

Abstract
Development and characterization of a simple microfluidic electrochemical flow cell that can be coupled with HPLC to enable dual absorbance/electrochemical detection is described. Coupling absorbance and electrochemical detection increases the information that can be gathered from a single injection, but a second (typically expensive) detection system is required. Here, an inexpensive, customizable microfluidic electrochemical detector is coupled in series with a commercial HPLC/UV system. The microfluidic device is made from poly(dimethylsiloxane) and contains carbon paste electrodes. To demonstrate the utility of this dual-detection system, the reaction products of the radical scavenging agent salicylic acid and hydroxyl radical generated by Fenton chemistry were analyzed. The dual-detection system was used to quantify 2,5-dihydroxybenzoic acid, 2,3-dihydroxybenzoic acid, and catechol produced by the addition of H2O2 to filter samples of welding fumes. Measurement recovery was high, with percent recoveries between 97-102%, 92-103%, and 95-103% for 2,5-dihydroxybenzoic acid, 2,3-dihydroxybenzoic acid, and catechol, respectively, for control samples. The methods described in this work are simple, reliable, and can inexpensively couple electrochemical detection to HPLC-UV systems.
Keywords: HPLC, Microfluidic electrochemical detector, Fenton chemistry, Oxidative load
1. Introduction
Exposure to airborne environmental pollutants is a major worldwide health problem, listed as the 9th leading cause of morbidity and mortality in 2010 by the World Health Organization (WHO).1 Common air pollutants include both molecular species and particulate matter http://en.wikipedia.org/wiki/Particulates. Reactive oxygen species (ROS) are present in and/or can be generated by aromatic compounds and transition metals found in particulate matter. ROS include H2O2, ·OH, RO2·, O2·-, and NO and are frequently monitored in air pollution due to their health effects.2-8 Among the ROS, hydroxyl free radical (·OH, HFR) is particularly concerning because it reacts with DNA, membrane lipids, proteins, and carbohydrates and can lead to arthritis, cancer, cardiovascular disorders, stroke, and neurodegenerative diseases such as Alzheimer's and Parkinson's.7-9 Measuring radical generation ability in PM is challenging because radicals are short lived and are often present in low abundance. One approach for measuring ·OH generation from PM is through radical trapping agents like salicylic acid. Salicylic acid reacts with ·OH to produce dihydroxybenzoic acid (DHBA) isomers that can be quantified using separation techniques such as high performance liquid chromatography (HPLC)9-17 or capillary electrophoresis (CE) coupled with either UV or electrochemical detection (ECD).18-19 Adding ECD to a HPLC-UV system results in a dual-detection system with more selectivity and specificity than either detection technique can provide alone.
HPLC is a common separation technique that has been used to analyze a wide range of organic and inorganic compounds. The most popular detectors used for HPLC separations are absorbance, fluorescence, refractive index, and conductivity detectors.20 For electrochemically active compounds, ECD is useful because of its sensitivity and selectivity.21-22 ECD can readily detect many molecular targets including phenols, aromatic amines,21 glutathione,22 reducing sugars,23 antioxidants24,25 and thiols26 by controlling electrode potential and/or electrode composition. Thus, electrochemistry has been widely demonstrated as a viable detection method for a variety of flow techniques including microfluidic applications. Microfluidic-ECDs have also been used in many applications ranging from microchip CE to microfluidic biosensors, covering a wide variety of clinical, pharmaceutical, immunosensor, biosensor, and measurement applications.2,27-38 Microfluidic technology is amenable for shorter analysis times, lower reagent and sample consumption, higher portability, and lower material costs than other bench top methods. Generally, commercial electrochemical detectors provide wide linearity (3-4 orders of magnitude), low noise levels (nA-pA), high sensitivity (nM detection limits), and low response to flow, pressure, and temperature variations. However, commercial ECDs are usually expensive.39 Therefore, it would be advantageous to fabricate inexpensive electrochemical flow cells whose geometry can be readily changed to reflect the specific application that can be directly coupled to HPLC instrumentation. Soft lithography coupled with inexpensive electrochemical detectors provides this opportunity.2,26 These advantages spurred us to create a custom microfluidic-ECD that could be coupled directly to a commercial HPLC-UV system to demonstrate the utility of this analytical method.
This work presents a novel dual-detection HPLC-UV/ECD system built using a custom-made microfluidic ECD. This microfluidic system can be coupled to a commercially available HPLC system for determination of ·OH generation from Fenton chemistry. Detection of Fenton reaction products using salicylic acid (SA) as the radical trapping agent was selected to demonstrate system applicability. The separation efficiency, sensitivity, and other analytical features of the custom ECD were studied as a function of the flow cell width and depth. Using the optimized system, ·OH from PM samples was analyzed. Welding fume samples collected on filters were mixed with H2O2 and salicylic acid. The extraction parameters of the welding fume samples were optimized, and a recovery study of the welding fume samples was performed using the dual-detection system.
