

Abstract
With successful antiretroviral therapy, HIV-1-infected subjects can achieve undetectable peripheral viral loads and immune homeostasis. However, in a subset of individuals on therapy, peripheral monocytes have a gene expression profile characteristic of a type 1 interferon α (IFN) response. This type 1 IFN response correlates with a number of pathogenic conditions including neural cell injury and in combination with HCV infection, cognitive impairment. Lessons from the non-human primate models of pathogenic and nonpathogenic SIV suggest that returning the initial IFN spike in acute SIV infection to normal allows the immune system to control infection and return to homeostasis. An IFN “alarm” signature, defined as monocyte activation with overexpression of the type1 IFN genes IF127 and SN, would be useful for identifying a subset of subjects with HIV-1 infection that could progress to a number of pathologies associated with immune activation including cognitive dysfunction. This strategy is being actively pursued for autoimmune diseases that are characterized by an IFN signature. Therapies to block the IFN signature are under investigation as a means to reset the immune system and in a subset of HIV-1-infected subjects may be an adjuvant to standard antiviral therapy to return cognitive function.
Keywords: HIV-1, HCV, impairment, interferon, cognition, monocyte
INTRODUCTION
Chronic immune activation plays a major role in HIV-1 neuropathogenesis and progression to AIDS. In spite of effective antiretroviral therapy (ART), cognitive impairment in chronically infected individuals has been reported to be up to 48% [1]. One model of immune activation is that lipopolysaccharide (LPS) translocation through a CD4 T cell-depleted gut is responsible for immune activation [2]. And yet recent evidence does not support this [3–5]. Interferons are key cytokine defense components of the antiviral response. Production of interferons results in the overexpression of Interferon Stimulated Genes (ISGs) [6]. Three distinct interferon types are recognized based on their receptors, type 1 (α/β), type II (γ) and type III (λ) [7, 8]. While all have antiviral activities, the type 1 IFN response is integral to innate immunity in HIV-1 and simian immunodeficiency virus (SIV) infections [9–11].
The major arms of the antiviral response are the natural killer cells and the production of type 1 interferon (IFNα/β) by plasmacytoid dendritic cells (pDCs) [12]. HIV-1 activates pDCs to produce IFN early in infection [13]. In HIV-1 and SIV studies, early IFN response is associated with acute illness and is important in retarding infection [14–16]. Our recent published data on monocyte gene expression from HIV-1-infected individuals who are on ART, showed that continued activation is predominantly due to IFNα and not LPS activation [3]. This mini review addresses the negative aspects of a sustained monocyte IFN signature on cognitive dysfunction and how this type 1 IFN “alarm” signature, defined as monocyte ISGs, IF127 and CD169 (SN, sialoadhesin), identifies subjects at risk for cognitive impairment in both HIV-1 infection and coinfection with hepatitis C virus (HCV) [3, 17].
LESSONS FROM NON-HUMAN PRIMATES IN CONTROL OF HIV-1 INFECTION
In the last decade, immune activation has been recognized as a more relevant indicator of T cell depletion and immunodeficiency than viral load. Contributing to this perspective are numerous studies using two closely related non-human primates, which exhibit two dramatically different outcomes following SIV infection. When rhesus macaques (RM), old-world Asian primates, are infected with SIV, the animals become viremic supporting virus replication that leads to chronic immune activation, T cell depletion and ultimately immune system collapse. This is similar to the disease course observed in humans and as a result, RM are used extensively to model human HIV-1-related immunology and neuropathology (Table 1).
Table 1.
SIV infection of non-human primates.
| Rhesus Macaques | Sootey Mangabeys |
|---|---|
| Pathogenic infection | Natural Host |
| Early interferon response continues | Early interferon response drops off |
| T cell proliferation continues | Early T cell proliferation drops off |
| T cell counts drop after 60 days | T cell count remains high |
| Plasma viral load stays high | Plasma viral load still high |
| CD169 increase on monocytes | CD169 decrease on monocytes |
| Chronic Activation | Reduced Activation |
In contrast, sooty mangabeys (SM), which are indigenous to Africa where SIV is endemic, exhibit a burst of immune activation during the acute infection phase but then down-regulate the immune response approaching pre-infection levels despite high SIV titers in the plasma (reviewed in [18]). Sooty mangabeys are natural hosts of SIV, tolerating high viral loads without immune activation while maintaining a normal immune system without T cell depletion and rarely develop AIDS. In fact, SIV-infected SM maintain high circulating CD4+ T cell levels that are similar to uninfected SM counterparts [19–21]. Importantly, during non-pathogenic SIV infection, SM are able to control and limit immune activation despite constant viral replication [20–23]. Acute infection of SM is associated with an increase in ISGs in the blood and lymph nodes that is rapidly resolved in spite of a continued HIV-1 viremia [24, 25]. A marker of monocyte IFNα activation, CD169, is markedly increased in RM and decreased in SM [26]. This suggests that SM have adapted to SIV infection by modifying their immune response and thereby avoided damage to their immune system caused by chronic activation. In HIV-1, monocyte CD169 expression closely follows viral load and can be suppressed with successful ART and an undetectable viral load [27].
In both human and non-human primates, elevated IFNα is associated with acute HIV-1 and SIV infections, respectively [14, 15], and in the chronic HIV-1 infection phase, serum IFNα levels correlated with disease progression and diminished benefit from ART [28]. So while an acute immune activation process is beneficial in stimulating the immune system and antiviral state necessary to contain viruses, the chronic activation state also driven by viral infection may lead to further pathogenic conditions.
