

Abstract
Background
Evidence point to vascular dysfunction and hypoperfusion as early abnormalities in Alzheimer's disease (AD); probing their mechanistic bases can lead to new therapeutic approaches. We tested the hypotheses that β-amyloid peptide induces endothelial dysfunction and oxidative stress in human microvasculature and that response will be similar between peripheral adipose and brain leptomeningeal arterioles.
New Method
Abdominal subcutaneous arterioles from living human subjects (n=17) and cadaver leptomeningeal arterioles (n=6) from rapid autopsy were exposed to Aβ1-42 (Aβ) for 1-hour and dilation response to acetylcholine/papaverine were measured and compared to baseline response. Adipose arteriole reactive oxygen species (ROS) production and nitrotyrosine content were measured.
Comparison with existing methods
Methods described allow direct investigation of human microvessel functional response that cannot be replicated by human noninvasive imaging or post-mortem histology.
Results
Adipose arterioles exposed to 2 μM Aβ showed impaired dilation to acetylcholine that was reversed by antioxidant polyethylene glycol superoxide dismutase (PEG-SOD) (Aβ-60.9±6%, control-93.2±1.8%, Aβ+PEGSOD-84.7±3.9%, both p<0.05 vs. Aβ). Aβ caused reduced dilation to papaverine. Aβ increased adipose arteriole ROS production and increased arteriole nitrotyrosine content. Leptomeningeal arterioles showed similar impaired response to acetylcholine when exposed to Aβ (43.0±6.2% versus 81.1±5.7% control, p<0.05).
Conclusion
Aβ exposure induced adipose arteriole endothelial and non-endothelial dysfunction and oxidative stress that were reversed by antioxidant treatment. Aβ-induced endothelial dysfunction was similar between peripheral adipose and leptomeningeal arterioles. Ex vivo living adipose and cadaver leptomeningeal arterioles are viable, novel and practical human tissue models to study Alzheimer's vascular pathophysiology.
Keywords: Alzheimer's disease, amyloid, endothelial function, microvessels
1. Introduction
Despite decades of research, there is still no viable treatment to prevent or reverse Alzheimer's disease (AD), a disease expected to affect 80 million people by 2040 (Ferri et al., 2005). It is possible that treatments developed from and that worked well in genetically engineered mouse models poorly translate to human pathophysiology due to fundamental interspecies differences in biophysical, biochemical, genomic and cellular pathology (Kokjohn and Roher, 2009; Seok et al., 2013). In addition, modifying the disease likely requires intervention at the earliest stages prior to onset of neuronal tissue damage and frank dementia (Iadecola, 2004). There is substantial clinical evidence that vascular dysfunction leading to cerebral hypoperfusion is an early pathologic event in AD (Claassen et al., 2009; Jobst et al., 1997; Johnson and Albert, 2000; Johnson et al., 1998), yet the mechanisms by which vascular disease contribute to AD remain poorly understood. Probing the mechanistic bases of early vascular dysfunction in AD in human vessels would be valuable in developing therapies for the early preclinical stages of AD. We tested the hypotheses that Aβ1-42 (Aβ), a major peptide derived from cleavage of amyloid precursor protein and implicated in AD pathology (Dietrich et al., 2010), causes human microvascular endothelial dysfunction through oxidative stress and that the dysfunction induced by Aβ to peripheral adipose arterioles will be similar to the effects on brain leptomeningeal arterioles. By successfully establishing acute microvascular toxicity of Aβ in both human abdominal adipose arterioles and brain leptomeningeal arterioles, we also aim to demonstrate the feasibility of using adipose tissue from living subjects and cadaver leptomeningeal arterioles as practical models to study AD microvascular pathophysiology in human tissue.
2. Materials and Methods
2.1 Human Subjects
A. Living subjects without known vascular disease, diabetes or AD scheduled to undergo elective abdominal surgery (herniorrhaphy) were recruited (n=17, 61±2.5 years, all males). After obtaining informed consent, abdominal subcutaneous adipose tissues from these subjects were collected during surgery. The collection and testing of living adipose tissue, the testing of cadaver tissue shared under a Materials Transfer agreement with Banner Sun Health Research Institute (BSHRI) and the research study were approved by the Phoenix Veterans Affairs Institutional Review Board. B. Post-mortem leptomeningeal arterioles were isolated from the cadavers of subjects (n=12, 85.8±2.2 years, 6 females) who while living provided informed consent for brain±body donation following death under an existing BSHRI Brain and Body Donation Program (www.brainandbodydonationprogram.org) (Beach et al., 2008). The operations of the Program have been approved by the Banner Sun Health Research Institute Institutional Review Board. When available, abdominal adipose tissues were collected. The BSHRI program involves 24-hour on-call pathology technicians who perform rapid autopsy of community-recruited participants within a few hours of declaration of death. Tissues were immediately placed in sterile 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (4°C, pH 7.4). This buffer contains in mM 10 HEPES acid, 138 NaCl, 4 KCl, 1.2 MgSO4, 1.3 CaCl2, 1.2 KH2PO4, 6 D-glucose, 0.02 EDTA.