2. Experimental
2.1 Chemicals
All chemicals used in this study were analytical reagent grade and are given in the following list: salicylic acid (SA, Acros, U.S.A.), 2,3-dihydroxy benzoic acid (2,3-DHBA, Sigma-Aldrich, USA), 2,5-dihydroxy benzoic acid, (2,5-DHBA, Sigma-Aldrich), acetic acid (Macron Chemicals, U.S.A.), sodium acetate (Sigma-Aldrich), citric acid (Fisher Scientific, U.S.A.), sodium citrate (Fisher), ferrous ammonium sulfate hexahydrate (Mallinckrodt, U.S.A.), 30% w/w hydrogen peroxide, H2O2 (Fisher), absolute ethanol (Sigma-Aldrich), pyro-catechol (Sigma-Aldrich), L-ascorbic acid (AA, Mallinckrodt), SU-8 negative photoresist (3050, 3025 and 2007) (Microchem, U.S.A.), 99% propylene glycol monomethyl ether acetate as developer agent (Sigma-Aldrich), Sylgard® 184 silicon elastomer base poly(dimethylsiloxane) (PDMS) monomer (Dow Corning, U.S.A.), Sylgard® 184 elastomer curing agent (Dow Corning), O122-1 heavy mineral oil (Fisher), graphite powder, grade 38 (Fisher), high-purity silver paint (SPI Suppliers, U.S.A.), tinned copper conductive wire (NTE Electronics, Inc., U.S.A.) and silicon wafers with 100 mm diameter and 500 ± 25 μm thickness (Silicon Inc., U.S.A.). All separations used HPLC grade solvents (EMD Chemicals Inc., Germany).
2.2 Microfluidic mold fabrication
Microfluidic chip fabrication was accomplished using standard soft lithography methods following established protocols.2,26 Briefly, two molds were made on 100 mm diameter silicon wafers, one for the electrode channels (500 μm width, 50 μm depth and 3.5 cm length) and one for the flow channel (250 μm width, 25 μm depth and 3.5 cm length). The silicon wafer was cleaned with acetone, methanol, and water, dried with compressed air, and plasma cleaned (MSC plasma generator, U.S.A.) at 150 W at 0.8 torr for 5 min. Next the wafer was placed in a spin coater (WS-650MZ-23NPP/Lite, Laurell Technologies Corporation, U.S.A.) and 3-5 g of SU-8 (3050) was deposited on the wafer and spun at 3000 rpm for 30 s to achieve an SU-8 thickness of 50 μm. The wafer was then put on a hotplate to pre-bake at 95 °C for 20 min. The desired mask pattern was placed over the photoresist on the wafer and exposed to UV light (OmniCure® Series 2000, Lumen Dynamics, Canada) at 50% intensity for 90 s. The post-exposure bake was performed at 95 °C for 5 min. The development stage was completed by soaking the wafer in developer agent for 10 min followed by rinsing with acetone and drying with compressed air. Finally, the wafer was baked at 195 °C for 1 hr. For the flow channel depth study, 25 and 12.5 μm channel depths used SU-8 3025 and SU-8 2007 (1000 rpm spin coating and 5 min pre-bake time) instead SU-8 3050 photoresists with the same procedure as previously described.
2.3 Electrode platform construction and assembly of the coupled HPLC-UV/ECD system
The electrodes were made from a three-channel pattern with channel dimensions of 500 μm width, 50 μm depth, and 3.5 cm length that were subsequently filled with electrode material.2,26 The channels were made by mixing PDMS at a ratio of 10:1 (w/w) of PDMS oligomer: cross linker followed by degassing with a vacuum pump (Vacuubrand, Germany) in a vacuum desiccator. The mixed PDMS was poured on the center of the mold surface slowly to prevent the addition of air bubbles and then baked at 80 °C for 30 min. The electrode material was prepared as previously described by mixing PDMS and heavy mineral oil in a 1:1 ratio and then the binding material was mixed with graphite powder to get the final carbon paste electrode material. The electrode paste was spread into the PDMS channels and excess carbon paste removed using Scotch tape. The electrodes were baked at 65 °C for 30 min. The electrode channels were refilled with carbon paste using the same procedure as stated above and then baked at 120 °C for 1 hr. Repeating this step improved electrode conductivity and thus the performance. The flow channels were fabricated using PDMS with the same procedure as described above. The flow channels had different dimensions of 12.5, 25, and 50 μm depths or 125, 250, 500 and 750 μm widths, and were all a length of 3.5 cm. The electrodes were exposed to air plasma at 150 W for 5 min. The flow channel and electrode layer were placed in 18-W plasma (PDC-32G Harrick, U.S.A.) for 20 s and then irreversibly sealed together by bringing the layers into conformal contact. Wires were connected to all electrodes using silver paint followed by covering the connections with epoxy to increase structural stability. A complete microfluidic electrode platform that can be coupled with the HPLC-UV system is shown in Fig. 1. To make the connection between the HPLC UV detector and the ECD microchip, a 200 mm length of 0.0625 in PEEK tubing was inserted into a 1 mm hole made in the ECD via a tissue biopsy punch. Once the tubing was in place, the connection was reinforced by adding uncured PDMS to the opening. Once cured, this created an impermeable seal that prevented leakage around the tubing.
