

Abstract
Mutations in the gene encoding NLRP3 cause a spectrum of autoinflammatory diseases known as the cryopyrin-associated periodic syndromes (CAPS)1. NLRP3 is a key component of one of several distinct cytoplasmic multiprotein complexes (inflammasomes) that mediate the maturation of the proinflammatory cytokine interleukin-1β (IL-1β) by activating caspase-1. Although several models for inflammasome activation, such as K+ efflux2, generation of reactive oxygen species3 and lysosomal destabilization4, have been proposed, the precise molecular mechanism of NLRP3 inflammasome activation, as well as the mechanism by which CAPS-associated mutations activate NLRP3, remain to be elucidated. Here we show that the murine calcium-sensing receptor (CASR) activates the NLRP3 inflammasome, mediated by increased intracellular Ca2+ and decreased cellular cyclic AMP (cAMP). Ca2+ or other CASR agonists activate the NLRP3 inflammasome in the absence of exogenous ATP, whereas knockdown of CASR reduces inflammasome activation in response to known NLRP3 activators. CASR activates the NLRP3 inflammasome through phospholipase C, which catalyses inositol-1,4,5-trisphosphate production and thereby induces release of Ca2+ from endoplasmic reticulum stores. The increased cytoplasmic Ca2+ promotes the assembly of inflammasome components, and intracellular Ca2+ is required for spontaneous inflammasome activity in cells from CAPS patients. CASR stimulation also results in reduced intracellular cAMP, which independently activates the NLRP3 inflammasome. cAMP binds to NLRP3 directly to inhibit inflammasome assembly, and downregulation of cAMP relieves this inhibition. The binding affinity of cAMP for CAPS-associated mutant NLRP3 is substantially lower than for wild-type NLRP3, and the uncontrolled mature IL-1β production from CAPS patients' peripheral blood mononuclear cells is attenuated by increasing cAMP. Taken together, these findings indicate that Ca2+ and cAMP are two key molecular regulators of the NLRP3 inflammasome that have critical roles in the molecular pathogenesis of CAPS.
NLRP3 inflammasome activation by extracellular ATP is mediated through the purinergic receptor, P2X7R, and is inhibited by high concentrations of extracellular K+, which may block K+ efflux5. However, ATP-driven NLRP3 inflammasome activation is also inhibited by other extracellular cations such as Ca2+ (ref. 6). Hence, we initially explored the role of extracellular cations in inflammasome activation to investigate the signalling pathway that links danger-associated molecular patterns to the NLRP3 inflammasome. As seen in previous reports, addition of K+ or Ca2+ with ATP to lipopolysaccharide (LPS)-primed bone marrow-derived macrophages (BMDMs) suppressed bioactive IL-1β secretion, and the same effect was also observed with Mg2+ (Fig. 1a). Surprisingly, we also found that in the absence of ATP, LPS-primed BMDMs secreted IL-1β when cultured with Ca2+ alone but not with Mg2+ or K+ (Fig. 1a and Supplementary Fig. 1a). The Ca2+-induced IL-1β secretion was independent of P2X7R, or the AIM2 (absent in melanoma 2) or NLRC4 (IPAF, ICE protease-activating factor) inflammasomes, but dependent on the NLRP3 inflammasome (Fig. 1b and Supplementary Fig. 1b, c). NLRP3 inflammasome activation was also induced by gadolinium (Gd3+), an agonist of CASR, or R-568, an allosteric agonist of CASR (Supplementary Fig. 1d). These results indicate that the recognition of extracellular Ca2+ through CASR activates the NLRP3 inflammasome.
Figure 1. Extracellular calcium and ATP activate the NLRP3 inflammasome through CASR.
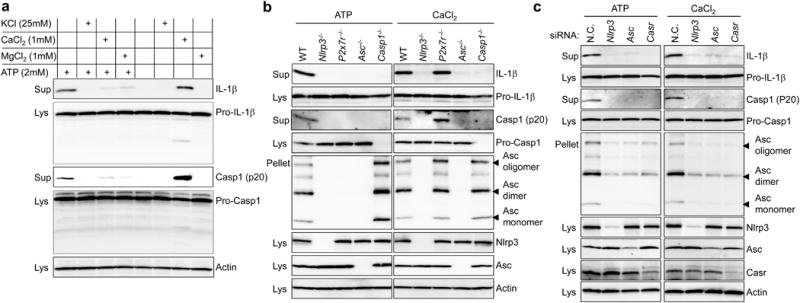
a, b, LPS-primed BMDMs from wild-type (WT) or designated knockout mice were stimulated in RPMI medium supplemented with the indicated reagents. c, BMDMs were transiently transfected with scrambled (N.C.), Nlrp3, Asc, or Casr siRNA, and then stimulated with 2 mM ATP or 1 mM CaCl2 after priming with LPS. Cell culture supernatants (Sup), cell lysates (Lys), and crosslinked pellets from whole-cell lysates were analysed by immunoblotting as indicated (a–c). All immunoblot data shown are representative of at least three independent experiments.
