

Abstract
We report the synthesis of a series of distance-matching aryl and vinylaryl cross-linkers for constructing stapled peptides containing cysteines at i,i+7 positions. Langevin dynamics simulation studies helped to classify these cross-linkers into two categories: the rigid cross-linkers with narrower S-S distance distribution and the flexible cross-linkers with wider S-S distance distribution. The stapled Noxa BH3 peptides with the flexible distance-matching cross-linkers gave the highest degree of helicity as well as the most potent inhibitory activity against Mcl-1. However, the stapled peptides with the highest hydrophobicity showed the most efficient cellular uptake. Together, this work illustrates the divergent nature of binding affinity and cellular uptake, and the vital importance of choosing appropriate cross-linkers in constructing stapled peptides with the drug-like properties.
Keywords: peptides, cross-linkers, stapled peptides, inhibitors, protein-protein interaction
1. Introduction
Constraining peptides via side chain cross-linking has served as an effective means to reinforce peptide helices. This side chain cross-linking strategy, also referred to as “peptide stapling”, has been successfully employed in designing peptide-based inhibitors of the protein-protein interactions that are historically considered undruggable.1,2 In stabilizing peptide helices, earlier non-covalent approaches relied on the use of the salt bridge,3 the π-π stacking,4 or the cation-π interaction, while the covalent ones were built upon the lactam ring6 or the disulfide bridge.7 More recently, other covalent linkages generated through olefin metathesis,8 1,3-dipolar cycloaddition reactions,9,10 and cysteine alkylation11,12 have renewed the interest in developing the side chain cross-linked peptides (“stapled peptides”) as potential drug candidates for the various biological targets.13–15 For example, we reported a simple side chain cross-linking chemistry based on the distance-matching biaryl cross-linkers,12 and successfully employed this strategy in designing the cell-permeable and proteolytically stable stapled Noxa BH3 peptides as potent Mcl-1 inhibitors,16 the stapled peptide-based dual inhibitors of Mdm2/Mdmx,17 and a dual-active stapled peptide that binds to the C-terminal domain of HIV capsid protein as well as the envelop glycoprotein gp120 and inhibits HIV-1 virus entry and assembly.18
Despite their success, the limited cell permeability of the stapled peptides remains to be a major challenge in targeting the intracellular protein-protein interactions. Several parameters such as helicity, amphipathicity, charge, and hydrophobicity have been shown to be important for peptide cell permeability.12,16,17,19,20 While the amino acid residues are crucial to the physicochemical properties of a stapled peptide including its cell permeability, the effect of the cross-linker structure has not been systematically investigated, particularly with regard to the length and flexibility of the cross-linkers. It has been shown that the rigid cross-linkers exhibited stronger effect in stabilizing helical conformation than the flexible ones.21 However, in many instances the flexible cross-linkers may be preferred because they can adapt to the subtle distance variation between the amino acid side chains, especially upon binding to its target. Importantly, a poor choice of a rigid cross-linker may constrain the stapled peptide in the inactive conformations. In our previous studies, we designed a pair of rigid distance-matching cross-linkers,12 namely, 4,4′-bisbromomethyl-biphenyl (Bph) and 6,6′-bis-bromomethyl-[3,3′] bipyridine (Bpy) and showed that the attachment of Bph and Bpy to the cysteines located at i, i+7 positions of the NoxaB-(75–93)-C75A peptide dramatically increased the biological activity and cellular uptake of the Noxa BH3 peptides.16 To probe the effect of cross-linker structure on physicochemical properties of the stapled Noxa BH3 peptides, here we synthesized a series of simple aryl and vinylaryl cross-linkers with varying length, rigidity, and hydrophobicity. The effect of cross-linker structure on stapled peptide conformation was investigated by circular dichroism. The inhibitory activity against Mcl-1 was determined using a competitive fluorescence polarization (FP) assay. The uptake of these stapled peptides into HeLa cells was analyzed by fluorescence activated cell sorting. The results indicated that the conformational flexibility of the cross-linkers correlated well with the inhibitory activity while the hydrophobicity correlated with the extent of cellular uptake.
