted for in these alternative models.
Our structural models have implications about the toxicity of oligomers. Several different oligomer preparations have been shown to have cytotoxicity (8–11). If different oligomers exert their toxicity through similar mechanisms, these oligomers may have similar structural features underlying their toxicity. This work and previous studies (15–19, 39) suggest that one common structural feature observed in different oligomers is the antiparallel β-sheet. Oligomer-specific polyclonal antibody A11 recognizes pore-forming bacterial toxin α-hemolysin and block its toxicity (71). The membrane-spanning core of α-hemolysin is a β-barrel consisting of antiparallel β structures (72). The antiparallel structure may explain the reactivity of our GU-Aβ42 oligomer with the A11 antibody. An alternative explanation to toxicity is that toxicity is associated with properties that are related to the aggregation process and are not specific to particular oligomer species (73). One candidate for such a property is hydrophobicity. In Aβ fibrils, due to symmetric packing of protofilament, the C-terminal hydrophobic region is packed inside the fibril core (50). In the oligomer model of this work (Fig. 9), the packing between adjacent β-sheets is face to back, so more hydrophobic residues are exposed to solvent compared with Aβ fibrils.
Structural conversion from oligomers to fibrils is a critical step in Aβ aggregation. In Fig. 12 we summarize several potential mechanisms for oligomer-fibril interconversion. The mechanism of strand rotation for oligomer-fibril interconversion has been proposed for Aβ based on biochemical studies of Aβ40 protein (74, 75). Here we provide direct structural evidence for strand rotation conversion in that (i) exhaustive EPR distance restraints dictate the model for prefibrillar oligomers and (ii) EPR mobility measurements demonstrate the essential flexibility of the turn connecting the two β-strands. Conversion between toxic oligomers and non-toxic fibrils may represent an attractive point of therapeutic intervention to treat Alzheimer disease. Understanding the conversion mechanisms is essential for rational design of potential therapeutics targeting this process.
FIGURE 12.

Schematic summary of oligomer-fibril interconversion mechanisms. An amyloid protein (e.g. Aβ42) is depicted as a U-turn of two β-strands. The green β-strand is N-terminal to the yellow β-strand. Oligomers are known to take many forms, two of which are represented here: β-cylindrin (77) (Oligomer 1, represented as a cylinder) and cross-β oligomers (Oligomer 2, represented as a stack of U-turns) (Stroud et al. (18) and this work). Fibril seeds have an amyloid spine made of a pair of β-sheets, represented as laminae upon which β-strands are superimposed). Unlike oligomers, which are limited in size, fibrils have either one or two growing ends onto which monomers may add indefinitely. A, in conversion by homogeneous nucleation, oligomers (shown here as a cylinder), must first dissociate before nucleating to form fibril seeds. B, in conversion by secondary nucleation, oligomers with fibril-like structure (as described in the present work) may add new monomers that form a fibril repeat different from the oligomer repeat. In the context of amyloid, such differences in repeats are known as polymorphisms. The example of polymorphism conversion by packing polymorphism is shown here. C, oligomers may convert to fibrils by nucleated conformational conversion (35). Two types of nucleated conformational conversion have been proposed: barrel unrolling and strand rotation. In conversion by barrel unrolling, an amyloid oligomer β-barrel (i.e. β-cylindrin) breaks open by dissolution of β-hydrogen bonding between one pair of β-strands to produce a linear β-sheet (77). To form a fibril seed, two such linear sheets must interact through lateral association. Strand rotation conversion, described in the present work and elsewhere (74, 75), entails the rotation of β-strands by ∼90° around their long axes. This rotation turns the β-sheets of the oligomer (running horizontally in the bottom panel) into fibril β-sheets (running vertically).
Acknowledgments
We thank Dr. David Eisenberg for providing access to the x-ray crystallography facilities, So Hui Won, Kyung-Soo Lee, Tiffany Y. Lin, Gregory J. Wong, Rohini Jain, Sherwin Tavakol for preparation of Aβ proteins, Dr. Wayne Hubbell and Dr. Christian Altenbach for providing EPR analysis programs.