It seemed paradoxical that high calcium buffers could inhibit ATP-induced IL-1β secretion while stimulating IL-1β secretion in the absence of ATP. The inhibitory effect of extracellular Ca2+ was confined to NLRP3 co-stimulation with ATP, as high calcium buffers did not inhibit inflammasome activation by other NLRP3 activators, double-stranded DNA (dsDNA), or flagellin (Supplementary Fig. 2a–c). In addition, although both high calcium and high magnesium buffers inhibited ATP-induced inflammasome activation, equivalent concentrations of the trivalent cation Gd3+ (which, like Ca2+ itself, is an agonist of CASR) did not inhibit ATP-induced inflammasome activation (Supplementary Fig. 2d). One possible explanation for these observations is the well-recognized interaction between divalent cations and ATP, in which unbound ATP acts as the agonist form7,8. Indeed, when BMDMs were treated with a given stimulatory concentration of Ca2+, varying ATP concentrations, and vice versa, we observed first decreasing IL-1β production as inactive Ca2+/ATP complexes form, and then eventually increasing IL-1β activation as the ATP or Ca2+ exceeded the available free Ca2+ or ATP, respectively (Supplementary Fig. 2e, f).
Next, to examine the role of CASR in NLRP3 inflammasome activation further, the Casr gene was knocked down in mouse BMDMs by short interfering RNA (siRNA; Supplementary Fig. 3a, b). Similar to Nlrp3 or Asc knockdowns, IL-1β secretion from the BMDMs with Casr knockdown was substantially diminished in response to extracellular Ca2+ (Fig. 1c). Furthermore, we also observed that downregulation of Casr reduced IL-1β secretion when BMDMs were stimulated with several other known NLRP3 inflammasome activators, but not with AIM2 or NLRC4 inflammasome activators (Fig. 1c and Supplementary Fig. 4). These findings suggest that CASR is an upstream gatekeeper of the NLRP3 inflammasome.
Intracellular Ca2+ signalling is important for NLRP3 inflammasome activation induced by extracellular ATP9. Elevation of extracellular Ca2+ concentrations or other CASR agonists commonly elicits intracellular Ca2+ signals through the interaction between CASR and phospholipase C (PLC)10,11. We observed a correlation between PLC activity and the presence of active IL-1β in the supernatant of LPS-primed BMDMs. Cells treated with 2,4,6-Trimethyl-N-[3-(trifluoromethyl)phenyl]benzenesulfonamide (m-3M3FBS), a direct activator of PLC (Fig. 2a), secreted IL-1β in the absence of any other exogenous stimulus, indicating a role for PLC-stimulated Ca2+ signals in inflammasome activation. In contrast, PLC inhibitors edelfosine or U73122 (but not U73343, an inactive analogue of U73122) blocked IL-1β secretion from cells treated with Ca2+ or ATP (Supplementary Fig. 5a and Fig. 2b). U73122 had no effect on AIM2 or NLRC4 inflammasome activation (Supplementary Fig. 5b), indicating that PLC activation is dispensable for triggering these inflammasomes.
Figure 2. PLC-InsP3-mediated calcium release from the ER triggers NLRP3 inflammasome activation.
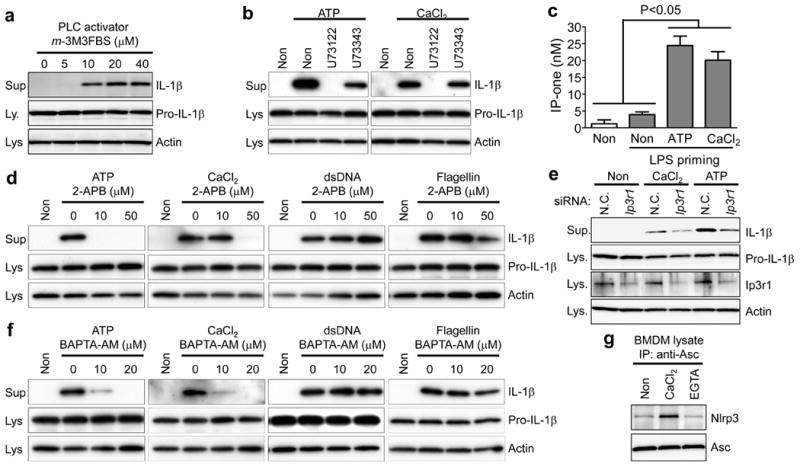
a–d, f, LPS-primed BMDMs were treated as indicated. Intracellular IP-one, [Author: Please give IP-one in full. inositol monophosphate]a surrogate for InsP3 levels, was measured (c). Data represent the mean ± s.e.m. from three independent experiments. e, BMDMs transiently transfected with scrambled (N.C.) or Ip3r1 siRNA were primed with LPS and stimulated with 5 mM ATP or 1 mM CaCl2. g, LPS-primed BMDMs were immunoprecipitated with anti-ASC antibody in buffer containing no additives (Non), 1 mM CaCl2, or 1 mM EGTA. Supernatants and lysates were analysed by immunoblotting. Data are representative of three independent experiments.