2. Results and Discussion
In designing cross-linkers for stapling reaction with the dicysteine-containing Noxa BH3 peptide, we decided to choose structures that contain the benzylic or allylic bromide groups because of their enhanced reactivity in the nucleophilic substitution reaction with the cysteine thiols compared to the alkyl bromides.22 In the Langevin dynamics simulation study, the S-S distances between the cysteines located at i,i+7 positions of the helical Noxa peptide exhibited a Gaussian distribution in the range of 9.0–13 Å with the median located at 11.2 Å (Figure 1).
Figure 1.
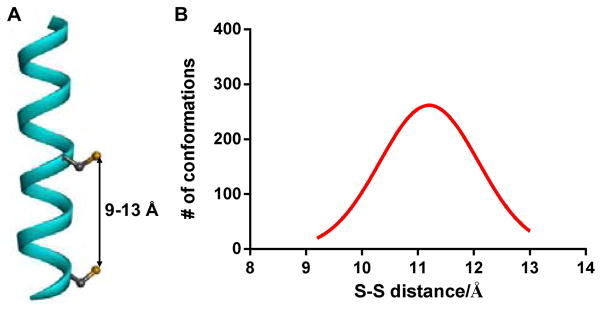
(A) Ribbon diagram of the Noxa BH3 peptide helix with the S-S distance between the two cysteines located at i,i+7 positions marked. (B) Calculated S-S distance distribution of the NoxaBH3 peptide using Langevin dynamics simulation.
Based on this result, we designed 6 new cross-linkers (3–8) whose end-to-end distances fall in this range in addition to Bph (1) and Bpy (2) reported previously12 (Figure 2A). Langevin dynamic simulations indicated that compared to Bph and Bpy, cross-linkers 3–5 exhibited narrower Gaussian distributions with the slightly longer median S-S distances (~12 Å) while cross-linker 6 showed a much broader distribution with the similar median S-S distance, indicating its high flexibility (Figure 2B). The S-S distance distribution of 7 was in a range significantly shorter than that of the BH3 peptide (compare Figure 2B to 1B). On the other hand, cross-linker 8 exhibited a narrower Gaussian distribution with the median S-S distance of 10.8 Å (Figure 2B), the closest to that of the BH3 peptide (Figure 1B).
Figure 2.

(A) Structures of cross-linkers 1–8. (B) Calculated S-S distance distributions of the S-methyl cross-linkers using the Langevin dynamics simulations.
Among six new cross-linkers, compound 7 is commercially available while the rest can be readily synthesized in short routes (Scheme 1). Briefly, cross-linker 3 was prepared in 5 steps with the key step to be the palladium-catalyzed reductive homo-coupling to generate the 1,1′-biphenyl intermediate 3c (Scheme 1a).23 The dihydrophenanthrene cross-linker 4 was obtained in 36% yield by treating dihydrophenanthrene with paraformaldehyde and a mixture of phorphoric acid, HBr and acetic acid (Scheme 1b). Subsequent oxidation of 4 by DDQ produced the phenanthrene cross-linker 5 (Scheme 1c). The p-phenylene-3,3′-bis(allylbromide) 6 was derived from dimethyl-1,4-phenylene-diacrylate through the functional group inter-conversions with an overall yield of 64% (Scheme 1d). Finally, the phenazine cross-linker 8 was prepared in two steps involving the reciprocal Buchwald-Hartwig amination reaction24 followed by bromination with NBS with an overall yield of 11% (Scheme 1e).
Scheme 1.

Synthesis of the distance-matching cross-linkers.
With the cross-linkers in hand, we then performed the stapling reactions with the dicysteine-containing Noxa BH3 peptide. Two conditions were employed depending on cross-linker solubility. A 1:1 mixture of acetonitrile/ammonium bicarbonate buffer, pH 8.5, was used for cross-linkers 1–3 and 7 while DMF was used for cross-linkers 4–6 and 8 together with DIEA as the base. Based on HPLC analysis, all cross-linking reactions reached completion within 2 hours. After HPLC purification, the identities of the stapled peptides were confirmed by ESI-MS analyses (Table 1).
Table 1.
