

Abstract
Purpose of review
Several decades of work by many investigators have elucidated the major signaling pathways responsible for platelet activation. Still to be fully understood is how these pathways are integrated into a single network and how changing conditions within a growing thrombus affect that network. In this review we will consider some of the recent studies that address these issues and describe a model that provides insights into platelet activation as it occurs in vivo.
Recent findings
Genetic and pharmacologic studies performed in vivo have demonstrated that platelet activation during hemostasis and thrombosis is heterogeneous. Those studies indicate that distinct platelet activation pathways are not merely redundant, but are coordinated in time and space to achieve an optimal response. This coordination is achieved at least in part by the evolving distribution of platelet agonists and changes in solute transport within a hemostatic plug.
Summary
Studies examining the coordination of platelet signaling in time and space continue to increase our understanding of hemostasis and thrombosis. In addition to helping to decipher platelet biology, the results have implications for the understanding of new and existing anti-platelet agents and their potential risks.
Keywords: Platelets, hemostasis, thrombosis, in vivo models, systems biology
Introduction
Platelet accumulation at a site of vascular injury is a dynamic process that integrates chemical and physical cues to promote cellular responses that are coordinated in time and space. Extensive studies of platelet intracellular signaling pathways over the past several decades have greatly enhanced our understanding of platelet function. In general, these signaling pathways start with an extracellular input acting on a platelet surface receptor. Inputs may either promote (e.g. thrombin) or dampen (e.g. prostacyclin) platelet activation, depending on the pathways involved and physiological context. Receptor activation leads to intracellular signaling, which in the case of platelet activation culminates in the release of intracellular Ca2+ stores, αIIbβ3 integrin activation, platelet aggregation, and granule exocytosis. The signaling pathways that are required have been reviewed extensively [1–4].
As many of the major players in platelet signaling have been identified, we and others have begun to try to understand how multiple signaling inputs are coordinated in time and space to achieve an optimal hemostatic response. An optimal response is defined as one sufficient to stop bleeding and promote wound healing, but not so robust that unwarranted vascular occlusion and ischemia occurs. In addition to traditional platelet signaling studies that are typically conducted in vitro, recent efforts have taken a systems approach, combining in vitro, computational and, most importantly, in vivo approaches that rely on animal models. Indeed, it is only by examining platelet function in the complex milieu of an intact vasculature that we can begin to understand how the platelet signaling network is integrated along with contributions from other cells, the vessel wall and local hemodynamic conditions in order to generate a hemostatic plug. This review summarizes some of those efforts and considers a model that arises from them. Observations of platelet response heterogeneity and the underlying mechanisms responsible for such heterogeneity will be discussed.
Heterogeneity of platelet activation during the hemostatic response
It has long been recognized that platelet activation within a hemostatic plug or thrombus is not necessarily uniform. Early evidence demonstrating this point comes from transmission electron microscopy studies of experimental or human pathological thrombi showing platelets with varying degrees of shape change and granule release [5–7]. As these studies were static in nature, it was impossible to determine whether platelets that appeared less activated represented a distinct subpopulation, or rather were merely newly arrived platelets that had simply not yet been fully activated. In recent years, the utilization of fluorescence intravital imaging, pioneered by the Furie laboratory [8–10] and others [11–13], has permitted the visualization of platelet accumulation and activation at sites of vascular injury in vivo in real-time. Studies using this approach in animal models, as well as flow chamber studies using human blood, have demonstrated that both the platelet and coagulation response to vascular injury are indeed heterogeneous in time and space. With regard to platelet activation in vivo, studies have visualized heterogeneity in shape change [14–16], calcium signaling [13,17,18], granule exocytosis [19*, 20–22] and phosphatidylserine exposure [23,24], at the level of individual platelets and platelet subpopulations. Further, thrombin activity [25*] and fibrin deposition [8,19,20,26] are not uniform throughout a platelet plug. The following is a description of some of the evidence demonstrating heterogeneity of platelet activation following vascular injury.
Discoid platelet aggregation
Platelet activation studies performed in vitro have long recognized that one of the initial platelet responses upon activation is shape change from a round or discoid morphology to one with extended filopods. Thus, it was somewhat surprising when investigators reported the accumulation of discoid platelets during the hemostatic response in vivo [14–16], since formation of platelet aggregates was assumed to require platelet activation. The formation of discoid platelet aggregates has been reported to depend on rheological factors (discussed more below), with some degree of platelet activation required as this aggregation was blocked by treating platelets with PGE1 or the PGI2 analog iloprost [15,27**].
Heterogeneity of intracellular Ca2+ mobilization
Multiple studies have examined intracellular Ca2+ mobilization at the level of individual platelets using both in vitro flow chamber systems and in vivo models. In vitro systems are best suited to tease out the Ca2+ response of individual platelets to distinct stimuli. For example, von Willebrand factor (VWF) engagement by GPIbα on the platelet surface results in a transient rise in intracellular Ca2+ [28,29], ADP signaling results in oscillatory Ca2+ signals [28,30–33] and collagen/GPVI signaling results in a sustained rise in intracellular Ca2+ [34]. The in vivo setting is more complex as multiple inputs are integrated. However, studies have observed heterogeneous Ca2+ signals in individual platelets within a platelet plug. For example, van Gestel et al. [13] found that platelets with a transient rise in Ca2+ are more likely to embolize, while stably adherent platelets exhibit a sustained elevation of intracellular Ca2+.