PLC hydrolyses phosphatidylinositol-4,5-bisphosphate into diacyl glycerol (DAG), which activates protein kinase C, and inositol trisphosphate (InsP3), which causes the cytosolic concentration of Ca2+ to increase by binding to InsP3 receptors (InsP3R) in the endoplasmic reticulum (ER). Indeed, we observed generation of InsP3 in BMDMs exposed to the NLRP3 inflammasome activators ATP or Ca2+ (Fig. 2c). When LPS-primed BMDMs were stimulated with ATP or Ca2+ in the presence of an InsP3R inhibitor (2-APB), little or no IL-1β was secreted (Fig. 2d), whereas IL-1β secretion was not suppressed in the presence of protein kinase C inhibitors (Supplementary Fig. 5c). Moreover, in BMDMs in which the InsP3 receptor (Ip3r; also called Itpr1) had been knocked down, IL-1β secretion induced by extracellular Ca2+ or ATP was substantially reduced (Fig. 2e). In addition, when wild-type cells were stimulated with ATP, CaCl2, or GdCl3, we observed increased intracellular Ca2+ levels, which were blocked by 2-APB or BAPTA-AM (Supplementary Fig. 6a–c and Supplementary Videos 1–9), and IL-1β secretion, but not IL-6 or TNF-α secretion, was markedly inhibited by chelating intracellular Ca2+ with BAPTA-AM but not the heavy metal chelator TPEN (Fig. 2f and Supplementary Fig. 7a, b). Conversely, inflammasome activation was observed in LPS-primed BMDMs by treatment with thapsigargin, which increases intracellular Ca2+ (Supplementary Fig. 7c). BAPTA-AM or 2-APB did not block IL-1β secretion induced by dsDNA or flagellin (Fig. 2d, f), which do not induce intracellular Ca2+ increases (data not shown). Taken together, these data indicate that activation of the AIM2 and NLRC4 inflammasomes is independent of InsP3R signalling and intracellular Ca2+, whereas signalling from CASR activates the NLRP3 inflammasome via InsP3-mediated intracellular Ca2+ release. Indeed, the NLRP3–ASC association in cell-free lysates from BMDMs increased in the presence of Ca2+ (Fig. 2g), whereas Ca2+ had no direct effect on the activity of caspase 1 (Supplementary Fig. 7d). Nevertheless, it remains to be elucidated whether the role of Ca2+ on NLRP3 inflammasome activation is direct or indirect.
CASR interacts not only with PLC but also with adenylate cyclase (ADCY), which results in inhibition of ADCY and subsequent reduction of cellular cAMP levels10. Indeed, addition of ATP or Ca2+ reduced the LPS-mediated cAMP increase in BMDMs12 (Fig. 3a). Thus, we also investigated a possible role for cAMP in inflammasome activation. Notably, an inverse correlation between the levels of cAMP and IL-1β secretion was observed in LPS-primed BMDMs. The increase of cAMP synthesis by ADCY activators (NKH477 or forskolin) or the decrease of cAMP hydrolysis by phosphodiesterase (PDE) 4 inhibitors (Ro-20-1724, (R)-(-)-rolipram, or zardaverine) suppressed IL-1β secretion, but not IL-6 or TNF-α secretion, and ASC oligomerization induced by ATP or Ca2+, but did not inhibit cytokine secretion induced by dsDNA or flagellin (Fig. 3b, c and Supplementary Fig. 8a–e). In contrast, an ADCY inhibitor (KH7) alone induced dose-dependent secretion of IL-1β and ASC oligomerization in wild-type BMDMs, but not in BMDMs from Nlrp3−/−, Asc−/− or Casp1−/− mice (Supplementary Fig. 8f and Fig. 3d). Using knockdown experiments, we found that ADCY and PLC are downstream of CASR in NLRP3 activation, and that these pathways are intact even after Casr knockdown (Supplementary Fig. 8g). We also observed that the NLRP3–ASC interaction in KH7-treated BMDM lysates increased, whereas the interaction in lysates from forskolin-treated cells decreased (Fig. 3e). Consistent with these data, spontaneous inflammasome activation was observed in BMDMs upon knockdown of the several adenylate cyclases (Fig. 3f) that are expressed in macrophages (ADCY3, ADCY6, ADCY7 and ADCY9; Supplementary Fig. 8h). Taken together, these results indicate that cAMP specifically suppresses NLRP3 inflammasome activation.
Figure 3. cAMP binds to NLRP3 and suppresses inflammasome activation.
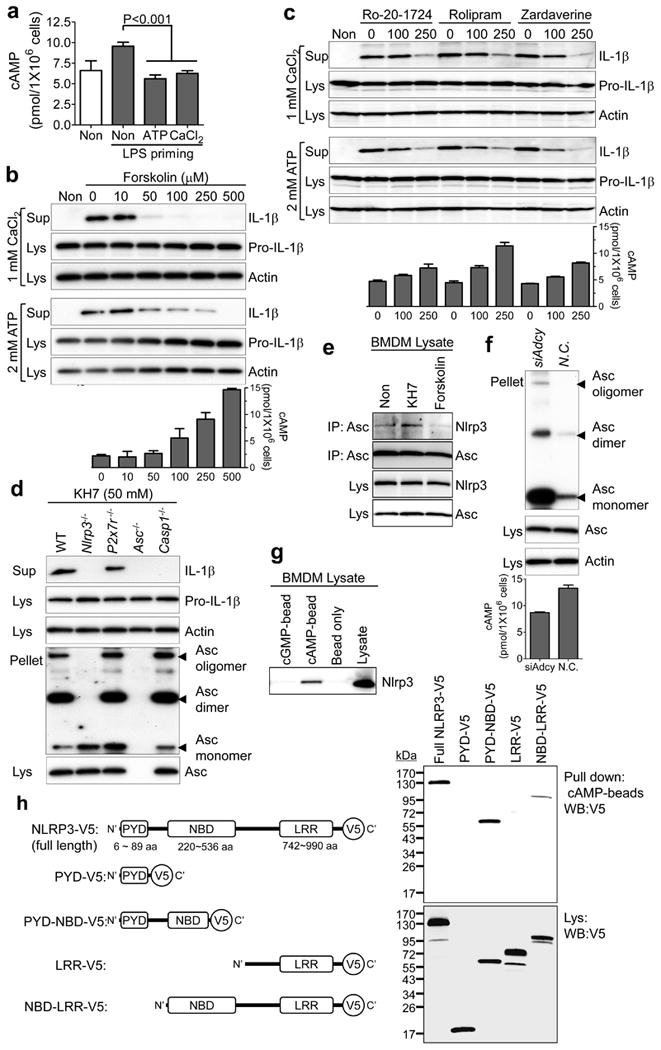
a–e, LPS-primed BMDMs were treated as indicated. IL-1β secretion (b–d), ASC–NLRP3 interaction (e), or intracellular cAMP levels (a–c) were analysed. Data represent the mean ± s.e.m. from five experiments. f, BMDMs transfected with scrambled or Adcy3 + Adcy6 + Adcy7 + Adcy9 siRNAs were analysed for ASC oligomerization after LPS priming. g, LPS-primed BMDMs were pulled down with indicated beads and analysed by immunoblot for NLRP3. h, Indicated NLRP3 proteins were expressed in 293T cells and pulled down with cAMP beads. LRR, leucine-rich repeats; NBD, nucleotide-binding domain; PYD, PYRIN domain. Data are representative of three independent experiments.