This work was supported by National Institutes of Health Grants P50AG016570 and R01GM110448, Alzheimer Association (Grant NIRG-09-133555), and BrightFocus Foundation (Grant A2010362).
- Aβ
- amyloid β
- SDSL
- site-directed spin labeling
- EPR
- electron paramagnetic resonance
- GU
- GroES-ubiquitin
- TEM
- transmission electron microscopy
- ThT
- thioflavin T
- CD
- circular dichroism
- FTIR
- Fourier transform infrared spectroscopy.
REFERENCES
- 1. Selkoe D. J. (2011) Alzheimer's Disease. Cold Spring Harb. Perspect. Biol. 3, a004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sipe J. D., Benson M. D., Buxbaum J. N., Ikeda S., Merlini G., Saraiva M. J., Westermark P. (2012) Amyloid fibril protein nomenclature: 2012 recommendations from the Nomenclature Committee of the International Society of Amyloidosis. Amyloid 19, 167–170 [DOI] [PubMed] [Google Scholar]
- 3. Klein W. L., Krafft G. A., Finch C. E. (2001) Targeting small Aβ oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci. 24, 219–224 [DOI] [PubMed] [Google Scholar]
- 4. Larson M. E., Lesne S. E. (2012) Soluble Aβ oligomer production and toxicity. J. Neurochem. 120, 125–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benilova I., Karran E., De Strooper B. (2012) The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357 [DOI] [PubMed] [Google Scholar]
- 6. Shankar G. M., Li S., Mehta T. H., Garcia-Munoz A., Shepardson N. E., Smith I., Brett F. M., Farrell M. A., Rowan M. J., Lemere C. A., Regan C. M., Walsh D. M., Sabatini B. L., Selkoe D. J. (2008) Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lesné S., Koh M. T., Kotilinek L., Kayed R., Glabe C. G., Yang A., Gallagher M., Ashe K. H. (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357 [DOI] [PubMed] [Google Scholar]
- 8. Lambert M. P., Barlow A. K., Chromy B. A., Edwards C., Freed R., Liosatos M., Morgan T. E., Rozovsky I., Trommer B., Viola K. L., Wals P., Zhang C., Finch C. E., Krafft G. A., Klein W. L. (1998) Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barghorn S., Nimmrich V., Striebinger A., Krantz C., Keller P., Janson B., Bahr M., Schmidt M., Bitner R. S., Harlan J., Barlow E., Ebert U., Hillen H. (2005) Globular amyloid β-peptide1–42 oligomer - a homogenous and stable neuropathological protein in Alzheimer's disease. J. Neurochem. 95, 834–847 [DOI] [PubMed] [Google Scholar]
- 10. Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- 11. Hoshi M., Sato M., Matsumoto S., Noguchi A., Yasutake K., Yoshida N., Sato K. (2003) Spherical aggregates of β-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3β. Proc. Natl. Acad. Sci. U.S.A. 100, 6370–6375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Glabe C. G. (2008) Structural classification of toxic amyloid oligomers. J. Biol. Chem. 283, 29639–29643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fändrich M. (2012) Oligomeric intermediates in amyloid formation: structure determination and mechanisms of toxicity. J. Mol. Biol. 421, 427–440 [DOI] [PubMed] [Google Scholar]
- 14. Chimon S., Shaibat M. A., Jones C. R., Calero D. C., Aizezi B., Ishii Y. (2007) Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer's β-amyloid. Nat. Struct. Mol. Biol. 14, 1157–1164 [DOI] [PubMed] [Google Scholar]
- 15. Cerf E., Sarroukh R., Tamamizu-Kato S., Breydo L., Derclaye S., Dufrêne Y. F., Narayanaswami V., Goormaghtigh E., Ruysschaert J. M., Raussens V. (2009) Antiparallel β-sheet: a signature structure of the oligomeric amyloid β-peptide. Biochem. J. 421, 415–423 [DOI] [PubMed] [Google Scholar]
