

Abstract
Introduction. Macrophage reprogramming is vital for resolution of acute inflammation. Parenteral vitamin C (VitC) attenuates proinflammatory states in murine and human sepsis. However information about the mechanism by which VitC regulates resolution of inflammation is limited. Methods. To examine whether physiological levels of VitC modulate resolution of inflammation, we used transgenic mice lacking L-gulono-γ-lactone oxidase. VitC sufficient/deficient mice were subjected to a thioglycollate-elicited peritonitis model of sterile inflammation. Some VitC deficient mice received daily parenteral VitC (200 mg/kg) for 3 or 5 days following thioglycollate infusion. Peritoneal macrophages harvested on day 3 or day 5 were examined for intracellular VitC levels, pro- and anti-inflammatory protein and lipid mediators, mitochondrial function, and response to lipopolysaccharide (LPS). The THP-1 cell line was used to determine the modulatory activities of VitC in activated human macrophages. Results. VitC deficiency significantly delayed resolution of inflammation and generated an exaggerated proinflammatory response to in vitro LPS stimulation. VitC sufficiency and in vivo VitC supplementation restored macrophage phenotype and function in VitC deficient mice. VitC loading of THP-1 macrophages attenuated LPS-induced proinflammatory responses. Conclusion. VitC sufficiency favorably modulates macrophage function. In vivo or in vitro VitC supplementation restores macrophage phenotype and function leading to timely resolution of inflammation.
1. Introduction
Resolution of inflammation typically follows an ordered series of events orchestrated by different cell types [1]. During the early stages of inflammation, leukocytes such as polymorphonuclear neutrophils (PMN) are the first immune cells to arrive at the site of inflammation. PMN are recruited by gradients of proinflammatory signals and usually reach peak numbers within 24–48 hrs. PMN have short half-lives and are normally cleared from sites of inflammation by undergoing apoptosis [2]. Mobilized monocyte-derived macrophages extravasate to inflammatory tissue sites and clear apoptotic PMN in a nonphlogistic fashion by the process of efferocytosis. Apoptotic PMN release “find-me” signals that are sensed by extravasated macrophages [3]. Following phagocytosis, apoptotic PMN provides resolution cues to macrophages by evoking distinct signaling events that block release of proinflammatory mediators thus allowing further engulfment of apoptotic cells. Mantovani et al. and Fleming and Mosser note that mobilized macrophages are divided into three groups based upon their activation states [4, 5]. These include the M1, M2, and the recently described regulatory macrophages (Mres). M1 macrophages, classically referred to as activated macrophages, secrete proinflammatory factors that mediate host defense against invading pathogens. M2 macrophages, termed alternatively activated macrophages, are considered to be anti-inflammatory [6, 7]. Finally, Mres macrophages secrete considerable amounts of anti-inflammatory cytokines that prevent inflammatory and autoimmune pathology [8, 9]. Mres macrophages also secrete various lipid mediators that play critical roles in resolution of inflammation (see below). Extensive new research has identified expression markers or phenotypic signatures for the various macrophage activation states in mice. They include gene expression changes in IL-1β, TNFα, and iNOS for classical activation and arginase-1 (Arg1), chitinase 3-like 3 (YM-1), and TGFβ for the alternatively activated macrophages [7].
Resolution of inflammation and restoration of normal tissue function prevent the development of “complications” of excessive inflammation, a process referred to as catabasis [10]. Catabasis is driven by synthesis and release of proresolution lipid mediators such as resolvins, protectins, and lipoxins [11]. Lipoxins [12] and protectins [13] are synthesized by lipoxygenase enzymes (such as 15-lipoxygenase (15-Lox)). Resolvins are derived from omega-3 polyunsaturated fatty acids such as docosahexaenoic acid and eicosapentaenoic acid [14]. They are products of metabolism involving 15-Lox and cyclooxygenase. Russell and Schwarze have reviewed the proresolution effects of proresolution mediators in a variety of inflammatory states [15]. However their regulation by vitamin C (VitC, ascorbic acid, AscA) has yet to be examined.
VitC readily functions as one or two electron-reducing agents for many oxidants and serves as a primary chemical antioxidant in most cell types. It modulates complex biochemical pathways that form an essential part of normal metabolism of immune cells [16]. Intracellular levels of VitC in cells differ significantly from circulating plasma levels. In particular, VitC accumulates in millimolar quantities in immune cells such as PMN and macrophages in which intracellular VitC concentrations are typically 40–60 fold higher than that present in circulation [17, 18]. In PMN, Vissers and Wilkie showed that intracellular VitC levels regulate neutrophil apoptosis [19]. Further, VitC contributes to the antioxidant defenses as well as normal PMN and macrophage function. Oberritter and colleagues showed that intracellular concentrations of VitC in macrophages are in the low millimolar range in freshly prepared peritoneal macrophages and in vivo or in vitro activation of peritoneal macrophages results in a significant decline in their VitC content [20]. Li et al. found that VitC deficiency worsens the inflammatory response following infection with the influenza virus [21]. Moreover, mice deficient in VitC generate excessive proinflammatory responses upon infection with the virulent bacterium Klebsiella pneumonia [22]. In humans, VitC levels are significantly reduced in critically ill patients and specifically in patients with poorly resolving proinflammatory states (e.g., sepsis, systemic inflammatory response syndrome) [23, 24]. Several studies performed in septic patients have found that plasma VitC levels correlate inversely with the incidence of organ failure and directly with survival [25, 26]. We recently showed that VitC attenuates inflammation and normalizes PMN function in septic mice [27, 28]. We further showed that parenteral VitC attenuates proinflammatory biomarkers and reduces mortality in human sepsis [29]. However information is limited regarding the mechanism by which VitC regulates the progression and eventual resolution of inflammatory states.
In the current study we examined the progression and resolution of inflammation using a murine thioglycollate (TG-)elicited peritonitis model in VitC sufficient and deficient mice. While humans lack L-gulono-γ-lactone oxidase (Gulo), the final enzyme in the biosynthesis pathway of VitC [30], mice express functional Gulo, resulting in cells and tissues generally maintaining high levels of VitC thereby complicating the translatability of VitC studies in mice, to humans. In order to establish the studies more relevant to humans, TG-induced peritonitis was performed in transgenic mice lacking Gulo (Gulo−/−). Our studies reveal that progression and resolution of TG-induced inflammation is significantly delayed in VitC deficient mice. In particular, the spatiotemporal profile of pro- and anti-inflammatory mediator production by TG-elicited macrophages was significantly different between the VitC sufficient and deficient mice. Further, macrophage function and phenotype, as well as the antioxidant capacity of VitC deficient macrophages, was significantly impaired by the decline in intracellular VitC levels. Infusion of parenteral VitC as ascorbic acid (AscA) partly restored macrophage phenotype and function in VitC deficient mice.
2. Materials and Methods
2.1. Animals
Gulo−/− mice were bred in-house from an established homozygous colony as previously described [27]. In order to maintain their plasma VitC levels similar to that observed in humans, VitC sufficient mice were fed ad libitum with regular chow and water supplemented with vitamin C (0.33 g/L) renewed twice per week. Gulo−/− mice were made VitC deficient by reducing VitC supplementation (0.033 g/L) for 1 week, followed by complete removal of dietary VitC for additional 2 weeks. We and others have shown that this reduced supplementation significantly decreases the concentration of VitC in immune cells, plasma, and organs [27, 31, 32].
2.2. Thioglycollate Induced Peritonitis and Isolation of Mouse Peritoneal Macrophages
Thioglycollate-mediated peritonitis was established by intraperitoneal (i.p.) injection of 1 mL aged, sterile 3% TG solution to 9–11-week old Gulo−/− mice. Thirty minutes following i.p. challenge, some VitC deficient mice were randomized to receive daily i.p. injection of VitC as AscA (200 mg/kg in saline) for a further 3 or 5 days. Untreated mice received i.p. saline instead of VitC. Mice were euthanized on day 3 or day 5, and the peritoneal cavity lavaged with 7 mL of Hanks' balanced salt solution (HBSS) containing 1% bovine serum albumin (BSA). The lavage was centrifuged and the resulting leukocyte pellet was washed with HBSS and resuspended in RPMI-1640 medium. Cell counts of the peritoneal lavage were performed using a Hemocytometer. Cytochemical staining of peritoneal cells was performed using HARLECO Hemacolor solution (EMD Millipore) [28]. PMNs were then separated from macrophages by adherence to a plastic dish as described previously [28, 33]. Peritoneal macrophages were plated at a density of 2 × 106 cells in 35 mm dishes in growth media (DMEM, 10% FBS). Media were changed after 2 h to remove floating cells prior to experimentation.