A major role of cAMP is activating protein kinase A (PKA), which could potentially suppress inflammasome activation. However, no IL-1β secretion was observed in the supernatant of LPS-primed BMDMs treated with PKA inhibitors or after knockdown of Pkaα (Supplementary Fig. 9a, b). To understand how cAMP might suppress NLRP3 inflammasome activation, we tested whether cAMP interacts directly with NLRP3. In a pull-down assay, we found an interaction of endogenous NLRP3 from BMDMs with cAMP but not with cGMP (Fig. 3g). We also found that this interaction was mediated by the nucleotide-binding domain (NBD, also known as NACHT) of NLRP3 (Fig. 3h), which has previously been shown to bind and hydrolyse ATP13. However, cAMP does not inhibit binding of ATP to NLRP3 (Supplementary Fig. 8i).
Because cAMP suppresses NLRP3 inflammasome activation and binds to the NBD of NLRP3, which is the most common location of CAPS-associated mutations1, we considered the possible effect of these mutations on binding to cAMP. The binding of cAMP to the disease-associated mutant NLRP3 (D303N, A352V and F523C) was substantially decreased relative to wild-type NLRP3 (Fig. 4a), which indicates that the spontaneous activation of the inflammasome in myeloid cells of CAPS patients might be due to reduced suppression of NLRP3 activation by cAMP. Thus, we investigated the effect of cAMP on IL-1β secretion from peripheral blood mononuclear cells (PBMCs) of CAPS (familial cold autoinflammatory syndrome, FCAS; Muckle–Wells syndrome, MWS; neonatal-onset multisystem inflammatory disease, NOMID) patients. As previously reported14, without any inflammasome activator, mutation-positive CAPS patients' PBMCs secreted IL-1β in response to LPS priming alone, whereas healthy control PBMCs did not secrete IL-1β (Supplementary Fig. 10a–c). However, IL-1β secretion from CAPS PBMCs was substantially blocked by activating ADCY or inhibiting PDE4 (Fig. 4b, c and Supplementary Fig. 10b–d). In contrast, we observed no inhibitory effect of ADCY activator or PDE4 inhibitors on IL-1β secretion from PBMCs of patients with familial Mediterranean fever (FMF) (Supplementary Fig. 10e), the prototypic autoinflammatory disease which is caused by mutations of pyrin that induce an NLRP3-independent inflammasome15. These results are consistent with the hypothesis that the inhibitory effect of cAMP on IL-1β secretion is specific to the NLRP3 inflammasome.
Figure 4. The role of cAMP and calcium in the pathogenesis of CAPS.
