

Summary
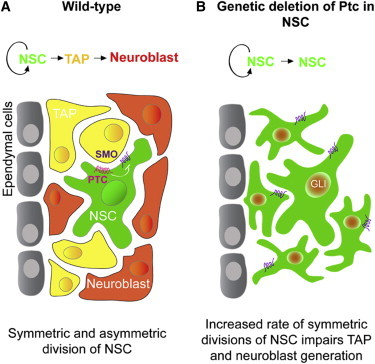
In the adult brain, self-renewal is essential for the persistence of neural stem cells (NSCs) throughout life, but its regulation is still poorly understood. One NSC can give birth to two NSCs or one NSC and one transient progenitor. A correct balance is necessary for the maintenance of germinal areas, and understanding the molecular mechanisms underlying NSC division mode is clearly important. Here, we report a function of the Sonic Hedgehog (SHH) receptor Patched in the direct control of long-term NSC self-renewal in the subependymal zone. We show that genetic conditional activation of SHH signaling in adult NSCs leads to their expansion and the depletion of their direct progeny. These phenotypes are associated in vitro with an increase in NSC symmetric division in a process involving NOTCH signaling. Together, our results demonstrate a tight control of adult neurogenesis and NSC renewal driven by Patched.
Graphical Abstract
Highlights
Ptc inactivation in the adult NSCs expands the NSC pool and depletes their progeny
Neurogenesis blockade is related to the increase of NSC symmetric division
PTC exerts a central role via NOTCH, in the regulation of adult NSC self-renewal
In the adult brain, self-renewal is essential for the persistence of neural stem cells throughout life. Traiffort, Ruat, and colleagues report that the Sonic Hedgehog receptor Patched controls long-term self-renewal of these cells in the subependymal zone by increasing their symmetric division in a process involving NOTCH signaling.
Introduction
Neural stem cells (NSCs) remain in specialized niches of the brain over the lifespan and give rise to new neurons that integrate neural circuits. They are proposed to enable regeneration of injured tissue. The subependymal zone (SEZ), one of the main neurogenic niches in the adult mammalian brain, is comprised of astrocyte-like NSCs corresponding to a heterogeneous cell population. Among the identified subpopulations, active NSCs (aNSCs) expressing the epidermal growth factor receptor (EGFR) give rise to transit amplifying progenitors (TAPs). Most of these cells generate neuroblasts, migrating along the rostral migratory stream (RMS) and differentiating into granule and periglomerular interneurons in the olfactory bulb (OB). Quiescent NSCs (qNSCs) do not express the EGFR and are resistant to antimitotic drugs or irradiation. They are implicated in SEZ neurogenesis replenishment through aNSCs and TAPs (Costa et al., 2011; Pastrana et al., 2011; Ponti et al., 2013). Establishing the identity and lineage of SEZ stem cells is under intense studies, but the regulatory mechanisms involved in their self-renewal and differentiation still need more investigation. Only a few signals determining these distinct behaviors have been discovered. For instance, bone morphogenetic protein signals and EGFR-mediated inactivation of NOTCH signaling in NSCs are required for progression of the NSC progeny toward the neurogenic lineage (Aguirre et al., 2010; Colak et al., 2008), whereas the pigment epithelium-derived factor was proposed to regulate the NSC expansion (Karpowicz et al., 2009).
The Sonic Hedgehog (SHH) pathway is active in the adult SEZ, where it has been proposed to regulate cell proliferation (Ruat et al., 2012; Ahn and Joyner, 2005; Machold et al., 2003) and to modulate the migration of neuroblasts exiting the niche (Angot et al., 2008). The mosaic inactivation of the Smoothened (SMO) receptor in cell types expressing the neuroepithelial marker NESTIN suggested the requirement of this transducer of SHH signal for maintenance of the NSC population (Balordi and Fishell, 2007). Patched (PTC) is the main SHH receptor and is considered an antagonist of the pathway (Briscoe and Thérond, 2013). Embryonic deletion of Ptc in multipotent stem cells of human glial fibrillary acidic protein (hGFAP)-Cre;PtcC/C mice results in medulloblastoma. The tumors do not manifest until the cells have committed to the neuronal lineage (Yang et al., 2008). However, the effects of Ptc inactivation in adult NSCs of the SEZ remain yet unknown.
Here, we used a tamoxifen-inducible Cre transgene under the control of the astrocyte-specific glutamate transporter (GLAST) expressed in astrocyte-like NSCs (Mori et al., 2006) and took advantage of a conditional Ptc knockout (Ptcfl/fl) mouse line that has loxP recombinase recognition sites within the Ptc gene (Uhmann et al., 2007). We show that Ptc inactivation in the adult NSCs leads to a dramatic decrease of the neurogenic process and to a marked expansion of NSCs in the SEZ. Neurogenesis blockade was related to a shift in NSC division mode from asymmetric to symmetric, leading to a decrease in the differentiation process and involving NOTCH signaling. Thus, we report a role for PTC in the regulation of adult NSC self-renewal mechanisms.
Results
Conditional Deletion of Ptc in GLAST-Expressing Cells Promotes Endogenous Activation of HH Signaling in the SEZ Niche
To investigate the role of PTC in NSCs of the adult SEZ niche, we used a genetic approach aimed at conditionally deleting this receptor in the astroglial population in which we formerly demonstrated its expression (Figure 1; Figure S1 available online). Ptc transcripts and proteins were evidenced in the SEZ niche. Confocal analysis using a specific PTC antiserum (Bidet et al., 2011; Figure S1) showed PTC expression in a subset of GFAP+ cells (36% ± 5%) (Figure 1A). Moreover, analysis of the Ptc+/LacZ mice (Goodrich et al., 1997) led to visualizing β-galactosidase (β-GAL+)/PTC+ cells coexpressing GLAST, a marker of astrocyte-like NSCs (Figure 1B). Therefore, we took advantage of the Glast-CreERT2 mouse line (Mori et al., 2006) expressing a tamoxifen-inducible Cre in the locus of Glast to induce Cre recombinase activity specifically in adult astroglia and NSCs. Glast-CreERT2 mice were crossed with the Rosa26 reporter line, R26R-YFP (yellow fluorescent protein) (Srinivas et al., 2001). In the Glast-CreERT2;PtcWT/WT;R26R-YFP offspring (so-called YFP-control), YFP expression is initiated upon tamoxifen-mediated activation of the Cre recombinase and permanently marks all progeny of the recombined cells. At 2 months after tamoxifen treatment, a high number of YFP+ cells are detected in the SEZ that coexpress the astrocyte markers GLAST and GFAP (Figure 1C). Confocal analysis of the SEZ niche indicated that YFP reporter is expressed in 85% ± 5% of GLAST+ cells. Recombination also occurred in neurogenic astroglial-like cells of the hippocampal subgranular zone and in other astrocyte populations throughout the brain as expected from Glast expression profile (Mori et al., 2006) (Figure S2A).
Figure 1.
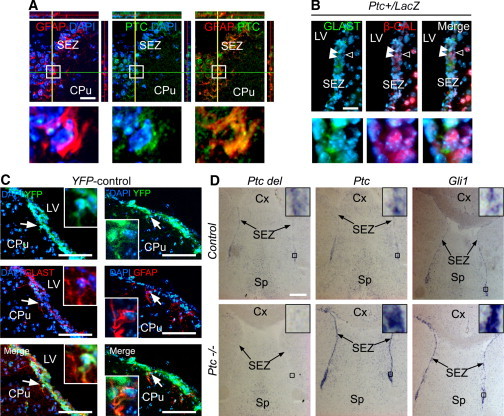
Inducible Deletion of Ptc in GLAST-Expressing Cells from the Adult Brain SEZ
(A) Immunohistofluorescence (IHF) indicating the presence of a population of PTC+ cells (green) coexpressing the astroglial marker GFAP (red) in the dorsal SEZ. Magnifications of the boxed areas are presented below each panel.