2.2 Arteriole Vasoreactivity
Living subject adipose arterioles were isolated, cannulated and pressurized to 60 mm Hg similar to previous protocol (Franco et al., 2012; Migrino et al., 2011; Truran et al., 2014). Baseline (control) dilator response was measured by videomicrometer after preconstriction with endothelin-1 followed by administration of acetylcholine (for endothelium-dependent dilation, 10−9-10−4M) and papaverine (for smooth muscle-dependent dilation, 10−4M), similar to methods validated by other investigators (Dharmashankar et al., 2012; Kizhakekuttu et al., 2012; Suboc et al., 2013). The vessels were preconstricted with gradually increasing doses of endothelin-1 and further administration of endothelin-1 was discontinued after the vessel crossed the threshold of having a diameter ~60% of baseline diameter. The doses of endothelin-1 administered are in Table 1. Dilator response was calculated as percentage change from the diameter achieved following endothelin-1 preconstriction. The method has been validated by other investigators to correlate closely with clinical/in vivo resistance arteriole response in patients (Dharmashankar et al., 2012; Kizhakekuttu et al., 2012; Suboc et al., 2013). After washing, arterioles were exposed for 1-hour to 2 μM Aβ1-42 (Anaspec, Fremont CA, Sigma-Aldrich, St. Louis MO or GenScript, Piscataway NJ) ± polyethylene glycol superoxide dismutase (PEGSOD, 250 U/mL, Sigma-Aldrich) or 0.2 μM Aβ. The dose of 2 μM was chosen because it is less than but close to the published concentration of Aβ42 in cortical tissue found in autopsies of patients with Alzheimer's disease (~30,000 ng/g tissue) from the BSHRI Brain and Body Donation program (Morawski et al., 2014) and it is close to the concentrations used in published preclinical studies that showed impaired vasomotor response (Dietrich et al., 2010; Thomas et al., 1997). In living adipose arterioles treated with Aβ 2 μM, Aβ was administered intraluminally in 6 arterioles and extraluminally in 9 arterioles. We found no significant difference in dilator responses to acetylcholine or papaverine by mode of administration so we combined the data for purpose of analysis. Aβ 0.2 μM and Aβ+PEGSOD were administered to the arterioles extraluminally.
Table 1.
Arteriole diameters and endothelin-1 dose.
| Maximum diameter (μM) | Pre-endothelin-1 diameter (μM)a | Post-endothelin-1 diameter (μM)a | Endothelin-1 dose (μM)a | |
|---|---|---|---|---|
| Adipose arterioles (living) | ||||
| Control (baseline) | 142.3±14.9 | 135.8±15.0 | 53.6±6.6 | 3.6±0.7 |
| Aβ | 124.9±10.5 | 52.7±6.8 | 7.6±2.8 | |
| Leptomeningeal arterioles | ||||
| Control (baseline) | 163.4±14.7 | 149.6±18.3 | 82.5±9.3 | 12.7±5.2 |
| Aβ | 150.0±19.0 | 88±9.9 | 62.5±28.5 | |
| Cadaver adipose arterioles | ||||
| Control (baseline) | 165.3±30.4 | 164.9±17.5 | 65.9±15.3 | 8.0±3.1 |
| Aβ | 151.0±28.6 | 62.3±13.4 | 7.6±3.1 |
p=NS baseline control versus Aβ
In similar fashion but in separate experiments, dilator responses to acetylcholine and papaverine following exposure to Aβ were tested in cadaver leptomeningeal arterioles (n=6) and cadaver adipose arterioles (n=5). A seventh leptomeningeal arteriole (subject 2 in Table 1) showed responsiveness to endothelin-1, acetylcholine (51.4%) and papaverine (100%) at baseline but Aβ was not administered following baseline response. Five leptomeningeal arterioles showed no responsiveness to endothelin-1. Aβ was administered intraluminally in 5 leptomeningeal arterioles and 3 cadaver adipose arterioles and extraluminally in 1 leptomeningeal arteriole and 2 cadaver arterioles; the intraluminal and extraluminal data were combined for purpose of analysis.