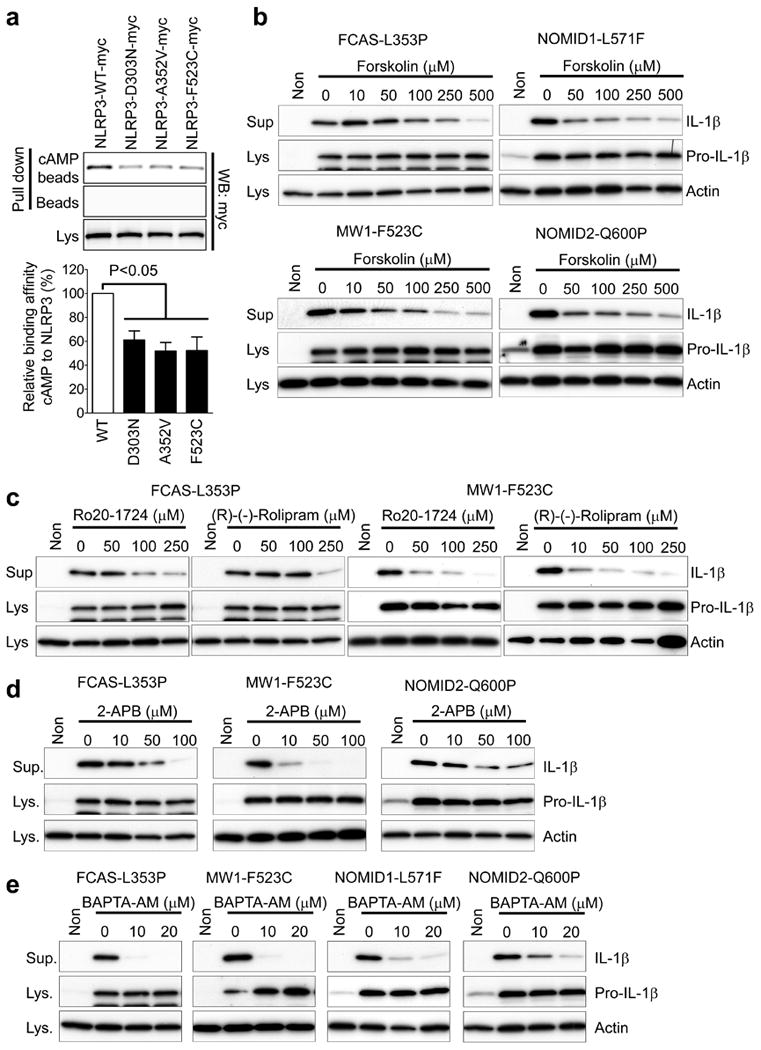
a, Wild-type or CAPS-associated mutant NLRP3 proteins were expressed in 293T cells and pulled down with cAMP beads. Densitometric analysis of cAMP-associated NLRP3 bands, normalized to the NLRP3 in lysates, is shown in the histogram. Data represent the mean ± s.e.m. of the seven experiments. b–e, PBMCs from CAPS patients with the designated mutations in NLRP3 were primed with LPS and treated with the indicated dose of forskolin (b), Ro-20-1724 or (R)-(-)-rolipram (c), 2-APB (d), or BAPTA-AM (e). Lysates and supernatants were analysed for IL-1β secretion by immunoblot.
In addition, we found that the constitutive IL-1β secretion from CAPS PBMCs was also substantially reduced by blockers of InsP3-mediated intracellular Ca2+ signalling pathways, 2-APB and BAPTA-AM, (Fig. 4d, e and Supplementary Fig. 10f, g), which can also inhibit NLRP3 inflammasome activation induced by ATP or extracellular Ca2+ (Fig. 2d, f). These data indicate an essential role for intracellular Ca2+ signalling in CAPS-associated mutant NLRP3 activation.
Using a combination of genetic, pharmacological and biochemical approaches, we provide evidence that CASR has a major role in NLRP3 inflammasome activation through its effects on intracellular Ca2+ and cAMP (summarized in Supplementary Fig. 11 and modelled in Supplementary Fig. 12), which also suggests a possible role for extracellular Ca2+ as a danger signal. Indeed, extracellular fluid at sites of injury and inflammation has been reported to contain high concentrations of Ca2+, attracting cells of the monocyte/macrophage lineage16. The CASR is a G-protein-coupled receptor that interacts directly not only with Gαq eliciting intracellular Ca2+ signals through PLC, but also with Giα, which results in the inhibition of ADCY and a reduction in cellular cAMP levels10. It has been widely recognized that in inflammatory cells, cAMP acts as a negative regulator of immune and inflammatory responses17,18. Our results clarify a molecular mechanism for how cAMP inhibits IL-1β maturation in macrophages. As a second messenger, cAMP exerts its role by direct binding to the regulatory unit or domain of target proteins, thereby inducing conformational changes19,20. The low binding affinity of cAMP with CAPS-associated mutant NLRP3 proteins and inhibition of IL-1β secretion from CAPS PBMCs by activation of ADCY or the inhibition of PDE4 is also consistent with an inhibitory effect of cAMP on NLRP3 inflammasome activation. Although IL-1 inhibition is currently being used as an effective therapy for CAPS, our data indicate a broader spectrum of potential targets for therapy of CAPS as well as other inflammatory conditions involving the NLRP3 inflammasome, including gout21, type 2 diabetes mellitus3,22, atherosclerosis23 and Alzheimer's disease24.
Methods Summary
Cell stimulation
BMDMs were obtained by differentiating bone marrow progenitors from tibial and femoral bone in Iscove's modified Dulbecco's medium (IMDM) containing 20 ng ml−1 of M-CSF for 7 days. Blood specimens from healthy controls, and CAPS and FMF patients were drawn after obtaining informed consent under a protocol approved by the NIAMS/NIDDK Institutional Review Board. Human PBMCs were isolated by LSM-lymphocyte separation medium. Inflammasome activation experiments were performed in two stages: initial LPS priming for 3 h and then inflammasome activation for 30–50 min by replacing the medium with RPMI 1640 medium supplemented with activators (ATP, 2 mM; CaCl2, 1 mM, resulting in a total [Ca2+] of 1.42 mM; GdCl3, 1 mM; R-586, 10 μM; nigericin, 20 μM; KH7, 50 μM; m-3M3FBS, 5–40 μM; MSU, 200 μg ml−1; CPPD, 200 μg ml−1; thapsigargin, 50 nM; dsDNA, 1 μg ml−1 with 2.5 μl ml−1 of Lipofectamine 2000; flagellin, 0.5 μg ml−1 with 25 μl ml−1 DOTAP). For the inhibition of inflammasome activation, LPS-primed BMDMs were treated with inhibitors [U73122, 10 μM; U73343, 10 μM; edelfosine, 4–100 μM; 2-APB, 10 or 50 μM; BAPTA-AM, 10 or 20 μM; forskolin, 10–500 μM; NKH477, 10–500 μM; Ro 20-1724, 50–250 μM; (R)-(-)-rolipram, 50–250 μM; zardaverine, 100 or 250 μM; GF 109203X, 50 μM; C-1, 50 μM] in the presence of each activator.