TRANSITION FROM ACUTE TO CHRONIC ACTIVATED MONOCYTE PHENOTYPE IN HIV-1 INFECTION
Blood monocytes contribute to innate immunity and migrate to tissues where they differentiate into macrophages. Along with T cells, macrophages are a target for HIV-1 infection whereas monocytes are somewhat refractory to infection. Susceptibility to HIV-1 infection is thought to increase with monocyte differentiation into a macrophage [29, 30] and while it is the monocyte/macrophage (M/Mϕ) that transmigrates into the brain, it is the activation of the monocyte by HIV-1 that induces soluble products that help differentiate the monocyte into a macrophage and contribute to cognitive impairment [31]. Before the era of ART, CD14+ peripheral monocytes in HIV-1 infection were activated and expressed high CD16 and TNFα cell markers as well as a CD69 monocyte subset that correlated with HIV-1 dementia [31, 32]. The peripheral monocyte activation phenotype in HIV-1 infection has markers normally present on macrophages that facilitate migration across the blood brain barrier as well as increase susceptibility to HIV-1 infection [33, 34]. In the era of ART, some individuals control viral load and others spike to high HIV-1 viral load with monocyte gene and protein expression increased for MCP-1, CD16 and CD169 suggesting a hybrid monocyte/macrophage (M/Mϕ) phenotype [33]. When monocytes from HIV-1-infected individuals were cultured to macrophages in vitro, the supernatants caused extensive neurotoxicity on human brain cells consistent with that seen in human brains [31]. The CD14+ low/CD16+ high subset, like the tissue macrophage, elaborates proinflammatory cytokines, chemokines and neurotoxins [35] and is more permissive to HIV-1 infection [36] and migration to the brain [34, 37]. Monocytes expressing CD163, a scavenger receptor binding hemoglobin: haptoglobin, have been associated with increased HIV-1 viral load and inversely with T cell count [38]. The CD16+/CD163+ subset has also been demonstrated in the blood and brain in the SIV model [39].
TYPE 1 IFN EXPRESSION ON PERIPHERAL MONOCYTES IN HIV-1 INFECTION
HIV-1-infected subjects treated with ART but with detectable viral loads, may continue to show monocyte activation with elevated expression of interferon-related genes [3]. When monocyte expression profiles were compared with in vitro IFN-treated monocytes, there was high correlation (r=0.789) and significance (p<0.001)in ISGs in spite of the inability to measure IFN protein in the periphery [3]. The proposal that microbial translocation is likely the source of activation is based on the finding that HIV-1-infected subjects, even those with very low viral loads, have significantly higher plasma LPS levels than HIV-1-negative subjects [2, 3]. However, when monocytegene expression profiles from healthy HIV-1 seronegative subjects were treated with LPS (1 ng/ml) or IFNα (100U/ml) in vitro for 48 hours and compared with monocyte gene expression profiles from HIV-1 seropositive subjects, the profiles from HIV-1 infected individuals did not have a corresponding inflammatory profile consistent with LPS activation but rather a type 1 IFN response (Table 2) [3].
Table 2.
Monocyte gene expression from LPS or IFNα – treatment or HIV-infected subjects.
| Gene | Fold Change | ||||||
|---|---|---|---|---|---|---|---|
| LPS/NT1 | IFNα/NT1 | HIV-1/C2 | |||||
| IP-10 (CXCL10) | 1.5 | 107.5 | 3.6 | ||||
| SN (CD169) | 2.3 | 49.2 | 15.9 | ||||
| CCL2 | 5.8 | 4.1 | 5.1 | ||||
| CXCL3 | 60.1 | 1.3 | 1.2 | ||||
| IL6 | 45 | 1.1 | 1.4 | ||||
| IL1β | 5.7 | 0.2 | 0.7 | ||||
Monocytes from HIV-1 seronegative subjects treated in vitro with LPS or IFNα vs non-treated monocytes (NT).
Subjects with HIV-1 viral load vs control HIV-1 seronegative controls (C).
Adapted from [3].
Three highly expressed genes in LPS-treated monocytes, CXCL3, IL-6 and IL-1β, were not elevated in control subjects (not shown), HIV-1-infected subjects or IFN-treated monocytes, whereas SN (CD169) and IP-10 (CXCL10) were highly elevated in both IFNα-treated and HIV-1-infected subject monocytes. IP-10 can also be measured in the plasma of HIV-1-infected subjects and when correlated with monocyte gene expression showed a significantly high correlation (Fig. 1) (adapted from [17]). These findings demonstrate that an IFNα response is present on monocytes from HIV-1-infected subjects in spite of increased peripheral LPS. Monocytes do not respond with classic LPS gene markers suggesting they might be desensitized to LPS activation. The source of the monocyte type 1 IFN activation profile in treated HIV-1 infection is unknown but several explanations have been put forward. One recent paper reported that HIV-1 Tat could activate a subset of ISGs including IP-10 through p38 MAP kinase and IRF7 pathways independent of IFN [40].
Fig. 1. Monocyte gene expression correlates with plasma concentration of IP-10 in HIV-1-infected subjects.

Monocyte gene expression significantly correlated with plasma levels. Spearman correlation coefficient (R) and p value is shown. Cross is the mean value for HIV-1 seronegative controls (± S.D.) (Adapted from [17]).
Other data show that IL27, an anti-HIV-1 cytokine, can also induce ISGs, similar to IFNα on mono nuclear cells via a type 1 IFN independent pathway [41, 42]. Alternatively, low levels of IFN induced by HIV-1 reactivation from reservoirs are possible even with effective therapy.