ESI-MS characterization of the cross-linked Noxa BH3 peptidesa
| Name | Sequence | Calculated mass | Found mass |
|---|---|---|---|
| Noxa-1 |
|
2375.3 | 2375.0 |
| Noxa-2 |
|
2377.3 | 2377.0 |
| Noxa-3 |
|
2435.4 | 2435.0 |
| Noxa-4 |
|
2401.4 | 2400.8 |
| Noxa-5 |
|
2399.3 | 2398.8 |
| Noxa-6 |
|
2351.3 | 2351.0 |
| Noxa-7 |
|
2375.3 | 2375.0 |
| Noxa-8 |
|
2401.3 | 2401.2 |
Peptides were acetylated at the N-termini and amidated at the C-termini. C1–8 denotes L-cysteine alkylated with cross-linker 1–8.
Next, we examined the secondary structures of the stapled Noxa BH3 peptides using far-UV circular dichroism (CD). The CD spectra displayed two minima at 222 nm (n→π* transition) and 199–204 nm (π→π* transitions), respectively (Figure 3A). While the minima at 222 nm ([θ]222) showed small variations, large changes in [θ]208 were observed (Figure 3). Since the ratio of [θ]222/[θ]208 can be used to determine the relative amount of 310- and α-helices in a conformational ensemble;25 a value close to 0.4 indicates a 310-helix while a value of 1.0 indicates an α-helix.26 In PBS buffer, Noxa-2 and -6 showed the highest [θ]222/[θ]208 ratios (Figure 3B), indicating these two peptides may have the highest α-helical content in the series.
Figure 3.

Circular dichroism analyses of the Noxa BH3 peptides. (A) CD spectra of the stapled Noxa peptides along with the linear peptide. (B) Table showing absorption minima, [θ]222, [θ]208, and [θ]222/[θ]208 values for all peptides. The peptides were dissolved in PBS buffer at 50 μM concentrations.
With the structural understanding, we then measured the inhibitory activities of the stapled Noxa BH3 peptides carrying the various cross-linkers against Mcl-1 and compared them to that of the linear peptide using a competitive FP assay (Table 2). All stapled peptides showed higher inhibitory activity than the linear one. Among them, Noxa-2 and -6 showed the highest inhibitory activity, consistent with their CD signatures displaying the highest [θ]222/[θ]208 ratios. Intriguingly, Noxa-7 also showed good inhibitory activity despite the fact that it deviates the most in the mean S-S distance in comparison to the native helical peptide (Figure 2B). This could be attributed to an additional hydrophobic interaction formed between the biphenyl group and the hydrophobic binding pocket of the Mcl-1.16
Table 2.
Inhibitory activity of the cross-linked Noxa BH3 peptidesa
| Name | Sequence | IC50 (nM) |
|---|---|---|
| Noxa | AACLRRIGDCVNLRQKLLN | 355 ± 96 |
| Noxa-1 |
|
71 ± 8 |
| Noxa-2 |
|
41 ± 1 |
| Noxa-3 |
|
183 ± 30 |
| Noxa-4 |
|
148 ± 3 |
| Noxa-5 |
|
71± 3 |
| Noxa-6 |
|
49 ± 8 |
| Noxa-7 |
|
78 ± 5 |
| Noxa-8 |
|
111 ± 23 |
Competitive FP assay was performed three times to obtain the average IC50 value along with the standard deviation.
To determine the hydrophobicity of the stapled Noxa peptides carrying various cross-linkers, we subjected these peptides to the reverse-phase HPLC and compared their retention times (Figure 4A). As expected, the stapled peptides carrying heteroatom-containing cross-linkers such as Noxa-2, -3, and -8 showed shorter retention times, whereas the stapled peptides containing pure phenyl and vinylphenyl cross-linkers such as Noxa-1 and -5–7 gave longer retention times, indicating higher hydrophobicity. Since peptide stapling generally leads to increased cellular uptake, we prepared the N-terminal fluorescein-conjugated stapled Noxa BH3 peptides, Fluo-Noxa-1–8, and measured their cellular uptake by fluorescence activated cell sorting (FACS) analysis.16 Among the cross-linked peptides, Fluo-Noxa-1, and -5–7 were taken into the HeLa cells most efficiently after 2 hour incubation at 37°C (Figure 4B). The uptake tread appears to correlate well with the hydrophobicity of the peptides (Figure 4A), and not with the peptide helicity (Figure 3B). While the conformation, charge and hydrophobicity are three key parameters in determining peptide permeability,27 the trend observed here is attributed to peptide hydrophobicity because of their identical sequences and small conformational variations. Similar phenomena have been observed in the literature; for example, the uptake of the cell penetrating peptide PFVYLI was driven by hydrophobicity,28 and the arginine-to-alanine substitutions in the stapled Noxa BH3 peptide resulted in the enhanced cellular uptake.16
Figure 4.