Fig. 1.
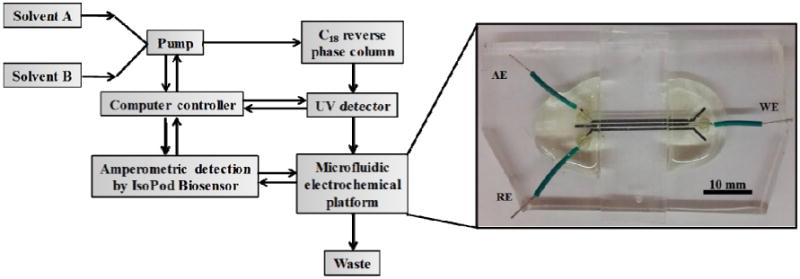
Scheme of the coupled microfluidic electrochemical detector with HPLC-UV, where AE = Auxiliary electrode, WE = Working electrode, and RE = Reference electrode.
The HPLC (Thermo Scientific, Dionex Ultimate 3000 model and Chromeleon 7 software, U.S.A.) was coupled with a Poroshell 120 EC-C18 column, 2.1×150 mm, 2.7 μm (Agilent Technologies, U.S.A.), a binary gradient solvent system, and dual-detection setup. The detection setup consisted of a commercial multi-wavelength UV/Vis detector coupled in series with the microfluidic electrochemical detector (Fig. 1). A portable potentiostat (EDAQ, EPU-352 model, IsoPod™ Biosensor with Pod-Vu software, Australia) was used for all electrochemical measurements. This detector was selected because of its low cost (<$800) and ease of connection to the HPLC control computer. All compounds were detected first by UV absorbance and then amperometry.
2.4 Application of the dual-detection system
In this work, Fenton chemistry was used to generate ·OH followed by trapping it using salicylic acid.9-18 In the first step in Reaction 1, Fe(II) or a similar transition metal present in welding fume reacts with added H2O2.
| (1) |
The resulting ·OH reacts with salicylic acid to produce three products9,16-18, shown schematically in Reaction 2. In these experiments 42% catechol, 24.5% 2,5-DHBA, and 33.5% 2,3-DHBA was generated.
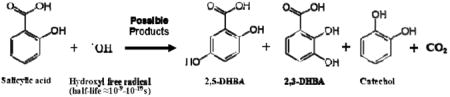 |
(2) |
We elected to study this reaction because of our ongoing interest in measuring the oxidative load associated with aerosol exposure. The reaction is well suited for dual absorbance and electrochemical detection given the high oxidation potential of salicylic acid (>800 mV) versus that of the reaction products (300 mV). Combining both detection methods allows us to follow both product formation and reagent consumption simultaneously. Gradient elution was performed using a binary solvent system (modified from E. DiStefano et. al.)17 of 25 mM, pH 3.5 citrate buffer (solvent A) and acetonitrile (CH3CN, ACN solvent B). For separation, a linear gradient from 5% (t = 0 min) to 50% (t = 5 min) ACN was used, followed by 50% ACN for 1 min. The column was re-equilibrated in 5% ACN for 10 min between each run. The optimal detection potential was determined using hydrodynamic voltammetry. When dual detectors were used, UV detection was performed at 280 nm and amperometric detection at +800 mV (vs carbon paste pseudo-reference electrode). The elution time of each compound, the effect of applied potential on signal-to-noise ratio (S/N), linear range, and calibration curves for each of the Fenton reaction products were investigated using the dual-detection setup. The separation and detection of an additional reducing compound, ascorbic acid (AA), was also studied as it is commonly used with ·OH generation studies.17 The AA is used to reduce the Fe(III) to Fe(II). To demonstrate a field application, welding samples were collected on filters from laboratory generated welding fumes and analyzed with the dual-detection system. Fumes were collected through mixed cellulose ester (MCE, Millipore, U.S.A.) membrane filters (0.8 μm pore size and 37 mm ø) at 4 L min-1 flow rate for 60 min. Samples No. 1-3 were collected through the MCE membrane with 4 L min-1 flow rate for 10 min while No. 4 was collected for 20 min. Then, a 5 mm diameter punch from each of the 4 samples was extracted by 500 μL DI water for 2 hrs.
