

Background: DNA end resection is a critical step in the homology-directed repair of DNA double strand breaks (DSBs).
Results: Human WRN helicase stimulates the DNA2-catalyzed resection of DNA ends and acts in concert with DNA2 to promote DSB repair by single strand annealing.
Conclusion: DNA2 cooperates with WRN or BLM to mediate the resection of DSBs in mammalian cells.
Significance: Defects in DNA end resection might, in part, account for the genomic instability phenotype of Werner syndrome.
Keywords: DNA Damage, DNA Helicase, DNA Recombination, DNA Repair, Genomic Instability, RecQ
Abstract
The 5′-3′ resection of DNA ends is a prerequisite for the repair of DNA double strand breaks by homologous recombination, microhomology-mediated end joining, and single strand annealing. Recent studies in yeast have shown that, following initial DNA end processing by the Mre11-Rad50-Xrs2 complex and Sae2, the extension of resection tracts is mediated either by exonuclease 1 or by combined activities of the RecQ family DNA helicase Sgs1 and the helicase/endonuclease Dna2. Although human DNA2 has been shown to cooperate with the BLM helicase to catalyze the resection of DNA ends, it remains a matter of debate whether another human RecQ helicase, WRN, can substitute for BLM in DNA2-catalyzed resection. Here we present evidence that WRN and BLM act epistatically with DNA2 to promote the long-range resection of double strand break ends in human cells. Our biochemical experiments show that WRN and DNA2 interact physically and coordinate their enzymatic activities to mediate 5′-3′ DNA end resection in a reaction dependent on RPA. In addition, we present in vitro and in vivo data suggesting that BLM promotes DNA end resection as part of the BLM-TOPOIIIα-RMI1-RMI2 complex. Our study provides new mechanistic insights into the process of DNA end resection in mammalian cells.
Introduction
DNA double strand breaks (DSBs)4 are a very dangerous form of DNA damage because they can cause cell death or chromosomal rearrangements, a hallmark of cancer (1). DSBs can occur accidentally during normal cellular metabolism or upon exposure of cells to exogenous agents such as ionizing radiation and radiomimetic drugs (2). There are also programmed DSBs that drive recombination events essential for physiological processes, such as meiosis and lymphocyte development (3, 4). In eukaryotic cells, DSBs are repaired by one of two major pathways: non-homologous end joining (NHEJ) and homologous recombination (HR). NHEJ involves religation of the broken DNA ends and is frequently associated with a short deletion or insertion of DNA at the break site (5). In contrast, HR restores the DNA integrity accurately because it uses sister chromatids or homologous chromosomes as a template for repair (6, 7). HR is initiated by resection of the broken DNA ends to generate 3′ single-stranded (ss) DNA tails that are utilized by the RAD51 recombinase for a homology search on the donor DNA molecule (6, 7). Genetic and biochemical studies in budding yeast have shown that broken DNA ends are resected in a two-step process (8–10). DNA end resection in yeast is initiated by the Mre11-Rad50-Xrs2 complex in conjunction with Sae2 (8, 9, 11). These proteins may initiate resection of the 5′ strand of the broken DNA to remove a stretch of about 100–200 nucleotides from the DNA end (8, 9, 11). The Mre11-Rad50-Xrs2 complex also recruits the components of the long-range resection pathways Exo1 or Dna2-Sgs1 (8–10, 12, 13). Exo1 is a dsDNA-dependent 5′-3′ exonuclease that preferentially degrades DNA substrates with a 3′ ssDNA tail in a reaction stimulated by the ssDNA-binding protein RPA (13). Dna2 is a ssDNA-specific nuclease and a DNA helicase that functions in conjunction with the RecQ family DNA helicase Sgs1 and RPA to catalyze long-range DNA end resection (10, 14). In this reaction, RPA stimulates DNA unwinding by Sgs1 and promotes degradation of the 5′-terminated strand by Dna2 while protecting the growing 3′ ssDNA tail (10). DNA end resection is also the initial step in two other DSB repair pathways, single strand annealing (SSA) and microhomology-mediated end joining (8, 15).
The molecular machinery of DNA end resection appears to be largely conserved between yeast and man (15–19). However, it remains a matter of debate which DNA helicase mediates DNA2-catalyzed resection in mammalian cells. Mammals possess five RecQ homologues: RECQ1, BLM, WRN, RECQ4, and RECQ5 (20). Biochemical studies have shown that human DNA2 can act in conjunction with the BLM helicase and RPA to mediate 5′-3′ resection of DNA ends in vitro (17). In agreement with these findings, it has been observed that cells depleted of both BLM and EXO1 show a reduction in the formation of RPA foci in response to DSBs and are defective in DSB repair by HR (16, 19). However, studies using Xenopus egg extracts and purified proteins have shown that Dna2 mediates DNA end resection together with WRN rather than BLM (21–23). This discrepancy prompted us to investigate the role of WRN in DNA end resection in human cells. Here we demonstrate that WRN helicase is capable of acting in concert with DNA2 and RPA to resect 5′-recessed DNA ends in vitro with a catalytic efficiency even higher than that of BLM. Moreover, our results show that human cells may employ either BLM or WRN to assist DNA2 in long-range DNA end resection. Finally, we present data suggesting that BLM acts in DNA end resection as part of the BLM-TOPOIIIα-RMI1-RMI2 (BTRR) complex.
EXPERIMENTAL PROCEDURES
Antibodies and siRNA
Primary antibodies used for immunoblotting were as follows: mouse monoclonal anti-WRN (BD Biosciences, catalog no. 611169), rabbit polyclonal anti-DNA2 (Abcam, catalog no. ab96488), rabbit polyclonal anti-BLM (Abcam, catalog no. ab476), rabbit polyclonal anti-TFIIH (Santa Cruz Biotechnology, catalog no. sc293), mouse monoclonal anti-FLAG (Sigma, catalog no. F1804), and rabbit polyclonal anti-RMI1 (Proteintech, catalog no. 14630-1-AP). Anti-FLAG M2 magnetic beads (Sigma) were used for immunoprecipitation. Primary antibodies used for immunofluorescence staining were as follows: mouse monoclonal anti-RPA2 (Abcam, catalog no. ab2175) and rabbit monoclonal anti-γ-H2AX (Cell Signaling Technology, catalog no. 9718S). Rabbit polyclonal anti-WRN antibody used for immunoprecipitation has been described previously (24).
All siRNA oligoduplexes used in this study were purchased from Microsynth. The sequences of the sense strands of these duplexes were as follows: siLuc, 5′-CGUACGCGGAAUACUUCGAdTdT-3′; siWRN, 5′-UAGAGGGAAACUUGGCAAAdTdT-3′; siBLM, 5′-CCGAAUCUCAAUGUACAUAGAdTdT-3′; siDNA2, 5′-UACCGCUUAAAUCUAAGUCAAdTdT-3′; siEXO1, 5′-CAGCCAUUCUUACUACGCUAAdTdT-3′; siMRE11, 5′-GAGCAUAACUCCAUAAGUAdTdT-3′ (25); siCtIP, 5′-UCCACAACAUAAUCCUAAUdTdT-3′ (26); and siRMI1, 5′-AGCCUUCACGAAUGUUGAUdTdT-3′ (27).
Plasmid Constructions
The human DNA2 (hDNA2) ORF was amplified by PCR without the initiation and stop codons to generate a fragment including ggatcc-hDNA2-ctcgag. After digestion with BamHI and XhoI, the hDNA2 fragment was cloned into pFLAG-CMV2 (Sigma) digested with BglII/SalI (pFLAG-CMV2-hDNA2). The human WRN (hWRN) ORF was inserted into pcDNA3.1/Hygro(−) (Invitrogen) via the NheI and DraI sites (pcDNA3.1-hWRN). The siRNA-resistant form of this construct was generated by changing four nucleotides in the siWRN-targeting region (T270C, A273G, G276C, and A279G) using the QuikChange site-directed mutagenesis kit (Stratagene).