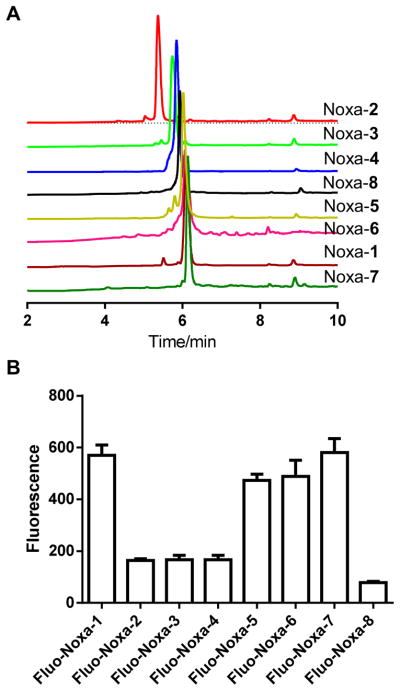
(A) HPLC traces of the stapled Noxa BH3 peptides. (B) FACS analysis of HeLa cells after treatment with 10 μM fluorescein-labeled stapled Noxa BH3 peptides at 37 °C for 2 hours. The normalized mean fluorescence is plotted in the graph.
3. Conclusion
In conclusion, we have designed and synthesized a series of distance-matching aryl and vinylaryl cross-linkers that can be employed to prepare stapled peptides containing cysteines at i,i+7 positions. The end-to-end distance distributions simulated using Langevin dynamics suggested that these cross-linkers can be classified into two categories: the rigid cross-linkers with narrower S-S distance distribution such as 3–5, and the flexible cross-linkers with wider S-S distance distribution such as 1–2 and 6–8. CD measurements indicated that stapled peptides carrying cross-linkers 2 and 6 gave the highest degree of α-helicity due to their closest match in mean S-S distance to the native helical peptide. Stapled peptides Noxa-2 and -6 also showed the most potent inhibitory activity against Mcl-1, presumably due to their highest helicity. On the other hand, Noxa-1 and -7 showed the most efficient cellular uptake, which was attributed to their highest hydrophobicity. Taken together, this work illustrates the vital importance of choosing appropriate cross-linkers in constructing stapled peptides with the desired physicochemical properties. Most notably, the divergence of in vitro inhibitory activity and cellular uptake may potentially make it difficult to predict the in vivo activity of the stapled peptides.
4. Experimental section
4.1. General
Solvents and chemicals were purchased from the commercial sources and used directly without further purification. Flash chromatography was performed with SiliCycle P60 silica gel (40–63 μm, 60 Å). 1H NMR spectra were recorded with Inova-300, −400 or −500 MHz spectrometers and chemical shifts were reported in ppm using either TMS or deuterated solvents as internal standards (TMS, 0.00; CDCl3, 7.26; CD3OD, 3.31; DMSO-d6, 2.50). Multiplicity was reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, brs = broad. Electrospray LC-MS analysis was performed on Finnigan LCQ Advantage IonTrap mass spectrometry coupled with a Surveyor HPLC system. Noxa peptide was purchased from GenScript (Piscataway, NJ). The fluorescein-labeled cross-linked peptides were synthesized as reported previously.16 Peptides were purified using a Gilson semi-preparative reverse-phase HPLC system equipped with Phenomenex C18 column with a flow rate of 5 mL/min and a gradient of 10–90% acetonitrile/H2O while monitoring at 220 and 254 nm. Analytical HPLC was performed using Kinetex C18 column (250 × 4.6 mm) with a flow rate of 1.0 mL/min and a gradient of 10–90% acetonitrile/H2O over 15 min with UV-vis detection wavelength set at 220 and 254 nm.