For the recovery study, three experimental sets of 250 μL sample extract (set A), 250 μL sample extract with 25 μM Fe(II) (set B), and a standard 25 μM Fe(II) (set C) were separately prepared in 20 mM salicylic acid, 2 mM H2O2 in acetate buffer pH 5 for a total 1,000 μL volume to perform the real Fenton reaction using 30 min reaction time. All solutions were injected into the HPLC for quantification. Fenton reaction product recovery was also investigated from the welding samples to support the validity of the proposed method.
3. Results and discussion
3.1 Optimization of separation and detection conditions
An optimal elution program was determined by injecting a mixture of ascorbic acid, 2,5-DHBA, 2,3-DHBA, catechol and salicylic acid (500 μM each). Isocratic elution was tested first using 5, 7.5 and 10% ACN, respectively. As expected, the elution time and resolution decreased with increasing ACN. Unfortunately, an optimized isocratic elution gave long separation times and significant peak tailing for salicylic acid (>20 min with 5% ACN isocratic elution). Therefore, a gradient elution was used for the separation. The optimal gradient started at 5% ACN and linearly increasing to 50% B within 5 min, held constant for 1 min before decreasing to 5% B in 1 min and held constant for 3 min using 10 min total running time. Using the optimized gradient elution, a baseline-resolved separation was possible in under 10 min. Fig. 2 shows the elution order and dual-detection results obtained using the optimized gradient. The cause of the peak width increase for the ECD is unknown but is likely a combination of dead volume at the chip interface and differences in tubing diameters between the UV and ECD detectors. In Fig. 2, there is not a peak for salicylic acid in the ECD since salicylic acid is not detected at the potential used in these experiments. The differences in relative sensitivities between the two methods is also noteworthy and exhibits one of the advantages of the dual-detection approach.
Fig. 2.
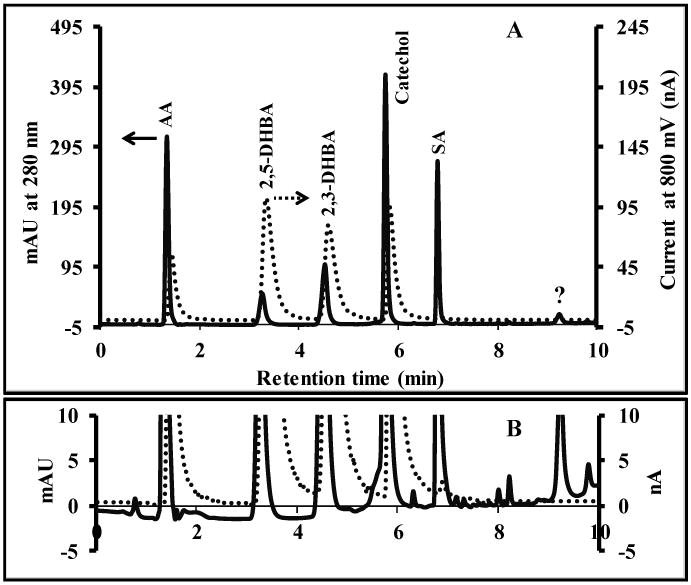
(A) Chromatogram of Fenton reaction products using the dual detection system. Dashed and solid lines represent amperometric detection and UV absorption signal respectively. (B) The differences in baseline between both detection methods. Separation conditions; column 2.7 μm Poroshell C18 (21×150 mm), injection volume of 10 μL, flow rate of 0.3 mLmin-1, mobile phase binary solvent system (A) 25 mM citrate buffer pH 3.5, and (B) CH3CN, gradient elution starting at 5% B and linearly increasing to 50% B within 5 minutes, held constant for 1 min before decreasing to 5% B in 1 minute and held constant for 3 minutes (10 min run time).
Next, the S/N for electrochemical detection was investigated as a function of applied potential (Fig. 3). From these results, the highest S/N ratio for all analytes except salicylic acid was in the range of 700-800 mV against a carbon paste pseudo-reference electrode. Higher applied potentials produced higher noise, thereby decreasing the S/N (Fig. 3 A). We selected 800 mV for amperometric detection to obtain the highest sensitivity for all of the analytes studied. Salicylic acid requires a basic medium to be electrochemically active, and therefore is not detectable under our conditions.40-42
Fig. 3.
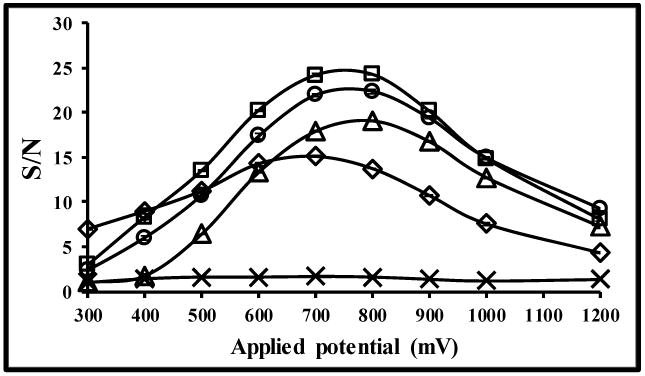
Voltage optimization study for all analytes, where (□) 2,5-DHBA, (○) catechol, (◊) ascorbic acid, (△) 2,3-DHBA and (×) salicylic acid. Separation conditions were the same as stated in Figure 2.