Granule exocytosis
One of the most commonly used markers of platelet activation in intravital imaging studies is P-selectin, a marker of α-granule secretion. In response to focal injuries in the microcirculation, expression of P-selectin on the platelet surface lags behind platelet accumulation temporally, propagates initially from the site of injury, and remains restricted to a subpopulation of platelets adjacent to the site of injury that is overlaid by a shell of P-selectin negative platelets [19*,20,21]. These observations (and others) led to the description of a hemostatic plug as having a heterogeneous architecture comprised of a stable core of fully activated, densely packed platelets with an outer shell of less-activated, loosely associated platelets (Figure 1A). The spatio-temporal localization of dense granule secretion during the hemostatic response in vivo is less well understood due to the lack of imaging reagents indicative of this event. However, the regulation of dense granule release may be inferred from studies of ADP signaling (discussed below), since dense granules represent the primary storage pool of ADP released during the hemostatic response.
Figure 1. A model for the interaction of local conditions with the platelet signaling network resulting in heterogeneous platelet plug architecture.
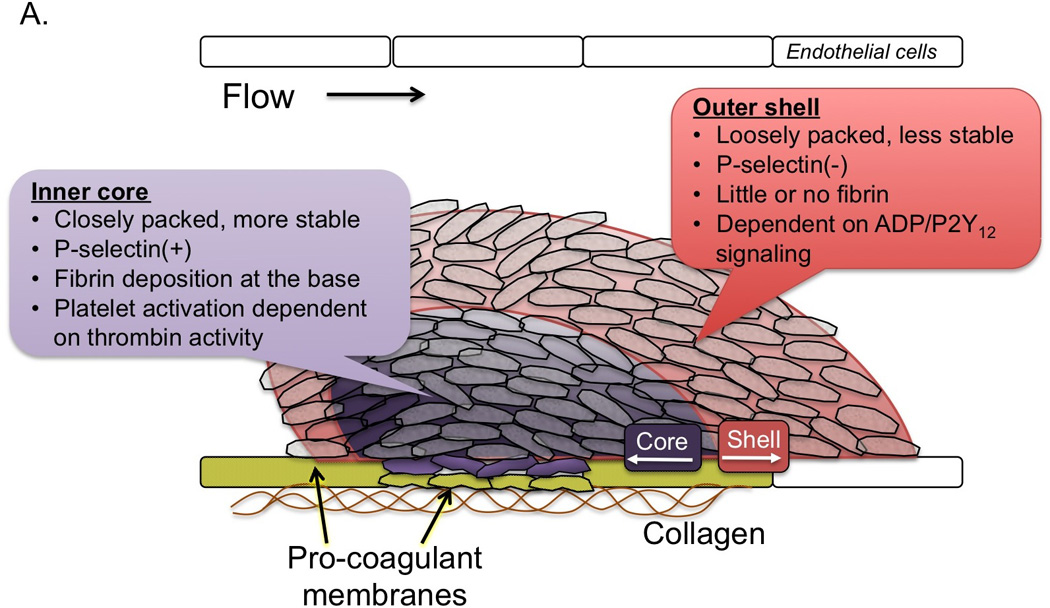
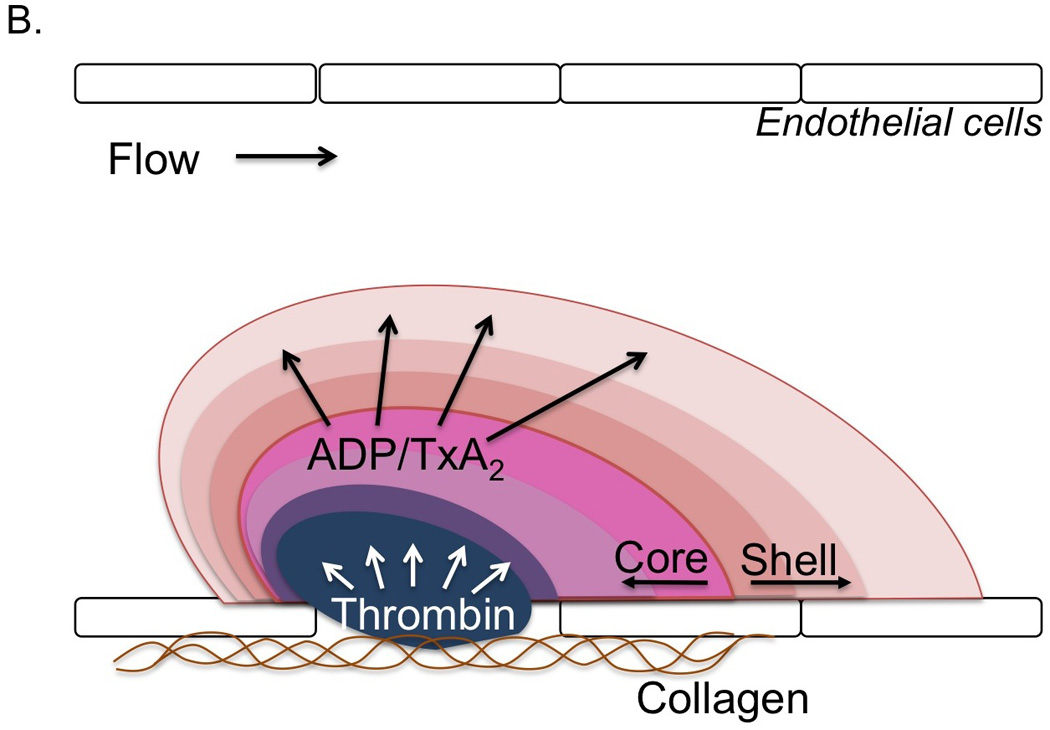
A) A description of the heterogeneous architecture of the platelet plug observed in intravital imaging studies following focal injuries in the microcirculation. The platelet mass is comprised of at least two distinct regions, termed the core and outer shell, identified based on the secretion of the α-granule marker P-selectin. Additional properties of these regions are also indicated. Recent observations suggest that the endothelium is an important source of procoagulant membranes (yellow). B) Data suggest that the development of the heterogeneous architecture described in A is the result of partially overlapping agonist gradients within the platelet mass emanating from the site of injury.
