

Abstract
Atypical hemolytic uremic syndrome (aHUS) has a high mortality rate if not detected and treated early. While in the past, it was associated with renal failure in children, today, it has become increasingly identified among adults. Due to recent advances in the pathogenesis of aHUS and other major thrombotic microangiopathies (TMA), diagnosing it has become a lot easier. We present a case of a 62-year-old man who was initially thought to have thrombotic thrombocytopenic purpura (TTP), but after further evaluation was diagnosed with aHUS. We will discuss how to distinguish aHUS from other major TMA and the role of eculizumab in the management of aHUS.
Introduction
Atypical hemolytic uremic syndrome is the rarest form of thrombotic microangiopathy (TMA) and affects people of all ages. It is caused by mutations in the alternative complement pathway resulting in the failure of regulators to inactivate C3b.1–3 Then overactive complements will attack not only foreign antigens but also normal host cells leading to endothelial cell injury, platelet activation and aggregation, coagulation, and multi-system microthrombosis. Eculizumab is a monoclonal antibody that blocks the formation of C5a and membrane attack complex (MAC). It was first approved for paroxysmal nocturnal hemoglobinuria (PNH), a related complement mediated disease, and now it is also used for aHUS. Multiple case studies have proven the effectiveness of eculizumab in reversing multi-organ dysfunction caused by TMA.4 Before the discovery of eculizumab, plasma exchange (PEX) was considered to be the first line therapy for all three major TMA syndromes: TTP, Shiga toxin-producing Escherichia coli (STEC-HUS), and aHUS. Patients with aHUS sometimes transiently respond to PEX. However, approximately 65% would progress to end-stage renal disease (ESRD) or die within a year of diagnosis.5 Clinical trials have shown that treatment with eculizumab improves and reverses the effect of TMA in 80% of patients with aHUS.5 Thus, it is very important to distinguish aHUS as a separate entity early in its course in order to implement appropriate therapy. This report will explain the distinguishing factors among the three major TMA and highlight the treatment of aHUS with eculizumab.
Case Report
A 62-year-old Filipino man with a history of chronic kidney disease stage 3 and diabetes mellitus type 2 experienced a decline in renal function with abdominal pain, arthritis, and palpable purpura a year prior to diagnosis of aHUS. Renal biopsy at the time revealed, on immunofluorescence microscopy, mesangial IgA deposits consistent with IgA vasculitis. He was treated with six months of steroid therapy with improvement of his symptoms and renal function back to baseline. Six months later he again presented with a one month history of progressively worsening dyspnea on exertion with orthopnea, disorientation, confusion, fatigue, abdominal pain, and loose stools. Labs showed acute and chronic renal failure with creatinine 8.0 mg/dL, BUN 115.0 mg/dL, sodium 131.0 mmol/L, potassium 6.2 mmol/L, chloride 98.0 mmol/L, bicarbonate 19.0 mmol/L. Complete blood count was significant for hemoglobin of 8.9 g/dL, hematocrit 28.5%, mean corpuscular volume 88 fL, and platelets 140 × 10(9)/L.
The patient was admitted to the intensive care unit for initiation of continuous renal replacement therapy and, thereafter, dialysis for volume overload, uremia, and hyperkalemia. Within 48 hours, a rapid fall of his hemoglobin and platelets was noted. Hemoglobin and platelets dropped to 6.7 g/dL and 66 × 10(9)/L respectively. Lactate dehydrogenase rose to 1155 IU/L and haptoglobin decreased to < 26 mg/dL. Review of his peripheral smear revealed extensive microangiopathic hemolytic anemia with thrombocytopenia (see Figure 1). PEX was initiated for possible TTP while further workup for TMA was performed. He was started on high dose steroid therapy. Further laboratory and imaging evaluations excluded antiphospholipid antibody syndrome, systemic lupus erythematosus, heparin-induced thrombocytopenia, malignant hypertension, autoimmune hemolytic anemia, infections, disseminated intravascular coagulation, toxin producing bacterial colitis, and malignancy. He transiently responded, minimally, to plasma exchange and high dose steroids. However, after PEX and steroids were stopped, his anemia and thrombocytopenia worsened. A therapeutic trial of rituximab was initiated and was unsuccessful. TTP was excluded and aHUS favored when the ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) level returned at 33% and TMA relapsed despite 14 days of PEX and steroids, and 1 month of rituximab. Since clinical evidence supported the diagnosis of aHUS, further investigation with complement genetic testing was not pursued. Eculizumab was started using published dosing guidelines for aHUS and we documented a rapid normalization of his hemoglobin, platelet, and lactate dehydrogenase levels within 2 weeks of therapy. While there was a rapid improvement in his hematologic parameters, his renal function did not recover and he remains on dialysis despite therapy.