3.2 Device optimization
The effect of channel width and depth was also investigated. The results are shown in Fig. 4 and 5. In agreement with the Levich equation, better device sensitivity was obtained by increasing channel width or decreasing channel depth for all compounds, likely from an increase in the number of molecules in contact with the electrode surface.43 Wider channels have a larger surface area for electrode contact and decrease the linear solution velocity (increasing residence time), and shallower channels decrease the average molecular diffusion distance to the electrode. The chromatographic separation efficiency, reported as plate number, N,44 was calculated for catechol as a function of channel width and depth. The resolution between analyte pairs 2,5-DHBA/2,3-DHBA and 2,3-DHBA/catechol were in the range of 2.52-3.20 and 2.78-3.78, respectively. For future studies the channel design will be selected based on the relative importance of sensitivity and separation efficiency.
Fig. 4.
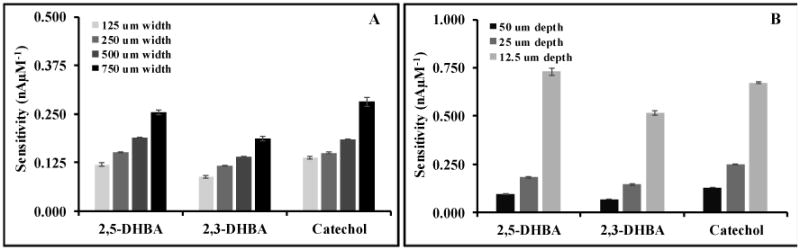
Effect of flow channel width using constant 25 μm depth (A) and flow channel depth with constant 250 μm width (B) on sensitivity of the analysis. Separation conditions were the same as stated in Figure 2.
Fig. 5.
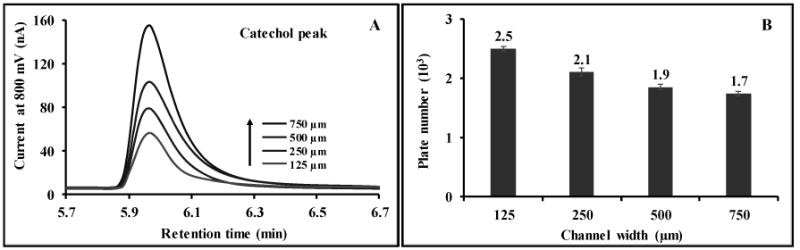
The observed peak shape for catechol as the ECD flow channel is increased in width (A). Peak tailing is attributed to dead volume between the UV and ECD. The calculated plate number as the flow channel in the ECD is increased (B). Separation conditions were the same as stated in Figure 2.
3.3 Analytical performance study
After determining the optimal conditions for the microfluidic electrochemical detector, we found that amperometric detection provided a slightly improved sensitivity relative to UV detection for the key components of the mixture. The results in Table 1 show the amperometric detector was able to measure the signal at the lowest concentration of 0.25 μM of catechol and both DHBA species, while UV absorbance was 0.50 μM. Conversely, UV detection gave a wider linear range for all compounds. We also investigated the theoretical detection limit45 of all analytes by measuring peak height at 0.50 μM all analytes from chromatogram by 10 replicates, three times of the standard deviation was divided by slope (3SD/Slope) from its calibration curve to obtain the limit of detection (LOD) while 10SD/Slope was used for calculating the limit of quantification (LOQ) from both UV absorption and amperometric signals (Table 1). For reproducibility, the relative standard deviation (%RSD) was calculated for 10 injections at 0.50 μM all analytes. It was found that both reproducibility and detection limits of all compounds obtained from ECD were better than the UV detector. The improvements gained by the ECD can be attributed to the baseline drift of the UV signal caused by the gradient elution (See Fig. 2B). The elution gradient did not cause a baseline drift in the electrochemical detector allowing it to achieve better performance metrics for this application. The S/N ratio for the ECD system increased as the exposed electrode area was increased as can be seen in Figure 4. The ECD could be used for >300 sample injections without cleaning before detrimental signal loss from electrode fouling was observed.
Table 1.