In spite of effective antiretroviral therapy, cognitive impairment persists in a subset of chronically infected individuals with HIV-1 infection. Investigators are actively trying to identify biomarkers for predicting and identifying cognitive impairment. The majority of studies have focused on the use of cerebrospinal fluid (CSF) for analyses. Recently, soluble CD163, shed from activated M/Mϕ into plasma and CSF was significantly elevated in the plasma of HIV-1-infected subjects with cognitive impairment compared to unimpaired infected subjects [43]. Another activation marker of macrophages is neopterin. It is produced by stimulation of macrophages with IFNγ, and to a lesser degree by IFNα, and is indicative of cellular immune activation [44]. Interestingly, neopterin elevation in CSF significantly correlated with cognitive impairment and also with elevated plasma sCD163 levels in controlled HIV-1 infection [43]. Several reports demonstrate that indices of cerebral inflammation are present in CSF including an increase in the interferon chemokine CXCL10 [45, 46]. In a recent report, a CSF cytokine profile of elevated G-CSF, IL-8, MCP-1 and CXCL10, strongly correlated with cognitive impairment [46]. Of added interest, CXCL10, an interferon marker, was higher in ART-treated subjects with cognitive impairment. In the periphery, only sCD163 has been found to correlate with cognitive impairment [43] although peripheral immune activation can be monitored by checking plasma interferon-stimulated products CXCL9 and CXCL1, sIL-2R and sCD14 [45, 47] and CXCL10 [17]. Looking at monocyte gene expression from HIV-1-infected individuals, 14 differentially expressed genes with greater than five fold increase in activation were identified and 7 of these genes correlated with a decrease in N-acetylaspartate (NAA) in the frontal white matter (FWM) using magnetic resonance spectroscopy (MRS) [17]. While the gene expression profile did not correlate with cognitive impairment, the decrease in NAA suggests neuronal injury. Elevated monocyte CD169 expression significantly correlated with a decrease in NAA (data not shown here) and was detected in the brains of HIV-1-infected subjects as a subset of infiltrating macrophages (Fig. 2) [17]. This links a peripheral type 1 IFN-induced monocyte marker with infiltrating macrophages in the brain.
Fig. 2. Immunohistochemistry staining for CD169-expressing macrophages in CNS.
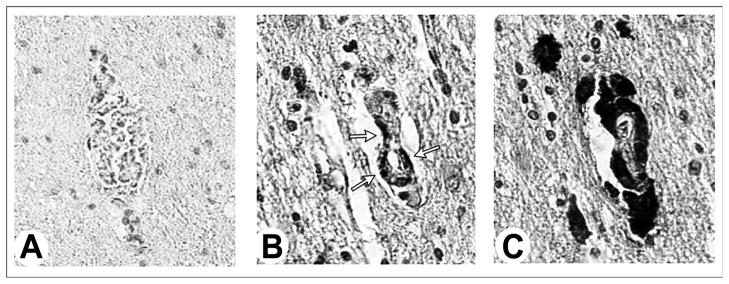
Section from the frontal cortex of an (A) HIV-1-seronegative subject and (B, C) sequential sections from an HIV-1 seropositive individual. CD169 is not present in the brain of an HIV-1-seronegative subject (A) and is a subset (B) of perivascular CD68 macrophages (C). Sections were immuno stained with antibodies to CD169 (A, B) and CD68 (C) and visualized using a DAB substrate.
Both exogenous administration and endogenous production of IFNα have been associated with, and in animal models causative for, neuronal dysfunction. IFNα used as a treatment strategy in a variety of conditions can cause cognitive dysfunction (reviewed in [48, 49]) as well as in healthy individuals [50]. Importantly, cognitive dysfunction associated with IFN therapy usually diminishes when therapy is terminated. In vitro studies show that IFNα can cause a loss of dendritic arborization that can be blocked by IFN-neutralizing antibody and partially by NMDA antagonists [51]. Using a SCID mouse model of HIV-1 encephalitis, blocking IFNα with neutralizing antibody significantly improved cognitive function [48, 51].
EFFECTS OF HCV COINFECTIONON HIV-1 AND MONOCYTE ACTIVATION: IMPLICATIONS FOR COGNITIVE DYSFUNCTION
A molecular process driven by a gene signature usually represents pathological conditions. This profile can be a prognostic biomarker of disease or a profile of clinical response to therapy. In a small study of HIV-1/HCV coinfected individuals with controlled HIV-1 and untreated HCV, 65% were cognitively impaired [52]. Looking at their monocyte gene expression signature, there was a distinct increase in interferon signaling with coinfection compared to HCV mono infected subjects and HIV-1-infected subjects having undetectable HIV-1 viral loads (Fig. 3) [53]. One would expect that the coinfected monocyte signature would mimic that seen in subjects with HCV infection alone since the subjects were not treated for their HCV infection and were on ART with an undetectable HIV-1 viral load.
Fig. 3. Canonical pathways for monocyte gene expression analyzed using Interactive Pathway Analysis (IPA, Ingenuity).
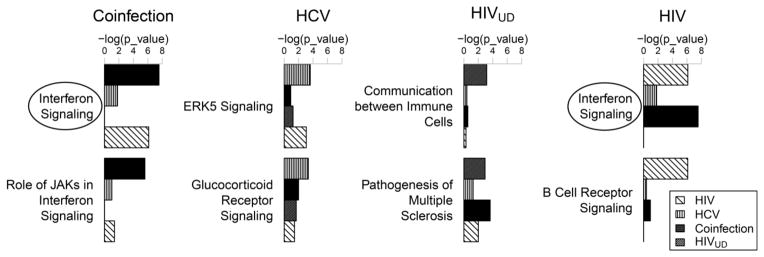
Differentially expressed genes from subjects with HIV-1/HCV (n=17) on ART, HCV (n=19), HIV-1UD, (undetectable viral load, n=14) and HIV-1with viral load on ART (n=22) relative to healthy controls (n=28) were analyzed using IPA. The top two most significant canonical pathways were shown. The Interferon Signaling pathway was the most significant pathway in both the Coinfection (−log(p)=7.55) and HIV groups (−log(p)=6.13). X axis (significance) indicates a negative natural log-transformed Fisher’s exact test right-tailed p value as calculated in IPA. (Adapted from [53]).