Phosphatidylserine exposure
Phosphatidylserine (PS) exposure on the outer platelet membrane surface has long been recognized as a heterogeneous response at the level of individual platelets, as only a subpopulation of platelets will bind the PS marker annexin V (AV) in response to physiologic agonists in vitro [35,36]. PS exposure is thought to be a critical event during hemostasis and thrombosis to facilitate the assembly coagulation factor complexes on PS+ procoagulant membranes for subsequent thrombin generation. A number of studies have demonstrated unique localization of PS+ platelets at the periphery of platelet aggregates formed under flow in vitro [24]. The spatio-temporal regulation of platelet PS exposure in vivo is less well understood and perhaps context dependent, although studies have shown AV positive platelets at the base of platelet plugs in the region of the thrombus core [23,24,37]. A recent study by Ivanciu and colleagues [38**] used fluorescently-labeled Factors Va and Xa rather than AV in order to specifically determine cites of procoagulant complex assembly. They observed FVa and Xa binding to membranes localized at the base of the thrombus near the site of injury as well as on surrounding endothelial cell surfaces in a microcirculation focal injury model [38**].
Spatio-temporal regulation of platelet signaling
The recognition that platelet activation is not uniform during the hemostatic response leads to the question of how such a heterogeneous architecture develops and whether there are implications for both the use of existing anti-platelet therapies and the development of new ones. One possible explanation is that platelets with different degrees of activation represent subpopulations of circulating platelets with distinct properties. While intriguing, there is at present little experimental evidence to support this hypothesis. Instead, the available evidence suggests that heterogeneity of platelet activation reflects non-uniformity in agonist distribution (Figure 1B). Here, we will discuss what is known about each of the major platelet agonist signaling pathways with regard to their contribution to the spatio-temporal regulation of platelet activation.
Thrombin
Thrombin is a key regulator of robust platelet activation in response to vascular injury. In mouse models, inhibition of thrombin generation/activity or genetic deletion of the platelet thrombin receptor (PAR-4 on mouse platelets) results in significantly impaired platelet accumulation and a near complete lack of intracellular calcium mobilization [17,18] and P-selectin expression [19*,20] as markers of platelet activation. Thus, thrombin activity is critical for development of a stable core composed of fully activated platelets. However, the localization of thrombin activity in time and space is limited. As described above, thrombin generation is localized to procoagulant membranes of platelets and endothelial cells at or immediately adjacent to the site if injury, thus limiting the distribution of thrombin within a platelet mass [23,24,38**]. Once generated, thrombin distribution is limited by its ability to diffuse away from the site of generation (discussed in more detail below) and by plasma-borne inhibitors that either directly inhibit its activity (e.g. antithrombin) or generation (e.g. tissue factor pathway inhibitor and activated protein C). This combination of factors limits thrombin activity to the core region of the hemostatic plug as demonstrated by studies using a fluorogenic thrombin sensor bound to the surface of platelets [25*] and studies showing the localization of fibrin formation restricted to the core [8,19*,20].
ADP
ADP is released from damaged cells at the site of injury as well as from dense granules of activated platelets. It acts on two platelet receptors, P2Y1 and P2Y12, to reinforce platelet activation in a paracrine or autocrine fashion [39–44]. The importance of P2Y12 in platelet activation is highlighted by the efficacy of P2Y12 antagonists in inhibiting platelet activation and protecting against thrombotic events in humans. This role for P2Y12 signaling is recapitulated in animal models, where deletion of P2Y12 [40,45] and the introduction of P2Y12 receptor antagonists have been shown to attenuate thrombus formation [46]. In general, these studies have ascribed a role for P2Y12 in regulating thrombus stability [45,47,48]. Viewed from the perspective of spatio-temporal regulation of platelet activation, this effect of P2Y12 signaling on thrombus stability is due to the importance of this signaling pathway in platelet recruitment and retention in the outer layers of a developing platelet plug, a region where thrombin activity rapidly declines. Indeed, we have shown that inhibition of P2Y12 activation greatly reduces platelet accumulation in the outer platelet shell, while a gain of function mutation in Gi2α, the principal G protein coupled to P2Y12 receptors, leads to an expansion of the shell [19*]. In contrast, a P2Y12 antagonist had no effect on robust platelet activation in the thrombus core, where thrombin activity is high [19*]. This latter finding may help to explain the relative safety of P2Y12 antagonists used clinically.
Thromboxane A2
Like ADP, thromboxane A2 (TxA2) generated and released by activated platelets acts to reinforce platelet activation in an autocrine and paracrine fashion. TxA2 is generated via the aspirin sensitive cyclooxygenase-1 (COX-1) pathway in platelets. Upon release, it binds its receptors (TPα and β) on the platelet surface to provide an activating signal. Its importance in platelet activation in vivo is demonstrated by a number of large clinical studies demonstrating the efficacy of aspirin treatment in the prevention of platelet-mediated cardiovascular events (i.e. myocardial infarction and stroke) [49]. Thrombus formation is also attenuated in TP-deficient mice [50]. The spatio-temporal distribution of TxA2 within a growing hemostatic plug is not yet well defined. As a highly diffusible molecule, its localization will primarily be determined by its source (activated platelets) and its rapid metabolism in plasma to inactive metabolites. However, additional studies are required to ascertain the precise contribution of TxA2 signaling to heterogeneous platelet activation.