The results of linearity, LOD, LOQ and reproducibility study using dual detectors.
| Chemicals | Amperometric detection (Peak current, nA) | UV absorption (Peak height, mAU) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||
| Linear (μM) | LOD (μM) | LOQ (μM) | %RSD | Sensitivity (nN/ μM) | Linear (μM) | LOD (μM) | LOQ (μM) | %RSD | Sensitivity (mAU/ μM) | |
| 2,5-DHBA | 0.25-125 | 0.11 | 0.37 | 0.77 | 0.4181 | 0.50-500 | 0.15 | 0.48 | 2.39 | 0.1068 |
| 2,3-DHBA | 0.25-125 | 0.18 | 0.59 | 1.07 | 0.3506 | 0.50-500 | 0.19 | 0.63 | 3.67 | 0.2038 |
| Catechol | 0.25-125 | 0.23 | 0.77 | 1.43 | 0.3735 | 1.25-500 | 0.24 | 0.79 | 3.35 | 0.9296 |
| SA | ND | ND | ND | ND | ND | 0.50-500 | 0.05 | 0.17 | 0.86 | 0.6182 |
ND = Not detected
3.4 Method application to Fenton reaction and welding fume sample analysis
Next, the dual-detection system was used to analyze the presence of ·OH produced by Fenton chemistry. 20 mM salicylic acid and 2 mM H2O2 in acetate buffer (pH 5) were mixed with various Fe(II) concentrations (0, 25, 50, 100 and 200 μM), and the resulting products monitored after a 30 min reaction time. As expected, analyte generation and ascorbic acid consumption were dependent on the Fe(II) concentration. The H2O2 should not be retained by the column and therefore it is likely the first small peak in the chromatogram. The peak current (pA) as a function of Fe(II) concentration (μM) showed a good linear relationship for 2,5-DHBA, 2,3-DHBA and catechol giving a linear calibration of y=3x+3464 (r2=0.9997), y=6x+3527 (r2=0.9968) and y=40x+3088 (r2=0.9998), respectively. These findings suggest that this technique can quantify ·OH generated by Fenton chemistry.
For real sample analysis, laboratory generated welding fumes were generated using an electrode-arc generator. The welding sample extract without ascorbic acid did not generate peaks for catechol, 2,3- or 2,5-DHBA. However, peaks did appear when 500 μM ascorbic acid was added to the reaction mixture, suggesting the metal content was already completely oxidized (predominately Fe(III)), and thus not reactive towards H2O2. The effect of extraction volume on extraction efficiency was also investigated (Table 2). The results showed that decreasing extraction volume increased current signal due to reduced dilution, while the signal of ascorbic acid also decreased simultaneously. The consumption of ascorbic acid had a linear relationship with the concentration of Fe(II). These results indicate that 500 μL DI water was optimal for sample extraction. Volumes of less than 500 μL were not tested because of the need to fully wet the filter membrane for extraction.
Table 2.
Effect of extraction volume on extraction efficiency of welding filtered sample.
| Current at 800 mV at various solvent volumes, Mean ± SD (n=3) | ||||
|---|---|---|---|---|
|
|
||||
| Chemicals | 1,500 μL | 1,000 μL | 750 μL | 500 μL |
| ascorbic acid | 31.49±1.26 | 19.62±0.77 | 14.42±0.31 | 8.96±0.05 |
| 2,5-DHBA | 4.35±0.20 | 5.79±0.16 | 7.55±0.31 | 9.28±0.04 |
| 2,3-DHBA | 5.96±0.15 | 8.05±0.41 | 10.38±0.27 | 12.23±0.01 |
| catechol | 7.46±0.25 | 12.93±0.79 | 16.06±0.57 | 17.47±0.66 |
The percent recovery of four welding fume samples was tested. Each analyte can be calculated its concentration in initial 500 μL extract while percent recovery was obtained as shown the results in Fig. 6. Highly accurate results for all analytes were obtained from four different welding fume samples, confirming the reliability of the results obtained from the proposed microfluidic ECD system.
Fig. 6.
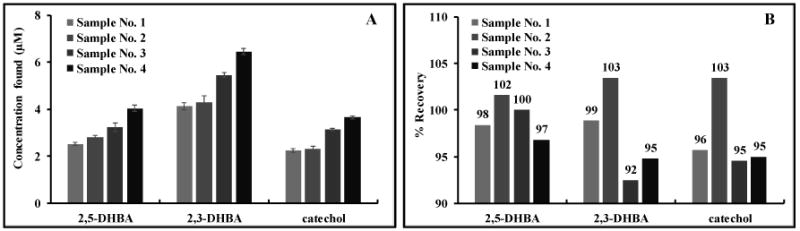
Welding fume samples analysis (A), and percent recovery study (B), (n=3).
4. Conclusions
The custom-built ECD described here provides desirable characteristics including high sensitivity, high reproducibility, and ease of integration into commercial HPLC systems. The dimensions of the microfluidic channel over the electrodes can be quickly and inexpensively changed for different applications. For the molecules in this study it was seen that the custom microfluidic ECD was more sensitive and had a higher reproducibility than the commercial UV detector on the HPLC system. Potential advantages of the dual-detection system include rapid peak identification, peak deconvolution, and rapid detector validation. In addition, further improvements in the detector design and electrode composition should make the ECD more sensitive than existing UV detectors for HPLC. This proposed system can be applied for confirming the peak position in the same run between both detectors in case of complicated sample analysis. For some applications, we can use amperometry instead of UV absorption if an electrochemically inactive species is unable to completely separate from other electrochemically active compounds. Furthermore, multiple microfluidic ECDs can be connected in series and different potentials can be applied to each set of electrodes for analyzing both oxidizable and reducible compounds in a single injection.