2.3 Adipose arteriole reactive oxygen species and nitrotyrosine
Reactive oxygen species production was assessed using 5 μM 2’, 7’-dichlorodihydrofluorescein diacetate (Invitrogen, Eugene OR), a cell-permeable redox-sensitive fluorophore that was previously used to measure light-chain induced cellular ROS production (Shi et al., 2010) through conversion to the highly fluorescent 2’, 7’-dichlorofluorescein (DCF). Living subject adipose arterioles were exposed for 1 hour to 2 μM Aβ ± PEGSOD (250 U/mL). At 45 minutes of treatment, acetylcholine 10−4M was administered and DCF was administered after 60 minutes of Aβ treatment. Fluorescence signal was measured using Olympus IX51 fluorescent microscope (excitation 495, emission 525 nM). Living adipose arteriole nitrotyrosine content was measured to assess reactive nitrogen species-induced nitrative stress (Mohiuddin et al., 2006) using Western blot (anti-3-nitrotyrosine antibody, Abcam, Cambridge MA) densitometry normalized to β-actin content. Vehicle or Aβ (2 μM) ± PEGSOD (250 U/mL) was administered to arterioles for 1 hour and acetylcholine 10−4M was administered at 45 minutes of treatment.
2.4 Thioflavin staining
To identify vessel amyloid deposits in leptomeningeal arterioles, three leptomeningeal arterioles were rinsed five times in deionized water, stained for 15 minutes with 0.5 mL of 1% aqueous thioflavin-S, rinsed 10 times with 70% ethanol followed by deionized water and then imaged on glass slides using a Leica DMLB epifluorescent microscope.
2.5 Data Analyses
Data are expressed as means±standard error of means, and significant p-value (2-sided) was set at p<0.05. For vasoreactivity, overall response to acetylcholine was assessed by computing effective dose of acetylcholine producing 50% dilator response (LogM EC50). This was calculated using nonlinear regression using variable slope (4 parameters) and least squares (ordinary fit), as described previously (Migrino et al., 2011). Control (baseline) response was compared to the post-treatment response in the same arteriole for each acetylcholine and papaverine dose using paired Student's t-test. Dilator responses to acetylcholine and papaverine were compared between different treatments (e.g. Aβ versus Aβ+PEGSOD) using unpaired Student's t-test. For DCF and nitrotyrosine assays, arteriole response to Aβ was compared separately to vehicle and Aβ+PEGSOD using paired Student's t-test of raw DCF and nitrotyrosine data. Prior to analysis, the DCF and nitrotyrosine data (in arbitrary units) were transformed (reciprocal) and the resulting paired Student's t-test passed test of normality of distribution of data. The representation of the data, however, was expressed as values relative to vehicle control (Figure 2). Statistical analyses were performed using GraphPad Prism 5.0 (San Diego CA) and SigmaStat 3.5 (Richmond CA).
Figure 2.

Arteriole fluorescence and nitrotyrosine. A. Adipose arterioles from living subjects exposed to Aβ showed increased DCF fluorescence compared to vehicle control and co-treatment with PEG-SOD prevented the increase in DCF fluorescence. B. Adipose arterioles from living subjects showed increased vessel nitrotyrosine content when exposed to Aβ. Although the nitrotyrosine level was different in arterioles treated with Aβ+PEGSOD, there was no significant difference when compared to Aβ treatment alone. C-E. Thioflavin staining showing amyloid deposits in the arteriole of subject 5 with AD (2C) whereas no amyloid deposit is seen in the arterioles of subjects 4 and 3 who had AD and dementia/with Lewy bodies (2D-E).
3. Results
3.1 Living subject adipose arterioles
The ex-vivo adipose arterioles had maximum diameters of 142.3±14.9 μM (Table 1). There was dose-dependent reduction in dilator response to acetylcholine in adipose arterioles exposed to 0.2 and 2 μM of Aβ (Figure 1A and 1B) with significant decrease in dilator response with 2 μM Aβ (10−4M acetylcholine: control 93.2±1.8% versus Aβ 60.9±6%, p<0.001). Co-treatment with antioxidant PEGSOD partially restored dilator response to acetylcholine (84.7±3.9%, p=0.03 versus Aβ). Similarly, 2 μM Aβ modestly reduced dilator response to papaverine (control 96.6±1.4%, Aβ 79.6±4.2%, p=0.002 versus control, Aβ+PEGSOD 93.5±2.7%). Treatment with scrambled Aβ did not show change in dilator response to acetylcholine or papaverine when compared to baseline control (Figure 1). However, there was a significant difference in dilator response to maximum acetylcholine dose between scrambled Aβ versus Aβ. There was no significant difference in dilator response to acetylcholine or papaverine when Aβ was administered intraluminally or extraluminally (acetylcholine EC50: −4.98±0.82 versus −5.1±0.35, p=NS; papaverine: 75.3±8.8% versus 82.5±4.8%, respectively).