- 16. Ahmed M., Davis J., Aucoin D., Sato T., Ahuja S., Aimoto S., Elliott J. I., Van Nostrand W. E., Smith S. O. (2010) Structural conversion of neurotoxic amyloid-β(1–42) oligomers to fibrils. Nat. Struct. Mol. Biol. 17, 561–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morgado I., Wieligmann K., Bereza M., Rönicke R., Meinhardt K., Annamalai K., Baumann M., Wacker J., Hortschansky P., Malešević M., Parthier C., Mawrin C., Schiene-Fischer C., Reymann K. G., Stubbs M. T., Balbach J., Görlach M., Horn U., Fändrich M. (2012) Molecular basis of β-amyloid oligomer recognition with a conformational antibody fragment. Proc. Natl. Acad. Sci. U.S.A. 109, 12503–12508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stroud J. C., Liu C., Teng P. K., Eisenberg D. (2012) Toxic fibrillar oligomers of amyloid-β have cross-β structure. Proc. Natl. Acad. Sci. U.S.A. 109, 7717–7722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eckert A., Hauptmann S., Scherping I., Meinhardt J., Rhein V., Dröse S., Brandt U., Fändrich M., Müller W. E., Götz J. (2008) Oligomeric and fibrillar species of β-amyloid (Aβ42) both impair mitochondrial function in P301L tau transgenic mice. J. Mol. Med. 86, 1255–1267 [DOI] [PubMed] [Google Scholar]
- 20. Mustata G. M., Shekhawat G. S., Lambert M. P., Viola K. L., Velasco P. T., Klein W. L., Dravid V. P. (2012) Insights into the mechanism of Alzheimer's β-amyloid aggregation as a function of concentration by using atomic force microscopy. Appl. Phys. Lett. 100 [Google Scholar]
- 21. Kayed R., Canto I., Breydo L., Rasool S., Lukacsovich T., Wu J., Albay R., 3rd, Pensalfini A., Yeung S., Head E., Marsh J. L., Glabe C. (2010) Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Aβ oligomers. Mol. Neurodegener. 5, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Margittai M., Langen R. (2008) Fibrils with parallel in-register structure constitute a major class of amyloid fibrils: molecular insights from electron paramagnetic resonance spectroscopy. Q. Rev. Biophys. 41, 265–297 [DOI] [PubMed] [Google Scholar]
- 23. Agopian A., Guo Z. (2012) Structural origin of polymorphism for Alzheimer's amyloid-β fibrils. Biochem. J. 447, 43–50 [DOI] [PubMed] [Google Scholar]
- 24. Gu L., Guo Z. (2013) Alzheimer's Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. J. Neurochem. 126, 305–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ngo S., Chiang V., Guo Z. (2012) Quantitative analysis of spin exchange interactions to identify β strand and turn regions in Ure2 prion domain fibrils with site-directed spin labeling. J. Struct. Biol. 180, 374–381 [DOI] [PubMed] [Google Scholar]
- 26. Ngo S., Gu L., Guo Z. (2011) Hierarchical organization in the amyloid core of yeast prion protein Ure2. J. Biol. Chem. 286, 29691–29699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jeschke G. (2012) DEER distance measurements on proteins. Annu. Rev. Phys. Chem. 63, 419–446 [DOI] [PubMed] [Google Scholar]
- 28. McHaourab H. S., Steed P. R., Kazmier K. (2011) Toward the fourth dimension of membrane protein structure: insight into dynamics from spin-labeling EPR spectroscopy. Structure 19, 1549–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liebman S. W., Chernoff Y. O. (2012) Prions in yeast. Genetics 191, 1041–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hashimoto T., Adams K. W., Fan Z., McLean P. J., Hyman B. T. (2011) Characterization of oligomer formation of amyloid-β peptide using a split-luciferase complementation assay. J. Biol. Chem. 286, 