2.3. Cell Culture
Human acute monocytic leukemia suspension cell line (THP-1) was obtained from ATCC (Manassas, VA). THP-1 cells were maintained in RPMI-1640 medium containing 10% FBS according to the instructions supplied. For induction of macrophages, PMA (100 nM) was added to the medium and cells were seeded at a density of 0.1 × 106 cells/cm2 into tissue culture dishes and maintained in a humidified atmosphere of 95% air and 5% CO2. Media containing PMA were replaced every 2 days, and experiments started after 5 days in culture, when the cells were phenotypically macrophage [34].
2.4. Vitamin C Analysis
Plasma and intracellular VitC levels of peritoneal macrophages seeded onto 35 mm dishes were measured using a fluorescence end-point assay adopted from Vislisel et al. [35]. Plasma was deproteinized as described previously [29]. Briefly, 0.2 mL of cold 20% trichloroacetic acid (TCA) and 0.2 mL of cold 0.2% dithiothreitol (DTT) were added to 0.1 mL of plasma, vortexed for 2 min, and centrifuged (10,000 g, 10 min, 4°C). Supernatants were aliquoted and frozen at −70°C for batch analysis. Peritoneal macrophages were similarly extracted with TCA and DTT and frozen at −70°C for batch analysis. Supernatant or AscA standards were transferred in triplicate to a 96-well plate. Assay buffer containing 1 M sodium acetate, pH 5.5, and 1 mM TEMPOL was added to each well and the plate was incubated for 10 minutes at room temperature. Freshly prepared o-phenylenediamine (OPDA) solution (5.5 mM OPDA in acetate buffer of pH 5.5) was then added. After a further 30 min incubation in the dark, fluorescence was measured at an emission wavelength of 425 nm following excitation at 345 nm and values determined after comparison to a standard curve. Intracellular AscA levels were estimated spectrophotometrically from the standard curve and the intracellular concentrations derived from the measured amount of AscA and the known macrophage cell volume [36].
2.5. RNA Isolation and Real-Time Quantitative PCR (QPCR) Analysis
Isolation of total RNA and real-time QPCR analyses were performed as described previously [37]. Briefly total RNA was extracted and purified using QIAshredders and RNeasy columns according to the manufacturer's specifications (Qiagen). Total RNA (1 μg) was reverse-transcribed into cDNA using the High Capacity cDNA Reverse Transcription kit. Complimentary DNA (cDNA) was diluted (1 : 500) and real time QPCR performed using POWER SYBR Green QPCR Master Mix. Primers were designed to anneal to sequences on separate exons or to span two exons. Primers used for QPCR are listed in Table 1. Cycling parameters were 95°C, 10 min, 40 cycles of 95°C, 15 sec, and 60°C, 1 min. A dissociation profile was generated after each run to verify specificity of amplification. All PCR assays were performed in triplicate. No template controls and no reverse transcriptase controls were included. The mRNA expression in macrophages from a “sufficient” mouse or a media well was set to “1.” The mRNA expression of all other samples was compared relative to this sample which was used as the baseline. 18S rRNA was used as housekeeping gene against which all the samples were normalized for differences in the amount of total RNA added to each cDNA reaction and for variation in the reverse transcriptase efficiency among the different cDNA reactions. Automated gene expression analysis was performed using the Comparative Quantitation module of MxPro QPCR Software (Agilent).
Table 1.
Primer sequences for real time quantitative PCR.
| Name | Sequences 5′ to 3′ |
|---|---|
| Murine IL-1β forward | CTGAACTCAACTGTGAAATGCC |
| Murine IL-1β reverse | CAGGTCAAAGGTTTGGAAGC |
|
| |
| Murine TNFα forward | GATGAGAAGTTCCCAAATGGC |
| Murine TNFα reverse | TTGGTGGTTTGCTACGACG |
|
| |
| Murine MCP-1 forward | TTCTGGGCCTGCTGTTCACAG |
| Murine MCP-1 reverse | CCAGCCTACTCATTGGGATCATCTTGC |
|
| |
| Murine YM1 forward | CAAGACTTGCGTGACTATGAAGC |
| Murine YM1 reverse | AGGTCCAAACTTCCATCCTCC |
|
| |
| Murine Arg1 forward | AGGAAAGCTGGTCTGCTGG |
| Murine Arg1 reverse | TTGAAAGGAGCTGTCATTAGGG |
|
| |
| Murine IL-10 forward | CAAGGAGCATTTGAATTCCC |
| Murine IL-10 reverse | ATTCATGGCCTTGTAGACACC |
|
| |
| Murine Gal1 forward | CAGCAACCTGAATCTCAAACC |
| Murine Gal1 reverse | AGTGTAGGCACAGGTTGTTGC |
|
| |
| Murine 15-Lox forward | TGGTGGCTGAGGTCTTTGC |
| Murine 15-Lox reverse | TCTCTGAGATCAGGTCGCTCC |
|
| |
| Human IL-6 forward | GGATTCAATGAGGAGACTTGCC |
| Human IL-6 reverse | TCTGCAGGAACTGGATCAGG |
|
| |
| Human IL-8 forward | GTGTGAAGGTGCAGTTTTGC |
| Human IL-8 reverse | GAGCTCTCTTCCATCAGAAAGC |
|
| |
| Human TNFα forward | CCTCTTCTCCTTCCTGATCG |
| Human TNFα reverse | CGAGAAGATGATCTGACTGCC |
2.6. Western Blot Analysis
Mouse macrophage and THP-I whole-cell extracts were isolated for western blot analysis as described previously [37]. Proteins were resolved by SDS polyacrylamide gel electrophoresis (4–20%) and electrophoretically transferred to polyvinylidene fluoride membranes (0.2 μm pore size). Immunodetection was performed using chemiluminescent detection with the Renaissance Western Blot Chemiluminescence Reagent Plus (Perkin Elmer Life Sciences Inc., Boston, MA). Blots were stripped using the Restore Western Blot Stripping Buffer (Pierce Biotechnology Inc., Rockford, IL) as described by the manufacturer. Purified rabbit polyclonal antibodies to phospho-NFκB p65 (Ser276, Cell Signaling), NFκB p65 (sc-109, Santa Cruz Biotechnology), iNOS (sc-650, Santa Cruz Biotechnology), and actin (sc-1616, Santa Cruz Biotechnology) were used. Optical densities of antibody-specific bands were determined using Quantity One acquisition and analysis software (Bio-Rad, Hercules, CA).
2.7. Flow Cytometry
Mouse peritoneal lavage obtained on day 3 and day 5 from VitC sufficient or deficient mice following induction of TG-induced peritonitis was pelleted by centrifugation at 4°C. Cells were resuspended in FACS buffer containing Fc receptor block (CD16/CD32 eBioscience) for 10 min at 4°C. Aliquots of the suspension were incubated at 4°C for 30 min (in the dark) with fluorescein isothiocyanate (FITC-)conjugated anti-mouse CD45 (eBioscience) and allophycocyanin (APC-)conjugated anti-mouse CD11b (eBioscience). Unstained and single color controls were employed for each experiment. Samples were then fixed with 1% formaldehyde for 20 min at room temperature. All runs were performed on a BD Accuri C6 Flow Cytometer (BD Accuri Cytometers, MI, USA) and analyzed using FlowJo software (Tree Star, Ashland, OR).
2.8. Fluorescence Microscopy
Fluorescence microscopy for evaluation of mitochondrial reactive oxygen species (ROS) in macrophages was performed using the cell-permeant probe MitoTracker Red CMXRos as described by the manufacturer. Briefly, macrophages from VitC sufficient or deficient mice were grown on Ibidi 6-channel IbiTreat μ-slide VI. Following treatments (H2O2, 18 hours) culture media were aspirated and cells were fixed in 3.7% paraformaldehyde in PBS for 10 minutes at 4°C. Fluorescence imaging was performed using an Olympus model IX70 inverted phase microscope (Olympus America, Melville, NY) outfitted with an IX-FLA fluorescence observation system equipped with a TRITC filter cube (530–560 nm excitation and 590–650 nm emission, Chroma Technology Corp. Brattleboro, VT) through an Uplan FI objective (40x). Fluorescence images were digitized and captured by a MagnaFire digital camera (Optronics, Goleta, CA).