Collagen
In contrast to soluble platelet agonists, collagen is an insoluble component of the vessel wall and extravascular tissue. As such, its direct contribution to platelet signaling via activation of the collagen receptor GPVI is restricted to platelets in contact with the damaged vessel wall and those that escape into the extravascular compartment. The contribution of GPVI signaling in experimental models is therefore highly dependent on the mechanism and extent of injury, as well as the amount of thrombin generated [51]. This likely explains varying reports of GPVI being a critical regulator of platelet accumulation and activation in some settings, yet completely dispensable in others [51–54]. Thus, while collagen is clearly a potent activator of platelets in vitro, its contribution to hemostasis and thrombosis in vivo is likely much more context dependent than other platelet agonists such as thrombin, ADP and TxA2.
Contact-dependent signaling
In addition to the contribution of traditional platelet agonists, recent studies have elucidated a role for multiple contact-dependent signaling pathways in regulating platelet activation. These include outside-in signaling via the major platelet integrin αIIbβ3, surface receptor/ligand pairs such as semaphorin 4D, ephrin/eph kinases and Gas6/TAM receptors, as well as junctional adhesion molecules such as PECAM, JAM-A and ESAM. It would be predicted that signaling pathways dependent on close contact between platelets would exert their effects primarily in the thrombus core, as this is where platelet packing density is highest, as compared to the loosely associated platelets within the less stable platelet shell. Of the pathways studied to date, this appears to be true, although not necessarily via the same mechanism in each case. For example, genetic deletion of semaphorin 4D significantly impairs full platelet activation in the core region of a platelet plug [19*], presumably due to its reinforcement of platelet activation via Syk tyrosine kinase activation [55]. Tyrosine phosphorylation of β3 integrin cytoplasmic tails following αIIbβ3 ligand binding instead regulates platelet retractile mechanisms [56]. Disruption of this signaling pathway in mouse platelets by mutating β3 integrin tyrosines to phenylalanine results in impaired platelet mass contraction, leading to diminished local thrombin activity and platelet activation in the core via effects on the molecular transport properties of the uncontracted platelet plug [57*].
Influence of local hemodynamics
Several investigators have demonstrated the importance of local hemodynamic conditions on platelet accumulation at sites of injury in vivo and in flow chambers ex vivo. In particular, changes in flow patterns around an obstruction or in a stenotic region, such as when blood flows over a developing platelet mass or through a stenotic atherosclerotic artery, lead to the development of shear deceleration zones [14,15,27**]. Platelet aggregation in these zones on the downstream side of a stenosis is dependent on vWF/GPIb interactions as well as αIIbβ3 integrin [14,15,27**]. This “shear-dependent” aggregation is also dependent on platelet activation, as it is blocked by inhibitors of TxA2 generation and ADP signaling [15,27**]. These studies highlight the importance of the interaction between platelet aggregates and the bulk flow in shaping the response to vascular injury. As platelets accumulate within the lumen of a vessel they alter the flow characteristics creating local microenvironments in which conditions are favorable for aggregation. Such a phenomenon could be particularly important at sites of stenosis due to atherosclerotic plaque formation where platelet accumulation may be exacerbated. Importantly, even in the case of severely stenosed blood vessels, platelet accumulation is not spontaneous but rather still requires an initial injury to the vessel wall [14]. This differs from the vWF-dependent, activation independent platelet aggregation observed at extreme shear rates (>20,000 s−1) in vitro [58].
The intrathrombus microenvironment shapes agonist distribution
As described above, agonist distribution within a platelet mass is a critical factor responsible for the observed differences in the extent of platelet activation (Figure 1B). The distribution of each agonist depends on the location of its source, its ability to move, and its stability over time. For example, collagen/GPVI signaling is limited to platelets in contact with the site of injury where collagen is exposed. The distribution of soluble agonists, on the other hand, is highly dependent on the physical architecture of the platelet mass as it evolves. Specifically, the movement of molecular species within and/or around a pile of platelets is dependent on the size of the pores (i.e. plasma volume) between platelets, the spatial distribution, connectivity and tortuosity of the pore space, the plasma velocity and the size of the solute. Several of these parameters have now been estimated using computational approaches based on experimental data, or have been directly measured either in vitro or in vivo where possible. For example, a mature platelet mass formed under flow conditions in vitro was found to have a permeability approaching that of an intact endothelial cell layer (2 × 10−14 cm2) that was regulated by both platelet retraction and fibrin formation [59]. Computational modeling based thrombus structures obtained in vivo showed that the core region has greatly reduced permeability (100-fold) and increased tortuosity compared to the shell [60]. Computational models also show that low permeability will greatly reduce plasma flow within the intrathrombus microenvironment compared to the lumen [60,61*,62,63]. As a consequence, the diffusibility of a solute becomes a dominant factor in its transport properties [59,60,63]. Solute diffusibility is determined in part by size, decreasing as size increases. Within the growing platelet plug, solute movement is increasingly hindered in the core region as gap sizes decrease and path tortuosity increases [60,63,64*]. Large solutes can even be excluded from the densely packed core region [19*]. As a result, the platelet plug microenvironment becomes a molecular sieve capable of restricting movement of solutes dependent upon their size [60,61*,63,64*]. Also, due to the heterogeneous nature of the platelet plug microenvironment solutes have different transport properties in different regions of the plug, with the core being more restrictive to protein transport than the shell [57,63,65*]. In aggregate, computational and in vivo studies show that the platelet mass microenvironment alters plasma velocity and solute diffusion to regulate solute transport within the platelet mass.