Figure 1.

Arteriole vasoreactivity. A. Response to acetylcholine in arterioles from cadaver and living human subjects show similar increased EC50 response following exposure to Aβ in adipose arterioles from living subjects and cadaver sources as well as cadaver leptomeningeal arterioles. Co-treatment with PEGSOD restored dilator response to acetylcholine. There was no significant difference from baseline response when adipose arterioles were exposed to scrambled Aβ. B. Dilator response to maximum acetylcholine dose showed similar patterns of response in leptomeningeal or adipose arterioles treated with Aβ with restoration of response when Aβ is co-treated with PEG-SOD. C. There was no significant difference in papaverine response in cadaver arterioles exposed to Aβ, although adipose arterioles from living subjects showed modest but significant impaired dilator response to papaverine. *p<0.05 versus control (C); +p<0.05 versus Aβ treatment; #baseline control data for acetylcholine (B) and papaverine (C) were combined in the graph to simplify data presentation; scr=scrambled.
Aβ increased adipose arteriole DCF fluorescence that was reversed by PEGSOD (Figure 2A). Aβ increased adipose arteriole nitrotyrosine content when compared with control (Figure 2B); the difference in nitrotyrosine content between arterioles treated with Aβ versus Aβ-PEGSOD did not reach statistical significance.
3.2 Cadaver leptomeningeal and adipose arterioles
Median post-mortem interval (time interval between declaration of death and collection of tissue) was 2.66 hours (range 2.08-4.5 hours) (Table 1). Of 12 leptomeningeal arterioles collected, 7 (58%) were vasoreactive to standard acetylcholine and papaverine concentrations. There was no significant difference in post-mortem intervals between leptomeningeal arterioles that were vasoreactive versus those that were not (2.81±0.31 versus 2.97±0.25 hours, p=NS). Similar to living adipose arterioles, Aβ-treated leptomeningeal arterioles showed reduced dilator response to acetylcholine while the difference in response to papaverine was not significant (Figure 1). We simultaneously collected adipose tissues from 7 out of 12 donors and 5 (71%) were found to be vasoreactive. Similar to leptomeningeal arterioles, cadaver abdominal adipose arterioles showed reduction in dilator response to acetylcholine when exposed to Aβ, but the response to papaverine was not significantly different from control response. The magnitudes of Aβ-induced reduction in dilation to acetylcholine were similar among cadaver leptomeningeal, cadaver adipose and living subject adipose arterioles (−38.1±6.2%, −36.2±5.4% and −32.0±7%, respectively, p=NS). Similarly, the magnitudes of Aβ-induced change in dilation to papaverine were not significantly different among cadaver leptomeningeal, cadaver adipose and living subject adipose arterioles (−6.6±4.4%, −21.9±11.9% and −17±4.8%, respectively, p=NS). Thioflavin-S staining done in three leptomeningeal arterioles was positive for amyloid in 1 AD subject (#5) and negative in 2 AD/dementia with Lewy bodies subjects (#4 and #3) (Figure 2); baseline maximal dilation to acetylcholine in these subjects were 70%, 97% and 87%, respectively. Dilator response to acetylcholine or papaverine at baseline or following Aβ exposure were not significantly different in subjects with Alzheimer's disease or those without (Figure 3).
Figure 3.
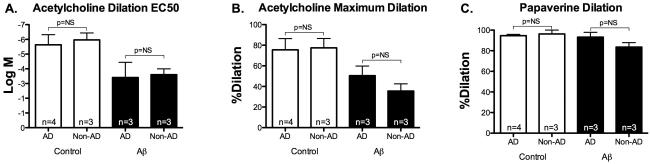
Leptomeningeal arteriole vasoreactivity in AD versus non-AD subjects. There were no significant differences in baseline and post-Aβ dilator responses to acetylcholine and papaverine following Aβ exposure in cadaver arterioles from AD or non-AD subjects.