2.9. Lipid Extraction and Analysis
Quantitative analysis of eicosanoids was performed as previously described by us with minor modifications [38–43]. Briefly, peritoneal lavage was clarified by centrifugation and 0.05% BHT and 10 ng of each internal standard added. The internal standards used were (d 4) 8-iso PGF2α, (d 11) 5-iso PGF2α-VI, (d 4) 6k PGF1α, (d 4) PGF2α, (d 4) PGE2, (d 4) PGD2, (d 4) LTB4, (d 5) Lipoxin A4, (d 5) Resolvin D2, (d 4) TXB2, (d 4) LTC4, (d 5) LTD4, (d 5) LTE4, (d 8) 5-hydroxyeicosatetranoic acid (5HETE), (d 8) 15-hydroxyeicosatetranoic acid (15HETE), (d 8) 14,15 epoxyeicosatrienoic acid, (d 8) arachidonic acid, and (d 5) eicosapentaenoic acid. The samples were mixed by vortexing and subjected to purification via solid phase extraction (SPE) using a 24 port vacuum manifold (Sigma-Aldrich). Strata-X SPE columns (Phenomenex) were washed with methanol (2 mL) and then dH2O (2 mL). The samples were applied to the column. Thereafter the sample vials were rinsed with 5% MeOH (2 mL), which was then used to wash the columns. Finally, the eicosanoids were eluted with isopropanol (2 mL). The eluents were then dried under vacuum and reconstituted in LCMS grade 50 : 50 EtOH : dH2O (100 μL) for eicosanoid quantitation via UPLC ESI-MS/MS analysis. A 14-minute reversed-phase LC method utilizing a Kinetex C18 column (100 × 2.1 mm, 1.7 μm) and a Shimadzu UPLC was used to separate the eicosanoids at a flow rate of 500 μL/min at 50°C. The column was first equilibrated with 100% Solvent A (acetonitrile : water : formic acid (20 : 80 : 0.02, v/v/v)) for two minutes and then 10 μL of sample was injected. 100% Solvent A was used for the first two minutes of elution. Solvent B (acetonitrile : isopropanol (20 : 80, v/v)) was increased in a linear gradient to 25% Solvent B to 3 minutes, to 30% by 6 minutes, to 55% by 6.1 minutes, to 70% by 10 minutes, and to 100% by 10.1 minutes. 100% Solvent B was held until 13 minutes and then was decreased to 0% by 13.1 minutes and held at 0% until 14 minutes. The eluting eicosanoids were analyzed using a hybrid triple quadrapole linear ion trap mass analyzer (ABSciex 6500 QTRAP) via multiple-reaction monitoring in negative-ion mode. Eicosanoids were monitored using species specific precursor → product MRM pairs. The mass spectrometer parameters used were curtain gas: 30; CAD: High; ion spray voltage: −3500 V; temperature: 300°C; Gas 1: 40; and Gas 2: 60; declustering potential, collision energy, and cell exit potential were optimized per transition.
2.10. Statistical Analysis
Statistical analysis was performed using SAS 9.3 and GraphPad Prism 6.0 (GraphPad Software, San Diego, CA, USA). Data are expressed as mean ± SE. Results were compared using one-way ANOVA and the post hoc Tukey test. Statistical significance was confirmed at a P value of <0.05.
3. Results
3.1. VitC Deficiency Alters the Progression of TG-Induced Peritoneal Inflammation
In order to make the Gulo−/− mice VitC deficient, supplementation of water with AscA was withdrawn as described in the Methods section. Within 3 weeks of removal of VitC supplementation, plasma VitC levels of Gulo−/− mice declined significantly (Figure 1). This decline was not associated with deleterious changes in weight or health status in the VitC deficient mice (data not shown). To determine whether VitC deficiency impacts the progression of peritoneal inflammation, VitC sufficient or deficient mice were injected with TG and the progression of inflammation was monitored on days 3 and 5 (as described in Section 2). Some VitC deficient mice were injected i.p. with AscA (200 mg/kg) prior to harvest of peritoneal lavage (see Section 2). Daily i.p. administration of ascorbate for 3 days restored circulating plasma VitC concentrations in these mice to levels observed in the VitC sufficient mice (Figure 1). In all 3 groups, the infiltration of inflammatory cells on day 1 was similar to that observed in wild type mice [44] and was in agreement with our previous observations (Table 2) [28]. As seen in Table 2, there was also no difference in the total number of cells elicited from the peritoneal exudation of day 3 and day 5. However, significant differences in the cellular composition of the lavage were evident on day 3 and day 5 between the 3 groups. In the VitC sufficient mice group, mononuclear cells were the predominant cell type on days 3 and 5 (Table 2). PMN numbers, which peaked on day 1 [28], returned to baseline by days 3 and 5. In contrast, significantly elevated numbers of PMNs persisted in the peritoneal exudates of VitC deficient mice on days 3 and 5 (Table 2). Infusion of AscA reduced PMN numbers by day 3 with a significant decline in PMN numbers to baseline similar to the VitC sufficient mice by day 5 Table 2.
Figure 1.
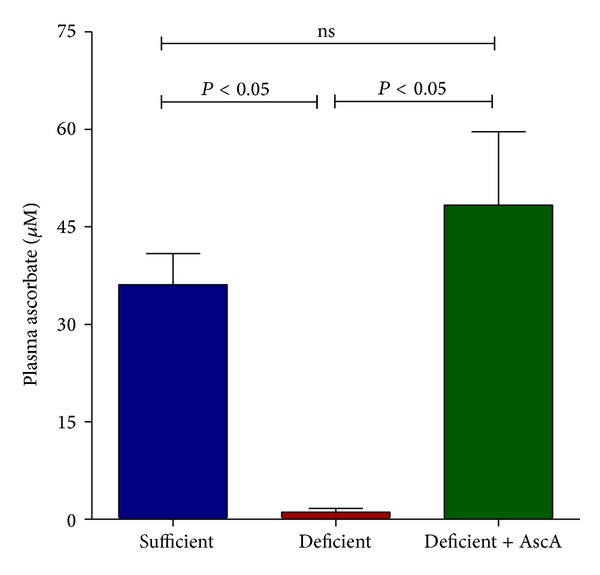
Vitamin C deficiency alters the progression of TG-induced peritoneal inflammation. Plasma VitC levels were measured in VitC sufficient and deficient Gulo−/− mice as well as in deficient mice treated daily with i.p. AscA for 3 days (N = 3–6 mice/group, ns = not significant).
Table 2.
Differential cell counts from peritoneal exudates following thioglycollate-induced peritonitis (N = 6–8 mice/group, n.d. = not determined).
| Sufficient | Deficient | Deficient + AscA | ||
|---|---|---|---|---|
| Day 1 | PMN (×106) | 20.02 ± 3.4 | 23.2 ± 2.6 | n.d. |
| M0 (×106) | 5.0 ± 0.8 | 3.9 ± 0.6 | n.d. | |
|
| ||||
| Day 3 | PMN (×106) | 1.8 ± 0.4 | 8.1 ± 1.9a | 3.8 ± 0.4 |
| M0 (×106) | 18.4 ± 3.2 | 13.1 ± 3.1 | 14.5 ± 3.2 | |
|
| ||||
| Day 5 | PMN (×106) | 1.0 ± 0.6 | 4.4 ± 1.1b | 0.4 ± 0.2c |
| M0 (×106) | 18.8 ± 5.6 | 22.1 ± 4.2 | 14.3 ± 4.1 | |
aSufficient versus deficient, P = 0.006.
bSufficient versus deficient, P = 0.02.
cDeficient versus deficient + AscA, P = 0.02.
3.2. Spatiotemporal Profiling of Inflammatory Mediators following TG-Induced Peritoneal Inflammation
We previously observed that TG-elicited PMN from VitC deficient mice (on day 1) demonstrated increased expression of the proinflammatory genes TNFα and IL-1β [28]. Here we examined the expression of multiple pro- and anti-inflammatory mediators originating from macrophages, the predominant cell type recruited to the inflamed peritoneum on days 3 and 5. As seen in Figure 2, significant differences were evident in the magnitude of pro- and anti-inflammatory mediator expression on days 3 and 5. On day 3, increased expression of the proinflammatory mediators (IL-1β, TNFα, and MCP-1) was observed in macrophages from VitC deficient mice when compared to macrophages from VitC sufficient mice (Figure 2(a), (A), (C), and (E)). Proinflammatory gene expression was significantly attenuated by i.p. infusion of AscA in the VitC deficient mice (Figure 2(a), (A), (C), and (E)). In contrast, anti-inflammatory gene expression (YM1 and Arg1, but not IL-10) was elevated in macrophages from VitC sufficient mice (Figure 2(a), (B), (D), and (F)). Daily AscA infusion induced YM1 expression in VitC deficient macrophages but failed to restore Arg1 expression. IL-10 expression on the other hand was significantly lowered by AscA infusion on day 3 (Figure 2(a), (B), (D), and (F)).