Why is it important to consider solute transport as a regulator of platelet activation? Transport has long been recognized as important for platelet mass formation as a mechanism for delivery of coagulation factors to the injury site, as well as elution of platelet agonists from within the platelet mass [66–68]. Coagulation is affected by solute transport as limited delivery of coagulation factors restrains thrombin generation, while limited transport of thrombin out of the platelet mass increases its effective concentration. Microfluidic assays have demonstrated that platelets create a physical barrier that covers up tissue factor surfaces and excludes plasma-borne coagulation factors altering the rate of fibrin deposition [68–70]. Increasing platelet packing density can lead to the retention of larger, less mobile solutes (e.g. thrombin), while small solutes (e.g. ADP) can more easily escape to the edges of the platelet mass [57,63,65*]. These effects tend to concentrate protein solutes within the core region of the hemostatic mass. Concentration of thrombin within the thrombus core helps to drive further platelet activation and greater fibrin accumulation [25,57,63,65*]. Interestingly, within the platelet mass formed in mice deficient in platelet retraction, solute transport within the core region is elevated resulting in reduced thrombin activity and platelet activation [57*]. These findings suggest that the reduced transport in the core is important for thrombin retention, as opposed to prevention of continued thrombin production.
Conclusion
In summary, consideration of platelet signaling pathways as an integrated network that responds to differences in agonist distribution, and the growing hemostatic mass as an obstacle to molecular transport, provides new insights into how an optimal hemostatic response is achieved. It also helps to better understand the safety and efficacy profiles of anti-platelet agents. Considerable work remains to be done, however. Much of what is currently known about platelet signaling mechanisms has been determined ex vivo with a focus on individual pathways, rather than with a view to understanding how different pathways are invoked and interact in time and space in vivo. Understanding the spatial and temporal relationships between the platelet signaling network and the altered transport environment within a growing hemostatic mass or thrombus is the realm of systems biology. We propose that the overall themes discussed in this review will be universally relevant, even if some of the details turn out to differ in different areas of the circulation or when pathologic thrombosis is compared to the hemostatic response. Much remains to be learned as new tools are brought to bear.
Key points.
Platelet activation at sites of vascular injury is heterogeneous
Heterogeneity of platelet activation arises from spatio-temporal regulation of agonist distribution within the evolving platelet mass
The physical characteristics of a platelet mass contribute to the development of agonist gradients
A systems approach is required to gain a better understanding of how multiple platelet signaling pathways are integrated to produce an optimal hemostatic
Acknowledgements
The authors would like to acknowledge research support from the American Heart Association (11SDG5720011 to TJS) and the National Heart, Lung and Blood Institute (R01 HL119070 to TJS and LFB, P01 HL40387 and R01 HL103419 to LFB). JDW is supported by NHLBI training grant T32-HL07439.
Conflict of interest
The authors (TJS and LFB) have received research funding from The Medicines Company (Parsippany, NJ).
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Stalker TJ, Newman DK, Ma P, et al. Platelet signaling. Handb Exp Pharmacol. 2012:59–85. doi: 10.1007/978-3-642-29423-5_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Versteeg HH, Heemskerk JW, Levi M, et al. New fundamentals in hemostasis. Physiol Rev. 2013;93:327–358. doi: 10.1152/physrev.00016.2011. [DOI] [PubMed] [Google Scholar]
- 3.Brass LF, Tomaiuolo M, Stalker TJ. Harnessing the platelet signaling network to produce an optimal hemostatic response. Hematol Oncol Clin North Am. 2013;27:381–409. doi: 10.1016/j.hoc.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brass LF, Newman DK, Wannemacher KM, et al. Signal transduction during platelet plug formation. In: Michelson AD, editor. Platelets. 3rd Edition. Boston, MA: Academic Press; 2013. pp. 367–398. [Google Scholar]
- 5.Jorgensen L, Rowsell HC, Hovig T, et al. Resolution and organization of platelet-rich mural thrombi in carotid arteries of swine. Am J Pathol. 1967;51:681–719. [PMC free article] [PubMed] [Google Scholar]
- 6.Stehbens WE, Biscoe TJ. The ultrastructure of early platelet aggregation in vivo. Am J Pathol. 1967;50:219–243. [PMC free article] [PubMed] [Google Scholar]
- 7.White JG. Platelet structural physiology: the ultrastructure of adhesion, secretion, and aggregation in arterial thrombosis. Cardiovasc Clin. 1987;18:13–33. [PubMed] [Google Scholar]
- 8.Falati S, Gross P, Merrill-Skoloff G, et al. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat Med. 2002;8:1175–1181. doi: 10.1038/nm782. [DOI] [PubMed] [Google Scholar]