4.2. Langevin Dynamics simulation
The Langevin dynamics simulation was carried out using Hyperchem 8.0. The structures were minimized using Amber 99 force field prior to dynamics simulation. The simulations were set up to last 1000 ps with 1-ps time step at 300 K. Both S-methylated peptide and the S-methylated cross-linkers were used in the simulation. The distance between the two sulfur atoms was monitored throughout the simulation. The unimodal distributions were observed for all structures and the number of conformations was plotted to the S-S distance. The median S-S distance for the cross-linkers and the peptide was calculated from the graph.
4.3. Synthesis of cross-linkers
2-Methoxy-4-methylaniline (3a)29
To a solution of 5-methyl-2-nitroanisole (1.0 g, 6 mmol) in 100 mL EtOH was added 0.1 g 10% Pd/C. The mixture was stirred at room temperature under a hydrogen balloon for 1.5 h. The reaction mixture was then filtered through a layer of Celite and the solvent was evaporated to give the titled compound (0.70 g, 85% yield): 1H NMR (400 MHz, CDCl3) δ 6.6 (m, 3H), 3.81 (s, 3H), 3.63 (brs, 2H), 2.25 (s, 3H).
2-Iodo-5-methylanisole (3b)29
To a solution of 3a (550 mg, 4 mmol) in 3 mL concentrated HCl at 0°C was added NaNO2 (320 mg, 4.6 mmol; dissolved in 5 mL H2O), and the mixture was stirred at 0 °C for 1 h. A solution of KI (950 mg, 5.7 mmol; dissolved in 10 mL H2O) was added dropwise at 0 °C, and the mixture was slowly warmed up to room temperature overnight with stirring. The mixture was extracted with ether and the combined organic layers were washed with brine, dried over anhydrous MgSO4, and evaporated under reduced pressure. The residue was purified by silica gel flash chromatography (hexane as eluent) to give the titled product as colorless oil (0.826 g, 84% yield): 1H MR (400 MHz, CDCl3) δ 7.62 (d, J = 7.9 Hz, 1H), 6.65 (s, 1H), 6.55 (d, J = 7.9 Hz, 1H), 3.87 (s, 3H), 2.33 (s, 3H).
2,2′-Dimethoxy-4,4′-dimethyl-l,l-biphenyl (3c)23
To a mixture of 10% Pd/C and Zn powder (100 mg, 1.2 mmol) in 5.2 mL water/acetone (1:1) was added 3b (100 mg, 0.4 mmol), and the resulting mixture was stirred at room temperature overnight. The desired product was obtained after silica gel flash chromatography as a white solid (45 mg, 46% yield): 1H NMR (400 MHz, CDCl3): δ 7.08 (d, J = 7.6 Hz, 2H), 6.77 (d, J = 7.6, 2H), 6.73 (s, 2H), 3.71 (s, 6H), 2.35 (s, 6H).
4,4′-Bis(bromomethyl)-2,2′-dimethoxy-1,1′-biphenyl (3)30
A mixture of 3c (50 mg, 0.2 mmol), NBS (71 mg, 0.4 mmol) and AIBN (6.5 mg, 0.04 mmol) in 2 mL CCl4 was refluxed for 8 h. After cooling down, the mixture was dissolved in CHCl3 and filtered. The filtrate was evaporated under reduce pressure, and the residue was recrystallized with CCl4/hexanes to give titled product as pale yellow crystals (25 mg, 31% yield): 1H NMR (400 MHz, CDCl3) δ 4.55 (s, 4H), 3.79 (s, 6H), 6.99 (s, 2H), 7.03 (d, J = 5.0 Hz, 2H), 7.19 (d, J = 5.0 Hz, 2H).