Figure 1.
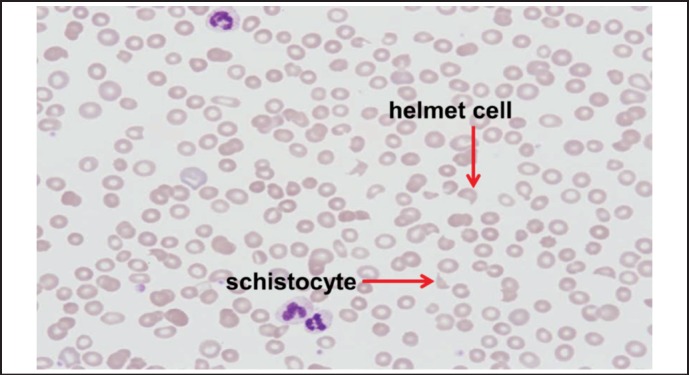
Patient's blood smear showing microangiopathic hemolytic anemia with red cell fragmentation and thrombocytopenia.
Discussion
The similarities in clinical features among the major TMAs have made it challenging to distinguish them apart. All of them, more or less, have microangiopathic hemolytic anemia, thrombocytopenia, and multi-organ system involvement. However, in the last two decades, there have been many discoveries in the pathogenesis of aHUS and TTP. For instance, an autoantibody-mediated deficiency of the von Willebrand factor (vWF) cleaving protease ADAMTS13 has been shown to be the leading cause of platelet aggregation and microvascular thrombi formation in TTP.6–8 On the contrary, aHUS is mediated by genetic dysfunction in the complement alternative pathway. Normally, in the alternative complement pathway, C3b is inactivated by complement regulators, thus preventing the formation of 5Ca and MAC (C5a-9). However, due to gene mutations or autoantibodies to these regulators, C3b is always active and goes on to generate 5Ca and MAC. These overactive complements then cause endothelial cell damage, inflammation, activation of the coagulation cascade, and thrombotic microangiopathy.2–3
The first step in diagnosing aHUS is to recognize TMA: schistocytes, elevated lactate dehydrogenase (typically > 600 IU/L), decreased haptoglobin, decreased hemoglobin, and thrombocytopenia (platelet count less than 150,000 or > 25% decrease from baseline).9 These lab abnormalities should also coincide with one or more of the following: neurological symptoms, acute renal failure, or gastrointestinal symptoms. Early initiation of PEX for TTP is indicated as the definitive evaluation is started given the increased mortality of untreated TTP. Etiologies that mimic TMA should be excluded and they include systemic infections, medications, disseminated malignancy, scleroderma, antiphospholipid antibody syndrome, systemic lupus erythematosus, heparin-induced thrombocytopenia, malignant hypertension, autoimmune hemolytic anemia, disseminated intravascular coagulation, and pregnancy-related syndromes such as preeclampsia and HELLP syndrome.10 AHUS can be distinguished from STEC-HUS by PCR or culture-based assays for the Shiga-toxin producing E. coli. TTP can be distinguished from aHUS by measuring ADAMTS13 level. Clinicians should be aware that ADAMTS13 activity alone could not be used to definitively exclude TTP. It is only an adjunct in the evaluation and diagnosis of aHUS and TTP. If ADAMTS13 is > 5% and the patient is resistant to plasma exchange, then the diagnosis is more likely to be aHUS than TTP.5,6,9,11 If the diagnosis is still elusive, then screening for complement mutations and antibodies should be performed. Unfortunately, laboratories that offer complement genetic testing are not widely available.6,11 Once aHUS is diagnosed, eculizumab may be used as first line therapy.