Immunoprecipitation and pull-down assay
The lysates from BMDMs or 293T cells transiently co-transfected with expression constructs for the wild type, CAPS-associated mutant, or various deleted forms of NLRP3 were immunoprecipitated with anti-Asc antibody followed by protein A agarose, or pulled down with pre-conjugated cAMP, cGMP or ATP beads.
Biochemical assays
Mouse IL-1β, IL-6 and TNF-α in cell culture supernatants and InsP3 and cAMP in cell lysates were measured using ELISA.
Methods
Reagents
Nigericin (catalogue no. N7143), GdCl3 (G7532), EGTA (E8145) and glyburide (G0639) were purchased from Sigma-Aldrich. Solutions of 2 M CaCl2 (351-033-721) and 1 M MgCl2 (351-130-721) were from Quality Biological, Inc. ATP (tlrl-atp), dsDNA (tlrl-patn), MSU (tlrl-msu), CPPD (tlrl-cppd), Z-VAD-FMK (tlrl-vad), ultra-pure flagellin (tlrl-pstfla), and ultra-pure LPS (tlrl-pelps) were obtained from InvivoGen. R-568 (3815), m-3M3FBS (1941), U73122 (1268), U73343 (4133), edelfosine (3022), 2-APB (1224), GF 109293X (0741), C-1 (0543), BAPTA-AM (2787), KH7 (3834), forskolin (1099), NKH477 (1603), KT5720 (1288), H89 (2910), Ro-20-1724 (0415), (R)-(-)-rolipram (1350) and zardaverine (1046) were from Tocris Bioscience.
Mice
P2x7r deficient mice were purchased from the Jackson Laboratory. Asc and Nlrp3 deficient mice were a gift from V. M. Dixit. Casp1 deficient mice were from R. Flavell. All animal studies were performed according to National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee of National Human Genome Research Institute.
Cell preparation
Unless otherwise indicated, all cells were cultured at 37 °C. BMDMs were obtained by differentiating bone marrow progenitors from the tibia and femur in Iscove's modified Dulbecco's medium (IMDM) containing 20 ng ml−1 of M-CSF (Peprotech), 10% heat-inactivated fetal bovine serum (FBS, Invitrogen), 1 mM sodium pyruvate, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (Invitrogen) for 7 days. BMDMs were then replated in 24-well plates 1 day before experiments. Blood specimens from healthy controls and CAPS and FMF patients were drawn after obtaining informed consent under a protocol approved by the NIAMS/NIDDK Institutional Review Board. Human PBMCs were isolated by LSM-Lymphocyte Separation Medium (50494, MP Biomedicals) from freshly drawn peripheral venous blood from healthy controls or patients.
Inflammasome activation or inhibition
Inflammasome activation experiments were performed in two stages: LPS priming for 3 h and inflammasome activation (within 1 h), to avoid any chemical inducing interference on the LPS priming stage. The duration of the activation phase was minimized to exclude the effects of secondary protein translation. BMDMs (1.0 × 106 cells per well) or PBMCs (2.0 × 106 cells per well) were plated in 12-well plates and then primed with 1 μg ml−1 LPS in RPMI 1640 (Invitrogen) containing 10% FBS, and antibiotics. For NLRP3 inflammasome activation, the medium was replaced with RPMI 1640 medium supplemented with ATP (2 mM), nigericin (20 μM), R-586 (10 μM), CaCl2 (1 mM, resulting in a total Ca2+ concentration of 1.42 mM, which is still in the physiological range), GdCl3 (1 mM), KH7 (50 μM), m-3M3FBS (5–40 μM), MSU (200 μg ml−1), [Author: ‘or’ in the correct place? No. corrected to the right place.] CPPD (200 μg ml−1), or thapsigargin (50 nM). For AIM2 or NLRC4 inflammasome activation, 1 μg ml−1 of dsDNA with 2.5 μl ml−1 of Lipofectamine 2000 (Invitrogen) or 0.5 μg ml−1 flagellin with 25 μl ml−1 DOTAP (Roche), respectively, were mixed in RPMI 1640 medium and incubated for 10 min before treatment of the cells. For the inhibition of inflammasome activation, LPS-primed BMDMs were treated with U73122 (10 μM), U73343 (10 μM), edelfosine (4–100 μM), 2-APB (10 or 50 μM), BAPTA-AM (10 or 20 μM), forskolin (10–500 μM), NKH477 (10–500 μM), Ro-20-1724 (50–250 μM), (R)-(-)-rolipram (50–250 μM), zardaverine (100 or 250 μM), GF 109203X (50 μM), or C-1 (50 μM) in the presence of each activator (ATP, CaCl2, dsDNA, or flagellin).
After 30–50 min of treatment, supernatants and cell lysates were collected for immunoblot analysis. For the ASC pyroptosome, pellets from whole-cell lysates were crosslinked with disuccinimidyl suberate (DSS) and analysed by immunoblotting for ASC. None of the reagents in these experiments induced cytotoxicity as confirmed by LDH assay (K311-400, BioVision).