(B) Double IHF performed on brain slices from the heterozygous Ptc+/LacZ mouse that expresses the LACZ reporter in one allele of Ptc. Anti-β-GAL (red) and GLAST (green) antibodies are used. Magnifications of the area indicated by arrowheads are presented below each panel and show β-GAL+ nuclei surrounded (white arrowheads) or not (black and white arrowhead) by GLAST+ signal.
(C) Fluorescent signals of YFP together with GLAST and GFAP in the SEZ of Glast-CreERT2;R26R-YFP (YFP-control) mice 2 months after tamoxifen administration. The white arrow indicates a cell coexpressing YFP with alternatively GLAST or GFAP markers and magnified in the inset. Nuclei are in blue (DAPI) in (A)–(C).
(D) In situ hybridization (ISH) of two HH target genes, Ptc and Gli1, in the SEZ of mice in which Ptc exons 8 and 9 are flanked by loxP sites. In Ptc−/− (Glast-CreERT2;Ptcfl/fl) mice, exons 8 and 9 were depleted by tamoxifen-mediated activation of Glast-CreERT2 that is expressed from the endogenous Glast locus. Tamoxifen-treated Ptcfl/fl mice served as controls (control). Two months after tamoxifen treatment, the animals were subjected to analysis. The Ptc del riboprobe encompasses nucleotides of Ptc exons 8 and 9 and thus detects only the WT Ptc transcripts. The Ptc riboprobe corresponds to half of the full-length Ptc transcript, including the C-terminal end. In the control mice, Ptc del, Ptc, and Gli1 allow the detection of the faint labeling previously reported in the SEZ with a stronger signal in the ventral niche. In Ptc−/− mice, strong Ptc and Gli1 signals are detected in the ventral and dorsal SEZ, whereas no signal is observed when the Ptc del riboprobe is used.
CPu, caudate putamen; Cx, cerebral cortex; LV, lateral ventricle; Sp, septal area. Scale bars, 50 μm (A and B), 100 μm (C), and 200 μm (D).
See also Figures S1 and S2.
Ptc was inactivated in GLAST+ cells by crossing alternatively the Glast-CreERT2 or YFP-control with Ptcfl/fl mouse lines allowing the excision of exons 8 and 9 encoding a domain involved in SHH binding (Uhmann et al., 2007). Adult Glast-CreERT2;Ptcfl/fl (so-called Ptc−/−) mice and littermate Ptcfl/fl controls (so-called control) were treated with tamoxifen and analyzed 10 days, or 2, 6, or 12 months later. Despite a loss of body weight in Ptc−/− animals treated with tamoxifen 1 year before, their brain was macroscopically indistinguishable from the brain of the controls. Notably, no tumor could be detected (data not shown). Analyses of Gli1 or Ptc transcription were used as readouts of Hedgehog (HH) pathway activation (Briscoe and Thérond, 2013). In Ptc−/− mice, both genes were highly upregulated in ventral and dorsal SEZs, indicating a sustained activation of HH signaling compared to controls. Additional control experiments demonstrated the specificity of both Gli1 and Ptc transcription in Ptc−/− animals (Figure 1D; see Figures S2B and S2C). Altogether, these results demonstrate that Cre-mediated recombination allows the efficient deletion of Ptc in GLAST+ cells, leading to the persistent activation of HH signaling in the adult SEZ niche.
Ptc Deletion Affects Both Fast and Slow Proliferation and Decreases Neurogenesis
To determine the functional meaning of Ptc deletion in GLAST+ cells in the SEZ, we first examined fast proliferating cells (TAPs and neuroblasts) by administrating to the animals two bromodeoxyuridine (BrdU) pulses at 2 hr intervals. Pulsing was done at various time points (10 days, and 2, 6, and 12 months) after tamoxifen injection. The animals were sacrificed after the last BrdU pulse (Figures 2A and 2B). In control animals, BrdU+ cell number decreases between 2 and 6 months after treatment, presumably reflecting a regular decline associated with aging as previously shown by Shook et al. (2012). Interestingly, BrdU+ cells were significantly decreased in the SEZ of Ptc−/− mice when compared to controls over the course of the study. At 10 days, the 27% decrease was already highly significant and reached 54% at 2 months. The drop in fast proliferating cells was still observed after 6 months and led to their almost complete disappearance at 1 year in Ptc−/− animals, likely related to both the observed phenotype and the regular decrease occurring in aging. We excluded cell survival decrease because the number of terminal deoxynucleotidyl transferase dUTP nick end labeling-positive cells was not significantly modified in Ptc−/− SEZs over the course of the study (Figure S3). Remarkably, the SEZ visualization of EGFR+ cells corresponding to TAPs and a low percentage of transitional stages (Pastrana et al., 2011; Ponti et al., 2013) revealed a dramatic 93% decrease of this cell population at 12 months in the Ptc mutants (Figures 2C and 2D). Polysialylated-neuronal cell adhesion molecule (PSA-NCAM+)-positive cell number was reduced by more than 90% in Ptc−/− mice, which is consistent with a high rate of Ptc excision in these animals, resulting in the quite complete absence of SEZ-migrating neuroblasts (Figures 2C and 2D). In contrast, the density of Gfap+ cells that correspond to astrocytes and NSCs in the SEZ showed a robust 62% increase in Ptc−/− animals analyzed at 12 months (Figures 2E and 2F).
Figure 2.
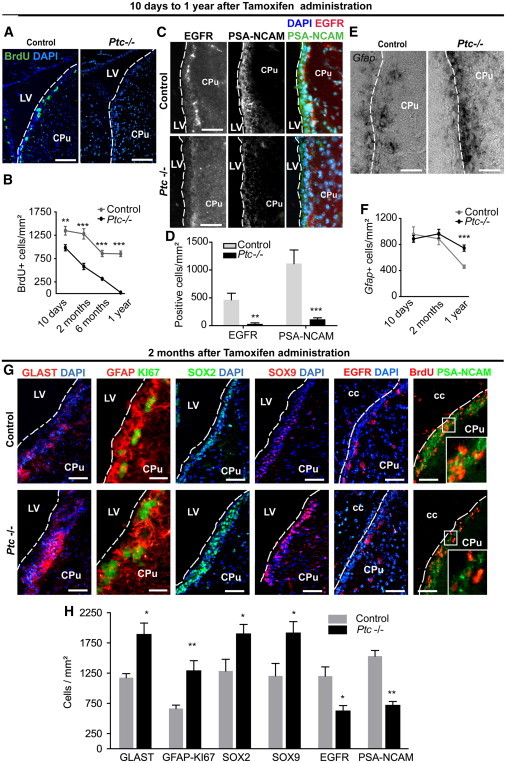
Alteration of Cell Proliferation and Phenotype in the Adult SEZ/RMS upon Ptc Deletion
(A) Detection of fast proliferating cells by IHF using anti-BrdU antibody in brain slices of Ptc−/− and control animals treated 1 year before with tamoxifen and analyzed 2 hr after short BrdU pulses.
(B) Histogram depicting the number of BrdU+ and GFAP+ cells quantified in the SEZ at the indicated time after tamoxifen treatment.
(C and D) IHF visualizing the expression of EGFR (red) and PSA-NCAM (green) in the SEZ of control and Ptc−/− mice 1 year after tamoxifen administration and quantification.
(E and F) ISH showing the Gfap gene expression in the SEZ of control and Ptc−/− mice 1 year after tamoxifen administration and quantification for the various time points.
(G) Fluorescent micrographs in the adult SEZ of control and Ptc−/− mice 2 months after tamoxifen administration. The micrographs depict immunostainings for GLAST and GFAP/KI67 as astrocyte and stem cell markers, SOX2 and SOX9 as transcription factors necessary for NSC formation, and EGFR and PSA-NCAM as markers of TAPs and migrating neuroblasts, respectively. In the last panels, BrdU labels the fast proliferating cell populations. The dashed lines delineate the wall of the lateral ventricle except for the last two panels on the right where they delineate the cc.