In one tissue, vasoreactivity was performed on the day of autopsy (D0) and a second performed the following day (D1) on a different arteriole. Dilation to acetylcholine was 51.4% on D0 and 44.4% on D1, while dilation to papaverine was 100% on D0 and 82.3% on D1, indicating continued vasoreactivity of the arteriole sample while kept on HEPES buffer (4°C) 24 hours post-collection. Leptomeningeal arteriole dilator response to acetylcholine (logMEC50) when Aβ was administered intraluminally (n=5) was −3.6±0.6 while it was −2.8 when Aβ was administered extraluminally (n=1); papaverine response was 89.7±4.9% (intraluminal) and 88.0% (extraluminal). Cadaver adipose arteriole dilator response to acetylcholine (logMEC50) when Aβ was administered intraluminally (n=3) was −3.3±0.6 while it was −3.6±0.8 when Aβ was administered extraluminally (n=2); papaverine response was 57.0±20% (intraluminal) and 75.1±2% (extraluminal).
4. Discussion
We present the following novel findings: 1) Acute exposure to Aβ1-42 peptide leads to significant endothelial dysfunction (and more modest smooth-muscle dependent dysfunction) in abdominal adipose arterioles of non-AD subjects that is associated with oxidative and nitrative stress. The endothelial dysfunction is partially reversed by antioxidant treatment. 2) Some, but not all, cadaver leptomeningeal and abdominal adipose arterioles obtained using rapid autopsy with median postmortem interval of 2.66 hours retain vasoreactivity to agonists (acetylcholine and papaverine). Aβ1-42-induced peripheral adipose arteriole endothelial dysfunction parallels the degree of dysfunction induced in leptomeningeal arterioles. The results support the practical feasibility of using living subject adipose arterioles and cadaver leptomeningeal arterioles as potential new human tissue models to study Alzheimer's disease vascular pathophysiology.
There remains a lack of viable treatment to prevent or reverse AD and therapeutic interventions that were effective in genetically engineered mouse models yielded disappointing results when tested in clinical trials. Accumulating evidence increasingly suggests that human disease is not fully recapitulated in mouse or rat models (Grande et al., 2012; Kokjohn and Roher, 2009; Seok et al., 2013). Testing therapies or probing signaling pathways on human tissue models can provide approaches with greater translational impact on human disease.
Vascular dysfunction occurs early in AD, possibly years or decades prior to overt clinical AD (Claassen et al., 2009; Iadecola, 2004; Johnson and Albert, 2000; Johnson et al., 1998), yet little is known about the mechanisms underlying the initiation and progression of vascular disease and the role it plays in cognitive impairment. Endothelial dysfunction involves impaired endothelium-dependent vasodilation from loss of vessel wall nitric oxide (NO) bioavailability (Cai and Harrison, 2000), as measured ex-vivo through vessel dilator response to agonists such as acetylcholine that act through NO-mediated transduction mechanisms. Endothelial dysfunction is an early event in vascular pathology (such as atherosclerosis, hypertension or diabetes) that eventually leads to consequential functional and structural abnormalities that can result in tissue ischemia, and is associated with adverse cardiovascular and cerebrovascular clinical events (Schachinger et al., 2000). Soluble Aβ acutely impairs cerebrovascular function in animal models of AD early in the disease (Dietrich et al.; Price et al., 1997). Similarly in two previous studies, Aβ peptides augmented endothelin-1-induced vasoconstriction in human cadaver vessels (Paris et al., 2003; Townsend et al., 2002), the first demonstration of the potential use of post-mortem vessels in studying Aβ vascular effects. These results together with our findings strongly suggest that soluble Aβ plays an important role in AD-associated vascular impairment. A mechanism by which Aβ induces vascular dysfunction was demonstrated in our study through induction of oxidative and nitrative stress, with partial restoration of endothelial function using antioxidant PEGSOD. This is consistent with previous reports that Aβ peptides including Aβ1-42 induce oxidative stress leading to neurotoxicity (Butterfield, 2002), although a prior report suggested that antioxidant treatment may not reverse Aβ neuronal toxicity (Pike et al., 1997) unlike our findings on human vascular tissue. The effects of Aβ shown in this study are similar to the endothelial dysfunction and oxidative/nitrative stress induced by immunoglobulin light chain amyloid proteins (Franco et al., 2012; Migrino et al., 2011), further bolstering the concept that misfolded amyloidogenic proteins, despite varying amino acid compositions, share common toxic pathophysiology possibly triggered by abnormal protein folding (Pastor et al., 2008; Schubert et al., 1995).
Importantly, our results demonstrate similarity of response to Aβ between leptomeningeal arterioles and peripheral adipose arterioles. The practical significance of this finding is that abdominal adipose arterioles from living subjects, e.g. those without disease, at-risk for disease or with early disease, may be used as an accessible surrogate to study Aβ effects on the microvasculature and to probe whether vessel response to Aβ is a biomarker of risk for AD initiation or progression. In addition, cadaver arterioles from well-characterized patients (by phenotype, clinical presentation and histopathology) with varying stages of disease or no disease may be used to study signaling pathways or test novel therapies, providing a high-throughput ex-vivo human model that could bridge the translational gap between genetically engineered mouse models and costly clinical trials while having the potential to discover unanticipated adverse effects prior to in-vivo human administration.