2,7-Bis(bromomethyl-9,10-dihydrophenanthrene (4)31
A mixture of dihydrophenanthrene (1.0 g, 5.5 mmol), paraformaldehyde (0.735 g, 24.5 mmol), 1.1 mL 85% phosphoric acid, 1.925 mL 48% HBr, and 2.2 mL 30% HBr in acetic acid was heated at 80 °C under nitrogen for 21 h. Afterwards, the reaction mixture was refluxed for 5 h before cooling down to room temperature. The gray solid was collected and washed with 5 mL acetone. The crude solid was recrystallized from benzene/hexanes to give the titled compound (360 mg, 36% yield): 1H NMR (400 MHz, CDCl3) δ 2.86 (s, 4H), 4.51 (s, 4H), 7.27 (s, 2H), 7.32 (d, J = 8.0 Hz, 2H), 7.70 (d, J = 8.0 Hz, 2H).
2,7-Bis(bromomethylphenanthrene (5)
A mixture of 4 (360 mg, 1.0 mmol), DDQ (315 mg, 1.4 mol) in 3 mL dry benzene was refluxed for 18 h. The solution was filtered through a layer of neutral alumina while still hot and rinsed with hot benzene. The filtrate was evaporated under reduced pressure and the residue was crystallized from benzene/hexanes to give the titled compound as pale-colored crystals (270 mg, 75% yield): 1H NMR (400 MHz, CDCl3) δ 8.64 (d, J = 8.6 Hz, 2H), 7.88 (d, J = 8.6 Hz, 2H), 7.67–7.73 (m, 4H), 4.72 (s, 4H).
p-Phenylene-3, 3′-bis(allylbromide) (6)32
To a solution of dimethyl-1,4-phenylenediacrylate (0.5 g, 2.0 mmol) in 10 mL THF at −78°C was added dropwise DIBAL (1.2 M in toluene, 10 mL), and the mixture was stirred overnight. The reaction was quenched by adding water followed by saturated ammonium chloride before extraction with ethyl acetate. The organic layer was separated, dried with MgSO4, and concentrated under reduced pressure to afford p-phenylene-3,3′-bis(allyl alcohol) as white flakes (330 mg, 85 % yield): 1H NMR (300 MHz, CDCl3) δ 4.21–4.23 (m, 4H), 6.33–6.40 (m, 2H), 6.56–6.61 (m, 2H), 7.35 (s, 4H). To a solution of p-phenylene-3,3′-bis(allyl alcohol) (15 mg, 0.08 mmol) in 2 mL anhydrous ether at 0 °C was added dropwise PBr3 (6 μL, 0.07 mmol), and the reaction mixture was stirred at 0 °C for 10 min and then at room temperature for 30 min. One mL of dichloromethane was added, and the organic layer was separated, washed with a saturated NaHCO3 solution, dried over anhydrous Na2SO4, and concentrated under reduced pressure to afford the titled compound (15 mg, 65% yield): 1H NMR (300 MHz, CDCl3) δ 4.17–4.19 (m, 4H), 6.37–6.42 (m, 2H), 6.59–6.65 (m, 2H), 7.35 (s, 4H).
Bis(bromomethyl)phenazine (8)
Phenazine derivative was synthesized through double Buchwald-Hartwig amination reaction as reported.24 Briefly, a mixture of bromoaniline (200 mg, 0.5 mmol), cesium carbonate (350 mg, 1.0 mmol), Pd(OAc)2 (6.0 mg, 0.025 mmol), and SPhos (20 mg, 0.084 mmol) in 5 mL anhydrous toluene was stirred at 120°C overnight. The mixture was then diluted with chloroform and filtered through a layer of Celite. The filtrate was concentrated to give bis(methyl)phenazine (60 mg, 54% yield): 1H NMR (500 MHz, CDCl3) δ 8.13 (d, J = 9.0 Hz, 2H), 8.00 (s, 2H), 7.68 (d, J = 9.0 Hz, 2H), 2.67 (s, 6H). A solution of bis(methyl)phenazine (50 mg, 0.24 mmol), NBS (84 mg, 0.48 mmol) and AIBN (8 mg, 0.2 equiv) in 3 mL CCl4 was refluxed overnight. After evaporating the solvent, the residue was subjected to silica gel flash chromatography using10% ethyl acetate/hexanes as eluent to afford the titled product (15 mg, 20% yield): 1H NMR (500 MHz, CDCl3) δ 8.26 (d, J = 8.5 Hz, 2H), 8.23 (d, J = 2.0 Hz, 2H), 7.91 (dd, J = 8.5, 2.0 Hz, 2H), 4.77 (s, 4H).