(H) Histograms describing the number of each cell type per square millimeter. Values are the mean ± SEM from three to four animals, two to four slices per animal.
CPu, caudate putamen; LV, lateral ventricle. ∗p ≤ 0.05; ∗∗p ≤ 0.005; ∗∗∗p ≤ 0.0005. Scale bars, 100 μm (A and G), 25 μm (GFAP/KI67 pictures in G), and 50 μm (C and E).
See also Figure S3.
Consistently, at 2 months, quantification of SEZ cell populations indicated that the Ptc deletion increases by almost 2-fold the number of GLAST+ cells corresponding to both niche astrocytes and NSCs, and the number of KI67+/GFAP+ NSCs. TAPs and neuroblasts were decreased by 2-fold as indicated by EGFR and PSA-NCAM stainings (Figures 2G and 2H). Moreover, in the absence of Ptc, we found a marked increase in the number of sex-determining region Y-box 2+ (SOX2+) and SOX9+ cells both expressed in NSCs and TAPs (Kazanis et al., 2010; Scott et al., 2010). Because the TAP population is reduced, the increase in SOX2+ and SOX9+ cells may consistently reflect the increase of NSC density. Together, these data show that the Ptc deletion is thus associated with an increase in the number of astrocyte-like NSCs in the SEZ and a decrease in the number of TAPs and migrating neuroblasts.
The progeny of the recombined cells was traced by following the expression of YFP in Glast-CreERT2;Ptcfl/fl;R26R-YFP (so-called YFP-Ptc−/−) and YFP-control mice treated with tamoxifen and analyzed 2 months later. As expected, strong YFP signals were detected in the SEZ-RMS-OB system of YFP-control mice (Figure 3A) and also in the corpus callosum (cc), where they reflect both the population of newly generated oligodendroglial cells derived from the SEZ (Gonzalez-Perez and Alvarez-Buylla, 2011) and the local GLAST-expressing astroglial cells present in the fiber tracts (Figure S4A). In YFP-Ptc−/− animals, YFP expression level was dramatically affected except in the SEZ, where the density of YFP+ cells was only slightly reduced without reaching significance (Figure 3B). The apparent divergence between the unaltered YFP+ cell density and the substantial reduction in the number of BrdU+ cells in the SEZ may be explained for the former by a compensatory mechanism between the increase in NSCs and the decrease in TAPs/neuroblasts and for the latter by the collapse of the dynamics of TAPs and neuroblasts. Indeed, TAPs have been proposed to divide three times before turning into neuroblasts and neuroblasts once or twice before leaving the SEZ (Ponti et al., 2013). In YFP-Ptc−/− animals, quantification of YFP+ cells in the OB granular (GrO) and glomerular (Gl) layers (Figure 3B; see also Figures S4A–S4C) and in the cc (Figures S4D–S4F) indicated a 69%, 36%, and 51% decrease, respectively. Interestingly, in the granular layer, YFP+ cells are decreased in both deep and superficial layers with, however, a slightly higher decrease in the deep layer. Thus, Ptc deletion affects both the ventral and dorsal parts of the SEZ known to contribute to the production of deep and superficial granule cells, respectively. This suggests that SHH signaling displays a broader effect than initially proposed alongside the whole SEZ (Ihrie et al., 2011). Moreover, the fluorescent signal was also dramatically reduced in the RMS connecting the SEZ to the OB (Figure 3A).
Figure 3.
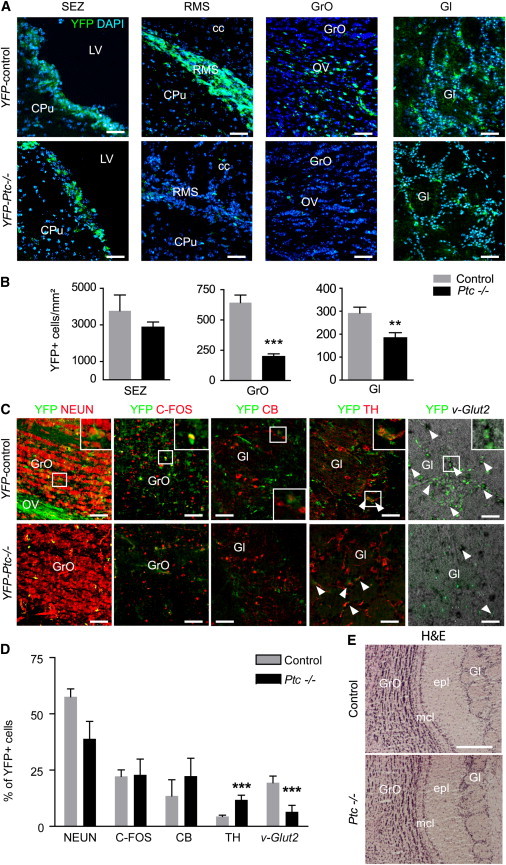
Cell Fate Analysis after Deletion of Ptc in the Adult SEZ
(A) Tracing of Glast+ cells in YFP-Ptc−/− and YFP-control mice treated with tamoxifen 2 months before. YFP is detected in the SEZ, RMS, and OB. A lower density of YFP+ cells is observed in the RMS and OB of YFP-Ptc−/− mice compared to YFP-controls. In contrast, no difference is observed in the SEZ.
(B) Quantification of the density of YFP+ cells in the SEZ and OB layers.
(C) Phenotype of the targeted cells at their final destination upon conditional Ptc deletion in the SEZ. IHF (NEUN, C-FOS, CB, and TH) or ISH (v-Glut2) in slices from YFP-control or YFP-Ptc−/− mice at the level of the OB is shown. White arrowheads indicate double-labeled cells.
(D) Histogram presenting the percentage of each cell phenotype within the YFP population.
(E) Brain slices performed at the level of the OB from control and Ptc−/− mice treated with tamoxifen 1 year before and stained with hematoxylin and eosin (H&E). No alteration of the multilayered cellular architecture of the OB is observed. Values are the mean ± SEM from three to four animals, two to four slices per animal.
∗∗p ≤ 0.005; ∗∗∗p ≤ 0.0005. Scale bars, 50 μm (A and C) and 200 μm (E).
See also Figure S4.
Because these results indicated a decrease in the generation of new cells in the SEZ, we further analyzed the phenotype of SEZ-derived cells at their final destinations: the OB (Figures 3C and 3D), and the cc (Figure S4C). In the GrO layer of the OB where the newly generated cells mature as GABAergic interneurons, the percentage of YFP+ cells expressing the neuronal marker NEUN or C-FOS, a transcription factor rapidly regulated by neuronal activity, was unchanged in Ptc−/− mice. These data indicated that there was no difference in the proportion of neurons fully integrated into the GrO layer. We then analyzed the percentage of YFP+ cells expressing tyrosine hydroxylase (TH), calbindin, or vesicular glutamate transporter 2 (vGlut2), which labels subsets of SEZ-derived new cells integrating the Gl layer (Brill et al., 2009). In the conditional Ptc mutants, the percentage of newly generated calbindin+ cells was not significantly modified. In contrast, the percentages of YFP+ cells coexpressing either TH or vGlut2 were 3-fold higher or lower, respectively. Remarkably, at 1 year post tamoxifen, the multilayered cellular architecture of the OB was not altered in Ptc−/− compared to control (Figure 3E). In the cc, a 2-fold lower proportion of YFP+ cells expressing OLIG2 was observed in YFP-Ptc−/− animals (Figure S4C), suggesting that Ptc deletion decreases the ability of precursor cells to give rise to oligodendrocytes. As a whole, these data establish that the generation of new precursors is highly decreased in the SEZ of the conditional Ptc mutants. Although these precursors still have the ability to differentiate into neurons and oligodendrocytes in the OB and cc, respectively, they nevertheless display discrete variations of fate determination.