Protein Purifications
Wild-type and mutant forms of WRN, BLM, EXO1, and RPA were produced and purified as described previously (28–31). The TOPOIIIα-RMI1-RMI2 (TRR) complex was a gift from Drs. Kata Sarlos and Ian Hickson (University of Copenhagen, Denmark). DNA2 was produced as a fusion with a His6 tag (N terminus) and a FLAG tag (C terminus) in Sf9 cells using the Bac-to-Bac baculovirus expression system (Invitrogen). The transfer vector for bacmid preparation was a gift from Dr. Judith L. Campbell (32). The transfer vectors for nuclease-deficient (D227A) and helicase-deficient (K654R) mutants of DNA2 were generated using the QuikChange site-directed mutagenesis kit (Stratagene). Sf9 cells expressing DNA2 fusion proteins were harvested 52 h after infection (typically a 800-ml culture) and washed with PBS. All subsequent steps were carried out at 4 °C. Pelleted cells were resuspended in lysis buffer (25 mm Tris-HCl (pH 7.5), 2 mm β-mercaptoethanol, 1× complete EDTA-free protease inhibitor (Roche), 1 mm phenylmethylsulfonyl fluoride, 30 μg/ml leupeptin, and 15 mm imidazole) and incubated for 20 min under continuous stirring. Subsequently, glycerol and 5 m NaCl were added slowly to final concentrations of 15% (v/v) and 300 mm, respectively, while mixing the sample. The cell suspension was then incubated for an additional 30 min under continuous stirring. The cell lysate was centrifuged at 55,000 × g for 30 min to obtain soluble extract, which was then incubated with 5 ml of nickel-nitrilotriacetic acid-agarose beads (Qiagen) for 1 h batchwise. The resin was washed extensively with lysis buffer containing 10% (v/v) glycerol and 1 m NaCl. The protein was eluted with lysis buffer supplemented with 10% (v/v) glycerol, 100 mm NaCl, and 250 mm imidazole. Fractions containing detectable amounts of protein, as measured by Bradford assay, were pooled, diluted 1:1 with TBS buffer (50 mm Tris-HCl (pH 7.5) and 150 mm NaCl) and incubated batchwise with 1 ml of anti-FLAG M2 affinity resin (Sigma) for 30 min. The resin was then transferred to a gravity flow column and washed with TBS-PI buffer (TBS buffer containing 1 mm β-mercaptoethanol and 5 μg/ml leupeptin). Elution of the protein was achieved by adding TBS-PI buffer supplemented with 200 μg/ml 3× FLAG peptide (Sigma). Fractions containing DNA2 were pooled, diluted with 0.5 volumes of water and 1 volume of AQ buffer (25 mm Tris-HCl (pH 7.5), 100 mm NaCl, 10% (v/v) glycerol, and 5 mm β-mercaptoethanol) and loaded onto a 1-ml HiTrap Q column (GE Healthcare) pre-equilibrated with AQ buffer. The column was washed with AQ buffer and DNA2 was eluted by AQ buffer supplemented with 600 mm NaCl. Fractions containing DNA2 were identified by SDS-PAGE, pooled, and stored at −80 °C. The activity of purified recombinant DNA2 proteins was tested using a Y structure oligonucleotide duplex with single-stranded arms (10). In agreement with previous reports, wild-type DNA2 was found to be capable of degrading both ssDNA arms of this structure (data not shown) (10, 17). In the presence of RPA, the cleavage of the 3′ ssDNA arm by DNA2 was inhibited, and DNA2 degraded preferentially the 5′ ssDNA arm (data not shown) (10, 17). The DNA2-D227A mutant did not contain any nuclease activity, which indicated that the nuclease activity of our wild-type DNA2 preparation was inherent to DNA2 (data not shown).
Nuclease and Helicase Assays
To test the activity of purified DNA2, we used a 31-bp forked duplex with 19-nt ssDNA arms, as described previously (10). The helicase activity of WRN and BLM was tested using a 29-bp forked duplex generated by annealing of the following oligonucleotides: f-9 (5′-ACTATCATTC AGTCATGTAA CCTAGTCAAT CTGCGAGCTC GAATTCACTG GAGTGACCT-3′) and f-10 (5′-GAGGTCACTC CAGTGAATTC GAGCTCGCAG TCAATGTCGA CATACCTAGT ACTTTACTCC-3′). Both DNA substrates were radiolabeled at the end of the 5′ ssDNA arm.
Nuclease and helicase assays were performed in buffer containing 25 mm Tris acetate (pH 7.5), 2 mm magnesium acetate, 1 mm dithiothreitol, 0.1 mg/ml BSA, 10.7 mm phosphocreatine, and 0.02 mg/ml creatine phosphokinase. Reactions (15 μl) contained 1 nm 32P-labeled forked DNA substrate and the indicated concentrations of DNA2 or WRN/BLM. Where indicated, RPA was present at a concentration of 6 nm. Reactions were assembled on ice and started by addition of ATP to a concentration of 1 mm. Reactions were incubated for 30 min at 37 °C. Termination of the reactions was achieved by adding 1/3 volume of stop solution (150 mm EDTA, 2% (w/v) SDS, 30% (v/v) glycerol, and 0.1% (w/v) bromphenol blue) and 1/15 volume of Proteinase K (10 mg/ml), followed by incubation at 37 °C for 15 min. The reaction products were separated by electrophoresis in a 10% Tris borate-EDTA polyacrylamide gel. Gels were dried on Whatman MM3 paper and analyzed by phosphorimaging using a Typhoon 9400 scanner (GE Healthcare). Images were quantified using ImageQuantTL software.
Construction of DNA Substrates for Resection Assays
The DNA substrates used in resection assays were derived from the plasmid pUC19 (2686 bp). The self-complementary oligonucleotide, 5′-AGCT GCTGAGG GCTGAGG GCTGAGG GCTGAGG AGGCCT CCTCAGC CCTCAGC CCTCAGC CCTCAGC-3′, was annealed to form a duplex that was cloned into the HindIII site of pUC19. This destroyed the HindIII site and inserted a single recognition sequence for StuI (AGGCCT) flanked on each side by four recognition sequences for the nickase Nt.BbvCI (CC*TCAGC; the cleavage position is indicated by the asterisk) that are oriented as an inverted repeat with respect to the StuI site. The resulting pOH-S plasmid allowed us to prepare a linear DNA substrate with 3′ overhangs of 26 nucleotides (nt) in length. A blunt-ended substrate was generated by digestion of pOH-S with StuI (New England Biolabs), followed by DNA purification using a Macherey Nagel NucleoSpin® gel and PCR cleanup kit. The substrate with 26-nt 3′ overhangs was generated as follows. After digestion of pOH-S with StuI and its heat inactivation, Nt.BbvCI (New England Biolabs) was added, and the reaction was incubated further for 2 h at 37 °C. Subsequently, the reaction mixture was diluted six times with water and incubated at 85 °C for 15 min. DNA purification was performed as described above. DNA concentration was determined using a NanoDrop ND-1000 spectrophotometer (Witec AG).
DNA End Resection Assays
DNA end resection reactions were carried out in a buffer containing 25 mm Tris acetate (pH 7.5), 2 mm magnesium acetate, 1 mm dithiothreitol, 0.1 mg/ml BSA, 10.7 mm phosphocreatine, 0.02 mg/ml creatine phosphokinase, and 1 mm ATP. Reactions contained 2 nm DNA substrate (molecules), 8 nm DNA2, 350 nm RPA (100% DNA strand coverage, assuming all DNA was single-stranded), and various concentrations of WRN or BLM as indicated. EXO1 was present at a concentration of 20 nm. The reactions were assembled on ice and initiated by the addition of ATP. Reaction mixtures (15 μl) were incubated at 37 °C for 60 min in the case of protein titration experiments. In time course experiments, 15-μl reaction aliquots were withdrawn at defined time points as indicated. Reactions were terminated as described for the helicase assays. The samples were subjected to electrophoresis in a 1% agarose gel run in 1× TAE buffer. Gels were post-stained with SYBR Gold (Invitrogen) and analyzed using MultiImage Light Cabinet (Alpha Innotech). To monitor resection by hybridization of radiolabeled oligonucleotide probes, terminated reactions (21 μl) were divided equally into two tubes. 5′ end-labeled oligonucleotide probes were then added to a final concentration of 5 nm. This mixture was heated in an oven to 75 °C for 5 min and then slowly cooled down to room temperature over 2.5 h. Reaction products were separated by electrophoresis in a 1% agarose gel. Gels were dried on DE81 anion exchange paper (Whatman) and subjected to phosphorimaging analysis using a Typhoon 9400 scanner (GE Healthcare). Images were quantified using ImageQuantTL software. The relative concentration of the resection products generated in WRN-DNA2 or BLM-DNA2 reactions was calculated as a percentage of the product generated in a reaction containing 20 nm EXO1 at the 2-min time point, which led to 100% resection within the region probed with radiolabeled oligonucleotides. Usually, the EXO1 reaction was loaded on each gel in triplicates. The following oligonucleotides were used for the preparation of the hybridization probes: oligo#224, 5′-GGCCGTCGTTTTACAACGTCGT-3′ (it anneals to the 3′-terminated strand; annealing position, 112–133 nt upstream of the StuI cleavage site; the complementary sequence is underlined); oligo#227, 5′-GGCATAGTTAAGCCAGCCCCGA-3′ (it anneals to the 3′-terminated strand; annealing position, 353–374 nt upstream of the StuI cleavage site); and oligo#237, 5′-GGTCGGGGCTGGCTTAACTATG-3′ (it anneals to the 5′-terminated strand; annealing position, 122–133 nt upstream of the StuI cleavage site). Oligonucleotides were 5′ end-labeled using [γ-32P]ATP and T4 polynucleotide kinase (New England Biolabs). The two non-complementary dG residues at the 5′ end of the oligonucleotides were added to ensure equal labeling efficiency.
Cell Culture and Transfection
U2OS and HEK293 cells were grown in DMEM (Sigma) supplemented with 10% fetal calf serum (Invitrogen) and streptomycin/penicillin (100 units/ml). Plasmid DNA was transfected using standard linear polyethyleneimine method. Lipofectamine RNAiMAX (Invitrogen) was used for siRNA transfection. To generate HEK293 clones stably expressing FLAG-DNA2, cells were cotransfected with pFLAG-CMV2-hDNA2 and pBABE-puro (Addgene) and subjected to puromycin 1 (μg/ml) selection. Puromycin-resistant clones were tested for expression of FLAG-DNA2 by Western blotting.