In controlled HIV-1 infection, the type 1 IFN monocyte signature did not correlate with a worsening global deficit score (GDS) but rather a decrease in NAA [17]. When looking at HIV-1/HCV-infected subjects, an IFN activation score of a composite of 6 highly overexpressed IFN genes on monocytes was determined [53]. This score strongly correlated with an increase in and worsening cognitive impairment (Fig. 4) suggesting that HCV in the presence of HIV-1 triggers monocyte activation in a subset of coinfected individuals. However, this does not appear to be dependent on HIV-1 viral load since it was undetectable in this cohort. While the explanation for increased cognitive impairment in HIV-1 controlled mono infection and coinfection remains elusive, it may imply ongoing intracerebral inflammation.
Fig. 4. IFN activation score correlates with worsening global deficit score (GDS) in HIV-1/HCV coinfection.
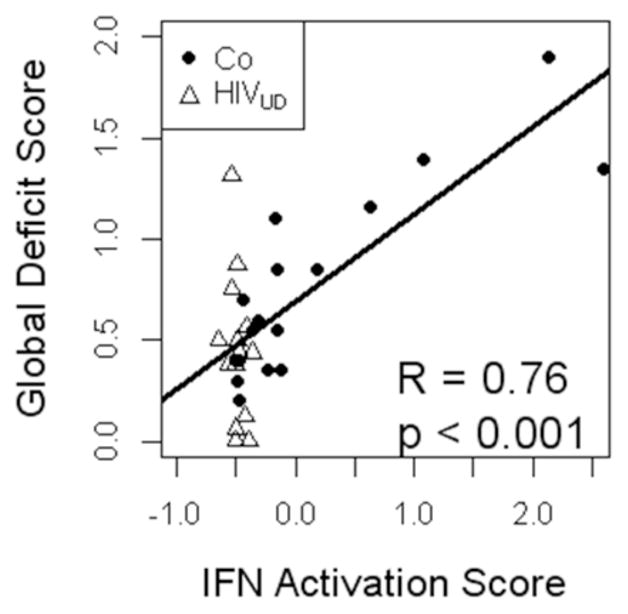
For each subject, 5 type 1 IFN monocyte genes (IF127, RSAD2, MX1, SIGLEC1 and LGALS3BP) were converted into a composite activation value. Expression was normalized to mean=0, sd=1. The composite activation score significantly correlated with worsening GDS in the HIV-1/HCV coinfected (Co) subjects (black circles) using a Spearman’s rank correlation. For reference, HIV-1UD (UD, undetectable viral load) subjects’ 6 gene activation score and GDS values are shown (open triangles). (Adapted from [53]).
TYPE I IFN EXPRESSION PROFILE AS AN “ALARM” SIGNATURE
Several reports review the neurotoxic effects of IFNα protein; however, in the case of controlled HIV-1 infection, monocyte genes IF127 and SN may be elevated in spite of low IFN protein levels. Unlike the beneficial therapeutic effects of IFN to treat viral infections and tumors, chronic expression of IFN genes on monocytes may carry deleterious side effects and constitute an “alarm” signature in the periphery. Several conditions that result in chronic endogenous production of IFN can give us clues as to how expression in HIV-1-infected subjects might influence disease progression including cognitive impairment.
Rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) area utoimmune diseases characterized by chronic inflammation and a common type 1 IFN gene expression response. Peripheral protein levels of IFN in these conditions do not necessarily agree with the IFN expression profiles [54]. In a recent study, a 5-gene type I IFN blood signature (IFI44, IFI44L, IFI27, RSAD2 and IFI6) was identified in five rheumatic diseases including a subset of RA and the majority of SLE patients. This study showed overexpression of type 1 IFN genes in both tissue-specific disease sites, such as skin in SLE and synovial fluid in RA and whole blood from the same subjects [54]. This interferon signature was considered a biomarker for active disease and severity [55]. In another report, a set of 6type I IFN genes was shown to represent a prognostic marker for poor response to rituximab treatment in a subset of RA subjects [56, 57]. Successful responders had a low or absent IFN signature before therapy [58]. In SLE, the IFN signature is relatively stable and cannot be used as a prognostic biomarker [55]. Cognitive impairment in RA has been reported to be in the range of 30–38% with decreases in attention and verbal fluency performance as the most common symptoms [59]. SLE is a more complicated disease than RA with multifactorial etiology and symptoms. Neuropsychiatric SLE is a broad umbrella of psychiatric symptoms with a range of neuropsychiatric symptoms including delirium, depression, anxiety, hallucinations as well as cognitive impairment, which are reported to be the most common manifestation of neuropsychiatric lupus having a prevalence of 15–66% (reviewed in [60–62]). CD169 was suggested as a peripheral monocyte biomarker for monitoring an IFN response in SLE, where inflammatory monocytes contribute to auto antibody formation [63]. To our knowledge there have not been any correlative studies to compare type I IFN expression with cognitive dysfunction in RA and SLE in spite of the high percentage of cognitive impairment in these conditions. However, there is active investigation into therapeutic agents including antibodies to IFNα that inhibit the type I IFN expression in SLE (reviewed in [64]).
While in HIV-1 infection a monocyte IFN “alarm” profile typically parallels viral load, in a subset of virally controlled individuals, it persists in spite of undetectable virus. In addition, the hyperactive immune state can contribute to a number of HIV-1-associated ageing complications including cardiovascular disease, liver and renal disease, osteoporosis as well as cognitive decline (reviewed in [65]). Chronic HCV coinfection synergistically unmasks an IFN inflammatory signature that correlates with cognitive decline even when HIV-1 is successfully suppressed with ART. In both HIV-1 mono and coinfection, elevated CD169 expression on monocytes is a leading IFN “alarm” biomarker [17, 27, 53]. Identifying those subjects with elevated monocyte CD169 would be a compelling risk factor for initiating treatment in coinfected subjects who otherwise are a difficult group to treat. In the new era of highly effective HCV protease inhibitors that come with significant costs, subjects with known progressive liver disease will likely be treated first. However, those with a monocyte IFN signature may have a compelling risk factor for cognitive impairment and may be prioritized for treatment.