Acknowledgments
This work was supported by research grants to T.P. from the Royal Thai government Science and Technology Ministry Scholarship, National Science and Technology Development Agency (NSTDA) and the Center for Innovation in Chemistry: Postgraduate Education and Research Program in Chemistry (PERCH-CIC), Department of Chemistry, Faculty of Science, Mahidol University, Thailand. We also thank to the grant from National Institute of Environmental Health Sciences (NIH Grant ES019264) to J. B. W. and D. M. C.
References
- 1.http://www.who.int/ceh/risks/cehair/en/
- 2.Sameenoi Y, Koehler K, Shapiro JJ, Boonsong K, Sun Y, Collett JJ, Volckens J, Henry CS. J Am Chem Soc. 2012;134:10562–10568. doi: 10.1021/ja3031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chevion M. Free Radical Biology & Medicine. 1988;5:27–37. doi: 10.1016/0891-5849(88)90059-7. [DOI] [PubMed] [Google Scholar]
- 4.Jiang M, Wei Q, Pabla N, Dong G, Wang CY, Yang T, Smith SB, Dong Z. Biochemical Pharmacology. 2007;73:1499–1510. doi: 10.1016/j.bcp.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zweier JL, Duke SS, Kuppusamy P, Sylvester JT, Gabrielson EW. Febs Letters. 1989;252(1,2):12–16. doi: 10.1016/0014-5793(89)80881-6. [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto H, Watanabe T, Mizuno H, Endo K, Hosokawa T, Kazusaka A, Gooneratne R, Fujita S. Free Radical Biology & Medicine. 2001;30(5):547–554. doi: 10.1016/s0891-5849(00)00496-2. [DOI] [PubMed] [Google Scholar]
- 7.Obata T. Toxicology Letters. 2002;132:83–93. doi: 10.1016/s0378-4274(02)00076-0. [DOI] [PubMed] [Google Scholar]
- 8.Ferger B, Teismann P, Earl CD, Kuschinsky K, Lertel WH. Pharmacology. Biochemistry and Behavior. 2000;65(3):425–431. doi: 10.1016/s0091-3057(99)00229-4. [DOI] [PubMed] [Google Scholar]
- 9.Application note “Measurement of The Hydroxyl Free Radical” ESA Analytical, Ltd. http://www.esainc.com/docs/spool/701749P_Hydroxyl_Free_Radical.pdf, 1995
- 10.McCabe DR, Maher TJ, Acworth IN. J of Chromatography B. 1997;691:23–32. doi: 10.1016/s0378-4347(96)00416-1. [DOI] [PubMed] [Google Scholar]
- 11.Sloot WN, Gramsbergen JBP. J of Neuroscience Methods. 1995;60:141–149. doi: 10.1016/0165-0270(95)00005-f. [DOI] [PubMed] [Google Scholar]
- 12.Jen JF, Leu MF, Yang TC. J of Chromatograph A. 1998;796:283–288. [Google Scholar]
- 13.Nappi AJ, Vass E. Biochim Biophys Acta. 1998;1380:55–63. doi: 10.1016/s0304-4165(97)00125-6. [DOI] [PubMed] [Google Scholar]
- 14.Nappi AJ, Vass E. Biochim Biophys Acta. 1998;1425:159–167. doi: 10.1016/s0304-4165(98)00062-2. [DOI] [PubMed] [Google Scholar]
- 15.Liu D, Liu J, Wen J. Free Radical Biology & Medicine. 1999;27:478–482. doi: 10.1016/s0891-5849(99)00073-8. [DOI] [PubMed] [Google Scholar]
- 16.Liu B, Wang H. J of Environmental Sciences. 2008;20:28–32. doi: 10.1016/s1001-0742(08)60003-0. [DOI] [PubMed] [Google Scholar]
- 17.DiStefano E, Fernandez AE, Delfino RJ, Sioutas C, Froines JR, Cho AK. Inhalation Toxicology. 2009;21(9):731–738. doi: 10.1080/08958370802491433. [DOI] [PubMed] [Google Scholar]
- 18.Coolen SAJ, Huf FA, Reijenga JC. J of Chromatography B. 1998;717:119–124. doi: 10.1016/s0378-4347(98)00289-8. [DOI] [PubMed] [Google Scholar]
- 19.Ai S, Wang Q, Li H, Jin L. J of Electroanalytical Chem. 2005;578:223–229. [Google Scholar]
- 20.Kats E, Eksteen R, Schoenmakers P, Miller N. Handbook of HPLC. Marcel Dekker, Inc.; New York, USA: 1998. [Google Scholar]