Immunoprecipitation
HEK293 cells were transfected with the pcDNA3.1-hWRN and/or pFLAG-CMV2-hDNA2 vectors. Cells were harvested to lysis buffer (50 mm Tris-HCl (pH 8.0), 120 mm NaCl, 20 mm NaF, 15 mm sodium pyrophosphate, and 0.5% (v/v) Nonidet P-40) supplemented before use with protease (Complete EDTA-free, Roche) and phosphatase (PhosSTOP, Roche) inhibitors, 2 mm MgCl2 and benzonase (50 units/ml). Cells were sonicated briefly, and lysates were clarified by centrifugation at 16,000 × g for 30 min. Cell extracts (1 mg of protein) were subjected to immunoprecipitation using anti-FLAG M2 magnetic beads (10 μl) or Protein A/G Plus UltraLink Resin (10 μl, Thermo Scientific) coated with rabbit polyclonal anti-WRN antibody (10 μg), which was carried out overnight at 4 °C. Immunoprecipitates were washed four times with lysis buffer. Bound proteins were eluted by Laemmli sample buffer and analyzed by SDS-PAGE and Western blotting. To test the interaction between purified WRN and DNA2 proteins, 500 ng of each protein was mixed in 200 μl of NET-N100 buffer (10 mm Tris-HCl (pH 8.0), 1 mm EDTA, 100 mm NaCl, and 0.5% (v/v) Nonidet P-40) and incubated at 4 °C for 4 h. As a control, DNA2 was incubated in the absence of WRN. The protein mixtures were subsequently subjected to immunoprecipitation using anti-WRN antibody (4 μg), which was carried out at 4 °C for 2 h. Immunoprecipitates were washed four times with NET-N100 buffer. Bound proteins were eluted by Laemmli sample buffer and analyzed by SDS-PAGE and Western blotting.
GST Pulldown Assay
GST-tagged fragments of WRN were produced in the Escherichia coli BL21-CodonPlus(DE3)-RIL strain (Stratagene) and bound to GSH Sepharose 4B (GE Healthcare) as described previously (24). As a control, beads were coated with GST protein only. The beads were incubated with 500 ng of purified His6-DNA2-FLAG protein in 400 μl of NET-N100 buffer at 4 °C for 2 h. After extensive washing with NET-N100 buffer, proteins bound to the beads were analyzed by Western blotting. Blots were first stained in Ponceau S solution (0.1% (w/v) Ponceau S and 5% (v/v) acetic acid) to visualize WRN fragments and subsequently probed with anti-FLAG antibody.
Reverse Transcription and Quantitative Real-time PCR
Total RNA was isolated from cells using the RNeasy mini kit (Qiagen). 200 ng of RNA was used for cDNA synthesis using a high-capacity cDNA reverse transcription kit (Applied Biosystems). The target gene expression level was determined by quantitative real-time PCR that was performed on a ABI Prism 7300 (Applied Biosystems) using SYBR Select Master Mix (Applied Biosystems). The following primer pairs were used to determine EXO1 mRNA levels: 5′-ACCTCTAAGG AACAAGGTTC-3′ (forward) and 5′-AGGAGGAAGC TTTTCAGAATC-3′ (reverse). The housekeeping gene RPLPO, used as a control, was amplified with the following primers: 5′-CCAGTCTGGA GAAACTGCTG-3′ (forward) and 5′-CAGCAGCTGG CACCTTATTGG-3′ (reverse). The Pfaffl equation was used for normalization and calculation of relative EXO1 expression levels in comparison with the control gene (33).
SA-GFP Reporter Assay
SA-GFP reporter assays were performed as described previously (34, 35). HEK293/SA-GFP cells were seeded in poly-l-lysine-coated 6-well plates at a density of 0.5 million cells/well. U2OS/SA-GFP cells were seeded in 6-well plates at a density of 0.25 million cells/well. The next day, cells were transfected with appropriate siRNA (40 nm) using Lipofectamine RNAiMAX (Invitrogen). After 24 h, siRNA-transfected cells were transferred into a 12-well plate, with 200,000 cells/well for HEK293/SA-GFP and 100,000 cells/well for U2OS/SA-GFP. 44 h after siRNA transfection, cells were transfected with 0.6 μg of the I-SceI expression vector pCBASce (36) using linear polyethyleneimine and, 6 h later, with appropriate siRNA (20 nm) using the standard calcium phosphate method. 52 h after I-SceI transfection, cells were harvested and subjected to flow cytometry analysis using LSRII (BD Biosciences) and FlowJo software to determine the percentage of GFP-positive cells. The mean values obtained with control siRNA (siLuc) samples were 0.9% for HEK293/SA-GFP cells and 2.0% for U2OS/SA-GFP cells. To test the effect of ectopic expression of WRN on SSA repair efficiency of WRN-depleted HEK293/SA-GFP cells, the mutant form of the pcDNA3.1-hWRN construct harboring silent mutations in the siWRN-targeting region (0.6 μg) was cotransfected with pCBASce (0.6 μg). The plasmid pcDNA3.1 was used as a control vector in these experiments. Cells were subjected to flow cytometry analysis at 52 h after plasmid transfection.
Immunofluorescence Assays
U2OS cells transfected with the indicated siRNAs were cultured on glass coverslips. 48 h after siRNA transfection, cells were treated with 1 μm camptothecin (CPT) for 1 h. After pre-extraction for 5 min on ice in 25 mm HEPES (pH 7.4) buffer containing 0.5% (v/v) Triton X-100, 50 mm NaCl, 1 mm EDTA, 3 mm MgCl2 and 0.3 m sucrose, cells were fixed with 4% (v/v) formaldehyde for 15 min at room temperature. Subsequently, cells were permeabilized by soaking in 0.2% (v/v) Triton X-100 for 5 min at room temperature. After blocking in PBS containing 10 mg/ml BSA for 30 min at room temperature, fixed cells were incubated for 2 h at room temperature with the indicated primary antibodies. The slides were washed with PBS and incubated for 1 h at room temperature with secondary antibodies diluted in blocking solution (Alexa Fluor 568-conjugated goat anti-rabbit IgG and Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen)). After washing with PBS, coverslips were mounted using Vectashield containing DAPI (Vector Laboratories). Automated image acquisition was performed using an Olympus IX70 microscope equipped with the Scan̂R imaging platform. A ×40/1.3 numerical aperture objective was used. 10 z stacks at a spacing of 0.3 μm were taken, and 100 images were acquired for each sample. Analysis was performed using Scan̂R analysis software. Nuclei were identified on the basis of the DAPI signal, and RPA foci were identified on the basis of edge-based subobject counts. At least 1000 cells were analyzed for each condition.
RESULTS
DNA2 Can Mediate DNA End Resection in Conjunction with WRN Helicase
To test whether the human WRN helicase can mediate resection of broken DNA ends in concert with DNA2, we purified these proteins to homogeneity and analyzed their activities in vitro (Fig. 1A). WRN and BLM unwind DNA in the 3′-5′ direction and require a 3′ ssDNA tail for loading onto the DNA substrate (37, 38). Therefore, we generated a derivative of the pUC19 plasmid in which a StuI site was flanked on each side by four recognition sites for the nicking endonuclease Nt.BbvCI. Cleavage of this pUC19 derivative with StuI and Nt.BbvCI resulted in a 2.7-kb-long linear DNA molecule ending with 3′ overhangs of 26 nt in length, whereas its cleavage by StuI alone gave rise to a linear DNA molecule with blunt ends. Processing of the DNA substrates was monitored by agarose gel electrophoresis followed by SYBR gold staining (Fig. 1B). In addition, 32P-labeled synthetic oligonucleotides complementary to the 3′-terminated strand were used as hybridization probes to detect ssDNA generated by resection at specific positions (Fig. 1B) (10). We found that WRN, together with DNA2 and RPA, could catalyze efficient 5′ end resection on the 3′-tailed substrate but not the blunt-ended substrate (Fig. 1C). As expected, no reaction products were detected with an oligonucleotide probe complementary to the 5′-terminated strand, indicating that the observed DNA resection activity is limited to the 5′ strand (Fig. 1D). Of note, the end product of the resection reaction on the 3′-tailed DNA substrate appeared as a discrete band on SYBR gold-stained gel that was clearly shifted with respect to the unprocessed dsDNA substrate, indicating extensive resection (Fig. 1C, top panel, compare lanes 2 and 7). In contrast, no gradual shift in the electrophoretic mobility of the resection product was apparent after annealing of the radioactive probes. This is most likely due to the fact that DNA2 nuclease generates short oligonucleotides that can reanneal to the resected DNA along with the radioactive probe, leading to a DNA molecule with an electrophoretic mobility similar to that of the DNA substrate. Together, these results clearly demonstrate that DNA2, in conjunction with WRN and RPA, can catalyze extensive 5′ end resection, providing that the DNA substrate contains a 3′ ssDNA overhang.
FIGURE 1.