- 9.Falati S, Gross PL, Merrill-Skoloff G, et al. In vivo models of platelet function and thrombosis: study of real-time thrombus formation. Methods Mol Biol. 2004;272:187–197. doi: 10.1385/1-59259-782-3:187. [DOI] [PubMed] [Google Scholar]
- 10.Celi A, Merrill-Skoloff G, Gross P, et al. Thrombus formation: direct real-time observation and digital analysis of thrombus assembly in a living mouse by confocal and widefield intravital microscopy. J Thromb Haemost. 2003;1:60–68. doi: 10.1046/j.1538-7836.2003.t01-1-00033.x. [DOI] [PubMed] [Google Scholar]
- 11.oude Egbrink MG, Tangelder GJ, Slaaf DW, et al. Thromboembolic reaction following wall puncture in arterioles and venules of the rabbit mesentery. Thromb Haemost. 1988;59:23–28. [PubMed] [Google Scholar]
- 12.Rosen ED, Raymond S, Zollman A, et al. Laser-induced noninvasive vascular injury models in mice generate platelet- and coagulation-dependent thrombi. Am J Pathol. 2001;158:1613–1622. doi: 10.1016/S0002-9440(10)64117-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Gestel MA, Heemskerk JW, Slaaf DW, et al. Real-time detection of activation patterns in individual platelets during thromboembolism in vivo: differences between thrombus growth and embolus formation. J Vasc Res. 2002;39:534–543. doi: 10.1159/000067208. [DOI] [PubMed] [Google Scholar]
- 14.Nesbitt WS, Westein E, Tovar-Lopez FJ, et al. A shear gradient-dependent platelet aggregation mechanism drives thrombus formation. Nat Med. 2009;15:665–673. doi: 10.1038/nm.1955. [DOI] [PubMed] [Google Scholar]
- 15.Maxwell MJ, Westein E, Nesbitt WS, et al. Identification of a 2-stage platelet aggregation process mediating shear-dependent thrombus formation. Blood. 2007;109:566–576. doi: 10.1182/blood-2006-07-028282. [DOI] [PubMed] [Google Scholar]
- 16.Nishimura S, Manabe I, Nagasaki M, et al. In vivo imaging visualizes discoid platelet aggregations without endothelium disruption and implicates contribution of inflammatory cytokine and integrin signaling. Blood. 2012;119:e45–e56. doi: 10.1182/blood-2011-09-381400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dubois C, Panicot-Dubois L, Gainor JF, et al. Thrombin-initiated platelet activation in vivo is vWF independent during thrombus formation in a laser injury model. J Clin Invest. 2007;117:953–960. doi: 10.1172/JCI30537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Gestel MA, Reitsma S, Slaaf DW, et al. Both ADP and thrombin regulate arteriolar thrombus stabilization and embolization, but are not involved in initial hemostasis as induced by micropuncture. Microcirculation. 2007;14:193–205. doi: 10.1080/10739680601139294. [DOI] [PubMed] [Google Scholar]
- 19. Stalker TJ, Traxler EA, Wu J, et al. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood. 2013;121:1875–1885. doi: 10.1182/blood-2012-09-457739. This study demonstrated the role of platelet agonist gradients in the spatio-temporal regulation of platelet activation following vascular injury in vivo.
- 20.Vandendries ER, Hamilton JR, Coughlin SR, et al. Par4 is required for platelet thrombus propagation but not fibrin generation in a mouse model of thrombosis. Proc Natl Acad Sci U S A. 2007;104:288–292. doi: 10.1073/pnas.0610188104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gross PL, Furie BC, Merrill-Skoloff G, et al. Leukocyte-versus microparticle-mediated tissue factor transfer during arteriolar thrombus development. J Leukoc Biol. 2005;78:1318–1326. doi: 10.1189/jlb.0405193. [DOI] [PubMed] [Google Scholar]
- 22.Hechler B, Nonne C, Eckly A, et al. Arterial thrombosis: relevance of a model with two levels of severity assessed by histologic, ultrastructural and functional characterization. J Thromb Haemost. 2010;8:173–184. doi: 10.1111/j.1538-7836.2009.03666.x. [DOI] [PubMed] [Google Scholar]
- 23.Hayashi T, Mogami H, Murakami Y, et al. Real-time analysis of platelet aggregation and procoagulant activity during thrombus formation in vivo. Pflugers Arch. 2008;456:1239–1251. doi: 10.1007/s00424-008-0466-9. [DOI] [PubMed] [Google Scholar]
- 24.Munnix IC, Kuijpers MJ, Auger J, et al. Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation: regulation by transient integrin activation. Arterioscler Thromb Vasc Biol. 2007;27:2484–2490. doi: 10.1161/ATVBAHA.107.151100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Welsh JD, Colace TV, Muthard RW, et al. Platelet-targeting sensor reveals thrombin gradients within blood clots forming in microfluidic assays and in mouse. J Thromb Haemost. 2012;10:2344–2353. doi: 10.1111/j.1538-7836.2012.04928.x. This paper used a fluorescent thrombin sensor to demonstrate localizatioin of thrombin activity to the core region of developing thrombi in vivo.
- 26.Kamocka MM, Mu J, Liu X, et al. Two-photon intravital imaging of thrombus development. J Biomed Opt. 2010;15:016020. doi: 10.1117/1.3322676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Westein E, van der Meer AD, Kuijpers MJ, et al. Atherosclerotic geometries exacerbate pathological thrombus formation poststenosis in a von Willebrand factor-dependent manner. Proc Natl Acad Sci USA. 2013;110:1357–1362. doi: 10.1073/pnas.1209905110. This study examined the impact of blood vessel stenosis on platelet accumulation using in vitro and in vivo approaches. The investigators found that platelet aggregation is enhanced in the outlet region of a stenosis via a vWF dependent mechanism.