NEUTRALIZING CHRONIC TYPE 1 IFN ACTIVATION
While a type 1 IFN response is essential in controlling viral infections, persistent activation of this pathway has detrimental consequences. In both HIV-1 and coinfection with HCV, sustained expression of an IFN “alarm” signature has an impact on neurological function, either as neuronal injury as described in HIV-1 or cognitive impairment in HIV-1 coinfection with HCV [17, 53]. Anti-IFNα immunization as an adjuvant to ART was previously used in several studies in Europe and Israel with results in HIV-1-infected subjects responding with increased levels of IFNα neutralizing antibodies and reduced disease progression [66–70]. These studies were performed before the year 2000 at a time when active discussions were ongoing about when to treat and with what combination of antivirals. With the present expectation in HIV-1 therapy of undetectable viral load and a reconstituted immune system, most individuals with HIV-1 infection will not maintain an IFN signature. However, for those that continue to have chronic activation, recent studies using a lymphocytic choriomeningitis virus (LCMV) model of infection show exciting results on blocking the IFN signaling pathway to reset the immune environment [71, 72]. A mouse model of persistent LCMV infection is accompanied by chronic type 1 IFN signaling, hyperactivation and disease progression [71, 72]. By blocking IFNα signaling using neutralizing antibody, immune activation was quenched, lymphoid tissue architecture was restored and the immune mediators necessary for viral clearance restored. While there are justifiable concerns that blocking the IFN response would exacerbate virus, the LCMV experiments suggest that this reset of the immune system can be achieved. The animal studies demonstrate that CD4 T cells were required for enhanced control after treatment. In HIV-1 infections, most subjects on ART have normal T cell counts although they may continue to have activated T cell compartments with elevated CD38/CD8 in spite of undetectable viral load [53]. Viral suppression usually parallels lower T cell and monocyte activation, although only monocyte activation is associated with cognitive impairment. Lessons from the persistent LCMV model demonstrate that ablating IFN signaling can reset the immune thermostat and in HIV-1 infection, the likelihood of improving cognitive dysfunction.
The use of IFN blocking strategies in humans has more recently been investigated for treatment of SLE (reviewed in [64]), where IFNα is strongly implicated in the pathogenesis. Strategies for anti-IFNα therapy include antibodies to IFNα, IFNα receptor and immunization to IFNα; clinical trials using these targets are in progress [73–75]. Alternatively, the use of general immunosuppressive drugs in combination with ART is another approach (reviewed in [76]). If results from these trials are successful in eliminating the chronic hyperimmune state caused by IFNα, identifying the subset of HIV-1-infected individuals with or without HCV coinfection having the IFN “alarm” signature would be important to add an immune modulatory component to standard antiviral therapy.
Acknowledgments
The author thanks Dr. Hans Rempel and Dr. Bing Sun for editorial comments. This publication was supported by NIH grant RO1 MH096673 (LP).
Footnotes
CONFLICT OF INTEREST
The author confirms that this article content has no conflicts of interest.
PATIENT CONSENT
Declared none.
HUMAN/ANIMAL RIGHTS
Declared none.
References
- 1.Harezlak J, Buchthal S, Taylor M, et al. Persistence of HIV-associated cognitive impairment, inflammation, and neuronal injury in era of highly active antiretroviral treatment. Aids. 2011;25(5):625–33. doi: 10.1097/QAD.0b013e3283427da7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ancuta P, Kamat A, Kunstman KJ, et al. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PloS one. 2008;3(6):e2516. doi: 10.1371/journal.pone.0002516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rempel H, Sun B, Calosing C, et al. Interferon-alpha drives monocyte gene expression in chronic unsuppressed HIV-1 infection. AIDS. 2010;24(10):1415–23. doi: 10.1097/QAD.0b013e32833ac623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Redd AD, Dabitao D, Bream JH, et al. Microbial translocation, the innate cytokine response, and HIV-1 disease progression in Africa. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(16):6718–23. doi: 10.1073/pnas.0901983106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Redd AD, Gray RH, Quinn TC. Is microbial translocation a cause or consequence of HIV disease progression? The Journal of infectious diseases. 2011;203(5):744–5. doi: 10.1093/infdis/jiq107. author reply 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Der SD, Zhou A, Williams BR, et al. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(26):15623–8. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Theofilopoulos AN, Baccala R, Beutler B, et al. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–36. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 8.Donnelly RP, Kotenko SV. Interferon-lambda: a new addition to an old family. J Interferon Cytokine Res. 2010;30(8):555–64. doi: 10.1089/jir.2010.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hartshorn KL, Neumeyer D, Vogt MW, et al. Activity of interferons alpha, beta, and gamma against human immunodeficiency virus replication in vitro. AIDS Res Hum Retroviruses. 1987;3(2):125–33. doi: 10.1089/aid.1987.3.125. [DOI] [PubMed] [Google Scholar]