27081–27091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bagriantsev S., Liebman S. (2006) Modulation of Aβ42 low-n oligomerization using a novel yeast reporter system. BMC Biol. 4, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim W., Hecht M. H. (2006) Generic hydrophobic residues are sufficient to promote aggregation of the Alzheimer's Aβ42 peptide. Proc. Natl. Acad. Sci. U.S.A. 103, 15824–15829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim W., Kim Y., Min J., Kim D. J., Chang Y. T., Hecht M. H. (2006) A high-throughput screen for compounds that inhibit aggregation of the Alzheimer's peptide. ACS Chem. Biol. 1, 461–469 [DOI] [PubMed] [Google Scholar]
- 34. Streltsov V. A., Varghese J. N., Masters C. L., Nuttall S. D. (2011) Crystal structure of the amyloid-β p3 fragment provides a model for oligomer formation in Alzheimer's disease. J. Neurosci. 31, 1419–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee J., Culyba E. K., Powers E. T., Kelly J. W. (2011) Amyloid-β forms fibrils by nucleated conformational conversion of oligomers. Nat. Chem. Biol. 7, 602–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shahnawaz M., Thapa A., Park I. S. (2007) Stable activity of a deubiquitylating enzyme (Usp2-cc) in the presence of high concentrations of urea and its application to purify aggregation-prone peptides. Biochem. Biophys. Res. Commun. 359, 801–805 [DOI] [PubMed] [Google Scholar]
- 37. Baker R. T., Catanzariti A. M., Karunasekara Y., Soboleva T. A., Sharwood R., Whitney S., Board P. G. (2005) Using deubiquitylating enzymes as research tools. Methods Enzymol. 398, 540–554 [DOI] [PubMed] [Google Scholar]
- 38. Ngo S., Guo Z. (2011) Key residues for the oligomerization of Aβ42 protein in Alzheimer's disease. Biochem. Biophys. Res. Commun. 414, 512–516 [DOI] [PubMed] [Google Scholar]
- 39. Gu L., Liu C., Guo Z. (2013) Structural insights into Aβ42 oligomers using site-directed spin labeling. J. Biol. Chem. 28, 18673–18683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Altenbach C., Oh K. J., Trabanino R. J., Hideg K., Hubbell W. L. (2001) Estimation of inter-residue distances in spin labeled proteins at physiological temperatures: experimental strategies and practical limitations. Biochemistry 40, 15471–15482 [DOI] [PubMed] [Google Scholar]
- 41. Brunger A. T. (2007) Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2, 2728–2733 [DOI] [PubMed] [Google Scholar]
- 42. Chandrasekhar G. N., Tilly K., Woolford C., Hendrix R., Georgopoulos C. (1986) Purification and properties of the GroES morphogenetic protein of Escherichia coli. J. Biol. Chem. 261, 12414–12419 [PubMed] [Google Scholar]
- 43. Zondlo J., Fisher K. E., Lin Z., Ducote K. R., Eisenstein E. (1995) Monomer-heptamer equilibrium of the Escherichia coli chaperonin GroES. Biochemistry 34, 10334–10339 [DOI] [PubMed] [Google Scholar]
- 44. Jahn T. R., Makin O. S., Morris K. L., Marshall K. E., Tian P., Sikorski P., Serpell L. C. (2010) The common architecture of cross-β amyloid. J. Mol. Biol. 395, 717–727 [DOI] [PubMed] [Google Scholar]
- 45. Maezawa I., Hong H. S., Liu R., Wu C. Y., Cheng R. H., Kung M. P., Kung H. F., Lam K. S., Oddo S., Laferla F. M., Jin L. W. (2008) Congo red and thioflavin-T analogs detect Aβ oligomers. J. Neurochem. 104, 457–468 [DOI] [PubMed] [Google Scholar]