4.4. Peptide Cross-Linking by 1, 2, 3 and 7
Cross-linking reactions were carried out by incubating Noxa peptide with 1.5 equiv of 1 or 2 or 3 or 7 in acetonitrile/50 mM NH4HCO3 (1:1), pH 8.5, at a final peptide concentration of 1 mM. The mixture was incubated at room temperature for 1.5–2 hours. The reaction was monitored by mass spectrometry. After completion, solvents were evaporated and excess amount of the cross-linkers was removed by washing with diethyl ether. The cross-linked peptides were purified by preparative HPLC.
4.5. Peptide Cross-Linking by 4, 5, 6 and 8
Cross-linking reactions were carried out in eppendorf tubes by incubating Noxa peptide with 1.5 equiv of 4, 5, 6, or 8 in DMF at a peptide concentration of 10 mM. 20 equivalents of DIEA were added and the reaction was allowed to continue for 1 hour. The reaction mixture was diluted in ether to precipitate the peptide. The cross-linked peptides were purified by preparative HPLC.
4.6. Circular dichroism spectroscopy
Circular dichroism spectra were recorded with a JASCO J-715 CD spectrometer at room temperature using a 0.1-cm path length cuvette. The spectra were recorded in the wavelength range of 185–250 nm and averaged over 3 scans with a resolution of 0.5 nm, a bandwidth of 1.0 nm and a response time of 4 s. The sensitivity and scan rate of the spectrometer were set to 100 mdeg and 50 nm/min, respectively. All peptides were dissolved in PBS to the final concentration of 50 μM. The mean residue ellipticity was plotted.
4.7. FP assay and FACS analysis
FP assays and FACS analyses were performed as described previously.16
Acknowledgments
We acknowledge the Pardee Foundation and the Oishei Foundation (to Q.L.), and the National Institutes of Health (CA82197 to H.G.W.) for financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Verdine GL, Walensky LD. Clin Cancer Res. 2007;13:7264–7270. doi: 10.1158/1078-0432.CCR-07-2184. [DOI] [PubMed] [Google Scholar]
- 2.Yin H, Lee GI, Hamilton AD. Drug Discovery Research. John Wiley & Sons, Inc; 2006. pp. 281–299. [Google Scholar]
- 3.Huyghues-Despointes BM, Scholtz JM, Baldwin RL. Protein Sci. 1993;2:80–85. doi: 10.1002/pro.5560020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albert JS, Hamilton AD. Biochemistry. 1995;34:984–990. doi: 10.1021/bi00003a033. [DOI] [PubMed] [Google Scholar]
- 5.Gallivan JP, Dougherty DA. Proc Nat Acad Sci U S A. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Condon SM, Morize I, Darnbrough S, Burns CJ, Miller BE, Uhl J, Burke K, Jariwala N, Locke K, Krolikowski PH, Kumar NV, Labaudiniere RF. J Am Chem Soc. 2000;122:3007–3014. [Google Scholar]
- 7.Jackson DY, King DS, Chmielewski J, Singh S, Schultz PG. J Am Chem Soc. 1991;113:9391–9392. [Google Scholar]
- 8.Blackwell HE, Grubbs RH. Angew Chem Int Ed. 1998;37:3281–3284. doi: 10.1002/(SICI)1521-3773(19981217)37:23<3281::AID-ANIE3281>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 9.Madden MM, Rivera Vera CI, Song W, Lin Q. Chem Commun. 2009:5588–5590. doi: 10.1039/b912094g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cantel S, Le Chevalier Isaad A, Scrima M, Levy JJ, DiMarchi RD, Rovero P, Halperin JA, D’Ursi AM, Papini AM, Chorev M. J Org Chem. 2008;73:5663–5674. doi: 10.1021/jo800142s. [DOI] [PubMed] [Google Scholar]
- 11.Flint DG, Kumita JR, Smart OS, Woolley GA. Chem Biol. 2002;9:391–397. doi: 10.1016/s1074-5521(02)00109-6. [DOI] [PubMed] [Google Scholar]