Figure 2.
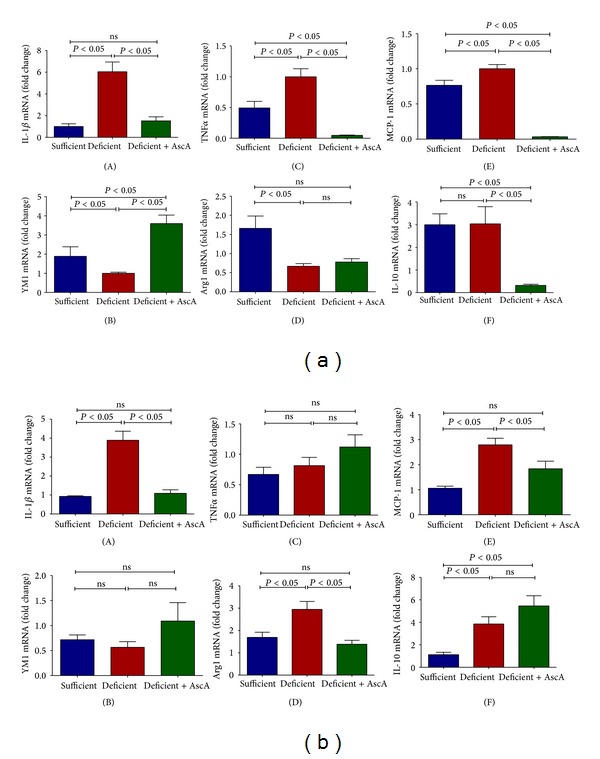
Spatiotemporal profiling of inflammatory mediators following TG-induced peritoneal inflammation. Real time QPCR for IL-1β, TNFα, MCP-1, YM1, Arg1, and IL-10 mRNA from peritoneal macrophages elicited on day 3 (a) and day 5 (b) following TG-induced peritonitis from VitC sufficient and deficient Gulo−/− mice. Following TG challenge, some VitC deficient mice were randomized to receive daily i.p. injection of VitC as AscA (200 mg/kg in saline) for a further 3 days (day 3, deficient + AscA) or 5 days (day 5, deficient + AscA) (N = 6 mice/group, ns = not significant).
On day 5 (Figure 2(b)), proinflammatory gene expression remained persistently elevated in macrophages from VitC deficient mice (IL-1β and MCP-1) but was attenuated by AscA infusion. In contrast, anti-inflammatory gene expression in VitC deficient macrophages was significantly higher when compared to macrophages from VitC sufficient mice (Arg1, IL-10). AscA infusion did not alter anti-inflammatory gene expression on day 5 although Arg1 levels were now similar to that observed in VitC sufficient mice (Figure 2(b), (B), (D), and (F)).
3.3. Ex Vivo Bacterial Lipopolysaccharide Differentially Activate Proinflammatory Gene Expression in Macrophages from VitC Sufficient and Deficient Mice
Canali et al. recently showed that in contrast with baseline physiological activation, exposure to a second “hit” such as an inflammatory stimulus results in a markedly different modulation of gene expression in human peripheral blood mononuclear cells in the presence or absence of VitC supplementation [45]. To examine whether peritoneal macrophages would exhibit an altered modulation of gene expression, we exposed day 3 peritoneal macrophages from VitC sufficient and deficient mice to bacterial lipopolysaccharide (LPS, 50 ng/mL). Some macrophages were incubated with AscA (3 mM, 16 hours) prior to LPS exposure. As seen in Figures 3(a) and 3(b), LPS exposure resulted in a robust increase in expression of proinflammatory markers (IL-1β, TNFα) in macrophages from VitC sufficient mice. Proinflammatory gene expression was also induced in macrophages from VitC deficient mice, but the magnitude of induction was significantly greater than that observed in the VitC sufficient macrophages (Figures 3(a) and 3(b)). Importantly, exposure of VitC deficient macrophages to AscA prior to LPS significantly attenuated IL-1β and TNFα expression. Increased NFκB activation (Figure 3(c)) and iNOS protein expression (Figure 3(d)) was observed upon exposure of VitC deficient macrophages to LPS (P < 0.05). AscA pretreatment attenuated NFκB activation and iNOS expression in VitC deficient macrophages.
Figure 3.
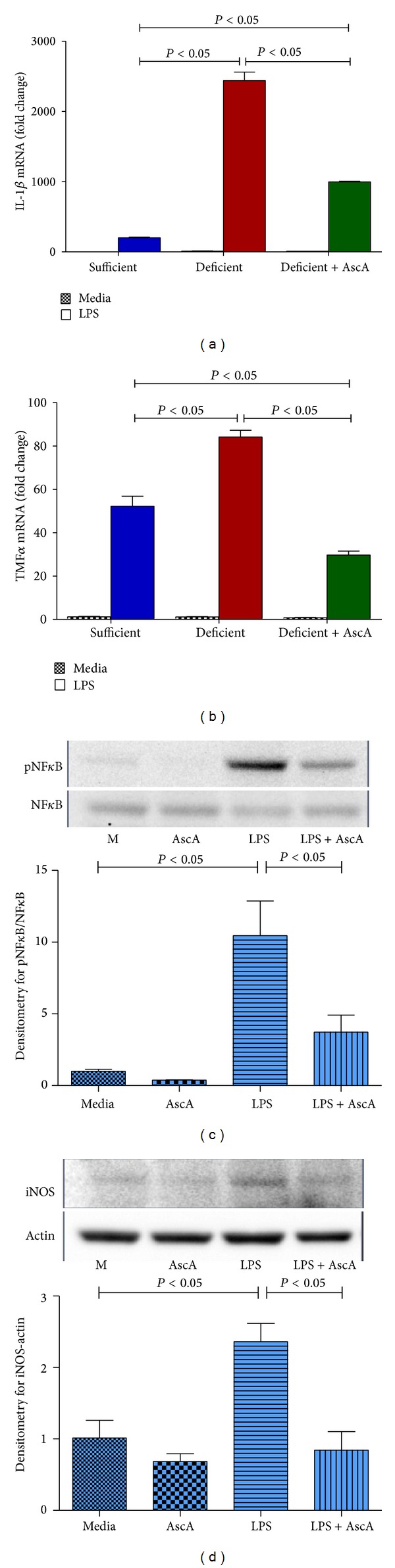
LPS differentially activates proinflammatory gene expression in macrophages from vitamin C sufficient and deficient mice. Peritoneal macrophages elicited on day 3 following TG-induced peritonitis from VitC sufficient and deficient Gulo−/− mice were exposed to LPS (50 ng/mL) for 4 hours. Macrophages from some VitC deficient mice were incubated with AscA (3 mM, 16 hours) prior to LPS exposure (deficient + AscA). Real time QPCR was performed for IL-1β (a) and TNFα (b) (N = 6/group). (c) Upper panel: representative western blot for expression of phospho-NFκB and NFκB from VitC deficient macrophages exposed to media alone (M), AscA (3 mM, 16 hours (AscA)), LPS (50 ng/mL) for 1 hour (LPS), or AscA for 16 hours followed by LPS for 1 hour (LPS + AscA). Lower panel: densitometry for normalized expression of phospho-NFκB from macrophages (N = 3/group). (d) Upper panel: representative western blot for expression of iNOS and actin from macrophages groups described in (c) and exposure to LPS (50 ng/mL) for 4 hour. Lower panel: densitometry for normalized expression of iNOS from macrophages (N = 3/group).