- 46. Bernstein S. L., Dupuis N. F., Lazo N. D., Wyttenbach T., Condron M. M., Bitan G., Teplow D. B., Shea J. E., Ruotolo B. T., Robinson C. V., Bowers M. T. (2009) Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer's disease. Nat. Chem. 1, 326–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boudker O., Todd M. J., Freire E. (1997) The structural stability of the co-chaperonin GroES. J. Mol. Biol. 272, 770–779 [DOI] [PubMed] [Google Scholar]
- 48. Love S. G., Muir T. W., Ramage R., Shaw K. T., Alexeev D., Sawyer L., Kelly S. M., Price N. C., Arnold J. E., Mee M. P., Mayer R. J. (1997) Synthetic, structural and biological studies of the ubiquitin system: synthesis and crystal structure of an analogue containing unnatural amino acids. Biochem. J. 323, 727–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hubbell W. L., Mchaourab H. S., Altenbach C., Lietzow M. A. (1996) Watching proteins move using site-directed spin labeling. Structure 4, 779–783 [DOI] [PubMed] [Google Scholar]
- 50. Tycko R. (2011) Solid-state NMR studies of amyloid fibril structure. Annu. Rev. Phys. Chem. 62, 279–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Olofsson A., Sauer-Eriksson A. E., Ohman A. (2006) The solvent protection of Alzheimer amyloid-β-(1–42) fibrils as determined by solution NMR spectroscopy. J. Biol. Chem. 281, 477–483 [DOI] [PubMed] [Google Scholar]
- 52. Lührs T., Ritter C., Adrian M., Riek-Loher D., Bohrmann B., Döbeli H., Schubert D., Riek R. (2005) 3D structure of Alzheimer's amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. U.S.A. 102, 17342–17347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yerbury J. J., Kumita J. R. (2010) Protein chemistry of amyloid fibrils and chaperones: implications for amyloid formation and disease. Curr. Chem. Biol. 4, 89–98 [Google Scholar]
- 54. Chirgadze Y. N., Nevskaya N. A. (1976) Infrared spectra and resonance interaction of amide-I vibration of the antiparallel-chain pleated sheet. Biopolymers 15, 607–625 [DOI] [PubMed] [Google Scholar]
- 55. Zandomeneghi G., Krebs M. R. H., McCammon M. G., Fandrich M. (2004) FTIR reveals structural differences between native β-sheet proteins and amyloid fibrils. Protein Sci. 13, 3314–3321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Barth A., Zscherp C. (2002) What vibrations tell us about proteins. Q. Rev. Biophys. 35, 369–430 [DOI] [PubMed] [Google Scholar]
- 57. Khurana R., Fink A. L. (2000) Do parallel β-helix proteins have a unique fourier transform infrared spectrum? Biophys. J. 78, 994–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Scheidt H. A., Morgado I., Huster D. (2012) Solid-state NMR reveals a close structural relationship between amyloid-β protofibrils and oligomers. J. Biol. Chem. 287, 22822–22826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ma B., Nussinov R. (2010) Polymorphic C-terminal β-sheet interactions determine the formation of fibril or amyloid β-derived diffusible ligand-like globulomer for the Alzheimer Aβ42 dodecamer. J. Biol. Chem. 285, 37102–37110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Strodel B., Lee J. W., Whittleston C. S., Wales D. J. (2010) Transmembrane structures for Alzheimer's Aβ(1–42) oligomers. J. Am. Chem. Soc. 132, 13300–13312 [DOI] [PubMed] [Google Scholar]
- 61. Pan J., Han J., Borchers C. H., Konermann L. (2011) Conformer-specific hydrogen exchange analysis of Aβ(1–42) oligomers by top-down electron capture dissociation mass spectrometry. Anal. Chem. 83, 5386–5393 [DOI] [PubMed] [Google Scholar]
- 62. Haupt C., Leppert J., Ronicke R., Meinhardt J., Yadav J. K., Ramachandran R., Ohlenschlager O., Reymann K. G., Gorlach M., Fandrich M. (2012) Structural basis of β-amyloid-dependent synaptic dysfunctions. Angew. Chem. Int. Ed. 51, 1576–1579 [DOI] [PubMed] [Google Scholar]