Ptc Deficiency in SEZ-Derived Neural Progenitors Changes Cell Differentiation Capacity in the Neurosphere Assay
To address the consequences of Ptc deficiency in SEZ progenitors, we used the neurosphere culture to analyze stem and progenitor cell properties between Ptc-deficient and control cells (Pastrana et al., 2011). We observed no difference in sphere formation of cells derived from Ptc−/− SEZs 2 months after tamoxifen injection, despite Gli1 upregulation observed in Ptc-deficient neurospheres (Figures 4A and 4B). The clonal expansion represented as the slope of the line indicated that Ptc deficiency does not alter the apparent capacity of self-renewal of the neurospheres. However, when we quantified the subpopulations of neurospheres according to their diameter, we observed a higher number of large neurospheres (≥100 μm) derived from the Ptc−/− SEZ (Figure 4C). This observation may suggest that more stem cells are present in Ptc-deficient spheres because it was previously proposed that aNSCs give rise to large spheres, whereas progenitors lead to smaller ones (Pastrana et al., 2011). This is also consistent with a decrease in TAPs in the SEZ of these animals as previously observed.
Figure 4.
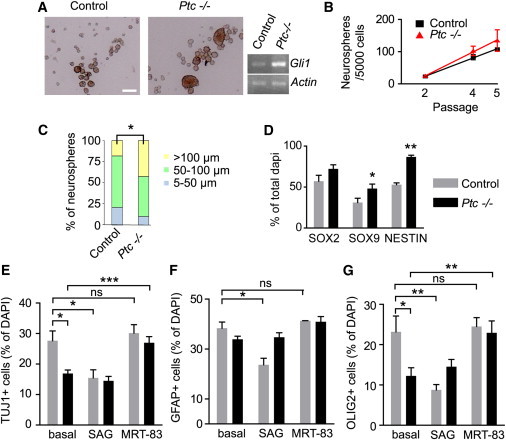
Sustained HH Signaling Activation in SEZ-Derived Neural Progenitors Maintains NSC Multipotency in the Neurosphere Assay
(A and B) SEZ neurospheres derived from Ptc−/− mice analyzed 2 months after tamoxifen administration can be expanded in a similar manner as controls and display Gli1 upregulation. The neurosphere numbers obtained at each passage were analyzed via a linear regression model. The rate of clonal expansion is represented as the slope of the lines (Ptc−/− 0.256 ± 0.047 versus control 0.226 ± 0.016; p > 0.05).
(C) Large sphere number (>100 μm in diameter) is slightly, but significantly, higher in Ptc−/− compared to controls (p = 0.03). Values are expressed as the percentage of the total number of spheres generated from 5,000 plated cells (total counted spheres: n = 138 for Ptc−/−, n = 98 for controls).
(D) Evaluation of the expression of progenitor cell markers (SOX2, SOX9, and NESTIN) in cells dissociated from neurospheres and then cultured for 12 hr in the absence of growth factors. Quantification of each cell type indicates a significant increase in the number of SOX9+ and NESTIN+ cells. The mean numbers of counted cells per marker are 227 ± 67 and 356 ± 147 for Ptc−/− and controls, respectively.
(E–G) Histograms depicting the differentiation potential of neurospheres from Ptc−/− and control mice cultured for 7 days without growth factors in the absence or the presence of pharmacological HH modulators, including the SMO agonist SAG (0.3 μM) and antagonist MRT-83 (3 μM). Differentiated cells were analyzed by IHF using TUJ1, GFAP, and OLIG2 antibodies to characterize neuronal (E), astroglial (F), and oligodendroglial (G) cells, respectively. In the absence of drugs, a significant decrease in the percentage of TUJ1+ and OLIG2+ cells is observed in Ptc−/− compared to control cells. The mean numbers of counted cells for each marker are 1,699 ± 309 and 3,822 ± 128 for Ptc−/− and controls, respectively. The data are expressed as the mean ± SEM of the percentage of DAPI+ nuclei, which express each marker and are derived from three independent experiments.
∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.005; ns, nonsignificant. Scale bar, 100 μm.
See also Figure S5.
When dissociated neurospheres were cultured for 12 hr in the absence of growth factors, significantly more cells were positive for SOX9 in the Ptc-deficient neurospheres when compared to the controls (Figure 4D; see also Figure S5A), consistent with our in vivo data and also with the upregulation of SOX9 observed upon HH activation (Scott et al., 2010). In contrast, SOX2 expression did not significantly differ between the groups, whereas the number of NESTIN+ neuroepithelial cells was increased by 1.6-fold, indicating that the Ptc deficiency has inhibited differentiation. To further evaluate the multipotency of Ptc−/− NSCs, the neurospheres were cultured for 7 days in the absence of growth factors to allow differentiation into neurons, oligodendrocytes, and astrocytes. Although Ptc−/− NSCs maintain their multipotency, their ability to differentiate was modified (Figures 4E–4G; see also Figure S5B). The percentage of TUJ1+ neuroblasts and OLIG2+ oligodendrocytes (only OLIG2+ GFAP− cells were counted to avoid including immature OLIG2+ GFAP+ astrocytes as previously reported in Marshall et al., 2005) was significantly reduced in Ptc-deficient cells, whereas the number of GFAP+ astrocytes was not modified by the Ptc deletion. Then, we addressed the question whether the effects observed above in Ptc-deficient cells could be modulated through SMO (Figures 4E–4G). In agreement with the involvement of SMO activation in Ptc−/− phenotype, the number of TUJ1+ neuroblasts, OLIG2+ oligodendrocytes, and GFAP+ astrocytes was reduced in cells derived from control mice upon addition of Smoothened agonist (SAG), a reference SMO activator (Chen et al., 2002). In contrast, application of the SMO antagonist MRT-83 (Roudaut et al., 2011) did not modify the generation of any cell types, suggesting that HH signaling is already antagonized by PTC. However, application of MRT-83 increased the capacity of Ptc−/− cells to differentiate into neuroblasts, oligodendrocytes, and astrocytes. Thus, the rescue of the Ptc−/− phenotype by SMO blockade is likely associated with the relief of PTC constitutive inhibitory effect on SMO. Importantly, Ptc deficiency decreases the total number of cells reaching the differentiated state, although it does not prevent cells from achieving terminal multilineage differentiation.
Ptc Deficiency in SEZ-Derived Neural Progenitors Promotes Symmetric GLAST+/EGFR− NSC Pairs through NOTCH Signaling and Reduces Cell-Cycle Exit of TAPs
The inactivation of Ptc in GLAST+ cells in the adult SEZ resulting in an increase of NSCs prompted us to investigate the potential crosstalk between HH and NOTCH signaling, which has been shown to regulate cell cycle to balance NSC maintenance during corticogenesis (Dave et al., 2011) and to be essential for the maintenance of NSCs (Aguirre et al., 2010; Imayoshi et al., 2013). First, we used the clonal pair cell assay adapted from the previously described protocol of Dave et al. (2011). We plated primary cells directly after their isolation from the SEZ, and we maintained them in culture 24–36 hr to allow cell division. We used GLAST and EGFR antibodies to label the stem cells and their direct progeny, respectively. SEZ-derived cells isolated from wild-type (WT) mice were treated with the SMO agonist SAG in the presence or absence of the inhibitor of γ-secretase, LY411575, which suppresses proteolysis-dependent activation of endogenous NOTCH receptors (Fauq et al., 2007). SAG significantly promotes symmetric GLAST+/EGFR− pairs at the expense of GLAST+/EGFR+ pairs compared to the basal condition. The SAG-mediated effect was counteracted by LY411575 treatment as well as by the SMO antagonist MRT-83 (Figures 5A and 5B). This indicates that HH-dependent generation of symmetric GLAST+/EGFR− pairs, likely corresponding to an activated state of NSCs different from those that are GLAST+/EGFR+, is also NOTCH dependent. Then, we performed semiquantitative RT-PCR analysis in SVZ-derived neurospheres from conditional Ptc−/− and control animals treated with tamoxifen for 2 months. Notch1 and two of its canonical effectors, the transcription factors Hes1 and Hes5 (Imayoshi et al., 2013), are upregulated when Ptc is conditionally deleted (Figure 5C). These data indicate that the NOTCH signaling pathway is activated upon Ptc deletion.