DNA end resection by DNA2 and WRN. A, SDS-PAGE analysis of purified WRN (0.7 μg) and DNA2 (0.4 μg) proteins. Gel was stained with Coomassie Brilliant Blue R-250. The molecular weights of protein standards are indicated on the left. B, schematic of the DNA end resection assay. The resection products were either left untreated or hybridized with synthetic 32P-labeled oligonucleotides complementary to the 3′-terminated strand. DNA species were separated by electrophoresis in a 1% agarose gel and visualized by SYBR gold staining and phosphorimaging, respectively. Probes complementary to the regions spanning nt positions 112–133 and 353–374 (relative to the 3′ end) were used in this study. Only a part of the DNA substrate is shown. C, time course of resection of 3′-tailed (26 nt) and blunt-ended DNA substrates by DNA2 and WRN. Reactions were carried out at 37 °C and contained 2 nm DNA, 350 nm RPA, 10 nm WRN, and 8 nm DNA2. Reaction products at the indicated time points were analyzed as outlined in B. The 112- to 133-nt probe was used in this analysis. Lane 1, heat-denatured substrate; lane 14, 3′-tailed substrate incubated with 20 nm EXO1 and 350 nm RPA for 2 min (R1); lane 15, blunt-ended substrate incubated with 20 nm EXO1 and 350 nm RPA for 2 min (R2). D, directionality of DNA end resection by WRN-DNA2. Reactions were carried out at 37 °C for 60 min and contained 2 nm 3′-tailed DNA substrate, 350 nm RPA, 8 nm DNA2, and 10 nm WRN. Resection products were annealed with radiolabeled oligonucleotide probes complementary to either 3′-terminated (position 353–374 nt relative to the 3′ end) or 5′-terminated (position 353–374 nt relative to 3′ end) strand and analyzed as in C. E, 5′ end resection of 3′-tailed DNA substrate by WRN-DNA2 is dependent on WRN concentration and the presence of ATP and RPA. Reactions were carried out at 37 °C for 60 min and contained, as indicated, 2 nm DNA, 350 nm RPA, 1 mm ATP, 8 nm DNA2, and different WRN concentrations. Resection products were detected using the 112–133-nt probe. F, dependence of WRN-DNA2-catalyzed resection of 3′-tailed substrate on WRN concentration. Resection at the positions of 112–133 nt and 353–374 nt from the 3′ end of the DNA substrate was monitored. Reactions were carried out as in E. Relative concentration of the resection product generated by WRN-DNA2 at each WRN concentration was calculated as a percentage of the product generated by 20 nm EXO1 after 2 min. Data are mean ± S.D. (n = 3).
To further characterize the DNA end resection reaction mediated by WRN-DNA2, reactions with the 3′-tailed substrate were carried out at various WRN concentrations, whereas DNA2 was kept at a concentration of 8 nm. We observed that the amount of resection product increased gradually with WRN concentration, reaching a plateau at about 10 nm (Fig. 1E, lanes 2–7, and Fig. 1F). Quantitative analysis of gel images revealed that about 35% of the DNA substrate was resected to the position of 133 nt from the 3′ end and that about 15% of the DNA substrate was resected to the position of 374 nt within 1 h of incubation (Fig. 1F). Interestingly, a small amount of resected product (1–2%) could also be detected in the absence of WRN, suggesting that DNA2 itself could slowly resect dsDNA ends, likely following RPA-mediated stabilization of ssDNA ends generated by thermal fraying (Fig. 1E, lane 2). In the absence of DNA2, WRN was only capable of DNA unwinding, as evident from the appearance of a fast-migrating band (Fig. 1E, lane 8). The resection process catalyzed by WRN and DNA2 was found to be dependent on the presence of ATP and RPA, as expected for a helicase-driven reaction (Fig. 1E, compare lanes 9–12).
WRN acts not only as a 3′-5′ DNA helicase, but it also possesses a dsDNA-dependent 3′-5′ exonuclease activity residing in a separate domain located in the N-terminal portion of the protein (39, 40). DNA2 functions as a 5′-3′ helicase and a ssDNA-specific endonuclease (32, 41). To define the functions of the enzymatic activities of WRN and DNA2 in DNA end resection, we carried out a set of resection reactions with the 3′-tailed pUC19 substrate where either WRN or DNA2 were substituted with catalytically inactive mutants. We found that the helicase-deficient mutant of WRN (K567M) failed to stimulate DNA resection by DNA2, whereas the nuclease-deficient mutant of WRN (E84A) behaved similarly as the wild-type WRN in this reaction (Fig. 2, lanes 2–4). Substitution of DNA2 with its nuclease-deficient mutant (D277A) completely abolished resection and stimulated unwinding of the plasmid substrate (Fig. 2, lane 6). In contrast, the helicase-deficient mutant of DNA2 (K654E) could resect the DNA substrate to the same degree as the wild-type protein (Fig. 2, compare lanes 3 and 7). These results indicate that DNA end resection mediated by DNA2, WRN, and RPA is dependent on the helicase activity of WRN and the endonuclease activity of DNA2.
FIGURE 2.
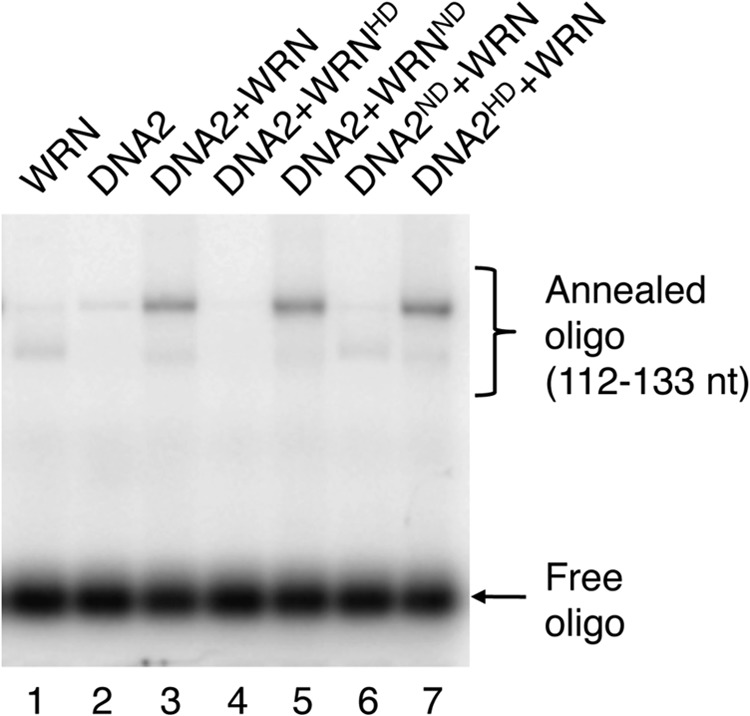
5′ end resection of 3′-tailed DNA substrate by WRN-DNA2 depends on the helicase activity of WRN and the nuclease activity of DNA2. Reactions were carried out at 37 °C for 60 min and contained 2 nm DNA, 350 nm RPA, 1 mm ATP, 8 nm DNA2, and 10 nm WRN. Resection products were detected using the 112–133 nt probe. WRNHD, helicase-deficient mutant of WRN (K567M); WRNND, nuclease-deficient mutant of WRN (E84A); DNA2HD, helicase-deficient mutant of DNA2 (K654R); DNA2ND, nuclease-deficient mutant of DNA2 (D277A).
DNA2 and WRN Interact Physically
Yeast Dna2 has been shown to interact physically with Sgs1 (10). Likewise, BLM forms a complex with human DNA2 (17). Therefore, we investigated whether human DNA2 interacts physically with WRN. To this end, HEK293 cells were transfected with plasmids expressing WRN and FLAG-tagged DNA2, respectively, and complex formation between these proteins was tested by immunoprecipitation using beads conjugated with anti-FLAG M2 antibody. We found that WRN coimmunoprecipitated with FLAG-DNA2, indicating that these proteins form a complex in vivo (Fig. 3A, lane 3). This interaction was specific because anti-FLAG beads did not immunoprecipitate WRN from an extract lacking FLAG-DNA2 (Fig. 3A, lane 1). To further investigate complex formation between WRN and DNA2, we generated a stable HEK293 cell line expressing FLAG-DNA2. By immunoprecipitation using anti-FLAG M2 beads or anti-WRN antibody, we found that FLAG-DNA2 formed a complex with endogenous WRN in these cells (Fig. 3, B and C). Western blot analysis indicated that the level of FLAG-DNA2 in these cells was only slightly higher than that of endogenous DNA2, suggesting that WRN and DNA2 form a complex under physiological conditions (Fig. 3B, top panel). Interaction between FLAG-DNA2 and endogenous BLM was also detected as expected (Fig. 3B) (17). The cellular concentration of these protein complexes was not altered when cells were subjected to treatment with CPT, which causes breakage of DNA replication forks (Fig. 3B, lanes 2–5) (42). This suggests that the interaction of DNA2 with WRN and BLM in the cell is not dependent on DNA damage.
FIGURE 3.