- 63. Hepler R. W., Grimm K. M., Nahas D. D., Breese R., Dodson E. C., Acton P., Keller P. M., Yeager M., Wang H., Shughrue P., Kinney G., Joyce J. G. (2006) Solution state characterization of amyloid β-derived diffusible ligands. Biochemistry 45, 15157–15167 [DOI] [PubMed] [Google Scholar]
- 64. Demuro A., Mina E., Kayed R., Milton S. C., Parker I., Glabe C. G. (2005) Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 280, 17294–17300 [DOI] [PubMed] [Google Scholar]
- 65. Ma B., Nussinov R. (2011) Polymorphic triple β-sheet structures contribute to amide hydrogen/deuterium (H/D) exchange protection in the Alzheimer amyloid β42 peptide. J. Biol. Chem. 286, 34244–34253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bertini I., Gonnelli L., Luchinat C., Mao J., Nesi A. (2011) A new structural model of Aβ40 fibrils. J. Am. Chem. Soc. 133, 16013–16022 [DOI] [PubMed] [Google Scholar]
- 67. Petkova A. T., Leapman R. D., Guo Z. H., Yau W. M., Mattson M. P., Tycko R. (2005) Self-propagating, molecular-level polymorphism in Alzheimer's β-amyloid fibrils. Science 307, 262–265 [DOI] [PubMed] [Google Scholar]
- 68. Kodali R., Williams A. D., Chemuru S., Wetzel R. (2010) Aβ(1–40) forms five distinct amyloid structures whose β-sheet contents and fibril stabilities are correlated. J. Mol. Biol. 401, 503–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Baldwin R. L., Rose G. D. (1999) Is protein folding hierarchic? I. Local structure and peptide folding. Trends Biochem. Sci. 24, 26–33 [DOI] [PubMed] [Google Scholar]
- 70. Qiang W., Yau W. M., Luo Y., Mattson M. P., Tycko R. (2012) Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 109, 4443–4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yoshiike Y., Kayed R., Milton S. C., Takashima A., Glabe C. G. (2007) Pore-forming proteins share structural and functional homology with amyloid oligomers. Neuromolecular Med. 9, 270–275 [DOI] [PubMed] [Google Scholar]
- 72. Song L., Hobaugh M. R., Shustak C., Cheley S., Bayley H., Gouaux J. E. (1996) Structure of staphylococcal α-hemolysin, a heptameric transmembrane pore. Science 274, 1859–1866 [DOI] [PubMed] [Google Scholar]
- 73. Ross C. A., Poirier M. A. (2005) What is the role of protein aggregation in neurodegeneration? Nat. Rev. Mol. Cell Biol. 6, 891–898 [DOI] [PubMed] [Google Scholar]
- 74. Hoyer W., Grönwall C., Jonsson A., Ståhl S., Härd T. (2008) Stabilization of a β-hairpin in monomeric Alzheimer's amyloid-β peptide inhibits amyloid formation. Proc. Natl. Acad. Sci. U.S.A. 105, 5099–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sandberg A., Luheshi L. M., Söllvander S., Pereira de Barros T., Macao B., Knowles T. P., Biverstål H., Lendel C., Ekholm-Petterson F., Dubnovitsky A., Lannfelt L., Dobson C. M., Härd T. (2010) Stabilization of neurotoxic Alzheimer amyloid-β oligomers by protein engineering. Proc. Natl. Acad. Sci. U.S.A. 107, 15595–15600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Petkova A. T., Ishii Y., Balbach J. J., Antzutkin O. N., Leapman R. D., Delaglio F., Tycko R. (2002) A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U.S.A. 99, 16742–16747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Laganowsky A., Liu C., Sawaya M. R., Whitelegge J. P., Park J., Zhao M., Pensalfini A., Soriaga A. B., Landau M., Teng P. K., Cascio D., Glabe C., Eisenberg D. (2012) Atomic view of a toxic amyloid small oligomer. Science 335, 1228–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]