Figure 5.
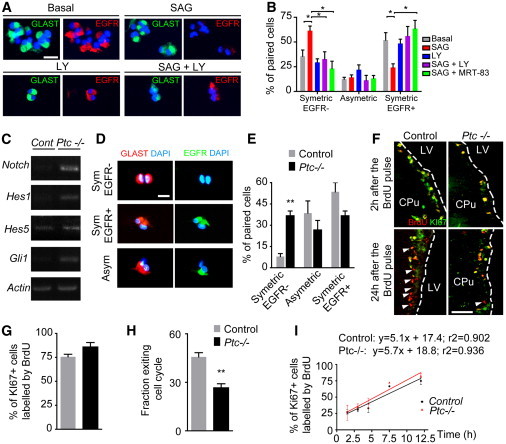
Ptc Deficiency Increases NSC Symmetric Division by Upregulating the NOTCH Pathway
(A) The clonal paired-cell assay is performed on SEZ-derived cells isolated from WT animals. The cells are cultured for 24–36 hr in the presence of the indicated drugs. SAG, LY, and MRT-83 were used at 0.3, 10, and 3 μM, respectively. The paired cells are labeled using GLAST (green) and EGFR (red) as markers of NSCs and TAPs, respectively. Nuclei are in blue (DAPI).
(B) Quantitative analysis of the percentage of each type of division indicates that SAG significantly increases the number of symmetric GLAST+/EGFR− pairs compared to the basal condition. This effect is counteracted by treatments with the SMO and NOTCH antagonists, MRT-83 and LY, respectively. ∗p ≤ 0.05.
(C) The upregulation of Gli1, Notch1, and the NOTCH effectors Hes1 and Hes5 is detected by semiquantitative RT-PCR in SEZ-derived progenitors isolated from adult Ptc−/− mice when compared to control animals. β-actin was used to assess input RNA levels.
(D) The clonal paired-cell assay performed on SEZ-derived precursors isolated from adult Ptc−/− and control mice indicates that GLAST-expressing cells from Ptc−/− more frequently generate two GLAST+ precursors after 24–36 hr when compared to control cultures.
(E) Quantitative analysis.
(F) BrdU (red) and KI67 (green) labeling of SEZ slices derived from Ptc−/− and control mice that had received tamoxifen 2 months before and were analyzed 2 or 24 hr after a single BrdU injection. The yellow fluorescence corresponds to double-labeled cells. In the bottom panels, the white arrowheads indicate the BrdU+KI67− cells.
(G and H) The histograms show the percentage of BrdU+ cells in the population of cycling cells 2 hr after the BrdU pulse (G) and the percentage of BrdU+ cells that left the cell cycle (KI67−) 24 hr after the BrdU pulse (H). Values are mean ± SEM from three to four animals, two to four slices per animal.
(I) Labeling index of cycling cells (i.e., the percentage of KI67+ cells that have incorporated BrdU) in control and Ptc−/− SEZ-derived cells. The equations of the linear regressions for each group indicate no significant difference in the slopes and consequently similar cell-cycle lengths. For cell cultures, values are mean ± SEM from three experiments.
∗∗p ≤ 0.01. Scale bars, 25 μm (A), 50 μm (D), and 100 μm (F). LY, LY411575.
We next tested if the above-reported effect on symmetric GLAST+/EGFR− NSCs observed upon pharmacological activation of SMO could be observed upon genetic deletion of Ptc in the adult NSCs. In cell cultures derived from Ptc−/− mice that received a tamoxifen injection 2 months before, a 4- to 5-fold increase of symmetric GLAST+/EGFR− pairs was detected at the expense of both the asymmetric pairs and symmetric GLAST+/EGFR+ pairs compared to the controls (Figures 5D and 5E). Thus, Ptc deficiency in adult SEZ-derived NSCs favors the generation of symmetric GLAST+/EGFR− NSCs. The absence of significant decrease in the percentage of symmetric GLAST+/EGFR+ pairs suggests that the pharmacological activation of SMO in WT neurospheres is not fully comparable to the genetic deletion of Ptc regarding the mechanism of self-renewal of GLAST+/EGFR− NSCs and aNSCs.
We determined the proportion of precursor cells labeled by a 2 hr pulse of BrdU in the population of cycling cells expressing KI67 in vivo in the SEZ of animals that received tamoxifen 2 months before. A 2 hr pulse of BrdU essentially reflects the proliferation of aNSCs, TAPs, and neuroblasts (Ponti et al., 2013). This protocol led us to determine that the labeling index was not modified by Ptc inactivation (Figures 5F and 5G). We also evaluated the number of cells exiting the cell cycle when animals were killed 24 hr after BrdU injection. The quantification of the number of proliferating cells remaining in the cell cycle 24 hr after BrdU incorporation (cells that are still KI67+) showed that a nearly 2-fold lower percentage of Ptc-deficient cells exit the cell cycle when compared to the control (Figure 5H). By using the BrdU cumulative approach, we also addressed the question of a potential difference in the cell-cycle length of NSCs when Ptc is deleted. We cultured SEZ-derived cells from animals treated with tamoxifen 5 months earlier. Taking into account our in vivo results, at that time point, SEZ cells of Ptc−/− animals correspond to a majority of aNSCs because TAPs and neuroblasts have collapsed. Under such conditions, a shortening of cell-cycle length resulting in an increase in NSC proliferation should be clearly detected because aNSCs were reported to display the shortest cell-cycle length compared to neuroblasts and TAPs with values close to 17, 18, and 18–25 hr, respectively (Ponti et al., 2013). The cell-cycle length obtained in Ptc−/− culture is slightly but nonsignificantly shorter (14.2 hr) compared to the control cultures (16.2 hr), thus suggesting that the NSC proliferation is likely not increased (Figure 5I). Altogether, these data indicate that Ptc deficiency likely lowers the number of fast proliferating cells exiting the cell cycle and, thus, their ability to differentiate but does not increase aNSC proliferation in vitro.
PTC Is Required for Replenishment of the SEZ Stem Cell Niche after In Vivo AraC Treatment
Next, we examined in the SEZ of control and Ptc−/− animals treated with tamoxifen 6 months before, the fate of NSCs after an infusion with AraC, an antimitotic reagent that kills dividing NSCs, TAPs, and proliferating neuroblasts while sparing qNSCs. After transient impairment of neurogenesis by AraC treatment, the damaged SEZ is then regenerated by astrocyte-like NSCs resulting in the increase of cell proliferation and the production of aNSCs and TAPs (Pastrana et al., 2011). Immediately after AraC treatment (day 0), cells that incorporated BrdU were mostly missing, whereas 10 days later (day 10), BrdU+ cell population returned to normal in the control SEZ when compared to vehicle treatment (Figures 6A and 6C; see also Figure S6). However, the number of BrdU+ cells in the Ptc−/− SEZ was 3-fold lower after AraC compared to vehicle treatment (Figures 6B and 6C). These data indicate that, upon AraC treatment, Ptc deficiency prevents qNSCs to be activated and to give rise to TAPs. Thus, PTC is required for effective regeneration after lesion in the SEZ.
Figure 6.