- 21.Parriott D. A Practical Guide to HPLC Detection. Academic press Inc.; California, USA: 1993. [Google Scholar]
- 22.Squellerio I, Caruso D, Porro B, Veglia F, Tremoli E, Cavalca V. J of Pharmaceutical and Biomedical Analysis. 2012;71:111–118. doi: 10.1016/j.jpba.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 23.Guan YG, Yu P, Yu SJ, Xu XB, Wu XL. J Dairy Sci. 2012;95:6379–6383. doi: 10.3168/jds.2012-5623. [DOI] [PubMed] [Google Scholar]
- 24.Skrinjar M, Kolar MH, Jelsek N, Hras AR, Bezjak M, Knez Z. J of Food Composition and Analysis. 2007;20:539–545. [Google Scholar]
- 25.Vovk T, Bogataj M, Roskar R, Kmetec V, Mrhar A. International J of Pharmaceutics. 2005;291:161–169. doi: 10.1016/j.ijpharm.2004.07.053. [DOI] [PubMed] [Google Scholar]
- 26.Sameenoi Y, Mensack MM, Boonsong K, Ewing R, Dungchai W, Chailapakul O, Cropek DM, Henry CS. Analyst. 2011;136:3177. doi: 10.1039/c1an15335h. [DOI] [PubMed] [Google Scholar]
- 27.Zimmerman WB. Chemical Engineering Science. 2011;66:1412–1425. [Google Scholar]
- 28.Kraly JR, Holcomb RE, Guan Q, Henry CS. Anal Chim Acta. 2009;653:23–35. doi: 10.1016/j.aca.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia CD, Henry CS. Anal Chim Acta. 2004;508:1–9. [Google Scholar]
- 30.Hong CC, Wang CY, Peng KT, Chu IM. Biosensors and Bioelectronics. 2011;26:3620–3626. doi: 10.1016/j.bios.2011.02.017. [DOI] [PubMed] [Google Scholar]
- 31.Mostowfi F, Czarnecki J, Masliyah J, Bhattacharjee S. J of Colloid and Interface Science. 2008;317:593–603. doi: 10.1016/j.jcis.2007.09.068. [DOI] [PubMed] [Google Scholar]
- 32.Weng CH, Yeh WM, Ho KC, Lee GB. Sensors and Actuators B. 2007;121:576–582. [Google Scholar]
- 33.Liang W, Li Y, Zhang B, Zhang Z, Chen A, Qi D, Yi W, Hu C. Biosensors and Bioelectronics. 2012;31:480–485. doi: 10.1016/j.bios.2011.11.021. [DOI] [PubMed] [Google Scholar]
- 34.Islam K, Jha SK, Chand R, Han D, Kim YS. Microelectronic Engineering. 2012;97:391–395. [Google Scholar]
- 35.Bertolino FA, De Vito IE, Messina GA, Fernandez H, Raba J. J of Electroanalytical Chemistry. 2011;651:204–210. [Google Scholar]
- 36.Yang J, Yu JH, Strickler JR, Chang WJ, Gunasekaran S. Biosensors and Bioelectronics. 2013;47:530–538. doi: 10.1016/j.bios.2013.03.051. [DOI] [PubMed] [Google Scholar]
- 37.Shiroma LY, Santhiago M, Gobbi AL, Kubota LT. Anal Chim Acta. 2012;725:44–50. doi: 10.1016/j.aca.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 38.Dawoud AA, Kawaguchi T, Markushin Y, Porter MD, Jankowiak R. Sensors and Actuators B. 2006;120:42–50. [Google Scholar]
- 39.Flanagan RJ, Perrett D, Whelpton R. Electrochemical Detection in HPLC; Analysis of drugs and poisons. The Royal Society of Chemistry; Cambridge, UK: 2005. [Google Scholar]
- 40.Wang Z, Ai F, Xu Q, Yang Q, Yu JH, Huang WH, Zhao YD. Colloids and Surfaces B: Biointerfaces. 2010;76:370–374. doi: 10.1016/j.colsurfb.2009.10.038. [DOI] [PubMed] [Google Scholar]
- 41.Zhang WD, Xu B, Hong YX, Yu YX, Ye JS, Zhang JQ. J Solid State Electrochem. 2010;14:1713–1718. [Google Scholar]
- 42.Gualandi I, Scavetta E, Zappoli S, Tonelli D. Biosensors and Bioelectronics. 2011;26:3200–3206. doi: 10.1016/j.bios.2010.12.026. [DOI] [PubMed] [Google Scholar]
- 43.Compton RG, Unwin PR. J Electroanal Chem. 1986;206:57–67. [Google Scholar]
- 44.Sewell AP, Clarke B. Chromatographic Separation. John Willey & Sons; London, UK: 1987. [Google Scholar]
- 45.Harris DC. Quantitative Chemical Analysis. 6th. W.H. Freeman and Company; New York, USA: 2003. [Google Scholar]