Physical interaction between DNA2 and WRN in vitro and in vivo. A, coimmunoprecipitation of WRN with DNA2 from human cells. HEK293 cells were transfected with vectors expressing FLAG-DNA2 and WRN as indicated. Cell extracts were immunoprecipitated (IP) with anti-FLAG antibody as described under “Experimental Procedures.” Blots were probed with the indicated antibodies. 5% of input material was loaded. B, effect of DNA damage on the formation of DNA2-WRN and DNA2-BLM complexes in human cells. HEK293 cells stably transfected with the FLAG-DNA2 construct (HEK293-D) were treated with 1 μm CPT. At the indicated time points, complex formation between FLAG-DNA2 and endogenous WRN and BLM, respectively, was tested by immunoprecipitation using anti-FLAG antibody. C, coimmunoprecipitation of DNA2 with WRN from human cells. Extracts from HEK293-D cells were subjected to immunoprecipitation with anti-WRN antibody or control IgG. The immunoprecipitates were tested for the presence of FLAG-DNA2 and WRN by Western blotting. As a control, a WRN immunoprecipitate from HEK293 cells was also analyzed (lane 3). D, coimmunoprecipitation of DNA2 with WRN from a mixture of purified proteins. DNA2 (500 ng) was incubated with or without WRN (500 ng) at 4 °C for 4 h. The mixtures were subjected to immunoprecipitation with anti-WRN antibody. E, domain organization of WRN. Exo, exonuclease domain; Zn, zinc-binding domain; WH, winged-helix domain; HRDC, helicase and RNaseD C-terminal domain. Black lines indicate WRN fragments used for mapping the DNA2-interaction site on WRN. F, GST pulldown assay. Glutathione beads coated with the indicated GST-tagged fragments of WRN were incubated with purified His6-DNA2-FLAG protein at 4 °C for 2 h, and bound proteins were analyzed by Western blotting as described under “Experimental Procedures.” 1% of input was loaded in B and C, whereas 10% of input was loaded in D and F.
To test whether WRN and DNA2 interact directly, purified proteins were mixed and incubated at 4 °C for 4 h. Complex formation between WRN and DNA2 was tested by immunoprecipitation using anti-WRN antibody. We found that DNA2 coimmunoprecipitated with WRN. DNA2 was not present in the immunoprecipitated material whether WRN was omitted, confirming a direct protein-protein interaction (Fig. 3D). To map the interaction site of DNA2 on WRN, we tested binding of purified His6-DNA2-FLAG protein to various WRN fragments covering the entire WRN polypeptide (Fig. 3E). The WRN fragments were produced in E. coli as fusions with a GST tag and isolated on GSH-Sepharose beads. Using a GST pulldown assay, we found that DNA2 bound specifically to a WRN fragment including the core helicase domain (helicase/Zn2+-binding domains) and the winged helix domain (Fig. 3F, compare lanes 1 and 4) a binding site of a number of other proteins shown to interact with WRN (43, 44). DNA2 was also bound to a fragment containing only the helicase core or to the C-terminal portion of WRN starting at the beginning of the winged helix domain (Fig. 3F, compare lanes 1, 3, 4, and 5). In contrast, DNA2 did not bind the N-terminal portion of WRN containing the exonuclease domain (Fig. 3F, lane 2). Collectively, these results suggest that there are at least two DNA2-interaction sites on WRN: one located in the central helicase domain and the other in the C-terminal region of WRN.
WRN-DNA2 Resects DNA Ends More Efficiently Than BLM-DNA2
Next, we set out to compare WRN and BLM with respect to their abilities to resect DNA ends in concert with DNA2 and RPA in vitro. Using a Y structure oligonucleotide duplex (29 bp) with single-stranded arms (30 nt each), we found that our preparations of WRN and BLM exhibited similar levels of specific helicase activity (Fig. 4A). For resection reactions, we used the 3′-tailed DNA substrate that was readily processed by WRN-DNA2 in the presence of RPA (Fig. 1C). The extent of DNA resection at various reaction time points was monitored by annealing of radiolabeled oligonucleotide probes. These experiments clearly showed that WRN-DNA2 resected the DNA substrate at a much higher rate compared with BLM-DNA2 (Fig. 4, B and C). Notably, WRN-DNA2-catalyzed resection to the position of 374 nt away from the 3′ end was faster than BLM-DNA2-catalyzed resection to the position of 133 nt (Fig. 4, B and C). We also compared the activities of WRN-DNA2 and BLM-DNA2 on blunt-ended DNA substrate in the presence of RPA. We found that this DNA substrate was largely refractory not only to processing by WRN-DNA2 but also to processing by BLM-DNA2 (Fig. 4D, compare lanes 4 and 5 to lanes 10 and 11). Taken together, we show that WRN-DNA2 resects DNA ends more efficiently than BLM-DNA2 in vitro.
FIGURE 4.

Comparison of DNA end resection activities of WRN-DNA2 and BLM-DNA2. A, comparison of helicase activities of WRN and BLM. Reactions contained 1 nm 32P-labeled forked DNA duplex (inset) and different concentrations of WRN or BLM. Reactions were incubated at 37 °C for 30 min, and reaction products were quantified as described under “Experimental Procedures.” Data are mean ± S.D. (n = 3). B, time course of resection of 3′-tailed DNA substrate catalyzed by WRN-DNA2 and BLM-DNA2, respectively. Reactions contained 2 nm DNA, 350 nm RPA, 8 nm DNA2, and 10 nm WRN/BLM. Reaction aliquots withdrawn at the indicated time points were subjected to electrophoresis on a 1% agarose gel after hybridization of radiolabeled probes complementary to 3′-terminated strand at the indicated positions. Radiolabeled DNA species were visualized by phosphorimaging. C, quantification of the reactions in B. Relative concentration of resection products generated at each time point was calculated as a percentage of the product generated by 20 nm EXO1 after 2 min. Data are mean ± S.D. (n = 3). D, processing of 3′-tailed (26 nt) and blunt-ended DNA substrates in reactions with indicated composition. Reactions were carried out at 37 °C for 60 min and contained 2 nm DNA, 350 nm RPA, and, where indicated, 8 nm DNA2, 20 nm WRN, and 20 nm BLM. Reaction products were analyzed as in Fig. 1C. Lane 1, heat-denatured substrate; lane 14, 3′-tailed substrate incubated with 20 nm EXO1 for 2 min (R1); lane 15, blunt-ended substrate incubated with 20 nm EXO1 for 2 min (R2).
Dissection of Pathways Involved in DNA End Resection in Human Cells
To assess whether WRN is involved in DNA end resection in vivo, we investigated the effect of its depletion on the efficiency of SSA-mediated repair of endonuclease-induced DSBs in cells that were either proficient or deficient for EXO1 and DNA2, respectively. For this epistasis analysis, we initially used the human embryonic kidney cell line HEK293 stably transfected with the SA-GFP reporter cassette consisting of two truncated GFP gene alleles (5′GFP and Sce3′GFP) that form a direct sequence repeat (280 bp) separated by a region of about 2.4 kb (Fig. 5A) (34, 45). SSA-mediated recombination between these homologous sequences triggered by a DSB generated in the distal GFP allele by the I-SceI endonuclease results in the formation of a functional GFP gene (Fig. 5A). This requires extensive DNA end resection to expose the complementary ssDNA regions for annealing. The proteins of interest were depleted from HEK293/SA-GFP cells by RNA interference. Cells were subsequently transfected with an I-SceI expression vector to create a DSB in the reporter cassette, and the percentage of GFP positive cells arising upon SSA-mediated repair was determined by flow cytometry 2 days after plasmid transfection. We found that cells depleted of either EXO1, WRN, or DNA2 exhibited a marked reduction in the frequency of SSA repair events (55, 65, and 75%, respectively) compared with mock-depleted cells (Fig. 5, B and C). In contrast, knockdown of BLM was found to be associated with a significant increase in SSA repair efficiency (140%) compared with control cells (Fig. 5, B and C). Of note, the SSA repair defect of WRN-depleted cells could be rescued by ectopic expression of the siRNA-resistant form of WRN, excluding an off-target effect of the WRN siRNA used in this study (Fig. 5D). Combined depletion of EXO1 and WRN or EXO1 and DNA2 further decreased the repair efficiency compared with the respective single depletions, whereas codepletion of DNA2 and WRN did not (Fig. 5, B and C). In addition, combined depletion of EXO1 and BLM had nearly the same effect on the SSA repair efficiency as EXO1 depletion (Fig. 5, B and C). Therefore, these findings suggest that HEK293 cells have at least two pathways for long-range resection of DSB ends: one mediated by EXO1 and the other dependent upon DNA2 and WRN.
FIGURE 5.

WRN and BLM interact epistatically with DNA2 to promote DSB repair by SSA in human cells. A, schematic of the SA-GFP reporter cassette. SSA-mediated repair of a DSB at the I-SceI-cutting site results in the formation of a functional GFP allele. B, efficiency of SSA-mediated repair of I-SceI-induced DSB in HEK293/SA-GFP cells treated with the indicated siRNAs. Cells were transfected with the appropriate siRNAs (40 nm) 2 days prior to transfection of the I-SceI-expressing plasmid. The percentage of GFP-positive cells in each sample was measured by flow cytometry 2 days after I-SceI plasmid transfection and taken as a measure of DSB repair efficiency. The plotted values represent the relative repair efficiency calculated as a percentage of repair efficiency measured in cells transfected with control siRNA (siLuc, 100%). Data are mean ± S.D. (n ≥ 3). C, Western blot analysis of extracts from HEK293/SA-GFP cells transfected with indicated siRNAs under the same conditions as for SA-GFP reporter assays. Blots were probed with the indicated antibodies. D, rescue of the SSA-repair defect of WRN-depleted HEK293/SA-GFP cells by expression of the siRNA-resistant variant of WRN. An SA-GFP reporter assay was performed as in B. The WRN plasmid (WRN) or empty vector (EV) were cotransfected with the I-SceI plasmid. E, efficiency of SSA-mediated repair of I-SceI-induced DSB in U2OS/SA-GFP cells treated with the indicated siRNAs. Experiments were performed as in B. F, Western blot analysis of extracts from U2OS/SA-GFP cells transfected with the indicated siRNAs. Blots were probed with the indicated antibodies. G, quantitative real-time PCR showing that EXO1 mRNA levels are down-regulated by specific siRNA. Data are mean ± S.D. (n = 3).