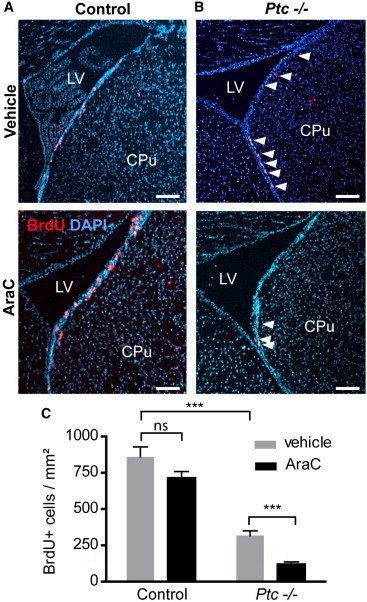
Altered Replenishment of the SEZ Stem Cell Niche in Conditional Ptc−/− Animals after AraC Injury In Vivo
(A and B) Coronal views of the SEZ showing BrdU-incorporating cells 10 days after pump removal in controls (A) and Ptc−/− (B) animals treated with vehicle or AraC. Animals had received tamoxifen 6 months before. The white arrowheads indicate BrdU+ nuclei. Scale bars, 100 μm.
(C) Quantification of BrdU-labeled cells in control and Ptc−/− animals. Values are the mean ± SEM from four animals/condition and two to four slices per animal. ∗∗∗p ≤ 0.0001; ns, nonsignificant.
See also Figure S6.
Discussion
Conditional Ptc inactivation in the adult NSCs allowed the study of HH signaling activation in a ligand-independent, cell-intrinsic fashion in the SEZ in vivo. This activation is likely due to the suspension of the inhibitory effect of PTC on SMO and may promote the generation of symmetric GLAST+/EGFR− pairs as observed in vitro. Moreover, our data suggest a likely alteration of the transition from aNSCs to TAPs, which might account for the high decrease of TAPs and subsequently of neuroblasts. The expression of NOTCH signaling and high-mobility group box transcription factors SOX2 and SOX9, all known to prevent neuronal differentiation (Scott et al., 2010; Ables et al., 2011), was also increased. Consistent with the alteration of the transition between aNSCs and TAPs, we also observed that the Ptc-deficient NSCs do not regenerate the SEZ properly after an antimitotic treatment. Nevertheless, our data do not allow excluding the potential existence of additional mechanisms such as, for instance, the alteration of TAP sensitivity to EGF, which would be consistent with the previously reported EGF-mediated induction of TAP proliferation (Doetsch et al., 2002).
Sustained HH signaling activation in the SEZ of deficient mice maintains multipotency but influences the fate of SEZ-derived precursors toward either the oligodendroglial phenotype in the cc or the glutamatergic and dopaminergic phenotypes, two populations of OB periglomerular interneurons, the specification of which may be of high interest in the context of brain repair (Brill et al., 2009; Lledo et al., 2006). The long-lasting high activation of HH signaling is still detected 1 year after the recombination. However, the absence of major alteration of OB morphology we observed in the present work is consistent with previous reports showing that adult neurogenesis in the SEZ allows only a partial turnover of newborn neurons in the OB (Imayoshi et al., 2008; Ninkovic et al., 2007).
Our results point out a likely interaction between HH and NOTCH signaling pathways in adult neurogenesis, suggesting that both should contribute to properly maintain adult NSCs, preventing them to become TAPs (Aguirre et al., 2010; Imayoshi et al., 2013; Kawaguchi et al., 2013). Such a crosstalk was proposed to be required for the regulation of neurogenic divisions and thus for proper corticogenesis in development (Dave et al., 2011). The constitutive activation of HH signaling through Ptc deletion in enteric neural crest cells also robustly activates NOTCH pathway via Hes1 upregulation and promotes premature gliogenesis, impairing these cells to form ganglia in the hindgut during enteric nervous system development (Ngan et al., 2011). In agreement with HH and NOTCH crosstalk in the adult SEZ, NSCs are both HH (Ahn and Joyner, 2005)- and NOTCH (Aguirre et al., 2010)-responding cells. If HES transcription factors are generally regarded as canonical NOTCH pathway effectors (Imayoshi et al., 2013), a NOTCH-independent HH/HES1 activity was reported in the progenitor cell proliferation of the retina (Wall et al., 2009). We do not know whether Hes1 is a direct target of HH signaling in the adult NSCs, but our results indicate that the effect of active HH signaling on NSCs is NOTCH dependent. TAPs were reported to regulate NSC self-renewal in vivo, and an interaction between EGFR and NOTCH pathways was proposed to have selective roles in this process with EGFR being an upstream regulator of NOTCH through a nonautonomous mechanism (Aguirre et al., 2010). Because the conditional Ptc deletion leads to a huge decrease in EGFR-expressing TAPs in the SEZ, the subsequent EGFR decrease might amplify NOTCH signaling induction and thus also participate in the observed effects. Likewise, SOX9 increase in the conditional Ptc mutant is consistent with data reporting that active HH signaling induces Sox9 transcription and results in the precocious ability of neuroepithelial cells to form neurospheres (Scott et al., 2010).
Despite PTC characterization as a tumor suppressor (Goodrich et al., 1997), we observed that its deletion in astrocyte-like NSCs increases the stemness of these cells without promoting tumor formation. Our data are consistent with the results observed upon the noninducible deletion of Ptc in GFAP-expressing NSCs, which was found to promote medulloblastomas only when NSCs have committed to granule neural precursors in the developing cerebellum (Yang et al., 2008). In contrast with the granule precursors, EGFR+ cells that comprise TAPs in the SEZ are not expanding upon Ptc deletion. One explanation is that PTC controls a major step of NSC maintenance in the SEZ: the lack of its antagonist activity on SMO results in a blockade of EGFR+ proliferative cells, presumably through increasing NSC symmetric divisions. Further work will have to address if small molecules, positive and negative modulators of SMO that have been developed (Amakye et al., 2013; Gorojankina et al., 2013), also regulate NSC stemness in vivo. Consistent with the present study, recent data have demonstrated that a major decrease in NSC number accounts for the progressive loss of neurogenesis in the SEZ of mice upon conditional ablation of Smo in postnatal GFAP-expressing cells (Petrova et al., 2013). By showing the existence of a PTC-mediated activity of SHH in the control of the symmetry or asymmetry of NSC divisions, our data now provide an answer to the question addressed by this study regarding the decision-making process between NSC self-renewal and progenitor generation. Likewise, though ectopic expression of Gli1 was reported to induce proliferation and transformation of numerous cell types (Dahmane et al., 1997; Nilsson et al., 2000), Gli1 upregulation is differently interpreted within self-renewing NSC context. Such behavior was previously described in vitro for hippocampus-derived NSCs (Galvin et al., 2008), which upon Gli1 upregulation, initiate cell-cycle arrest and apoptosis, interpreted as a defense mechanism against hyperproliferation.
As a whole, our work characterized PTC-mediated signals as implicated in the physiological regulation of the generation of NSCs required for sustaining the normal rate of neurogenesis. Instead of promoting precursor cell expansion, Ptc deletion was associated with a blockade of EGFR+ proliferating cells, leading to an almost disappearance of neurogenesis while preserving NSCs. Thus, Glast-CreERT2;Ptcfl/fl mice should be valuable tools for further investigating molecular regulation and transduction mechanisms associated with NSCs in the SEZ in normal and pathological conditions. These mice should also be of interest for evaluating the effect of PTC in NSC maintenance in the subgranular zone of the hippocampus (Traiffort et al., 2010) and in astroglia (Sirko et al., 2013).
Experimental Procedures
Complete Experimental Procedures are available online (see the Supplemental Experimental Procedures).