3.4. VitC Regulates Macrophage Function during the Resolution of Inflammation
Macrophages undergo reprogramming to adopt a variety of functional phenotypes upon receiving differentiation cues from their surrounding environment [46]. It was recently shown that macrophage reprogramming is vital for resolution of acute inflammation [47]. We examined whether macrophage VitC sufficiency or deficiency could influence macrophage function during resolution of acute inflammation. Macrophages were isolated on day 3 following TG-mediated peritonitis from VitC sufficient or deficient mice and intracellular concentrations of VitC measured (as described in Section 2). Some VitC deficient mice were injected daily with i.p. AscA (200 mg/kg) prior to harvest of peritoneal lavage (see Section 2). As seen in Figure 4(a), macrophages from VitC sufficient mice have high intracellular VitC concentrations. In contrast, intracellular ascorbate levels were significantly depleted in macrophages from VitC deficient mice. Daily i.p. administration of AscA for 3 days also restored macrophage intracellular concentrations to levels observed in the VitC sufficient mice (Figure 4(a)).
Figure 4.
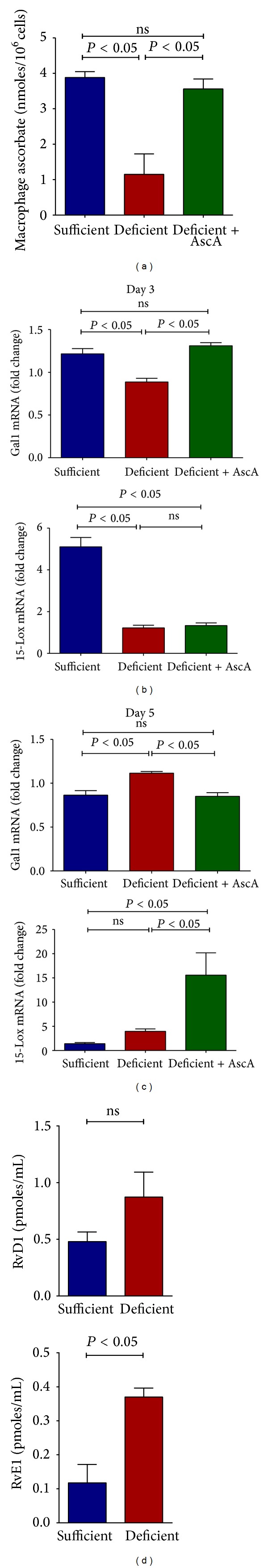
Vitamin C regulates macrophage function during the resolution of inflammation. (a) Macrophages were isolated on day 3 following TG-mediated peritonitis from VitC sufficient or deficient mice as well as in deficient mice treated daily with i.p. AscA for 3 days and intracellular concentrations of VitC measured (N = 3–10 mice/group, ns = not significant). (b) Real time QPCR for Gal1 and 15-Lox from peritoneal macrophages elicited on day 3 following TG-induced peritonitis from VitC sufficient and deficient Gulo−/− mice. Thirty minutes following TG challenge, some VitC deficient mice were randomized to receive i.p. injection of VitC as AscA (200 mg/kg in saline) for a further 3 days (deficient + AscA). (N = 6 mice/group, ns = not significant). (c) Real time QPCR for Gal1 and 15-Lox from peritoneal macrophages elicited on day 5 following TG-induced peritonitis from VitC sufficient and deficient Gulo−/− mice. Thirty minutes following TG challenge, some VitC deficient mice were randomized to receive i.p. injection of VitC as AscA (200 mg/kg in saline) for a further 5 days (Deficient + AscA) (N = 6 mice/group, ns = not significant). (d) UPLC ESI-MS/MS quantification of resolvin D1 (RvD1) and E1 (RvE1) in peritoneal lavage on day 5 following TG-induced peritonitis from VitC sufficient and deficient Gulo−/− mice (N = 3-4 mice/group, ns = not significant).
Gal-1 and 15-Lox expression is induced in macrophages during peritonitis. Their expression is associated with generation of proresolving lipid mediators [11] and successful resolution of inflammation [48]. Therefore we examined Gal-1 and 15-Lox expression in day 3 and day 5 macrophages from VitC sufficient or deficient mice. As seen in Figure 4(b), Gal-1 and 15-Lox expression was significantly induced in macrophages from VitC sufficient mice on day 3 when compared to macrophages from VitC deficient mice. AscA infusion restored Gal-1 expression in VitC deficient macrophages but did not affect 15-Lox expression on day 3 (Figure 4(b)). In contrast, Gal-1 expression in VitC deficient macrophages was delayed and observed to be higher on day 5 following TG-induced peritonitis (Figure 4(c)). 15-Lox expression was induced by AscA infusion on day 5 and was higher than that observed in macrophages from VitC sufficient or deficient mice. In agreement with the expression data seen above, resolvin (Figure 4(d)) production was higher on day 5 in VitC deficient mice indicating delayed resolution of inflammation.
3.5. VitC Influences Macrophage Phenotype during Resolution of Inflammation
Rostoker et al. recently showed that Gal-1 was selectively expressed in CD11bhigh macrophages, and its expression declined significantly once these cells converted toward a CD11blow phenotype [49]. Moreover, CD11blow macrophages are the predominant subtype to depart the peritoneum [49]. To determine whether VitC regulated reprogramming of peritoneal macrophages to proresolution CD11blow phenotype we used flow cytometry to examine the distribution of CD11bhigh and CD11blow population on macrophages isolated on day 3 and day 5 following TG-induced peritonitis in VitC sufficient or deficient mice. As seen in Figure 5, there was a significant transition from CD11bhigh to a CD11blow phenotype observed from day 3 to day 5 in the VitC sufficient macrophages. This was not evident in the macrophages from VitC deficient mice indicative of a delay in the resolution of TG-induced peritonitis in these mice.
Figure 5.
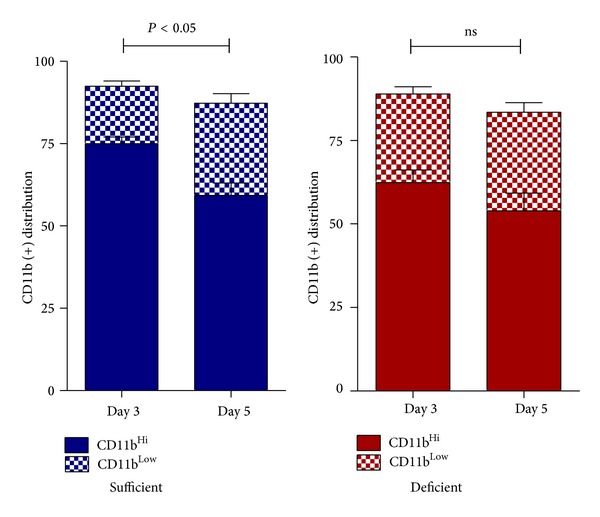
Vitamin C influences macrophage phenotype during resolution of inflammation. Flow cytometry for distribution of CD11bhigh and CD11blow population from macrophages isolated on day 3 and day 5 following TG-induced peritonitis in VitC sufficient or deficient mice (N = 5 mice/group, P < 0.05, CD11blow day 3 versus day 5, ns = not significant).
3.6. Macrophages Deficient in VitC Have Reduced Antioxidant Capacity
Activated macrophages potentially generate mitochondria-damaging deleterious reactive oxygen species (ROS). Release of large amounts of ROS during activation exposes macrophages themselves to oxidant stresses not encountered by most other cell types [50]. To test whether VitC deficiency affected mitochondrial function in macrophages, we exposed peritoneal macrophages (day 3) from VitC sufficient or deficient mice to varying concentrations of H2O2 for 18 hours and stained the cells with MitoTracker Red CMXRos as described in Section 2. This probe is selectively retained by mitochondria, where it is oxidized to its fluorescent form. As seen in Figures 6(a) and 6(e), control macrophages from VitC sufficient or deficient mice were stained brightly with the probe. Oxidative stress from exposure to H2O2 decreased fluorescent staining in macrophages from both VitC sufficient and VitC deficient mice. However, the magnitude of decrease was significantly greater in macrophages from VitC deficient mice (Figures 6(f)–6(h)). This decrease was partially reversed by pretreatment of VitC deficient macrophages with AscA (Figures 6(j) and 6(k)). These studies indicate that VitC deficient macrophages sustain greater mitochondrial dysfunction when challenged with ROS.
Figure 6.

Macrophages deficient in Vitamin C have reduced antioxidant capacity. Peritoneal macrophages elicited on day 3 following TG-induced peritonitis from VitC sufficient ((a)–(d)) and deficient Gulo−/− ((e)–(h)) mice were exposed to 12.5, 25, and 50 μM H2O2 for 18 hours and probed with MitoTracker Red CMXRos. Macrophages from some VitC deficient mice were incubated with AscA (3 mM, 16 hours) prior to exposure to H2O2 followed by staining with MitoTracker Red CMXRos (Deficient + AscA, ((i)–(l))).