- 10.Witwer KW, Gama L, Li M, et al. Coordinated regulation of SIV replication and immune responses in the CNS. PloS one. 2009;4(12):e8129. doi: 10.1371/journal.pone.0008129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alammar L, Gama L, Clements JE. Simian immunodeficiency virus infection in the brain and lung leads to differential type I IFN signaling during acute infection. J Immunol. 2011;186(7):4008–18. doi: 10.4049/jimmunol.1003757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siegal FP, Kadowaki N, Shodell M, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284(5421):1835–7. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 13.Fitzgerald-Bocarsly P, Jacobs ES. Plasmacytoid dendritic cells in HIV infection: striking a delicate balance. Journal of leukocyte biology. 2010;87(4):609–20. doi: 10.1189/jlb.0909635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.von Sydow M, Sonnerborg A, Gaines H, et al. Interferon-alpha and tumor necrosis factor-alpha in serum of patients in various stages of HIV-1 infection. AIDS Res Hum Retroviruses. 1991;7(4):375–80. doi: 10.1089/aid.1991.7.375. [DOI] [PubMed] [Google Scholar]
- 15.Khatissian E, Tovey MG, Cumont MC, et al. The relationship between the interferon alpha response and viral burden in primary SIV infection. AIDS Res Hum Retroviruses. 1996;12(13):1273–8. doi: 10.1089/aid.1996.12.1273. [DOI] [PubMed] [Google Scholar]
- 16.Tilton JC, Johnson AJ, Luskin MR, et al. Diminished production of monocyte proinflammatory cytokines during human immunodeficiency virus viremia is mediated by type I interferons. J Virol. 2006;80(23):11486–97. doi: 10.1128/JVI.00324-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pulliam L, Rempel H, Sun B, et al. A peripheral monocyte interferon phenotype in HIV infection correlates with a decrease in magnetic resonance spectroscopy metabolite concentrations. AIDS. 2011;25(14):1721–6. doi: 10.1097/QAD.0b013e328349f022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chahroudi A, Bosinger SE, Vanderford TH, et al. Natural SIV hosts: showing AIDS the door. Science. 2012;335(6073):1188–93. doi: 10.1126/science.1217550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirsch VM. What can natural infection of African monkeys with simian immunodeficiency virus tell us about the pathogenesis of AIDS? AIDS reviews. 2004;6(1):40–53. [PubMed] [Google Scholar]
- 20.Silvestri G, Sodora DL, Koup RA, et al. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited by stander immunopathology despite chronic high-level viremia. Immunity. 2003;18(3):441–52. doi: 10.1016/s1074-7613(03)00060-8. [DOI] [PubMed] [Google Scholar]
- 21.Sumpter B, Dunham R, Gordon S, et al. Correlates of preserved CD4(+) T cell homeostasis during natural, nonpathogenic simian immunodeficiency virus infection of sooty mangabeys: implications for AIDS pathogenesis. J Immunol. 2007;178(3):1680–91. doi: 10.4049/jimmunol.178.3.1680. [DOI] [PubMed] [Google Scholar]
- 22.Chakrabarti LA, Lewin SR, Zhang L, et al. Normal T-cell turnover in sooty mangabeys harboring active simian immunodeficiency virus infection. J Virol. 2000;74(3):1209–23. doi: 10.1128/jvi.74.3.1209-1223.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silvestri G, Fedanov A, Germon S, et al. Divergent host responses during primary simian immunodeficiency virus SIVsm infection of natural sooty mangabey and nonnatural rhesus macaque hosts. J Virol. 2005;79(7):4043–54. doi: 10.1128/JVI.79.7.4043-4054.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bosinger SE, Li Q, Gordon SN, et al. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. The Journal of clinical investigation. 2009;119(12):3556–72. doi: 10.1172/JCI40115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manches O, Bhardwaj N. Resolution of immune activation defines nonpathogenic SIV infection. The Journal of clinical investigation. 2009;119(12):3512–5. doi: 10.1172/JCI41509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaroenpool J, Rogers KA, Pattanapanyasat K, et al. Differences in the constitutive and SIV infection induced expression of Siglecs by hematopoietic cells from non-human primates. Cellular immunology. 2007;250(1–2):91–104. doi: 10.1016/j.cellimm.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rempel H, Calosing C, Sun B, et al. Sialoadhesin expressed on IFN-induced monocytes binds HIV-1 and enhances infectivity. PloS one. 2008;3(4):e1967. doi: 10.1371/journal.pone.0001967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stylianou E, Aukrust P, Bendtzen K, et al. Interferons and interferon (IFN)-inducible protein 10 during highly active anti-retroviral therapy (HAART)-possible immunosuppressive role of IFN-alpha in HIV infection. Clinical and experimental immunology. 2000;119(3):479–85. doi: 10.1046/j.1365-2249.2000.01144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fear WR, Kesson AM, Naif H, et al. Differential tropism and chemokine receptor expression of human immunodeficiency virus type 1 in neonatal monocytes, monocyte-derived macrophages, and placental macrophages. J Virol. 1998;72(2):1334–44. doi: 10.1128/jvi.72.2.1334-1344.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuitemaker H, Kootstra NA, Koppelman MH, et al. Proliferation-dependent HIV-1 infection of monocytes occurs during differentiation into macrophages. The Journal of clinical investigation. 1992;89(4):1154–60. doi: 10.1172/JCI115697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pulliam L, Herndier BG, Tang NM, et al. Human immunodeficiency virus-infected macrophages produce soluble factors that cause histological and neurochemical alterations in cultured human brains. The Journal of clinical investigation. 1991;87(2):503–12. doi: 10.1172/JCI115024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pulliam L, Gascon R, Stubblebine M, et al. Unique monocyte subset in patients with AIDS dementia. Lancet. 1997;349(9053):692–5. doi: 10.1016/S0140-6736(96)10178-1. [DOI] [PubMed] [Google Scholar]