Animals
Eight- to 12-week-old animals were used unless indicated. The Glast-CreERT2 mouse line (Mori et al., 2006) was received on a mixed 129 × C57BL/6 background and backcrossed three times to C57BL/6. The Ptc floxed/floxed mouse line (Uhmann et al., 2007) was received and maintained on C57BL/6 background. The reporter mouse line R26R-YFP/+ was a gift from Drs. F. Costantini and T.M. Jessell (Srinivas et al., 2001) kept on C57Bl/6 background. The Glast-CreERT2 mouse line was crossed to the Ptc floxed/floxed mouse line. The progeny was then crossed to R26R-YFP/+ mice genotyped as described by Soriano (1999). All procedures were carried out according to the European Communities Council Directive (86/806/EEC) and protocols approved by Institutional Animal Care and Use Committee.
Tamoxifen and BrdU Administration
Tamoxifen (Sigma-Aldrich) was dissolved in corn oil (Sigma-Aldrich) at 10 mg/ml, and 1 mg was injected intraperitoneally (i.p.) twice a day for 5 consecutive days. Animals were killed 10 days, or 2, 6, or 12 months after the end of tamoxifen application. In order to label fast proliferating cells, mice received an i.p. injection of BrdU (Sigma-Aldrich; 100 mg/kg of body weight) 4 and 2 hr before sacrifice (n = 5–6/group). For the determination of cell-cycle exit, animals received two BrdU i.p. injections at 2 hr intervals and were sacrificed 24 hr after the last injection.
Osmotic Minipump Infusion
To kill all fast proliferating cells in the SEZ, AraC (2%; Sigma-Aldrich) in vehicle (0.9% saline) was infused into the lateral ventricles in control and Ptc−/− mice that had been injected with tamoxifen 6 months earlier. We used miniosmotic pumps (ALZET). After 7 days of infusion, the pumps were removed, and mice were killed 10 days after the end of AraC infusion (n = 4 per condition). Cannulas were implanted stereotaxically into the lateral ventricle at the following coordinates: anteroposterior, 0.2 mm; mediolateral, 0.8 mm; and dorsoventral, −2.5 mm (relative to bregma).
Acknowledgments
We thank Professor M. Götz (Helmholtz Zentrum, Munich) for generously providing us with the Glast-CreERT2, Drs. P. Brulet and S. Picaud (NED, Gif/Yvette) for providing the initial breeding mice, and Professor T. Jessell (Columbia University) for R26R-YFP/+ mouse lines. We thank Professor O. Kiehn and Drs. L. Borgius and J. Bouvier (Karolinska Institute, Stockholm) for providing us with the vGlut2 probe. We are very grateful to E. Desale and T. Durand (TAAM, CNRS-UPS44, Orléans) for excellent technical assistance. LY411575 was obtained as a gift from Chemical Synthesis Core Facility (Mayo Clinic Jacksonville, Jacksonville). J.F. is supported in part by fellowships from the Direction Générale des Armées, the Association pour la Recherche sur le Cancer, and CNRS. This work was supported by the French Agence Nationale de la Recherche (Grant 07-physio-010-04 to E.T.) and La Ligue contre le Cancer, Comité de l’Essonne (n°M20632) et des Yvelines and Fondation ARC (n°20131200300 to M.R.).
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Contributor Information
Martial Ruat, Email: ruat@inaf.cnrs-gif.fr.
Elisabeth Traiffort, Email: elisabeth.traiffort@inserm.fr.
Supplemental Information
References
- Ables J.L., Breunig J.J., Eisch A.J., Rakic P. Not(ch) just development: Notch signalling in the adult brain. Nat. Rev. Neurosci. 2011;12:269–283. doi: 10.1038/nrn3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre A., Rubio M.E., Gallo V. Notch and EGFR pathway interaction regulates neural stem cell number and self-renewal. Nature. 2010;467:323–327. doi: 10.1038/nature09347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S., Joyner A.L. In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature. 2005;437:894–897. doi: 10.1038/nature03994. [DOI] [PubMed] [Google Scholar]
- Amakye D., Jagani Z., Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013;19:1410–1422. doi: 10.1038/nm.3389. [DOI] [PubMed] [Google Scholar]
- Angot E., Loulier K., Nguyen-Ba-Charvet K.T., Gadeau A.P., Ruat M., Traiffort E. Chemoattractive activity of sonic hedgehog in the adult subventricular zone modulates the number of neural precursors reaching the olfactory bulb. Stem Cells. 2008;26:2311–2320. doi: 10.1634/stemcells.2008-0297. [DOI] [PubMed] [Google Scholar]
- Balordi F., Fishell G. Mosaic removal of hedgehog signaling in the adult SVZ reveals that the residual wild-type stem cells have a limited capacity for self-renewal. J. Neurosci. 2007;27:14248–14259. doi: 10.1523/JNEUROSCI.4531-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidet M., Joubert O., Lacombe B., Ciantar M., Nehmé R., Mollat P., Brétillon L., Faure H., Bittman R., Ruat M., Mus-Veteau I. The hedgehog receptor patched is involved in cholesterol transport. PLoS ONE. 2011;6:e23834. doi: 10.1371/journal.pone.0023834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brill M.S., Ninkovic J., Winpenny E., Hodge R.D., Ozen I., Yang R., Lepier A., Gascón S., Erdelyi F., Szabo G. Adult generation of glutamatergic olfactory bulb interneurons. Nat. Neurosci. 2009;12:1524–1533. doi: 10.1038/nn.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe J., Thérond P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013;14:416–429. doi: 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]
- Chen J.K., Taipale J., Young K.E., Maiti T., Beachy P.A. Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. USA. 2002;99:14071–14076. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak D., Mori T., Brill M.S., Pfeifer A., Falk S., Deng C., Monteiro R., Mummery C., Sommer L., Götz M. Adult neurogenesis requires Smad4-mediated bone morphogenic protein signaling in stem cells. J. Neurosci. 2008;28:434–446. doi: 10.1523/JNEUROSCI.4374-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M.R., Ortega F., Brill M.S., Beckervordersandforth R., Petrone C., Schroeder T., Götz M., Berninger B. Continuous live imaging of adult neural stem cell division and lineage progression in vitro. Development. 2011;138:1057–1068. doi: 10.1242/dev.061663. [DOI] [PubMed] [Google Scholar]
- Dahmane N., Lee J., Robins P., Heller P., Ruiz i Altaba A. Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature. 1997;389:876–881. doi: 10.1038/39918. [DOI] [PubMed] [Google Scholar]
- Dave R.K., Ellis T., Toumpas M.C., Robson J.P., Julian E., Adolphe C., Bartlett P.F., Cooper H.M., Reynolds B.A., Wainwright B.J. Sonic hedgehog and notch signaling can cooperate to regulate neurogenic divisions of neocortical progenitors. PLoS ONE. 2011;6:e14680. doi: 10.1371/journal.pone.0014680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F., Petreanu L., Caille I., Garcia-Verdugo J.M., Alvarez-Buylla A. EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron. 2002;36:1021–1034. doi: 10.1016/s0896-6273(02)01133-9. [DOI] [PubMed] [Google Scholar]
- Fauq A.H., Simpson K., Maharvi G.M., Golde T., Das P. A multigram chemical synthesis of the gamma-secretase inhibitor LY411575 and its diastereoisomers. Bioorg. Med. Chem. Lett. 2007;17:6392–6395. doi: 10.1016/j.bmcl.2007.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvin K.E., Ye H., Erstad D.J., Feddersen R., Wetmore C. Gli1 induces G2/M arrest and apoptosis in hippocampal but not tumor-derived neural stem cells. Stem Cells. 2008;26:1027–1036. doi: 10.1634/stemcells.2007-0879. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Perez O., Alvarez-Buylla A. Oligodendrogenesis in the subventricular zone and the role of epidermal growth factor. Brain Res. Brain Res. Rev. 2011;67:147–156. doi: 10.1016/j.brainresrev.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich L.V., Milenković L., Higgins K.M., Scott M.P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Gorojankina T., Hoch L., Faure H., Roudaut H., Traiffort E., Schoenfelder A., Girard N., Mann A., Manetti F., Solinas A. Discovery, molecular and pharmacological characterization of GSA-10, a novel small-molecule positive modulator of Smoothened. Mol. Pharmacol. 2013;83:1020–1029. doi: 10.1124/mol.112.084590. [DOI] [PubMed] [Google Scholar]