Our study is a proof-of-concept initial report. Limitations of our study include small sample size and our model by its own nature can only probe acute effects of Aβ, since ex-vivo arterioles are not viable for long-term experiments. Teasing apart acute versus chronic effects using this model can, however, be approached by comparing baseline leptomeningeal vascular response at varying stages of disease (implicating chronic exposure) and testing additional dysfunction following experimental acute exposure to Aβ. Indeed, our preliminary results show a trend for a worse baseline dilatory response to acetylcholine in the AD patient with arteriole amyloid deposits compared with the AD subjects without evidence for amyloid, and worsening of endothelial function following Aβ exposure in all arterioles regardless of underlying neurologic disease. Peripheral adipose arterioles, especially if obtained from living subjects, represent the more practical and extensively available source of human vascular tissue for future studies. Although we demonstrated similar responses to vasodilators between peripheral adipose arterioles and leptomeningeal arterioles, further studies are needed to fully define the physiologic extent and limitations of adipose arterioles as surrogate tissue to study cerebrovascular arteriole responses. In the cardiovascular field, the adipose arteriole model is already an established model to study signaling pathways underlying coronary artery disease (Kizhakekuttu et al., 2012; Migrino et al., 2011; Phillips et al., 2006) pointing to the potential of the use of peripheral arterioles to study human central (coronary or cerebrovascular) microcirculatory pathologic mechanisms that may have enhanced translational relevance to human disease.
Highlights.
The lack of effective treatment for Alzheimer's disease emphasize the need for finding novel models to study its pathophysiology.
Leptomeningeal arterioles isolated from rapid autopsy organ donors show variable ability to respond to vasodilators.
Aβ1-42 peptide induces endothelial dysfunction to similar degrees in leptomeningeal and adipose arterioles of cadaver subjects and adipose arterioles from living subjects.
Aβ1-42 peptide induces human arteriole oxidative and nitrative stress.
Use of human adipose and leptomeningeal arterioles may be a useful model to study vascular dysfunction in Alzheimer's disease.
Table 2.
Donor demographic information
| Subject | Age (years) |
Gender | Postmortem Interval (hours) |
Neurologic Clinical or Pathologic Diagnoses |
Cause of death | Cardiovascular Conditions/Risks |
Leptomeningeal arteriole vasoreactivity |
|---|---|---|---|---|---|---|---|
| 1 | >85 | male | 2.25 | Parkinson's disease | Respiratory failure, cardiac arrest | CAD, hyperlidemia | Yes |
| 2 | >85 | female | 2.08 | Parkinson's disease/Alzheimer's disease | End-stage Parkinson's disease | hyperlipidemia, HTN | Yes |
| 3 | 82 | female | 2.75 | Alzheimer's disease/dementia with Lewy bodies | Sepsis, failure to thrive | CAD, HTN, PVD, hyperlipidemia | Yes |
| 4 | 76 | male | 3.18 | Alzheimer's disease/dementia with Lewy bodies | Failure to thrive | smoker | Yes |
| 5 | 76 | female | 2.58 | Alzheimer's disease | Myocardial infarction | CAD | Yes |
| 6 | >85 | female | 4.5 | Cognitively normal | Pancreatic cancer | HTN, PVD | Yes |
| 7 | >85 | female | 2.33 | Cognitively normal | Cardiorespiratory arrest | HTN, hyperlipidemia | Yes |
| 8 | 80 | male | 2.83 | Parkinson's disease | Failure to thrive, chronic obstructive pulmonary disease, Parkinson's disease | CAD, hyperlipidemia, smoker | No |
| 9 | >85 | male | 2.5 | Cognitively normal | Prostate cancer | PVD, smoker | No |
| 10 | 86 | male | 3.5 | Dementia probable AD | Cardiorespiratory arrest, lymphoma | smoker | No |
| 11 | 77 | male | 3.62 | Dementia probable AD | Cardiorespiratory arrest | CAD, HTN, hyperlipidemia | No |
| 12 | >85 | female | 2.4 | Probable AD | Natural causes | HTN | No |
Legend: CAD-coronary artery disease, HTN-hypertension, PVD-peripheral vascular disease
Acknowledgements and Grants
We would like to thank the Phoenix VA Surgery Service faculty (Howard Bourdages, Lillian Dawes, William Dolan, Maher Huttam, John Pyeatt III) and staff, John Hatfield, Wuqiong Ma and Juraj Koska for their assistance.