Gene knockdown assay
siRNAs targeting Nlrp3, Asc, Aim2, Nlrc4, Pkaα, Ip3r1, Adcy3, Adcy6, Adcy7 and Adcy9, and scrambled siRNA were purchased from Applied Biosystems, and siRNA targeting Casr was purchased from Invitrogen. The three siRNAs for Nlrp3 knockdown were 5′-UCUCAAGUCUAAGCACCAATT-3′ (s103710), 5′-CAUCAAUGCUGCUUCGACATT-3′ (s103711) and 5′-CAGUGACAAUACUCUGGGATT-3′ (s103712), which were used in a mixture. The three siRNAs for Asc knockdown were 5′-GAGCAGCUGCAAACGACUATT-3′ (s205578), 5′-GCUACUAUCUGGAGUCGUATT-3′ (s83918) and 5′-CCUGGAACCUGACCUGCAATT-3′ (s83919), which were used in a mixture. The three siRNAs for Casr knockdown were 5′-CACGAGCUGGAAGACGAAAUCAUCU-3′ (MSS202652, Casr siRNA no. 1), 5′-GCGAUGGCUGACAUUAUCGAGUAUU-3′ (MSS202653, Casr siRNA no. 2) and 5′-CCAUCCCAGGAAGUCUGUCCACAAU-3′ (MSS202654, Casr siRNA no. 3). Aim2 siRNA is 5′-GAUAGAGUACUGUAUGGUATT-3′ (s234106). Nlrc4 siRNA is 5′-GGGUGAAGAUAUCGACAUATT-3′ (s114184). Pkaα siRNA is 5′-GAAGCUCCCUUCAUACCAATT-3′ (s71681). Ip3r1 siRNA is 5′-CCUUAGGCUUGGUUGAUGATT-3′ (s68515). Adcy3 siRNA is 5′-GCGCAUAGGCAUGAACAAATT-3′ (s98221). Adcy6 siRNA is 5′-GACUUUGACGAGAUCAUCATT-3′ (s61992). Adcy7 siRNA is 5′-GCAUUGCGCUCAUCAGCAUTT-3′ (s61993). Adcy9 siRNA is 5′-GCACGGCAAAGAUCUGGAATT-3′ (s62001). For siRNA gene knockdown experiments, BMDMs (0.5 × 106 cells per well) were replated in 12-well plates and transfected the next day with 60 pmol siRNA and 3 μl Lipofectamine 2000 according to the manufacturer's instructions. After 48 h, the siRNA-transfected cells were analysed for inflammasome activation.
Immunoprecipitation and pull-down assay
The lysates from BMDMs were immunoprecipitated with anti-ASC antibody (antibody sc-22514, Santa Cruz Biotechnology) followed by protein A plus agarose (Thermo Scientific). After washing, bound proteins were eluted from the beads and analysed by immunoblot for NLRP3 or ASC.
LPS-primed BMDMs or 293T cells transiently co-transfected with expression constructs for the wild type, CAPS-associated mutant, or various deleted forms of NLRP3 were lysed and incubated with pre-conjugated cAMP (AC-147 or AC-146, Jena Bioscience), cGMP (AC-148, Jena Bioscience), ATP (AC-127, Jena Bioscience), or empty/unconjugated beads for 90 min at room temperature. After washing, bound proteins were eluted by 2× SDS sample buffer and analysed by immunoblot for NLRP3 with anti-V5 or anti-MYC antibodies.
Immunoblots
Immunoblots were prepared with Novex Tris-Glycine Gel Systems (Invitrogen) and probed overnight at 4 °C with anti-human IL-1β antibody (AF-201-NA, R&D Systems); anti-mouse IL-1β antibody (AF-401-NA, R&D Systems); anti-caspase-1 p20 antibody (06-503, Millipore); anti-NLRP3 antibody (ALX-804-881, Enzo Life Sciences); anti-CASR antibody (ab18200, Abcam); anti-Ip3R1 antibody (sc-28614, Santa Cruz Biothechnology); anti-ASC antibody (sc-22514, Santa Cruz Biotechnology); anti-caspase-1 antibody (sc-622, Santa Cruz Biotechnology); anti-actin antibody (sc-1615, Santa Cruz Biotechnology); anti-MYC antibody (sc-40, Santa Cruz Biotechnology); or anti-V5 antibody (Invitrogen).
Biochemical assays
Mouse IL-1β, IL-6 and TNF-α in cell culture supernatants were measured using mouse IL-1β/IL-1F2 Quantikine ELISA kit, mouse IL-6 Quantikine ELISA kit, and mouse TNF-α Quantikine ELISA kit (MLB00C, M6000B, MTA00B, R&D Systems), respectively. Intracellular cAMP was measured in cell lysates by an ELISA-based competitive immunoassay (K371-100, BioVision). PLC activity was tested by IP-one ELISA (72IP1PEA, Cisbio), in which BMDMs were stimulated with LPS and then cell culture medium was replaced by fresh medium without or with ATP (2 mM) or CaCl2 (1 mM). Intracellular IP-one, a measure of the degradation products of InsP3 and a surrogate for InsP3 levels, was measured after LiCl (50 mM) treatment to prevent the degradation of IP-one into myo-inositol.