- Ihrie R.A., Shah J.K., Harwell C.C., Levine J.H., Guinto C.D., Lezameta M., Kriegstein A.R., Alvarez-Buylla A. Persistent sonic hedgehog signaling in adult brain determines neural stem cell positional identity. Neuron. 2011;71:250–262. doi: 10.1016/j.neuron.2011.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I., Sakamoto M., Ohtsuka T., Takao K., Miyakawa T., Yamaguchi M., Mori K., Ikeda T., Itohara S., Kageyama R. Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat. Neurosci. 2008;11:1153–1161. doi: 10.1038/nn.2185. [DOI] [PubMed] [Google Scholar]
- Imayoshi I., Shimojo H., Sakamoto M., Ohtsuka T., Kageyama R. Genetic visualization of notch signaling in mammalian neurogenesis. Cell. Mol. Life Sci. 2013;70:2045–2057. doi: 10.1007/s00018-012-1151-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpowicz P., Willaime-Morawek S., Balenci L., DeVeale B., Inoue T., van der Kooy D. E-Cadherin regulates neural stem cell self-renewal. J. Neurosci. 2009;29:3885–3896. doi: 10.1523/JNEUROSCI.0037-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi D., Furutachi S., Kawai H., Hozumi K., Gotoh Y. Dll1 maintains quiescence of adult neural stem cells and segregates asymmetrically during mitosis. Nat Commun. 2013;4:1880–1892. doi: 10.1038/ncomms2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanis I., Lathia J.D., Vadakkan T.J., Raborn E., Wan R., Mughal M.R., Eckley D.M., Sasaki T., Patton B., Mattson M.P. Quiescence and activation of stem and precursor cell populations in the subependymal zone of the mammalian brain are associated with distinct cellular and extracellular matrix signals. J. Neurosci. 2010;30:9771–9781. doi: 10.1523/JNEUROSCI.0700-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lledo P.M., Alonso M., Grubb M.S. Adult neurogenesis and functional plasticity in neuronal circuits. Nat. Rev. Neurosci. 2006;7:179–193. doi: 10.1038/nrn1867. [DOI] [PubMed] [Google Scholar]
- Machold R., Hayashi S., Rutlin M., Muzumdar M.D., Nery S., Corbin J.G., Gritli-Linde A., Dellovade T., Porter J.A., Rubin L.L. Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron. 2003;39:937–950. doi: 10.1016/s0896-6273(03)00561-0. [DOI] [PubMed] [Google Scholar]
- Marshall C.A.G., Novitch B.G., Goldman J.E. Olig2 directs astrocyte and oligodendrocyte formation in postnatal subventricular zone cells. J. Neurosci. 2005;25:7289–7298. doi: 10.1523/JNEUROSCI.1924-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T., Tanaka K., Buffo A., Wurst W., Kühn R., Götz M. Inducible gene deletion in astroglia and radial glia—a valuable tool for functional and lineage analysis. Glia. 2006;54:21–34. doi: 10.1002/glia.20350. [DOI] [PubMed] [Google Scholar]
- Ngan E.S., Garcia-Barceló M.M., Yip B.H., Poon H.C., Lau S.T., Kwok C.K., Sat E., Sham M.H., Wong K.K., Wainwright B.J. Hedgehog/Notch-induced premature gliogenesis represents a new disease mechanism for Hirschsprung disease in mice and humans. J. Clin. Invest. 2011;121:3467–3478. doi: 10.1172/JCI43737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson M., Undèn A.B., Krause D., Malmqwist U., Raza K., Zaphiropoulos P.G., Toftgård R. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc. Natl. Acad. Sci. USA. 2000;97:3438–3443. doi: 10.1073/pnas.050467397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninkovic J., Mori T., Götz M. Distinct modes of neuron addition in adult mouse neurogenesis. J. Neurosci. 2007;27:10906–10911. doi: 10.1523/JNEUROSCI.2572-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastrana E., Silva-Vargas V., Doetsch F. Eyes wide open: a critical review of sphere-formation as an assay for stem cells. Cell Stem Cell. 2011;8:486–498. doi: 10.1016/j.stem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova R., Garcia A.D., Joyner A.L. Titration of GLI3 repressor activity by sonic hedgehog signaling is critical for maintaining multiple adult neural stem cell and astrocyte functions. J. Neurosci. 2013;33:17490–17505. doi: 10.1523/JNEUROSCI.2042-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponti G., Obernier K., Alvarez-Buylla A. Lineage progression from stem cells to new neurons in the adult brain ventricular-subventricular zone. Cell Cycle. 2013;12:1649–1650. doi: 10.4161/cc.24984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roudaut H., Traiffort E., Gorojankina T., Vincent L., Faure H., Schoenfelder A., Mann A., Manetti F., Solinas A., Taddei M., Ruat M. Identification and mechanism of action of the acylguanidine MRT-83, a novel potent Smoothened antagonist. Mol. Pharmacol. 2011;79:453–460. doi: 10.1124/mol.110.069708. [DOI] [PubMed] [Google Scholar]
- Ruat M., Roudaut H., Ferent J., Traiffort E. Hedgehog trafficking, cilia and brain functions. Differentiation. 2012;83:S97–S104. doi: 10.1016/j.diff.2011.11.011. [DOI] [PubMed] [Google Scholar]
- Scott C.E., Wynn S.L., Sesay A., Cruz C., Cheung M., Gomez Gaviro M.V., Booth S., Gao B., Cheah K.S., Lovell-Badge R., Briscoe J. SOX9 induces and maintains neural stem cells. Nat. Neurosci. 2010;13:1181–1189. doi: 10.1038/nn.2646. [DOI] [PubMed] [Google Scholar]
- Shook B.A., Manz D.H., Peters J.J., Kang S., Conover J.C. Spatiotemporal changes to the subventricular zone stem cell pool through aging. J. Neurosci. 2012;32:6947–6956. doi: 10.1523/JNEUROSCI.5987-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirko S., Behrendt G., Johansson P.A., Tripathi P., Costa M., Bek S., Heinrich C., Tiedt S., Colak D., Dichgans M. Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog. [corrected] Cell Stem Cell. 2013;12:426–439. doi: 10.1016/j.stem.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Srinivas S., Watanabe T., Lin C.S., William C.M., Tanabe Y., Jessell T.M., Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 2001;1:4–11. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traiffort E., Angot E., Ruat M. Sonic Hedgehog signaling in the mammalian brain. J. Neurochem. 2010;113:576–590. doi: 10.1111/j.1471-4159.2010.06642.x. [DOI] [PubMed] [Google Scholar]
- Uhmann A., Dittmann K., Nitzki F., Dressel R., Koleva M., Frommhold A., Zibat A., Binder C., Adham I., Nitsche M. The Hedgehog receptor Patched controls lymphoid lineage commitment. Blood. 2007;110:1814–1823. doi: 10.1182/blood-2007-02-075648. [DOI] [PubMed] [Google Scholar]
- Wall D.S., Mears A.J., McNeill B., Mazerolle C., Thurig S., Wang Y., Kageyama R., Wallace V.A. Progenitor cell proliferation in the retina is dependent on Notch-independent Sonic hedgehog/Hes1 activity. J. Cell Biol. 2009;184:101–112. doi: 10.1083/jcb.200805155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.J., Ellis T., Markant S.L., Read T.A., Kessler J.D., Bourboulas M., Schüller U., Machold R., Fishell G., Rowitch D.H. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell. 2008;14:135–145. doi: 10.1016/j.ccr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.