To substantiate these findings, we performed a similar set of experiments using U2OS/SA-GFP cells (35). This analysis indicated that combined depletion of EXO1 and DNA2 almost completely abolished (reduced by 91%) SSA-mediated DSB repair in U2OS/SA-GFP cells, as did depletion of MRE11 (by 89%) or CtIP (by 82%), suggesting that long-range DNA end resection in U2OS cells is largely dependent on EXO1 and DNA2 (Fig. 5, E–G). However, in contrast to the results obtained with HEK293/SA-GFP cells, we observed a significant reduction in SSA repair efficiency not only after depletion of WRN (by 57%) but also after depletion of BLM (by 59%) (Fig. 5, E and F). Codepletion of BLM and WRN further decreased the repair efficiency to a level comparable with that in DNA2-depleted (by 73%) cells (Fig. 5, E and F). Moreover, combined depletion of DNA2 with either BLM or WRN had nearly the same inhibitory effect on SSA repair as DNA2 depletion (Fig. 5, E and F). On the contrary, codepletion of EXO1 with either WRN or BLM caused a much higher reduction in repair efficiency than depletion of DNA2 alone, and triple depletion of EXO1, BLM, and WRN brought repair efficiency down to the level measured in cells depleted of EXO1 and DNA2 (Fig. 5, E and F). Collectively, these data suggest that, in U2OS cells, both WRN and BLM assist DNA2 to mediate long-range resection of broken DNA ends.
To bolster our conclusion that DNA2, WRN, and BLM have an epistatic relationship in DSB end resection, we extended our analysis to measurement of RPA focus formation in U2OS cells treated with CPT. As expected, 1 h after addition of CPT, RPA formed numerous foci in γ-H2AX-positive cells, which were dependent on the presence of CtIP (Fig. 6). Depletion of DNA2 resulted in a marked reduction in the number of RPA foci per cell compared with mock-depleted cells (Fig. 6). Cells depleted of BLM or WRN displayed a mild decrease in RPA focus frequency compared with mock-depleted cells (Fig. 6). In contrast, combined depletion of BLM and WRN caused approximately the same reduction in RPA focus frequency as depletion of DNA2 alone. Moreover, cells depleted of DNA2 and BLM or DNA2 and WRN displayed an RPA foci frequency comparable with that of DNA2-depleted cells (Fig. 6). These data further support the conclusion that DNA2, WRN, and BLM operate in the same DNA end resection pathway.
FIGURE 6.
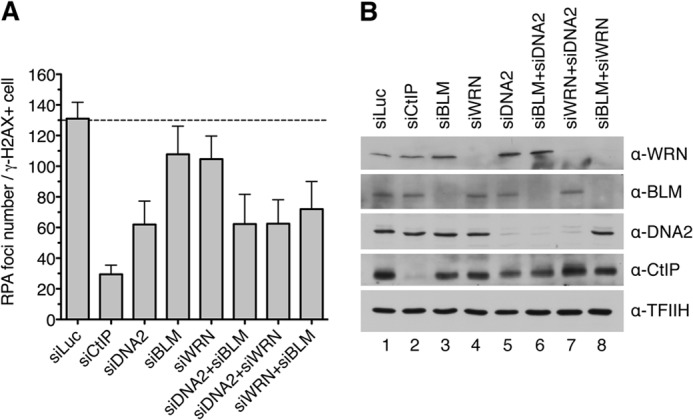
DNA2, WRN, and BLM act in the same pathway of DSB end resection. A, frequency of camptothecin-induced RPA foci in nuclei of U2OS cells depleted of the indicated proteins. Cells were transfected with appropriate siRNAs and, 48 h later, treated with 1 μm camptothecin for 1 h. Cells were then detergent-extracted and fixed with formaldehyde. RPA and γ-H2AX (a marker of DNA damage) were visualized by indirect immunofluorescence. DAPI was used to stain nuclei. The average number of RPA foci per γ-H2AX-positive cell was determined for each sample using an Olympus Scan̂R screening station. The data points are mean ± S.D. (n = 3). B, Western blot analysis of extracts from U2OS cells transfected with indicated siRNAs. Blots were probed with the antibodies indicated on the right.
Role of the BLM-TOPOIIIα-RMI1-RMI2 Complex in DNA End Resection
In human cells, BLM exists in a complex with TOPOIIIα, RMI1, and RMI2, which is known to catalyze double Holliday junction dissolution during HR (46–49). Studies in yeast have shown that Top3α and Rmi1 are also required for DNA-end resection in vivo and stimulate DNA end resection by Sgs1-Dna2 in vitro by promoting the helicase activity of Sgs1 (9, 10, 14). Our study revealed that BLM-DNA2 resects DNA ends less efficiently than WRN-DNA2 in vitro, whereas in vivo, at least in U2OS cells, BLM and WRN appeared to contribute equally to promote DNA end resection (Fig. 5E). Therefore, we investigated whether BLM requires TOPOIIIα, RMI1, and RMI2 (TRR) to efficiently support DNA end resection by DNA2. To this end, we first investigated the effect of a purified TRR complex on DNA end resection by BLM-DNA2 in vitro (Fig. 7A). We found that TRR enhanced resection of the 3′-tailed pUC19 substrate by BLM-DNA2 (Fig. 7, B and C, lanes 3–6). On the contrary, the TRR complex had no effect on DNA end resection by WRN-DNA2 (data not shown). Moreover, it could not enhance DNA end resection by DNA2 in the absence of BLM (Fig. 7, B and C, compare lanes 2 and 7).
FIGURE 7.

Involvement of TOPOIIIα, RMI1, and RMI2 in DNA end resection. A, SDS-PAGE analysis of purified TRR complex (1.5 μg) and BLM (0.5 μg). The gel was stained with Coomassie Brilliant Blue R-250. The molecular weights of protein standards are indicated on the left. B, stimulation of BLM-DNA2-catalyzed DNA end resection by the TRR complex. Reactions contained 2 nm 3′-tailed pUC19 substrate, 8 nm DNA2, 10 nm BLM, 350 nm RPA, and varying concentrations of TRR. BLM and TRR were preincubated for 5 min on ice prior to addition to the reaction. Reaction products were analyzed as in Fig. 1C using the 112–133-nt probe. C, quantification of the product of reactions in B. Data are mean ± S.D. (n = 3). The data are normalized to the amount of product in the reaction containing only BLM and DNA2 (100%). D, RMI1 acts epistatically with BLM and DNA2 to promote DSB repair by SSA in human cells. Efficiency of SSA-mediated repair of I-SceI-induced DSB in U2OS/SA-GFP cells transfected with indicated siRNAs was measured as in Fig. 5E. E, Western blot analysis of extracts from U2OS/SA-GFP cells transfected with indicated siRNAs under the same conditions as for SA-GFP reporter assays. Blots were probed with the indicated antibodies.
Next we tested the effect of depletion of RMI1 on the efficiency of SSA-mediated repair of I-SceI-induced DSBs in U2OS/SA-GFP cells. We found that RMI1 depletion reduced the repair efficiency to the level displayed by BLM- or DNA2-depleted cells (Fig. 7, D and E). Importantly, codepletion of RMI1 with BLM or DNA2 did not further reduce the repair efficiency compared with single depletions of these proteins, suggesting that RMI1, BLM, and DNA2 act in the same pathway (Fig. 7, D and E). Collectively, these results suggest that, in human cells, BLM promotes long-range DNA end resection as part of the BTRR complex.
DISCUSSION
Here we present evidence suggesting that human DNA2 acts in conjunction with either WRN or BLM to mediate long-range resection of broken DNA ends in vivo. Moreover, we show that WRN helicase can cooperate with DNA2 and RPA to catalyze resection of DNA ends in vitro, generating long 3′-terminated ssDNA tails. Our study also reveals that both WRN-DNA2 and BLM-DNA2 require a 3′ ssDNA overhang to efficiently initiate DNA end resection in vitro, which is in agreement with the “two-step” resection model in which the initial 5′ end trimming is carried out by the MRE11-RAD50-NBS1/Xrs2 complex in conjunction with CtIP/Sae2 (8, 9, 11, 19). In addition, we present evidence that BLM and DNA2 interact epistatically with RMI1 to mediate DNA end resection in vivo. Moreover, we show that the TRR complex stimulates DNA end resection by BLM-DNA2 in vitro. These data suggest that, in cells, BLM mediates DNA end resection as part of the BTRR complex.