3.7. VitC Attenuates Proinflammatory Gene Expression in Human Monocyte/Macrophages
To address whether the modulatory activities of VitC are effective in human monocyte/macrophages, we exposed THP-1 cells to bacterial LPS and examined the mRNA expression of the proinflammatory genes IL-6, IL-8, and TNFα. Since the culture medium in which THP-1 cells are grown contains no VitC, we increased intracellular concentrations of VitC by loading cells with AscA prior exposure to LPS. As seen in Figure 7(a), exposure of THP-1 cells to LPS resulted in a robust activation of mRNA for IL-6, IL-8, and TNFα. Loading cells with AscA did not affect baseline proinflammatory gene expression. However LPS exposure of AscA loaded cells resulted in significant attenuation of mRNA expression of these proinflammatory genes. Attenuation of mRNA expression was likely achieved by reduction in activation of the transcription factor NFκB following LPS exposure (Figure 7(b)).
Figure 7.
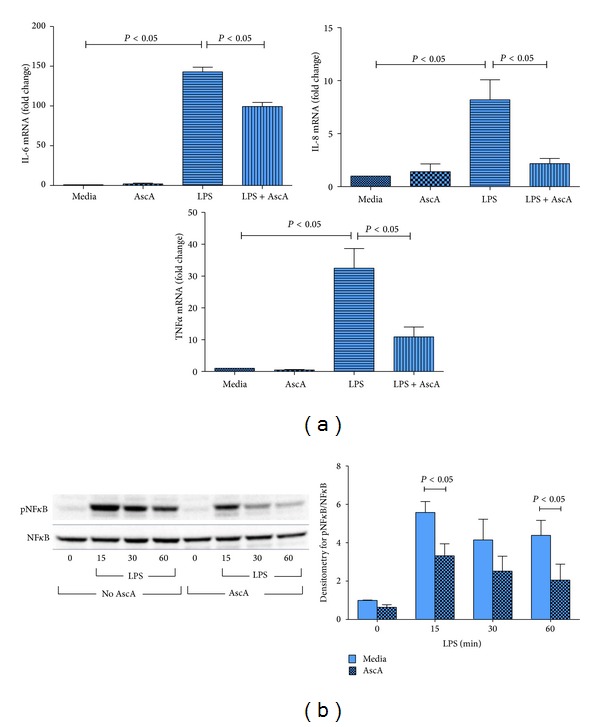
Vitamin C attenuates proinflammatory gene expression in human THP-1 monocyte/macrophages. THP-1 macrophages were exposed to media alone (Media), AscA (3 mM, 16 hours (AscA)), and LPS (50 ng/mL) for 4 hour (LPS) or AscA for 16 hours followed by LPS for 4 hour (LPS + AscA). (a) Real time QPCR for IL-6, IL-8, and TNFα was performed as described in Section 2 (N = 4/group; P < 0.05, Media versus LPS and LPS versus LPS + AscA). (b) Left panel: representative western blot for expression of phospho-NFκB and NFκB from THP-1 groups described above following exposure to LPS (50 ng/mL) for 0, 15, 30, and 60 minutes. Right panel: densitometry for normalized expression of phospho-NFκB from THP-1 (N = 4/group, P < 0.05, LPS versus LPS + AscA).
4. Discussion
In this study, we examined the mechanism by which VitC regulates the resolution of sterile inflammation. Using mice lacking the ability to synthesize VitC, we show that subnormal cellular VitC levels negatively impact the progression and resolution of sterile inflammation. In particular, our results demonstrate that low circulating VitC levels are associated with significant delays in the timing of resolution of inflammation. This apparent VitC-dependent process primarily occurs due to failure of macrophages to transition from a proinflammatory to a proresolving phenotype.
The initial response to sterile inflammation was identical in VitC sufficientanddeficient mice. During the early proinflammatory phase no differences in the cell numbers or cell types were observed. However, by days 3 and 5, VitC deficient mice exhibited significant numbers of PMN in peritoneal exudates (Table 2). Spatiotemporal mRNA profiling of macrophage-derived inflammatory mediators revealed dramatic differences in the magnitude of pro- and anti-inflammatory mediator gene expression (Figure 2). Macrophages from VitC sufficient mice displayed prominent anti-inflammatory phenotypes, while VitC deficient macrophages persistently expressed mRNA for IL-1β, TNFα, and MCP-1, findings characteristic of a proinflammatory phenotype. LPS activation of day 3 macrophages from VitC deficient mice led to proinflammatory gene expression that was significantly greater in magnitude than that observed in VitC loaded macrophages (Figure 3). LPS stimulation was characterized by enhanced NFκB activation and iNOS induction in VitC deficient macrophages (Figure 3). Importantly, on day 3, VitC sufficient macrophages demonstrated cues for reprogramming into resolution type macrophages, a vital step required for resolution of inflammation. In day 3 macrophages from VitC sufficient mice, expression of Gal-1 and 15-Lox mRNA was robust (Figure 4). In contradistinction, enhanced Gal-1 and 15-Lox mRNA expression was delayed to day 5 in VitC deficient macrophages. The delays in resolution we observed in VitC deficient mice were confirmed by quantification of resolvins in peritoneal exudates; increases of which were present only on day 5 (Figure 4). Further confirmation of altered spatiotemporal relationships was achieved by studying macrophage phenotypic changes by examining the distribution of CD11b on macrophages from VitC sufficient or deficient mice on day 3 and day 5 following TG-induced peritonitis (Figure 5). Phenotypic changes in macrophages were accompanied by alterations in macrophage function as demonstrated by the increased susceptibility of VitC deficient macrophages to mitochondrial dysfunction when exposed to reactive oxygen species (Figure 6). In final studies, we employed the human monocyte/macrophage cell line THP-1, which lacks VitC in culture medium when cultured under standard conditions. We demonstrated increased proinflammatory gene expression in THP-1 when exposed to LPS under VitC-deprived conditions. Loading THP-1 cells with AscA significantly attenuated mRNA expression of proinflammatory genes via a mechanism likely involving reduced activation of the transcription factor NFκB. VitC loading was effective both in vitro and in vivo since daily AscA infusion following induction of peritonitis significantly restored macrophage phenotype and function in the VitC deficient mice.
Few studies have examined the role of VitC in resolution of sterile inflammation. Ganguly et al. initially reported that VitC deficiency affected migration of guinea pig macrophages under in vitro conditions [51]. They further showed that addition of exogenous VitC partially restored the migratory response. May et al. showed that activated macrophages use ascorbate to lessen self-generated oxidant stress [18]. They later showed that ascorbate deficient peritoneal macrophages were more susceptible to H2O2-induced mitochondrial dysfunction and apoptosis [52]. However no studies to date have examined macrophage function during resolution of inflammation in mice lacking the ability to synthesize their own VitC. Our observation of persistence of PMN at the site of inflammation in VitC deficient mice is in agreement with our previous results [28] and those of Vissers and Wilkie who used a similar TG model of peritonitis to show impairment in PMN apoptosis and clearance [19]. It has been suggested that the engulfment of apoptotic cells is generally anti-inflammatory or immunologically silent [53] due to the fact that it sequesters dying cells thus preventing release of potentially toxic cell contents into the local environment. Based on the observations that PMN persists for up to 5 days in the peritoneum of VitC deficient mice (Table 2), it is therefore possible that the apoptosis-resistant PMN can cause strong proinflammatory responses from the macrophages that extravasate to sites of inflammation. Indeed strong and persistent proinflammatory responses were evident in VitC deficient macrophages elicited on day 3 and even day 5 (Figure 2).
Efferocytosis, a process by which dead and/or dying cells are being engulfed and removed by other cells, has been reported to induce production of anti-inflammatory mediators from macrophages that suppress inflammation thereby silently clearing apoptotic cells and thus dampening proinflammatory responses [54]. VitC sufficient mice exhibited anti-inflammatory mediator expression in macrophages early (day 3) in the post TG-induced inflammatory process, a phenomenon indicative of functional efferocytosis. In contrast, VitC deficient macrophages failed to upregulate anti-inflammatory mediator production until day 5 (Figure 2).