Grant support was provided by the VA Merit BLRD I01BX007080 (VA ORD), Amyloidosis Foundation, National Institute of Neurological Disorders and Stroke (U24NS072026), the National Institute of Aging (P30AG19610, RO1AG019795), the Arizona Department of Health Services (contract 211002), the Arizona Biomedical Research Commission (4001, 0011, 05-901 and 1001), Michael J. Fox Foundation for Parkinson's Research and American Heart Association Summer Student Scholarship grant. The study was supported by US Veterans Affairs employment. The contents do not represent the views of the US Veterans Affairs or the US government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beach TG, Sue LI, Walker DG, Roher AE, Lue L, Vedders L, Connor DJ, Sabbagh MN, Rogers J. The Sun Health Research Institute Brain Donation Program: description and experience, 1987-2007. Cell Tissue Bank. 2008;9:229–45. doi: 10.1007/s10561-008-9067-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA. Amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer's disease brain. A review. Free Radic Res. 2002;36:1307–13. doi: 10.1080/1071576021000049890. [DOI] [PubMed] [Google Scholar]
- Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–4. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- Claassen JA, Diaz-Arrastia R, Martin-Cook K, Levine BD, Zhang R. Altered cerebral hemodynamics in early Alzheimer disease: a pilot study using transcranial Doppler. J Alzheimers Dis. 2009;17:621–9. doi: 10.3233/JAD-2009-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmashankar K, Welsh A, Wang J, Kizhakekuttu TJ, Ying R, Gutterman DD, Widlansky ME. Nitric oxide synthase-dependent vasodilation of human subcutaneous arterioles correlates with noninvasive measurements of endothelial function. Am J Hypertens. 2012;25:528–34. doi: 10.1038/ajh.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich HH, Xiang C, Han BH, Zipfel GJ, Holtzman DM. Soluble amyloid-beta, effect on cerebral arteriolar regulation and vascular cells. Mol Neurodegener. 2010;5:15. doi: 10.1186/1750-1326-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112–7. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco DA, Truran S, Burciu C, Gutterman DD, Maltagliati A, Weissig V, Hari P, Migrino RQ. Protective role of clusterin in preserving endothelial function in AL amyloidosis. Atherosclerosis. 2012;225:220–3. doi: 10.1016/j.atherosclerosis.2012.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grande G, Nilsson E, Edvinsson L. Comparison of responses to vasoactive drugs in human and rat cerebral arteries using myography and pressurized cerebral artery method. Cephalalgia. 2012;33:152–9. doi: 10.1177/0333102412468340. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5:347–60. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Jobst KA, Barnetson LP, Shepstone BJ. Accurate prediction of histologically confirmed Alzheimer's disease and the differential diagnosis of dementia: the use of NINCDS-ADRDA and DSM-III-R criteria, SPECT, X-ray CT, and APO E4 medial temporal lobe dementias. The Oxford Project to Investigate Memory and Aging. Int Psychogeriatr. 1997;9(Suppl 1):191–222. discussion 47-52. [PubMed] [Google Scholar]
- Johnson KA, Albert MS. Perfusion abnormalities in prodromal AD. Neurobiol Aging. 2000;21:289–92. doi: 10.1016/s0197-4580(00)00137-8. [DOI] [PubMed] [Google Scholar]
- Johnson KA, Jones K, Holman BL, Becker JA, Spiers PA, Satlin A, Albert MS. Preclinical prediction of Alzheimer's disease using SPECT. Neurology. 1998;50:1563–71. doi: 10.1212/wnl.50.6.1563. [DOI] [PubMed] [Google Scholar]