Our discovery of the involvement of WRN in DNA end resection is consistent with the findings that WRN interacts physically with the MRN complex and accumulates at sites of DSBs in human cells (50, 51). Moreover, it has been demonstrated that WRN depletion leads to a marked reduction in the frequency of RPA and BrdU/ssDNA foci formed in response to ionizing radiation, indicative of a resection defect (52). A similar phenotype has been observed in DNA2-depleted cells (18). Although the previous studies did not address the relationship between WRN and DNA2, they demonstrated that these enzymes act synergistically with EXO1 to promote DNA end resection in human cells (18, 52). A role for WRN as a critical DNA end resection factor is also consistent with the cellular phenotype of Werner syndrome, a severe premature aging disorder caused by inherited mutations in the WRN gene (53). Cells derived from Werner syndrome patients are characterized by non-homologous chromosome exchanges, termed variegated translocation mosaicism, and large chromosomal deletions that may result from aberrant DSB repair by NHEJ as a consequence of a defect in DNA end resection (54–56). Indeed, it is becoming clear that NHEJ accounts for most chromosomal translocations in humans (57). Moreover, a role for DNA end resection as the critical determinant of DSB repair pathway choice is well established (58). Accumulating evidence suggests that defects in homology-directed repair pathways, which are dependent on DNA end resection, result in overuse of NHEJ for repair, leading to accumulation of chromosomal rearrangements (57). However, it should be noted that WRN is also known to promote DSB repair by the classical Ku-dependent NHEJ (C-NHEJ) pathway to suppress microhomology-mediated end joining (59, 60). This alternative end joining pathway is capable of producing chromosomal translocations, particularly when Ku-dependent NHEJ is deficient (57). Moreover, WRN has been shown to be involved in the resolution HR intermediates (61, 62). Therefore, it seems that the genomic instability in Werner syndrome is a consequence of multiple defects in DNA repair pathways.
Our finding that the TRR complex stimulates DNA end resection by BLM-DNA2 in vitro is consistent with previous reports showing that the association of BLM with TOPOIIIα and RMI1 enhances its DNA unwinding activity, which drives the BLM-DNA2-catalyzed resection reaction (17, 63). Similarly, RMI1 and RMI2 have been shown to enhance the efficiency of the BLM-TOPOIIIα-mediated double Holliday junction dissolution reaction (48, 49, 64). It has also been shown that RMI1 forms a complex with RPA and that this interaction is essential for the stimulatory effect of RPA on double Holliday junction dissolution by the BTRR complex (65). Therefore, it is possible that a physical interaction between RMI1 and RPA loaded on the 3′-terminated DNA strand during DNA2-catalyzed resection might enhance the DNA unwinding processivity of the BTRR complex and, hence, increase the efficiency of the resection reaction. However, it should be noted that the stimulatory effect of the TRR complex on DNA end resection by BLM-DNA2 in vitro was rather modest under our experimental conditions. On the contrary, RMI1 depletion in U2OS/SA-GFP cells reduced the efficiency of SSA-mediated DSB repair to levels displayed by BLM- or DNA2-deficient cells, suggesting that BLM requires RMI1 to promote DNA end resection in vivo. Of note, it has been shown that silencing of RMI1 or RMI2 expression by RNA interference destabilizes both BLM and TOPOIIIα (47, 49). Therefore, it is evident that, in addition to being important for the functional attributes of the BTRR complex, RMI1 and RMI2 are indispensable for the structural integrity of its components in vivo.
Although BLM depletion compromised SSA-mediated DSB repair in U2OS/SA-GFP cells, it had an opposite effect on SSA in HEK293/SA-GFP cells. Similarly, the efficiency of SSA-mediated DSB repair in HEK293/SA-GFP cells was elevated significantly upon depletion of RMI1 (data not shown). These findings suggest that, in HEK293 cells, the BTRR complex might act as an SSA suppressor, most likely through unwinding of the annealed intermediate formed following DNA end resection. Strikingly, we found that BLM concentration in HEK293 cells was much higher than in U2OS cells (data not shown). Therefore, it appears that the BTRR complex exerts an inhibitory effect on SSA when its concentration in the cell exceeds certain threshold.
Acknowledgments
We thank Kata Sarlos and Ian D. Hickson for the purified TRR complex, Judith L. Campbell for the transfer vector for preparation of the bacmid expressing His6-hDNA2-FLAG, Jeremy M. Stark for the HEK293/SA-GFP and U2OS/SA-GFP cell lines, Stefano Ferrari and Stephanie Bregenhorn for help with protein purification, and Christiane Koenig for technical assistance.
This work was supported by Swiss National Science Foundation Grants 31003A-129747 and 31003A_146206, by Czech Science Foundation Grant GAP305/10/0281, and by the Stiftung zur Krebsbekämpfung. This work was also supported by Swiss National Science Foundation Grant PP00P3 133636 (to P.C.) and by Forschungskredit of the University of Zurich Grant FK-13-098 (to A. S.).
- DSB
- double strand break
- NHEJ
- non-homologous end joining
- HR
- homologous recombination
- ssDNA
- single-stranded DNA
- SSA
- single strand annealing
- BTRR
- BLM-TOPOIIIα-RMI1-RMI2
- nt
- nucleotide(s)
- CPT
- camptothecin.
REFERENCES
- 1. Jackson S. P., Bartek J. (2009) The DNA-damage response in human biology and disease. Nature 461, 1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Khanna K. K., Jackson S. P. (2001) DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 27, 247–254 [DOI] [PubMed] [Google Scholar]
- 3. Longhese M. P., Bonetti D., Guerini I., Manfrini N., Clerici M. (2009) DNA double-strand breaks in meiosis: checking their formation, processing and repair. DNA Repair 8, 1127–1138 [DOI] [PubMed] [Google Scholar]
- 4. Soulas-Sprauel P., Rivera-Munoz P., Malivert L., Le Guyader G., Abramowski V., Revy P., de Villartay J. P. (2007) V(D)J and immunoglobulin class switch recombinations: a paradigm to study the regulation of DNA end-joining. Oncogene 26, 7780–7791 [DOI] [PubMed] [Google Scholar]
- 5. Lieber M. R. (2010) The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. San Filippo J., Sung P., Klein H. (2008) Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 77, 229–257 [DOI] [PubMed] [Google Scholar]
- 7. Heyer W. D., Ehmsen K. T., Liu J. (2010) Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 44, 113–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mimitou E. P., Symington L. S. (2008) Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455, 770–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhu Z., Chung W. H., Shim E. Y., Lee S. E., Ira G. (2008) Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134, 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cejka P., Cannavo E., Polaczek P., Masuda-Sasa T., Pokharel S., Campbell J. L., Kowalczykowski S. C. (2010) DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 467, 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nicolette M. L., Lee K., Guo Z., Rani M., Chow J. M., Lee S. E., Paull T. T. (2010) Mre11-Rad50-Xrs2 and Sae2 promote 5′ strand resection of DNA double-strand breaks. Nat. Struct. Mol. Biol. 17, 1478–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shim E. Y., Chung W. H., Nicolette M. L., Zhang Y., Davis M., Zhu Z., Paull T. T., Ira G., Lee S. E. (2010) Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J. 29, 3370–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cannavo E., Cejka P., Kowalczykowski S. C. (2013) Relationship of DNA degradation by Saccharomyces cerevisiae exonuclease 1 and its stimulation by RPA and Mre11-Rad50-Xrs2 to DNA end resection. Proc. Natl. Acad. Sci. U.S.A. 110, E1661–E1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Niu H., Chung W. H., Zhu Z., Kwon Y., Zhao W., Chi P., Prakash R., Seong C., Liu D., Lu L., Ira G., Sung P. (2010) Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature 467, 108–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Truong L. N., Li Y., Shi L. Z., Hwang P. Y., He J., Wang H., Razavian N., Berns M. W., Wu X. (2013) Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 110, 7720–7725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gravel S., Chapman J. R., Magill C., Jackson S. P. (2008) DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 22, 2767–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nimonkar A. V., Genschel J., Kinoshita E., Polaczek P., Campbell J. L., Wyman C., Modrich P., Kowalczykowski S. C. (2011) BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 25, 350–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Karanja K. K., Cox S. W., Duxin J. P., Stewart S. A., Campbell J. L. (2012) DNA2 and EXO1 in replication-coupled, homology-directed repair and in the interplay between HDR and the FA/BRCA network. Cell Cycle 11, 3983–3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shibata A., Moiani D., Arvai A. S., Perry J., Harding S. M., Genois M. M., Maity R., van Rossum-Fikkert S., Kertokalio A., Romoli F., Ismail A., Ismalaj E., Petricci E., Neale M. J., Bristow R. G., Masson J. Y., Wyman C., Jeggo P. A., Tainer J. A. (2014) DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 53, 7–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bernstein K. A., Gangloff S., Rothstein R. (2010) The RecQ DNA helicases in DNA repair. Annu. Rev. Genet. 