Calcium analysis by confocal microscopy
BMDMs were plated on 4-chambered coverglass dishes (155383, NUNC) at a density of 0.1 × 106 cells per well and incubated with fluo-4/AM. Cells were pre-treated with or without the InsP3 receptor blocker 2-APB (50 μM) or intracellular calcium chelator BAPTA-AM (10 μM). Images of untreated cells were acquired (t = 0), then cells were treated with added 1 mM ATP, 1 mM CaCl2, or 1 mM GdCl3 in RPMI 1640 (which already contains 0.42 mM Ca2+). Cells were imaged for 30 min with acquisition at 15 s intervals. At the end of the imaging session (at 30 min), ionomycin was added to the medium to a final concentration of 5 μM and cells were further imaged for 10 min. Images were acquired on a Leica SP5 Confocal Imaging System using the 488-nm laser and emission in the range of 500–600. Images were analysed using Imaris 7.3.1 software by creating surfaces to encompass the volume of each cell. Absolute intensity for all cells in a field at different time points was obtained, and normalized to time ‘0’ to obtain the fold increase in intensity. Data are displayed as the relative intensity of all cells in a field (20–25 cells per field) and are representative of three independent fields.
Statistical analyses
Statistical analyses were performed with the t-test for two groups or one-way ANOVA for multiple groups using GraphPad Prism (GraphPad Software).
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Programs of the NIAMS, NHGRI, and NIAID, NIH. We thank E. Remmers for discussion and a thorough review of this manuscript.
Footnotes
Supplementary Information is available in the online version of the paper.
Author Contributions: G.-S.L., D.L.K. and J.J.C. designed the research; G.-S.L., N.S., A.I.K. and J.J.C. performed the experiments; G.-S.L., N.S., I.A., D.B.S., R.N.G., D.L.K. and J.J.C. analysed the results; R.G.-M., I.A. and D.L.K. provided patient samples; G.-S.L., J.J.C. and D.L.K. wrote the paper; N.S., I.A., R.G.-M., D.B.S. and R.N.G. edited and commented on the manuscript.
Author Information: Reprints and permissions information is available at www.nature.com/reprints. Readers are welcome to comment on the online version of the paper.
The authors declare no competing financial interests.
References
- 1.Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol. 2009;27:621–668. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kahlenberg JM, Dubyak GR. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am J Physiol Cell Physiol. 2004;286 doi: 10.1152/ajpcell.00494.2003. [DOI] [PubMed] [Google Scholar]
- 3.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 4.Hornung V, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petrilli V, et al. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14:1583–1589. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 6.Brough D, et al. Ca2+ stores and Ca2+ entry differentially contribute to the release of IL-1 beta and IL-1 alpha from murine macrophages. J Immunol. 2003;170:3029–3036. doi: 10.4049/jimmunol.170.6.3029. [DOI] [PubMed] [Google Scholar]
- 7.Cockcroft S, Gomperts BD. Activation and inhibition of calcium-dependent histamine secretion by ATP ions applied to rat mast cells. J Physiol. 1979;296:229–243. doi: 10.1113/jphysiol.1979.sp013002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ross PE, Ehring GR, Cahalan MD. Dynamics of ATP-induced calcium signaling in single mouse thymocytes. J Cell Biol. 1997;138:987–998. doi: 10.1083/jcb.138.5.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murakami T, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci USA. 2012 doi: 10.1073/pnas.1117765109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hofer AM, Brown EM. Extracellular calcium sensing and signalling. Nat Rev Mol Cell Biol. 2003;4:530–538. doi: 10.1038/nrm1154. [DOI] [PubMed] [Google Scholar]
- 11.Xi YH, et al. The functional expression of calcium-sensing receptor in the differentiated THP-1 cells. Mol Cell Biochem. 2010;342:233–240. doi: 10.1007/s11010-010-0489-3. [DOI] [PubMed] [Google Scholar]
- 12.Osawa Y, Lee HT, Hirshman CA, Xu D, Emala CW. Lipopolysaccharide-induced sensitization of adenylyl cyclase activity in murine macrophages. Am J Physiol Cell Physiol. 2006;290:C143–151. doi: 10.1152/ajpcell.00171.2005. [DOI] [PubMed] [Google Scholar]
- 13.Duncan JA, et al. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci USA. 2007;104:8041–8046. doi: 10.1073/pnas.0611496104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gattorno M, et al. Pattern of interleukin-1beta secretion in response to lipopolysaccharide and ATP before and after interleukin-1 blockade in patients with CIAS1 mutations. Arthritis Rheum. 2007;56:3138–3148. doi: 10.1002/art.22842. [DOI] [PubMed] [Google Scholar]
- 15.Chae JJ, et al. Gain-of-function pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity. 2011;34:755–768. doi: 10.1016/j.immuni.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Menkin V. Biochemical mechanisms in inflammation. Charles Thomas Publisher: 1981. [Google Scholar]
- 17.Peters-Golden M. Putting on the brakes: cyclic AMP as a multipronged controller of macrophage function. Sci Signal. 2009;2:pe37. doi: 10.1126/scisignal.275pe37. [DOI] [PubMed] [Google Scholar]
- 18.Torphy TJ. Phosphodiesterase isozymes: molecular targets for novel antiasthma agents. Am J Respir Crit Care Med. 1998;157:351–370. doi: 10.1164/ajrccm.157.2.9708012. [DOI] [PubMed] [Google Scholar]
- 19.Kim C, Cheng CY, Saldanha SA, Taylor SS. PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell. 2007;130:1032–1043. doi: 10.1016/j.cell.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 20.Bos JL. Epac: a new cAMP target and new avenues in cAMP research. Nat Rev Mol Cell Biol. 2003;4:733–738. doi: 10.1038/nrm1197. [DOI] [PubMed] [Google Scholar]
- 21.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 22.Vandanmagsar B, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duewell P, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halle A, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.