- Kizhakekuttu TJ, Wang J, Dharmashankar K, Ying R, Gutterman DD, Vita JA, Widlansky ME. Adverse alterations in mitochondrial function contribute to type 2 diabetes mellitus-related endothelial dysfunction in humans. Arterioscler Thromb Vasc Biol. 2012;32:2531–9. doi: 10.1161/ATVBAHA.112.256024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokjohn TA, Roher AE. Amyloid precursor protein transgenic mouse models and Alzheimer's disease: understanding the paradigms, limitations, and contributions. Alzheimers Dement. 2009;5:340–7. doi: 10.1016/j.jalz.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migrino RQ, Truran S, Gutterman DD, Franco DA, Bright M, Schlundt B, Timmons M, Motta A, Phillips SA, Hari P. Human microvascular dysfunction and apoptotic injury induced by AL amyloidosis light chain proteins. Am J Physiol Heart Circ Physiol. 2011;301:H2305–12. doi: 10.1152/ajpheart.00503.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohiuddin I, Chai H, Lin PH, Lumsden AB, Yao Q, Chen C. Nitrotyrosine and chlorotyrosine: clinical significance and biological functions in the vascular system. J Surg Res. 2006;133:143–9. doi: 10.1016/j.jss.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Morawski M, Schilling S, Kreuzberger M, Waniek A, Jager C, Koch B, Cynis H, Kehlen A, Arendt T, Hartlage-Rubsamen M, Demuth HU, Rossner S. Glutaminyl cyclase in human cortex: correlation with (pGlu)-amyloid-beta load and cognitive decline in Alzheimer's disease. J Alzheimers Dis. 2014;39:385–400. doi: 10.3233/JAD-131535. [DOI] [PubMed] [Google Scholar]
- Paris D, Humphrey J, Quadros A, Patel N, Crescentini R, Crawford F, Mullan M. Vasoactive effects of A beta in isolated human cerebrovessels and in a transgenic mouse model of Alzheimer's disease: role of inflammation. Neurol Res. 2003;25:642–51. doi: 10.1179/016164103101201940. [DOI] [PubMed] [Google Scholar]
- Pastor MT, Kummerer N, Schubert V, Esteras-Chopo A, Dotti CG, Lopez de la Paz M, Serrano L. Amyloid toxicity is independent of polypeptide sequence, length and chirality. J Mol Biol. 2008;375:695–707. doi: 10.1016/j.jmb.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Phillips SA, Hatoum OA, Gutterman DD. The Mechanism of Flow-Induced Dilation in Human Adipose Arterioles Involves Hydrogen Peroxide During Cad. Am J Physiol Heart Circ Physiol. 2006 doi: 10.1152/ajpheart.00819.2006. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Ramezan-Arab N, Cotman CW. Beta-amyloid neurotoxicity in vitro: evidence of oxidative stress but not protection by antioxidants. J Neurochem. 1997;69:1601–11. doi: 10.1046/j.1471-4159.1997.69041601.x. [DOI] [PubMed] [Google Scholar]
- Price JM, Sutton ET, Hellermann A, Thomas T. beta-Amyloid induces cerebrovascular endothelial dysfunction in the rat brain. Neurol Res. 1997;19:534–8. doi: 10.1080/01616412.1997.11740853. [DOI] [PubMed] [Google Scholar]
- Schachinger V, Britten MB, Zeiher AM. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation. 2000;101:1899–906. doi: 10.1161/01.cir.101.16.1899. [DOI] [PubMed] [Google Scholar]
- Schubert D, Behl C, Lesley R, Brack A, Dargusch R, Sagara Y, Kimura H. Amyloid peptides are toxic via a common oxidative mechanism. Proc Natl Acad Sci U S A. 1995;92:1989–93. doi: 10.1073/pnas.92.6.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, Lopez CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110:3507–12. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Guan J, Jiang B, Brenner DA, Del Monte F, Ward JE, Connors LH, Sawyer DB, Semigran MJ, Macgillivray TE, Seldin DC, Falk R, Liao R. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38alpha MAPK pathway. Proc Natl Acad Sci U S A. 2010;107:4188–93. doi: 10.1073/pnas.0912263107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suboc TM, Dharmashankar K, Wang J, Ying R, Couillard A, Tanner MJ, Widlansky ME. Moderate Obesity and Endothelial Dysfunction in Humans: Influence of Gender and Systemic Inflammation. Physiol Rep. 2013 doi: 10.1002/phy2.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T, McLendon C, Sutton ET, Thomas G. Cerebrovascular endothelial dysfunction mediated by beta-amyloid. Neuroreport. 1997;8:1387–91. doi: 10.1097/00001756-199704140-00014. [DOI] [PubMed] [Google Scholar]
- Townsend KP, Obregon D, Quadros A, Patel N, Volmar C, Paris D, Mullan M. Proinflammatory and vasoactive effects of Abeta in the cerebrovasculature. Ann N Y Acad Sci. 2002;977:65–76. doi: 10.1111/j.1749-6632.2002.tb04799.x. [DOI] [PubMed] [Google Scholar]
- Truran S, Weissig V, Ramirez-Alvarado M, Franco DA, Burciu C, Georges J, Murarka S, Okoth WA, Schwab S, Hari P, Migrino RQ. Nanoliposomes protect against AL amyloid light chain protein-induced endothelial injury. J Liposome Res. 2014;24:69–73. doi: 10.3109/08982104.2013.838258. [DOI] [PMC free article] [PubMed] [Google Scholar]