- 28.Mazzucato M, Pradella P, Cozzi MR, et al. Sequential cytoplasmic calcium signals in a 2-stage platelet activation process induced by the glycoprotein Ibalpha mechanoreceptor. Blood. 2002;100:2793–2800. doi: 10.1182/blood-2002-02-0514. [DOI] [PubMed] [Google Scholar]
- 29.Nesbitt WS, Kulkarni S, Giuliano S, et al. Distinct glycoprotein Ib/V/IX and integrin alpha IIbbeta 3-dependent calcium signals cooperatively regulate platelet adhesion under flow. J Biol Chem. 2002;277:2965–2972. doi: 10.1074/jbc.M110070200. [DOI] [PubMed] [Google Scholar]
- 30.Mazzucato M, Cozzi MR, Pradella P, et al. Distinct roles of ADP receptors in von Willebrand factor-mediated platelet signaling and activation under high flow. Blood. 2004;104:3221–3227. doi: 10.1182/blood-2004-03-1145. [DOI] [PubMed] [Google Scholar]
- 31.Nesbitt WS, Giuliano S, Kulkarni S, et al. Intercellular calcium communication regulates platelet aggregation and thrombus growth. J Cell Biol. 2003;160:1151–1161. doi: 10.1083/jcb.200207119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heemskerk JW, Willems GM, Rook MB, et al. Ragged spiking of free calcium in ADP-stimulated human platelets: regulation of puff-like calcium signals in vitro and ex vivo. J Physiol. 2001;535:625–635. doi: 10.1111/j.1469-7793.2001.00625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heemskerk JW, Hoyland J, Mason WT, et al. Spiking in cytosolic calcium concentration in single fibrinogen-bound fura-2-loaded human platelets. Biochem J. 1992;283(Pt 2):379–383. doi: 10.1042/bj2830379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heemskerk JW, Vuist WM, Feijge MA, et al. Collagen but not fibrinogen surfaces induce bleb formation, exposure of phosphatidylserine, and procoagulant activity of adherent platelets: evidence for regulation by protein tyrosine kinase-dependent Ca2+ responses. Blood. 1997;90:2615–2625. [PubMed] [Google Scholar]
- 35.Munnix IC, Cosemans JM, Auger JM, et al. Platelet response heterogeneity in thrombus formation. Thromb Haemost. 2009;102:1149–1156. doi: 10.1160/TH09-05-0289. [DOI] [PubMed] [Google Scholar]
- 36.Thiagarajan P, Tait JF. Collagen-induced exposure of anionic phospholipid in platelets and platelet-derived microparticles. J Biol Chem. 1991;266:24302–24307. [PubMed] [Google Scholar]
- 37.Kuijpers MJ, Munnix IC, Cosemans JM, et al. Key role of platelet procoagulant activity in tissue factor-and collagen-dependent thrombus formation in arterioles and venules in vivo differential sensitivity to thrombin inhibition. Microcirculation. 2008;15:269–282. doi: 10.1080/10739680701653517. [DOI] [PubMed] [Google Scholar]
- 38. Ivanciu L, Krishnaswamy S, Camire RM. New insights into the spatio-temporal localization of prothrombinase in vivo. Blood. 2014 doi: 10.1182/blood-2014-03-565010. in press. This study utilized fluorescently labeled coagulation factors Va and Xa to determine the localization of procoagulant membranes at a site of injury in vivo. The results show that damaged vascular endothelium around the site of injury is a major binding site for coagulation factor complex assembly.
- 39.Daniel JL, Dangelmaier C, Jin J, et al. Molecular basis for ADP-induced platelet activation. I. Evidence for three distinct ADP receptors on human platelets. J Biol Chem. 1998;273:2024–2029. doi: 10.1074/jbc.273.4.2024. [DOI] [PubMed] [Google Scholar]
- 40.Foster CJ, Prosser DM, Agans JM, et al. Molecular identification and characterization of the platelet ADP receptor targeted by thienopyridine antithrombotic drugs. J Clin Invest. 2001;107:1591–1598. doi: 10.1172/JCI12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hollopeter G, Jantzen HM, Vincent D, et al. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature. 2001;409:202–207. doi: 10.1038/35051599. [DOI] [PubMed] [Google Scholar]
- 42.Jin J, Daniel JL, Kunapuli SP. Molecular basis for ADP-induced platelet activation. II. The P2Y1 receptor mediates ADP-induced intracellular calcium mobilization and shape change in platelets. J Biol Chem. 1998;273:2030–2034. doi: 10.1074/jbc.273.4.2030. [DOI] [PubMed] [Google Scholar]
- 43.Jin J, Kunapuli SP. Coactivation of two different G protein-coupled receptors is essential for ADP-induced platelet aggregation. Proc Natl Acad Sci U S A. 1998;95:8070–8074. doi: 10.1073/pnas.95.14.8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang FL, Luo L, Gustafson E, et al. ADP is the cognate ligand for the orphan G protein-coupled receptor SP1999. J Biol Chem. 2001;276:8608–8615. doi: 10.1074/jbc.M009718200. [DOI] [PubMed] [Google Scholar]
- 45.Andre P, Delaney SM, LaRocca T, et al. P2Y12 regulates platelet adhesion/activation, thrombus growth, and thrombus stability in injured arteries. J Clin Invest. 2003;112:398–406. doi: 10.1172/JCI17864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gachet C. The platelet P2 receptors as molecular targets for old and new antiplatelet drugs. Pharmacol Ther. 2005;108:180–192. doi: 10.1016/j.pharmthera.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 47.Nergiz-Unal R, Cosemans JM, Feijge MA, et al. Stabilizing role of platelet P2Y(12) receptors in shear-dependent thrombus formation on ruptured plaques. PLoS One. 2010;5:e10130. doi: 10.1371/journal.pone.0010130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stolla M, Stefanini L, Roden RC, et al. The kinetics of alphaIIbbeta3 activation determines the size and stability of thrombi in mice: implications for antiplatelet therapy. Blood. 2011;117:1005–1013. doi: 10.1182/blood-2010-07-297713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lauer MS. Clinical practice. Aspirin for primary prevention of coronary events. N Engl J Med. 2002;346:1468–1474. doi: 10.1056/NEJMcp012672. [DOI] [PubMed] [Google Scholar]