44, 393–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yan H., McCane J., Toczylowski T., Chen C. (2005) Analysis of the Xenopus Werner syndrome protein in DNA double-strand break repair. J. Cell Biol. 171, 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liao S., Toczylowski T., Yan H. (2008) Identification of the Xenopus DNA2 protein as a major nuclease for the 5′→3′ strand-specific processing of DNA ends. Nucleic Acids Res. 36, 6091–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liao S., Guay C., Toczylowski T., Yan H. (2012) Analysis of MRE11's function in the 5′→3′ processing of DNA double-strand breaks. Nucleic Acids Res. 40, 4496–4506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saydam N., Kanagaraj R., Dietschy T., Garcia P. L., Peña-Diaz J., Shevelev I., Stagljar I., Janscak P. (2007) Physical and functional interactions between Werner syndrome helicase and mismatch-repair initiation factors. Nucleic Acids Res. 35, 5706–5716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adams K. E., Medhurst A. L., Dart D. A., Lakin N. D. (2006) Recruitment of ATR to sites of ionising radiation-induced DNA damage requires ATM and components of the MRN protein complex. Oncogene 25, 3894–3904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sartori A. A., Lukas C., Coates J., Mistrik M., Fu S., Bartek J., Baer R., Lukas J., Jackson S. P. (2007) Human CtIP promotes DNA end resection. Nature 450, 509–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang J., O'Donnell L., Durocher D., Brown G. W. (2012) RMI1 promotes DNA replication fork progression and recovery from replication fork stress. Mol. Cell Biol. 32, 3054–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Orren D. K., Brosh R. M., Jr., Nehlin J. O., Machwe A., Gray M. D., Bohr V. A. (1999) Enzymatic and DNA binding properties of purified WRN protein: high affinity binding to single-stranded DNA but not to DNA damage induced by 4NQO. Nucleic Acids Res. 27, 3557–3566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kanagaraj R., Saydam N., Garcia P. L., Zheng L., Janscak P. (2006) Human RECQ5β helicase promotes strand exchange on synthetic DNA structures resembling a stalled replication fork. Nucleic Acids Res. 34, 5217–5231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. El-Shemerly M., Janscak P., Hess D., Jiricny J., Ferrari S. (2005) Degradation of human exonuclease 1b upon DNA synthesis inhibition. Cancer Res. 65, 3604–3609 [DOI] [PubMed] [Google Scholar]
- 31. Henricksen L. A., Umbricht C. B., Wold M. S. (1994) Recombinant replication protein A: expression, complex formation, and functional characterization. J. Biol. Chem. 269, 11121–11132 [PubMed] [Google Scholar]
- 32. Masuda-Sasa T., Imamura O., Campbell J. L. (2006) Biochemical analysis of human Dna2. Nucleic Acids Res. 34, 1865–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bennardo N., Cheng A., Huang N., Stark J. M. (2008) Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 4, e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gunn A., Stark J. M. (2012) I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol. Biol. 920, 379–391 [DOI] [PubMed] [Google Scholar]
- 36. Richardson C., Moynahan M. E., Jasin M. (1998) Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocations. Genes Dev. 12, 3831–3842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karow J. K., Chakraverty R. K., Hickson I. D. (1997) The Bloom's syndrome gene product is a 3′-5′ DNA helicase. J. Biol. Chem. 272, 30611–30614 [DOI] [PubMed] [Google Scholar]
- 38. Brosh R. M., Jr., Waheed J., Sommers J. A. (2002) Biochemical characterization of the DNA substrate specificity of Werner syndrome helicase. J. Biol. Chem. 277, 23236–23245 [DOI] [PubMed] [Google Scholar]
- 39. Shen J. C., Gray M. D., Oshima J., Kamath-Loeb A. S., Fry M., Loeb L. A. (1998) Werner syndrome protein: I: DNA helicase and DNA exonuclease reside on the same polypeptide. J. Biol. Chem. 273, 34139–34144 [DOI] [PubMed] [Google Scholar]
- 40. Kamath-Loeb A. S., Shen J. C., Loeb L. A., Fry M. (1998) Werner syndrome protein: II: characterization of the integral 3′ → 5′ DNA exonuclease. J. Biol. Chem. 273, 34145–34150 [DOI] [PubMed] [Google Scholar]
- 41. Kim J. H., Kim H. D., Ryu G. H., Kim D. H., Hurwitz J., Seo Y. S. (2006) Isolation of human DNA2 endonuclease and characterization of its enzymatic properties. Nucleic Acids Res. 34, 1854–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Avemann K., Knippers R., Koller T., Sogo J. M. (1988) Camptothecin, a specific inhibitor of type I DNA topoisomerase, induces DNA breakage at replication forks. Mol. Cell Biol. 8, 3026–3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee J. W., Harrigan J., Opresko P. L., Bohr V. A. (2005) Pathways and functions of the Werner syndrome protein. Mech. Ageing Dev. 126, 79–86 [DOI] [PubMed] [Google Scholar]
- 44. Kanagaraj R., Parasuraman P., Mihaljevic B., van Loon B., Burdova K., König C., Furrer A., Bohr V. A., Hübscher U., Janscak P. (2012) Involvement of Werner syndrome protein in MUTYH-mediated repair of oxidative DNA damage. Nucleic Acids Res. 40, 8449–8459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stark J. M., Pierce A. J., Oh J., Pastink A., Jasin M. (2004) Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol. Cell Biol. 24, 9305–9316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu L., Hickson I. D. (2003) The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426, 870–874 [DOI] [PubMed] [Google Scholar]
- 47. Yin J., Sobeck A., Xu C., Meetei A. R., Hoatlin M., Li L., Wang W. (2005) BLAP75, an essential component of Bloom's syndrome protein complexes that maintain genome integrity. EMBO J. 24, 1465–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu L., Bachrati C. Z., Ou J., Xu C., Yin J., Chang M., Wang W., Li L., Brown G. W., Hickson I. D. (2006) BLAP75/RMI1 promotes the BLM-dependent dissolution of homologous recombination intermediates. Proc. Natl. Acad. Sci. U.S.A. 103, 4068–4073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu D., Guo R., Sobeck A., Bachrati C. Z., Yang J., Enomoto T., Brown G. W., Hoatlin M. E., Hickson I. D., Wang W. (2008) RMI, a new OB-fold complex essential for Bloom syndrome protein to maintain genome stability. Genes Dev. 22, 2843–2855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cheng W. H., von Kobbe C., Opresko P. L., Arthur L. M., Komatsu K., Seidman M. M., Carney J. P., Bohr V. A. (2004) Linkage between Werner syndrome protein and the Mre11 complex via Nbs1. J. Biol. Chem. 279, 21169–21176 [DOI] [PubMed] [Google Scholar]
- 51. Lan L., Nakajima S., Komatsu K., Nussenzweig A., Shimamoto A., Oshima J., Yasui A. (2005) Accumulation of Werner protein at DNA double-strand breaks in human cells. J. Cell Sci. 118, 4153–4162 [DOI] [PubMed] [Google Scholar]
- 52. Tomimatsu N., Mukherjee B., Deland K., Kurimasa A., Bolderson E., Khanna K. K., Burma S. (2012) Exo1 plays a major role in DNA end resection in humans and influences double-strand break repair and damage signaling decisions. DNA Repair 11, 441–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu C. E., Oshima J., Fu Y. H., Wijsman E. M., Hisama F., Alisch R., Matthews S., Nakura J., Miki T., Ouais S., Martin G. M., Mulligan J., Schellenberg G. D. (1996) Positional cloning of the Werner's syndrome gene. Science 272, 258–262 [DOI] [PubMed] [Google Scholar]
- 54. Salk D., Au K., Hoehn H., Martin G. M. (1981) Cytogenetics of Werner's syndrome cultured skin fibroblasts: variegated translocation mosaicism. Cytogenet. Cell Genet. 30, 92–107 [DOI] [PubMed] [Google Scholar]
- 55. Fukuchi K., Martin G. M., Monnat R. J., Jr. (1989) Mutator phenotype of Werner syndrome is characterized by extensive deletions. Proc. Natl. Acad. Sci. U.S.A. 86, 5893–5897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Melcher R., von Golitschek R., Steinlein C., Schindler D., Neitzel H., Kainer K., Schmid M., Hoehn H. (2000) Spectral karyotyping of Werner syndrome fibroblast cultures. Cytogenet. Cell Genet. 91, 180–185 [DOI] [PubMed] [Google Scholar]
- 57. Bunting S. F., Nussenzweig A. (2013) End-joining, translocations and cancer. Nat. Rev. Cancer 13, 443–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Symington L. S., Gautier J. (2011) Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271 [DOI] [PubMed] [Google Scholar]
- 59. Chen L., Huang S., Lee L., Davalos A., Schiestl R. H., Campisi J., Oshima J. (2003) WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell 2, 191–199 [DOI] [PubMed] [Google Scholar]
- 60. Perry J. J., Yannone S. M., Holden L. G., Hitomi C., Asaithamby A., Han S., Cooper P. K., Chen D. J., Tainer J. A. (2006) WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat. Struct. Mol. Biol. 13, 414–422 [DOI] [PubMed] [Google Scholar]
- 61. Saintigny Y., Makienko K., Swanson C., Emond M. J., Monnat R. J., Jr. (2002) Homologous recombination resolution defect in Werner syndrome. Mol. Cell Biol. 22, 6971–6978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Swanson C., Saintigny Y., Emond M. J., Monnat R. J., Jr. (2004) The Werner syndrome protein has separable recombination and survival functions. DNA Repair 3, 475–482 [DOI] [PubMed] [Google Scholar]
- 63. Bussen W., Raynard S., Busygina V., Singh A. K., Sung P. (2007) Holliday junction processing activity of the BLM-Topo IIIα-BLAP75 complex. J. Biol. Chem. 282, 31484–31492 [DOI] [PubMed] [Google Scholar]
- 64. Raynard S., Bussen W., Sung P. (2006) A double Holliday junction dissolvasome comprising BLM, topoisomerase IIIα, and BLAP75. J. Biol. Chem. 281, 13861–13864 [DOI] [PubMed] [Google Scholar]
- 65. Xue X., Raynard S., Busygina V., Singh A. K., Sung P. (2013) Role of replication protein A in double Holliday junction dissolution mediated by the BLM-Topo IIIα-RMI1-RMI2 protein complex. J. Biol. Chem. 288, 14221–14227 [DOI] [PMC free article] [PubMed] [Google Scholar]