Eculizumab is a recombinant, fully humanized monoclonal antibody, which is effective in treating aHUS due to its high binding affinity for C5 thus preventing the formation of C5a and C5a-9. It was recently approved as first line therapy for aHUS in 2011.8 In 2013, Legendre, et al, studied the outcome of 37 patients after 26 weeks of eculizumab therapy in 2 clinical trials. Patients included in the trials were > 12 years of age, had clinical evidence of hemolysis, had previous treatment with PEX, and had impaired renal function. They were excluded from the trials if they had documented STEC infection, ADAMTS 13 activity < 5%, and previous treatment with eculizumab. Eighty percent of patients treated with eculizumab had improvement in thrombocytopenia, anemia, and renal function resulting in eventual cessation of dialysis.5 The trials also showed that patients had a significantly greater improvement in their estimated glomerular filtration rates (GFR) if eculizumab was initiated earlier. Given that 90% of patients relapse during the first year after an aHUS episode, long term therapy with eculizumab is encouraged.4 The optimal duration of therapy thereafter is yet to be determined.
Our patient experienced complete hematologic response and tremendous improvement in fatigue, gastrointestinal symptoms, and neurological symptoms. Unfortunately, he remains dialysis dependent and this is likely due to previous damage to his kidneys from concomitant IgA vasculitis, hypertension, and diabetes. We also suspect that he may have had an unknown duration of undiagnosed aHUS prior his initial presentation to us which may have contributed to this irreversible renal function.
Conclusion
TMAs and TMA-mimicking diseases should be excluded before making a diagnosis of aHUS. Eculizumab is a complement inhibitor that has been shown to be effective in reversing the TMA effect in patients with aHUS, especially when it is used early on in the course of the disease. While early administration of eculizumab has been shown to be successful in treating aHUS, further studies are needed to determine the optimal duration of therapy.
Footnotes
Disclaimer: The views expressed in this document are those of the authors and do not reflect the official policy or position of the Department of the Army, Department of Defense, or the US Government.
Conflict of Interest
None of the authors identify a conflict of interest.
References
- 1.Cataland SR, Wu HM. Diagnosis and management of complement mediated thrombotic microangiopathies. Blood Reviews. 2014;28:67–74. doi: 10.1016/j.blre.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:1676–1687. doi: 10.1056/NEJMra0902814. [DOI] [PubMed] [Google Scholar]
- 3.Zuber J, Fakhouri F, Roumenina LT, Loirat C, Fremeaux-Bacchi V. Use of eculizumab for atypical hemolytic uremic syndrome and C3 glomerulopathies. Nat Rev Nephol. 2012;8:643–657. doi: 10.1038/nrneph.2012.214. [DOI] [PubMed] [Google Scholar]
- 4.Fakhouri F, Fremeaux-Bacchi V, Loirat C. Atypical hemolytic uremic syndrome: from the rediscovery of complement to targeted therapy. Eur J Intern Med. 2013;24(6):492–495. doi: 10.1016/j.ejim.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 5.Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368:2169. doi: 10.1056/NEJMoa1208981. [DOI] [PubMed] [Google Scholar]
- 6.Cataland SR, Wu HM. Atypical hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: clinically differentiating the thrombotic microangiopathies. Eur J Intern Med. 2013;24(6):486–491. doi: 10.1016/j.ejim.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 7.Shah N, Sarode R. Thrombotic Thrombocytopenic Purpura-What is new? Journal of Clinical Apheresis. 2013;28:30–35. doi: 10.1002/jca.21264. [DOI] [PubMed] [Google Scholar]
- 8.FDA approval for eculizumab in the treatment of atypical hemolytic uremic syndrome. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm272990.htm.
- 9.Laurence J. Atypical hemolytic uremic syndrome (aHUS): making the diagnosis. Clin Adv Hematol Oncol. 2012;10(10 Suppl 17):1–12. [PubMed] [Google Scholar]
- 10.George JN, Charania RS. Evaluation of patients with microangiopathic hemolytic anemia and thrombocytopenia. Semin Thromb Hemost. 2013;39:153–160. doi: 10.1055/s-0032-1333538. [DOI] [PubMed] [Google Scholar]
- 11.Cataland SR, Wu HM. How I treat: the clinical differentiation and initial treatment of adult patients with atypical hemolytic uremic syndrome. Blood. 2014;123:2478–2484. doi: 10.1182/blood-2013-11-516237. [DOI] [PubMed] [Google Scholar]