Gal-1 and 12/15-Lox play vital roles in resolution of inflammation. Rostoker et al. have shown that Gal-1 promotes the generation of M2-like macrophages, which then favors tissue repair during early resolution of inflammation [49]. Ariel and Timor demonstrated that Gal-1 promotes generation of Mres from M2 macrophages, which generates proresolving lipid mediators. This phenotype change promotes macrophage departure from peritoneal cavities with resolving inflammation, thus allowing return of tissue to homeostasis [55]. Moreover, Gal-1 expression, which is enhanced in CD11bhigh macrophages, declines sharply as cells revert to the CD11blow phenotype. CD11blow macrophage phenotypes, as noted previously, promote departure from peritoneal cavities with resolving inflammation [49]. Our findings (Figures 4 and 5) which agree with the above studies implicate VitC as a critical regulator of macrophage transition during resolution of inflammation. Expression and function of 12/15-Lox produce key mediators (e.g., lipoxins, resolvins, protectins, and maresins) that promote resolution of proinflammatory pathologies [56]. In particular, human and murine monocytes/macrophages expression of 15-Lox is upregulated by efferocytosis with production of mediators such as RvD1, a mediator shown to promote the resolution of murine peritonitis [47, 57]. Further, Gal-1 directly promotes 15-lipoxygenase expression and activity in macrophages during the inflammatory and resolving phases of peritonitis [49]. The earlier increases in Gal-1 and 15-Lox mRNA expression in VitC sufficient macrophages (Figures 4(b) and 4(c)) and the delayed resolvin production in the VitC deficient macrophages (Figure 4(d)) indicate for the first time that VitC influences multiple processes leading to the resolution of inflammation.
5. Conclusions
The findings in this mouse model have significant human relevance since VitC levels are subnormal in multiple human inflammatory disease states including sepsis, systemic inflammatory response syndrome (SIRS), trauma, and cancer, among others. In a recently completed Phase I trial (ClinicalTrials.gov identifier NCT01434121) of intravenous AscA in critically ill patients with severe sepsis, we showed that septic patients exhibited abnormally low VitC plasma levels and that intravenous AscA infusion could significantly increase circulating VitC levels [29]. Further, AscA infusion significantly reduced the proinflammatory biomarkers C-reactive protein and procalcitonin as well as multiple organ dysfunction [29]. Our findings here add a previously unrecognized element to our understanding of the machinery that governs the resolution of inflammation.
Acknowledgments
This research was supported by grants from The Aubery Sage Mac Farlane Endowment for Acute Lung Injury Research, The AD Williams Research Foundation (NR), The Jeffress Memorial Trust (NR), and The Victoria Johnson Center for Critical Care and Pulmonary Research. This work was also supported by research Grants from the Veteran's Administration (VA Merit Award BX001792 to C.E.C. and a Research Career Scientist Award 13F-RCS-002 to C.E.C.); from the National Institutes of Health via HL072925 (C.E.C.); and from the US-Israel Binational Science Foundation via BSF#2011380 (C.E.C), NH1C06-RR17393 (to Virginia Commonwealth University for renovation), NIH/NCI Cancer Center Support Grant P30 CA016059 (to Massey Cancer Center), a National Research Service Award-T32 Post-Doctoral Fellowship in Wound Healing GM008695 (DSW), and a Career Development Award (CDA1) from the Department of Veterans Affairs (DSW).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Laurin L-P, Brissette M-J, Lepage S, Cailhier JF. Regulation of experimental peritonitis: a complex orchestration. Nephron Experimental Nephrology. 2012;120(1):e41–e46. doi: 10.1159/000334169. [DOI] [PubMed] [Google Scholar]
- 2.McGrath EE, Marriott HM, Lawrie A, et al. TNF-related apoptosis-inducing ligand (TRAIL) regulates inflammatory neutrophil apoptosis and enhances resolution of inflammation. Journal of Leukocyte Biology. 2011;90(5):855–865. doi: 10.1189/jlb.0211062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. Journal of Cell Biology. 2010;189(7):1059–1070. doi: 10.1083/jcb.201004096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends in Immunology. 2002;23(11):549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 5.Fleming BD, Mosser DM. Regulatory macrophages: setting the threshold for therapy. European Journal of Immunology. 2011;41(9):2498–2502. doi: 10.1002/eji.201141717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Frontiers in Bioscience. 2008;13(2):453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 7.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 8.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nature Reviews Immunology. 2011;11(11):723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A. IL-10 inhibits cytokine production by activated macrophages. The Journal of Immunology. 1991;147(11):3815–3822. [PubMed] [Google Scholar]
- 10.Bannenberg GL, Chiang N, Ariel A, et al. Molecular circuits of resolution: formation and actions of resolvins and protectins. Journal of Immunology. 2005;174(7):4345–4355. doi: 10.4049/jimmunol.174.7.4345. [DOI] [PubMed] [Google Scholar]
- 11.Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. 2007;447(7146):869–874. doi: 10.1038/nature05877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serhan CN. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukotrienes and Essential Fatty Acids. 2005;73(3-4):141–162. doi: 10.1016/j.plefa.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 13.Serhan CN, Gotlinger K, Hong S, et al. Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: assignments of dihydroxy-containing docosatrienes. Journal of Immunology. 2006;176(3):1848–1859. doi: 10.4049/jimmunol.176.3.1848. [DOI] [PubMed] [Google Scholar]
- 14.Serhan CN, Hong S, Gronert K, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. Journal of Experimental Medicine. 2002;196(8):1025–1037. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russell CD, Schwarze J. The role of pro-resolution lipid mediators in infectious disease. Immunology. 2014;141(2):166–173. doi: 10.1111/imm.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bail SS, Weindruch R, Walford RL. Free radicals, ageing and degenerative diseases. In: Johnson JE Jr., Walford IL, Harman D, Liss MJ, editors. Antioxidants and the Immune Response. New York, NY, USA: Alan R. Liss; 1996. pp. 427–756. [Google Scholar]
- 17.Bergsten P, Amitai G, Kehrl J, Dhariwal KR, Klein HG, Levine M. Millimolar concentrations of ascorbic acid in purified human mononuclear leukocytes. Depletion and reaccumulation. The Journal of Biological Chemistry. 1990;265(5):2584–2587. [PubMed] [Google Scholar]
- 18.May JM, Li L, Qu Z-C, Huang J. Ascorbate uptake and antioxidant function in peritoneal macrophages. Archives of Biochemistry and Biophysics. 2005;440(2):165–172. doi: 10.1016/j.abb.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 19.Vissers MC, Wilkie RP. Ascorbate deficiency results in impaired neutrophil apoptosis and clearance and is associated with up-regulation of hypoxia-inducible factor 1α . Journal of Leukocyte Biology. 2007;81(5):1236–1244. doi: 10.1189/jlb.0806541. [DOI] [PubMed] [Google Scholar]
- 20.Oberritter H, Glatthaar B, Moser U, Schmidt KH. Effect of functional stimulation on ascorbate content in phagocytes under physiological and pathological conditions. International Archives of Allergy and Applied Immunology. 1986;81(1):46–50. doi: 10.1159/000234106. [DOI] [PubMed] [Google Scholar]
- 21.Li W, Maeda N, Beck MA. Vitamin C deficiency increases the lung pathology of influenza virus-infected gulo-/- mice. Journal of Nutrition. 2006;136(10):2611–2616. doi: 10.1093/jn/136.10.2611. [DOI] [PubMed] [Google Scholar]
- 22.Gaut JP, Belaaouaj A, Byun J, et al. Vitamin C fails to protect amino acids and lipids from oxidation during acute inflammation. Free Radical Biology and Medicine. 2006;40(9):1494–1501. doi: 10.1016/j.freeradbiomed.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 23.Long CL, Maull KI, Krishnan RS, et al. Ascorbic acid dynamics in the seriously ill and injured. Journal of Surgical Research. 2003;109(2):144–148. doi: 10.1016/s0022-4804(02)00083-5. [DOI] [PubMed] [Google Scholar]