- 33.Pulliam L, Sun B, Rempel H. Invasive chronic inflammatory monocyte phenotype in subjects with high HIV-1 viral load. J Neuroimmunol. 2004;157(1–2):93–8. doi: 10.1016/j.jneuroim.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 34.Buckner CM, Calderon TM, Willams DW, et al. Characterization of monocyte maturation/differentiation that facilitates their transmigration across the blood-brain barrier and infection by HIV: implications for NeuroAIDS. Cellular immunology. 2011;267(2):109–23. doi: 10.1016/j.cellimm.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ziegler-Heitbrock L. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. Journal of leukocyte biology. 2007;81(3):584–92. doi: 10.1189/jlb.0806510. [DOI] [PubMed] [Google Scholar]
- 36.Ellery PJ, Tippett E, Chiu YL, et al. The CD16+ monocyte subset is more permissive to infection and preferentially harbors HIV-1 in vivo. J Immunol. 2007;178(10):6581–9. doi: 10.4049/jimmunol.178.10.6581. [DOI] [PubMed] [Google Scholar]
- 37.Fischer-Smith T, Croul S, Sverstiuk AE, et al. CNS invasion by CD14+/CD16+ peripheral blood-derived monocytes in HIV dementia: perivascular accumulation and reservoir of HIV infection. Journal of neurovirology. 2001;7(6):528–41. doi: 10.1080/135502801753248114. [DOI] [PubMed] [Google Scholar]
- 38.Fischer-Smith T, Bell C, Croul S, et al. Monocyte/macrophage trafficking in acquired immunodeficiency syndrome encephalitis: lessons from human and nonhuman primate studies. Journal of neurovirology. 2008;14(4):318–26. doi: 10.1080/13550280802132857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim WK, Alvarez X, Fisher J, et al. CD163 identifies perivascular macrophages in normal and viral encephalitic brains and potential precursors to perivascular macrophages in blood. The American journal of pathology. 2006;168(3):822–34. doi: 10.2353/ajpath.2006.050215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim N, Kukkonen S, del Martinez-Viedma MP, et al. Tat engagement of p38 MAP kinase and IRF7 pathways leads to activation of interferon-stimulated genes in antigen-presenting cells. Blood. 2013;121(20):4090–100. doi: 10.1182/blood-2012-10-461566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imamichi T, Yang J, Huang DW, et al. IL-27, a novel anti-HIV cytokine, activates multiple interferon-inducible genes in macrophages. Aids. 2008;22(1):39–45. doi: 10.1097/QAD.0b013e3282f3356c. [DOI] [PubMed] [Google Scholar]
- 42.Chen Q, Swaminathan S, Yang D, et al. Interleukin-27 is a potent inhibitor of cis HIV-1 replication in monocyte-derived dendritic cells via a type I interferon-independent pathway. PloS one. 2013;8(3):e59194. doi: 10.1371/journal.pone.0059194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burdo TH, Weiffenbach A, Woods SP, et al. Elevated sCD163 is a marker of neurocognitive impairment in HIV infection. Aids. 2013 doi: 10.1097/QAD.0b013e32836010bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huber C, Batchelor JR, Fuchs D, et al. Immune response-associated production of neopterin. Release from macrophages primarily under control of interferon-gamma. J Exp Med. 1984;160(1):310–6. doi: 10.1084/jem.160.1.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kamat A, Lyons JL, Misra V, et al. Monocyte activation markers in cerebrospinal fluid associated with impaired neurocognitive testing in advanced HIV infection. Journal of acquired immune deficiency syndromes. 2012;60(3):234–43. doi: 10.1097/QAI.0b013e318256f3bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan L, Qiao L, Wei F, et al. Cytokines in CSF correlate with HIV-associated neurocognitive disorders in the post-HAART era in China. Journal of neurovirology. 2013;19(2):144–9. doi: 10.1007/s13365-013-0150-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamat A, Misra V, Cassol E, et al. A plasma biomarker signature of immune activation in HIV patients on antiretroviral therapy. PloS one. 2012;7(2):e30881. doi: 10.1371/journal.pone.0030881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fritz-French C, Tyor W. Interferon-alpha (IFNalpha) neurotoxicity. Cytokine & growth factor reviews. 2012;23(1–2):7–14. doi: 10.1016/j.cytogfr.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 49.Myint AM, Schwarz MJ, Steinbusch HW, et al. Neuropsychiatric disorders related to interferon and interleukins treatment. Metabolic brain disease. 2009;24(1):55–68. doi: 10.1007/s11011-008-9114-5. [DOI] [PubMed] [Google Scholar]
- 50.Smith A, Tyrrell D, Coyle K, et al. Effects of interferon alpha on performance in man: a preliminary report. Psychopharmacology. 1988;96(3):414–6. doi: 10.1007/BF00216072. [DOI] [PubMed] [Google Scholar]
- 51.Sas AR, Bimonte-Nelson H, Smothers CT, et al. Interferon-alpha causes neuronal dysfunction in encephalitis. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29(12):3948–55. doi: 10.1523/JNEUROSCI.5595-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun B, Abadjian L, Rempel H, et al. Differential cognitive impairment in HCV coinfected men with controlled HIV compared to HCV monoinfection. Journal of acquired immune deficiency syndromes. 2013;62(2):190–6. doi: 10.1097/QAI.0b013e31827b61f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rempel H, Sun B, Calosing C, et al. Monocyte Activation in HIV/HCV Coinfection Correlates with Cognitive Impairment. PloS one. 2013;8(2):e55776. doi: 10.1371/journal.pone.0055776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Higgs BW, Liu Z, White B, et al. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Annals of the rheumatic diseases. 2011;70(11):2029–36. doi: 10.1136/ard.2011.150326. [DOI] [PubMed] [Google Scholar]