- 12.Muppidi A, Wang Z, Li X, Chen J, Lin Q. Chem Commun. 2011;47:9396–9398. doi: 10.1039/c1cc13320a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Azzarito V, Long K, Murphy NS, Wilson AJ. Nat Chem. 2013;5:161–173. doi: 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]
- 14.Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH, Olson KA, Kesavan K, Gangurde P, Mukherjee A, Baker T, Darlak K, Elkin C, Filipovic Z, Qureshi FZ, Cai H, Berry P, Feyfant E, Shi XE, Horstick J, Annis DA, Manning AM, Fotouhi N, Nash H, Vassilev LT, Sawyer TK. Proc Nat Acad Sci U S A. 2013;110:E3445–E3454. doi: 10.1073/pnas.1303002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walensky LD, Bird GH. J Med Chem. 2014 doi: 10.1021/jm4011675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muppidi A, Doi K, Edwardraja S, Drake EJ, Gulick AM, Wang HG, Lin Q. J Am Chem Soc. 2012;134:14734–14737. doi: 10.1021/ja306864v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muppidi A, Li X, Chen J, Lin Q. Bioorg Med Chem Lett. 2011;21:7412–7415. doi: 10.1016/j.bmcl.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muppidi A, Zhang H, Curreli F, Li N, Debnath AK, Lin Q. Bioorg Med Chem Lett. 2014;24:1748–1751. doi: 10.1016/j.bmcl.2014.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bird GH, Gavathiotis E, Labelle JL, Katz SG, Walensky LD. ACS Chem Biol. 2014;9:831–837. doi: 10.1021/cb4003305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madden MM, Muppidi A, Li Z, Li X, Chen J, Lin Q. Bioorg Med Chem Lett. 2011;21:1472–1475. doi: 10.1016/j.bmcl.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang F, Sadovski O, Xin SJ, Woolley GA. J Am Chem Soc. 2007;129:14154–14155. doi: 10.1021/ja075829t. [DOI] [PubMed] [Google Scholar]
- 22.Jo H, Meinhardt N, Wu Y, Kulkarni S, Hu X, Low KE, Davies PL, DeGrado WF, Greenbaum DC. J Am Chem Soc. 2012;134:17704–17713. doi: 10.1021/ja307599z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Venkatraman S, Li CJ. Org Lett. 1999;1:1133–1135. [Google Scholar]
- 24.Winkler JD, Twenter BM, Gendrineau T. Heterocycles. 2012;84 doi: 10.3987/COM-11-S(P)76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toniolo C, Polese A, Formaggio F, Crisma M, Kamphuis J. J Am Chem Soc. 1996;118:2744–2745. [Google Scholar]
- 26.Bezer S, Matsumoto M, Lodewyk MW, Lee SJ, Tantillo DJ, Gagne MR, Waters ML. Org Biomol Chem. 2014;12:1488–1494. doi: 10.1039/c3ob41421c. [DOI] [PubMed] [Google Scholar]
- 27.Madani F, Lindberg S, Langel U, Futaki S, Graslund A. J Biophys. 2011;2011 doi: 10.1155/2011/414729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watkins CL, Brennan P, Fegan C, Takayama K, Nakase I, Futaki S, Jones AT. J Control Release. 2009;140:237–244. doi: 10.1016/j.jconrel.2009.04.028. [DOI] [PubMed] [Google Scholar]
- 29.Coleman RS, Guernon JM, Roland JT. Org Lett. 2000;2:277–280. doi: 10.1021/ol991238z. [DOI] [PubMed] [Google Scholar]
- 30.Gruber J, Li RWC. Eur Polymer J. 2000;36:923–928. [Google Scholar]
- 31.Khalaf AI, Pitt AR, Scobie M, Suckling CJ, Urwin J, Waigh RD, Fishleigh RV, Young SC, Wylie WA. Tetrahedron. 2000;56:5225–5239. doi: 10.1021/jm990620e. [DOI] [PubMed] [Google Scholar]
- 32.Itoh T, Kanbara M, Ohashi M, Hayase S, Kawatsura M, Kato T, Miyazawa K, Takagi Y, Uno H. J Fluoro Chem. 2007;128:1112–1120. [Google Scholar]