- 50.Thomas DW, Mannon RB, Mannon PJ, et al. Coagulation defects and altered hemodynamic responses in mice lacking receptors for thromboxane A2. J Clin Invest. 1998;102:1994–2001. doi: 10.1172/JCI5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mangin P, Yap CL, Nonne C, et al. Thrombin overcomes the thrombosis defect associated with platelet GPVI/FcRgamma deficiency. Blood. 2006;107:4346–4353. doi: 10.1182/blood-2005-10-4244. [DOI] [PubMed] [Google Scholar]
- 52.Dubois C, Panicot-Dubois L, Merrill-Skoloff G, et al. Glycoprotein VI-dependent and -independent pathways of thrombus formation in vivo. Blood. 2006;107:3902–3906. doi: 10.1182/blood-2005-09-3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Massberg S, Gawaz M, Gruner S, et al. A crucial role of glycoprotein VI for platelet recruitment to the injured arterial wall in vivo. J Exp Med. 2003;197:41–49. doi: 10.1084/jem.20020945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bender M, Hagedorn I, Nieswandt B. Genetic and antibody-induced glycoprotein VI deficiency equally protects mice from mechanically and FeCl(3) -induced thrombosis. J Thromb Haemost. 2011;9:1423–1426. doi: 10.1111/j.1538-7836.2011.04328.x. [DOI] [PubMed] [Google Scholar]
- 55.Wannemacher KM, Zhu L, Jiang H, et al. Diminished contact-dependent reinforcement of Syk activation underlies impaired thrombus growth in mice lacking Semaphorin 4D. Blood. 2010;116:5707–5715. doi: 10.1182/blood-2010-04-279943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Law DA, DeGuzman FR, Heiser P, et al. Integrin cytoplasmic tyrosine motif is required for outside-in alphaIIbbeta3 signalling and platelet function. Nature. 1999;401:808–811. doi: 10.1038/44599. [DOI] [PubMed] [Google Scholar]
- 57. Stalker TJ, Welsh JD, Tomaiuolo M, et al. A systems approach to hemostasis:3. Thrombus consolidation regulates intrathrombus solute transport and local thrombin activity. Blood. 2014 doi: 10.1182/blood-2014-01-550319. In press. This study demonstrates the role of platelet retraction in regulating intrathrombus solute transport and localization of thrombin activity.
- 58.Ruggeri ZM, Orje JN, Habermann R, et al. Activation-independent platelet adhesion and aggregation under elevated shear stress. Blood. 2006;108:1903–1910. doi: 10.1182/blood-2006-04-011551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muthard RW, Diamond SL. Blood clots are rapidly assembled hemodynamic sensors: flow arrest triggers intraluminal thrombus contraction. Arterioscler Thromb Vasc Biol. 2012;32:2938–2945. doi: 10.1161/ATVBAHA.112.300312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Voronov RS, Stalker TJ, Brass LF, et al. Simulation of intrathrombus fluid and solute transport using in vivo clot structures with single platelet resolution. Ann Biomed Eng. 2013;41:1297–1307. doi: 10.1007/s10439-013-0764-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kim OV, Xu Z, Rosen ED, et al. Fibrin networks regulate protein transport during thrombus development. PLoS Comput Biol. 2013;9:e1003095. doi: 10.1371/journal.pcbi.1003095. This study used a combination of in vitro and computational approaches to examine the impact of a fibrin network on protein transport within a thrombus.
- 62.Leiderman K, Fogelson AL. Grow with the flow: a spatial-temporal model of platelet deposition and blood coagulation under flow. Math Med Biol. 2011;28:47–84. doi: 10.1093/imammb/dqq005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tomaiuolo M, Stalker TJ, Welsh JD, et al. A systems approach to hemostasis: 2. Computational analysis of molecular transport in the thrombus microenvironment. Blood. 2014 doi: 10.1182/blood-2014-01-550343. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Leiderman K, Fogelson AL. The influence of hindered transport on the development of platelet thrombi under flow. Bull Math Biol. 2013;75:1255–1283. doi: 10.1007/s11538-012-9784-3. This computational study showed that hindered transport of protein solutes within the intrathrombus microenvironment profoundly affects coagulation factor distribution, which may ultimately limit thrombus size.
- 65. Welsh JD, Stalker TJ, Voronov RA, et al. systems approach to hemostasis: 1. The interdependence of thrombus architecture and agonist movements in the gaps between platelets. Blood. 2014 doi: 10.1182/blood-2014-01-550335. In press. This series of papers characterizes for the first time intrathrombus solute transport in vivo and demonstrates how the distribution of soluble agonists is regulated by the internal architecture of a platelet mass.
- 66.Fogelson AL, Tania N. Coagulation under flow: the influence of flow-mediated transport on the initiation and inhibition of coagulation. Pathophysiol Haemost Thromb. 2005;34:91–108. doi: 10.1159/000089930. [DOI] [PubMed] [Google Scholar]
- 67.Hathcock JJ, Nemerson Y. Platelet deposition inhibits tissue factor activity: in vitro clots are impermeable to factor Xa. Blood. 2004;104:123–127. doi: 10.1182/blood-2003-12-4352. [DOI] [PubMed] [Google Scholar]
- 68.Neeves KB, Illing DA, Diamond SL. Thrombin flux and wall shear rate regulate fibrin fiber deposition state during polymerization under flow. Biophys J. 2010;98:1344–1352. doi: 10.1016/j.bpj.2009.12.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Colace TV, Muthard RW, Diamond SL. Thrombus growth and embolism on tissue factor-bearing collagen surfaces under flow: role of thrombin with and without fibrin. Arterioscler Thromb Vasc Biol. 2012;32:1466–1476. doi: 10.1161/ATVBAHA.112.249789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Okorie UM, Denney WS, Chatterjee MS, et al. Determination of surface tissue factor thresholds that trigger coagulation at venous and arterial shear rates: amplification of 100 fM circulating tissue factor requires flow. Blood. 2008;111:3507–3513. doi: 10.1182/blood-2007-08-106229. [DOI] [PMC free article] [PubMed] [Google Scholar]