- 55.Ronnblom L, Eloranta ML. The interferon signature in autoimmune diseases. Current opinion in rheumatology. 2013;25(2):248–53. doi: 10.1097/BOR.0b013e32835c7e32. [DOI] [PubMed] [Google Scholar]
- 56.Raterman HG, Vosslamber S, de Ridder S, et al. The interferon type I signature towards prediction of non-response to rituximab in rheumatoid arthritis patients. Arthritis research & therapy. 2012;14(2):R95. doi: 10.1186/ar3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Verweij CL, Vosslamber S. New insight in the mechanism of action of rituximab: the interferon signature towards personalized medicine. Discovery medicine. 2011;12(64):229–36. [PubMed] [Google Scholar]
- 58.Thurlings RM, Boumans M, Tekstra J, et al. Relationship between the type I interferon signature and the response to rituximab in rheumatoid arthritis patients. Arthritis and rheumatism. 2010;62(12):3607–14. doi: 10.1002/art.27702. [DOI] [PubMed] [Google Scholar]
- 59.Appenzeller S, Bertolo MB, Costallat LT. Cognitive impairment in rheumatoid arthritis. Methods and findings in experimental and clinical pharmacology. 2004;26(5):339–43. doi: 10.1358/mf.2004.26.5.831324. [DOI] [PubMed] [Google Scholar]
- 60.Sciascia S, Bertolaccini ML, Baldovino S, et al. Central nervous system involvement in systemic lupus erythematosus: Overview on classification criteria. Autoimmunity reviews. 2013;12(3):426–9. doi: 10.1016/j.autrev.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 61.Kozora E, Hanly JG, Lapteva L, et al. Cognitive dysfunction in systemic lupus erythematosus: past, present, and future. Arthritis and rheumatism. 2008;58(11):3286–98. doi: 10.1002/art.23991. [DOI] [PubMed] [Google Scholar]
- 62.Popescu A, Kao AH. Neuropsychiatric systemic lupus erythematosus. Current neuropharmacology. 2011;9(3):449–57. doi: 10.2174/157015911796557984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Biesen R, Demir C, Barkhudarova F, et al. Sialic acid-binding Ig-like lectin 1 expression in inflammatory and resident monocytes is a potential biomarker for monitoring disease activity and success of therapy in systemic lupus erythematosus. Arthritis and rheumatism. 2008;58(4):1136–45. doi: 10.1002/art.23404. [DOI] [PubMed] [Google Scholar]
- 64.Kirou KA, Gkrouzman E. Anti-interferon alpha treatment in SLE. Clinical immunology. 2013 doi: 10.1016/j.clim.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 65.High KP, Brennan-Ing M, Clifford DB, et al. HIV and aging: state of knowledge and areas of critical need for research. A report to the NIH Office of AIDS Research by the HIV and Aging Working Group. Journal of acquired immune deficiency syndromes. 2012;60(Suppl 1):S1–18. doi: 10.1097/QAI.0b013e31825a3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gringeri A, Santagostino E, Mannucci PM, et al. A randomized, placebo-controlled, blind anti-AIDS clinical trial: safety and immunogenicity of a specific anti-IFN alpha immunization. Journal of acquired immune deficiency syndromes. 1994;7(9):978–88. [PubMed] [Google Scholar]
- 67.Fall LS, M’Bika JP, Le Coq H, et al. Biological effect of active anti-IFN alpha immunization in HIV-infected patients. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 1995;49(9):422–8. doi: 10.1016/0753-3322(96)82679-5. [DOI] [PubMed] [Google Scholar]
- 68.Gringeri A, Santagostino E, Cusini M, et al. Absence of clinical, virological, and immunological signs of progression in HIV-1-infected patients receiving active anti-interferon-alpha immunization: a 30-month follow-up report. Journal of acquired immune deficiency syndromes and human retrovirology: official publication of the International Retrovirology Association. 1996;13(1):55–67. doi: 10.1097/00042560-199609000-00009. [DOI] [PubMed] [Google Scholar]
- 69.Gringeri A, Musicco M, Hermans P, et al. Active anti-interferon-alpha immunization: a European-Israeli, randomized, double-blind, placebo-controlled clinical trial in 242 HIV-1--infected patients (the EURIS study) Journal of acquired immune deficiency syndromes and human retrovirology: official publication of the International Retrovirology Association. 1999;20(4):358–70. doi: 10.1097/00042560-199904010-00006. [DOI] [PubMed] [Google Scholar]
- 70.Zagury D, Lecoq H, Gervi I, et al. Anti-IFN alpha immunization raises the IFN alpha-neutralizing capacity of serum--an adjuvant to antiretroviral tritherapy. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 1999;53(2):90–2. doi: 10.1016/s0753-3322(99)80065-1. [DOI] [PubMed] [Google Scholar]
- 71.Wilson EB, Yamada DH, Elsaesser H, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013;340(6129):202–7. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Teijaro JR, Ng C, Lee AM, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science. 2013;340(6129):207–11. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Petri M, Wallace DJ, Spindler A, et al. Sifalimumab, a human anti-interferon-alpha monoclonal antibody, in systemic lupus erythematosus: a phase I randomized, controlled, dose-escalation study. Arthritis and rheumatism. 2013;65(4):1011–21. doi: 10.1002/art.37824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ding HJ, Gordon C. New biologic therapy for systemic lupus erythematosus. Current opinion in pharmacology. 2013 doi: 10.1016/j.coph.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 75.Lauwerys BR, Hachulla E, Spertini F, et al. Down-regulation of interferon signature in systemic lupus erythematosus patients by active immunization with interferon alpha-kinoid. Arthritis and rheumatism. 2013;65(2):447–56. doi: 10.1002/art.37785. [DOI] [PubMed] [Google Scholar]
- 76.d’Ettorre G, Paiardini M, Ceccarelli G, et al. HIV-associated immune activation: from bench to bedside. AIDS Res Hum Retroviruses. 2011;27(4):355–64. doi: 10.1089/aid.2010.0342. [DOI] [PubMed] [Google Scholar]