- 24.Schorah CJ, Downing C, Piripitsi A, et al. Total vitamin C, ascorbic acid, and dehydroascorbic acid concentrations in plasma of critically ill patients. The American Journal of Clinical Nutrition. 1996;63(5):760–765. doi: 10.1093/ajcn/63.5.760. [DOI] [PubMed] [Google Scholar]
- 25.Borrelli E, Roux-Lombard P, Grau GE, et al. Plasma concentrations of cytokines, their soluble receptors, and antioxidant vitamins can predict the development of multiple organ failure in patients at risk. Critical Care Medicine. 1996;24(3):392–397. doi: 10.1097/00003246-199603000-00006. [DOI] [PubMed] [Google Scholar]
- 26.Galley HF, Davies MJ, Webster NR. Ascorbyl radical formation in patients with sepsis: effect of ascorbate loading. Free Radical Biology and Medicine. 1996;20(1):139–143. doi: 10.1016/0891-5849(95)02022-5. [DOI] [PubMed] [Google Scholar]
- 27.Fisher BJ, Kraskauskas D, Martin EJ, et al. Attenuation of sepsis-induced organ injury in mice by vitamin C. Journal of Parenteral and Enteral Nutrition. 2013 doi: 10.1177/0148607113497760. [DOI] [PubMed] [Google Scholar]
- 28.Mohammed BM, Fisher BJ, Kraskauskas D, et al. Vitamin C: a novel regulator of neutrophil extracellular trap formation. Nutrients. 2013;5(8):3131–3151. doi: 10.3390/nu5083131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fowler AA, III, Syed AA, Knowlson S, et al. Phase I safety trial of intravenous ascorbic acid in patients with severe sepsis. Journal of Translational Medicine. 2014;12, article 32 doi: 10.1186/1479-5876-12-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishikimi M, Fukuyama R, Minoshima S, Shimizu N, Yagi K. Cloning and chromosomal mapping of the human nonfunctional gene for L- gulono-γ-lactone oxidase, the enzyme for L-ascorbic acid biosynthesis missing in man. The Journal of Biological Chemistry. 1994;269(18):13685–13688. [PubMed] [Google Scholar]
- 31.Kim H, Bae S, Yu Y, et al. The analysis of vitamin C concentration in organs of gulo−/− mice upon vitamin C withdrawal. Immune Network. 2012;12(1):18–26. doi: 10.4110/in.2012.12.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vissers MCM, Bozonet SM, Pearson JF, Braithwaite LJ. Dietary ascorbate intake affects steady state tissue concentrations in vitamin C-deficient mice: tissue deficiency after suboptimal intake and superior bioavailability from a food source (kiwifruit) American Journal of Clinical Nutrition. 2011;93(2):292–301. doi: 10.3945/ajcn.110.004853. [DOI] [PubMed] [Google Scholar]
- 33.Tsurubuchi T, Aratani Y, Maeda N, Koyama H. Retardation of early-onset PMA-induced apoptosis in mouse neutrophils deficient in myeloperoxidase. Journal of Leukocyte Biology. 2001;70(1):52–58. [PubMed] [Google Scholar]
- 34.Ghosh S. Cholesteryl ester hydrolase in human monocyte/macrophage: cloning, sequencing, and expression of full-length cDNA. Physiol Genomics. 2000;2(1):1–8. doi: 10.1152/physiolgenomics.2000.2.1.1. [DOI] [PubMed] [Google Scholar]
- 35.Vislisel JM, Schafer FQ, Buettner GR. A simple and sensitive assay for ascorbate using a plate reader. Analytical Biochemistry. 2007;365(1):31–39. doi: 10.1016/j.ab.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Melmed RN, Karanian PJ, Berlin RD. Control of cell volume in the J774 macrophage by microtubule disassembly and cyclic AMP. Journal of Cell Biology. 1981;90(3):761–768. doi: 10.1083/jcb.90.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fisher BJ, Kraskauskas D, Martin EJ, et al. Mechanisms of attenuation of abdominal sepsis induced acute lung injury by ascorbic acid. American Journal of Physiology: Lung Cellular and Molecular Physiology. 2012;303(1):L20–L32. doi: 10.1152/ajplung.00300.2011. [DOI] [PubMed] [Google Scholar]
- 38.Wijesinghe DS, Brentnall M, Mietla JA, et al. Ceramide kinase is required for a normal eicosanoid response and the subsequent orderly migration of fibroblasts. The Journal of Lipid Research. 2014;55(7):1298–1309. doi: 10.1194/jlr.M048207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wijesinghe DS, Chalfant CE. Systems-level lipid analysis methodologies for qualitative and quantitative investigation of lipid signaling events during wound healing. Advances in Wound Care. 2013;2(9):538–548. doi: 10.1089/wound.2012.0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simanshu DK, Kamlekar RK, Wijesinghe DS, et al. Non-vesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoids. Nature. 2013;500(7463):463–467. doi: 10.1038/nature12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khanna S, Rink C, Ghoorkhanian R, et al. Loss of miR-29b following acute ischemic stroke contributes to neural cell death and infarct size. Journal of Cerebral Blood Flow and Metabolism. 2013;33(8):1197–1206. doi: 10.1038/jcbfm.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mietla JA, Wijesinghe DS, Hoeferlin LA, et al. Characterization of eicosanoid synthesis in a genetic ablation model of ceramide kinase. Journal of Lipid Research. 2013;54(7):1834–1847. doi: 10.1194/jlr.M035683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Norton SK, Wijesinghe DS, Dellinger A, et al. Epoxyeicosatrienoic acids are involved in the C 70 fullerene derivative-induced control of allergic asthma. Journal of Allergy and Clinical Immunology. 2012;130(3):761.e2–769.e2. doi: 10.1016/j.jaci.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qureshi R, Jakschik BA. The role of mast cells in thioglycollate-induced inflammation. Journal of Immunology. 1988;141(6):2090–2096. [PubMed] [Google Scholar]
- 45.Canali R, Natarelli L, Leoni G, et al. Vitamin C supplementation modulates gene expression in peripheral blood mononuclear cells specifically upon an inflammatory stimulus: a pilot study in healthy subjects. Genes & Nutrition. 2014;9(3, article 390) doi: 10.1007/s12263-014-0390-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annual Review of Immunology. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 47.Schif-Zuck S, Gross N, Assi S, Rostoker R, Serhan CN, Ariel A. Saturated-efferocytosis generates pro-resolving CD11blow macrophages: modulation by resolvins and glucocorticoids. European Journal of Immunology. 2011;41(2):366–379. doi: 10.1002/eji.201040801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gil CD, Cooper D, Rosignoli G, Perretti M, Oliani SM. Inflammation-induced modulation of cellular galectin-1 and -3 expression in a model of rat peritonitis. Inflammation Research. 2006;55(3):99–107. doi: 10.1007/s00011-005-0059-4. [DOI] [PubMed] [Google Scholar]
- 49.Rostoker R, Yaseen H, Schif-Zuck S, Lichtenstein RG, Rabinovich GA, Ariel A. Galectin-1 induces 12/15-lipoxygenase expression in murine macrophages and favors their conversion toward a pro-resolving phenotype. Prostaglandins and Other Lipid Mediators. 2013;107:85–94. doi: 10.1016/j.prostaglandins.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 50.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105(9):1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 51.Ganguly R, Durieux MF, Waldman RH. Macrophage function in vitamin C deficient guinea pigs. American Journal of Clinical Nutrition. 1976;29(7):762–765. doi: 10.1093/ajcn/29.7.762. [DOI] [PubMed] [Google Scholar]
- 52.Babaev VR, Whitesell RR, Li L, Linton MF, Fazio S, May JM. Selective macrophage ascorbate deficiency suppresses early atherosclerosis. Free Radical Biology and Medicine. 2011;50(1):27–36. doi: 10.1016/j.freeradbiomed.2010.10.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Henson PM. Dampening inflammation. Nature Immunology. 2005;6(12):1179–1181. doi: 10.1038/ni1205-1179. [DOI] [PubMed] [Google Scholar]
- 54.Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nature Reviews Immunology. 2007;7(12):964–974. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- 55.Ariel A, Timor O. Hanging in the balance: endogenous anti-inflammatory mechanisms in tissue repair and fibrosis. The Journal of Pathology. 2013;229(2):250–263. doi: 10.1002/path.4108. [DOI] [PubMed] [Google Scholar]
- 56.Serhan CN. Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? American Journal of Pathology. 2010;177(4):1576–1591. doi: 10.2353/ajpath.2010.100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levy BD, Romano M, Chapman HA, Reilly JJ, Drazen J, Serhan CN. Human alveolar macrophages have 15-lipoxygenase and generate 15(S)-hydroxy-5,8,11-cis-13-trans-eicosatetraenoic acid and lipoxins. Journal of Clinical Investigation. 1993;92(3):1572–1579. doi: 10.1172/JCI116738. [DOI] [PMC free article] [PubMed] [Google Scholar]