

Summary
WldS (slow Wallerian degeneration) is a remarkable protein that can suppress Wallerian degeneration of axons and synapses [1] but how it exerts this effect remains unclear [2]. Here, using Drosophila and mouse models, we identify mitochondria as a key site of action for WldS neuroprotective function. Targeting the NAD+ biosynthetic enzyme Nmnat to mitochondria was sufficient to fully phenocopy WldS, and WldS was specifically localized to mitochondria in synaptic preparations from mouse brain. Axotomy of live wild type axons induced a dramatic spike in axoplasmic Ca2+ and termination of mitochondrial movement—WldS potently suppressed both of these events. Surprisingly, WldS also promoted increased basal mitochondrial motility in axons before injury, and genetically suppressing mitochondrial motility in vivo dramatically reduced the protective effect of WldS. Intriguingly, purified mitochondria from WldS mice exhibited enhanced Ca2+ buffering capacity. We propose that the enhanced Ca2+ buffering capacity of WldS+ mitochondria leads to increased mitochondrial motility, suppression of axotomy-induced Ca2+ elevation in axons, and thereby suppression of Wallerian degeneration.
Results and Discussion
Mitochondria as a key site of Wlds neuroprotective function
Remarkably, the distal fragments of severed axons survive for weeks after axotomy in the Wlds mouse [3–6]. The Wlds mutation resulted from the fusion of two neighboring genes and lead to the production of a novel hybrid protein (Wlds) composed of the 70 NH-terminal amino acids of the E4 ubiqutin ligase Ube4b, a novel 18 amino acid linker domain, and the NAD+ biosynthetic enzyme Nmnat1 [7]. We previously found that expression of mouse Nmnat3 in Drosophila olfactory receptor neuron (ORN) axons provided protection equivalent to WldS 5 days after axotomy [8]. Recent work has shown that Nmnat3 expression in mouse neurons also robustly protects axons [9]. We co-expressed mouse UAS-Nmnat3::Myc and the mitochondrial marker UAS-mito::GFP in Drosophila ORNs. We found Nmnat3::Myc localized in a punctate pattern in ORN axons that precisely overlapped with mito::GFP (Figure 1A), indicating that Nmnat3 localized predominantly, if not exclusively, to mitochondria. We next assayed ORN axon preservation at 10, 20, and 50 days after axotomy. We found that Nmnat3 protected axons at levels indistinguishable from WldS at all time points tested (Figure 1B). Thus the N70 and W18 domains of WldS are dispensable for axon protection if Nmnat activity is targeted to mitochondria. By contrast, expression of Nmnat2 in ORN axons failed to suppress Wallerian degeneration, despite the fact that Nmnat2::Myc was localized throughout the axonal compartment (Supplementary Figure 1A).
Figure 1. Mitochondria as a focal point for WldS-mediated axon protection.
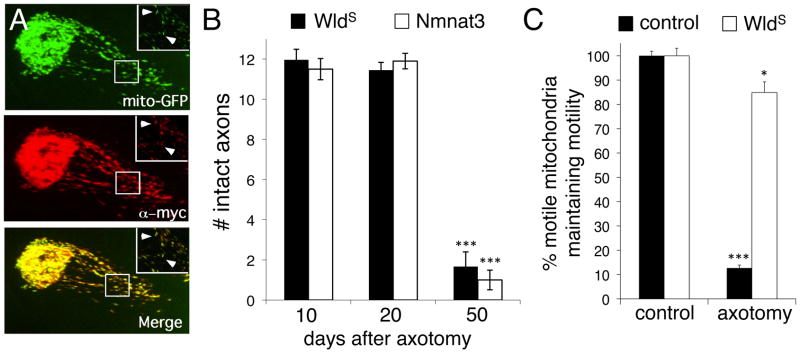
(A) Mouse Nmnat3::Myc localizes to mitochondria in Drosophila axons. 22a-Gal4 was used to drive UAS-Nmnat3::Myc and UAS-mito::GFP. Insets show boxed region.
(B) Mitochondrial Nmnat3 fully mimics WldS in axon protective function. 22a-Gal4 was used to drive UAS-Nmnat3 or UAS-WldS in a background where axons were labeled with membrane-tethered GFP (UAS-mCD8::GFP). n≥20 antennal lobes for each. *** indicates p<0.001. Error bars represent ±SEM.
(C) WldS-suppresses axotomy-induced termination of mitochondrial motility. Mitochondrial movement was assessed in live open-filet preparations of third instar Drosophila larvae immediately after axotomy for 5 minutes. Axotomy was induced by severing axons with a Micropoint laser ablation system and confirmed by a breakage of mCD8::mCherry-labeled axons. n≥10 live samples for each genotype and time point. ***, p<0.001.
Recently Yahata et al. (9) reported that in mouse neurons WldS protein is located in mitochondria, cytosol, perosixome/lysosome, and ER/Golgi-enriched cell fractions. We revisited Wlds localization in isolated mouse striatum from control and Wlds mice by separating the tissues into 3 fractions: non-synaptic striatal tissue, striatal synaptosomes without mitochondria, and synaptic mitochondria. We found WldS was detectable in the non-synaptic fraction, as would be expected from its predominantly nuclear localization. In addition, we detected WldS in synaptic mitochondria, but not in mitochondria-free synaptic preparations (Supplementary Figure 1B). These data are consistent with a primarily mitochondrial localization of WldS in axons and synapses in vivo in mouse brain.
Wlds suppresses termination of mitochondrial motility after injury and axotomy-induced increases in axonal Ca2+
We assayed mitochondrial dynamics in live Drosophila axons using the Tdc2-Gal4 driver, which is expressed in only 3 axons per segment of larval peripheral nerve, by driving UAS-mCD8::mCherry (to label axonal membranes) and UAS-mito::GFP (to label mitochondria; Figure 3A). In uninjured axons we found no differences in the total number of mitochondria, mitochondrial morphology, or mitochondrial size when we compared control and WldS-expressing axons (Supplementary Figure 1A,B). In control animals we found ~35% of mitochondria were motile before injury, but all motility terminated after laser axotomy (Supplementary Movie 1). In striking contrast, we found that laser axotomy of WldS-expressing axons had no effect on mitochondrial movement (Supplementary Movie 2; Figure 1C).
Figure 3. Wlds increases mitochondrial flux, which is essential for neuroprotective function.

(A) Mitochondrial flux was assayed in tdc2-Gal4-expressing neurons in live preparations of third instar larvae. Representative kymographs of mitochondrial movement are shown for control and WldS-expressing axons. Anterograde is to the right, retrograde is to the left.
(B) Mitochondrial flux was assayed in axons expressing each of the following molecules: Nmnat1, DN16-WldS, WldS-dead, WldS, N16-Nmnat1, and Nmant3. n≥10 live samples for each genotype. *, p<0.05; ***, p<0.001.
(C) Quantification of the movement of individual mitochondrial by binning into mobile, docked, or pausing/releasing during a 5 minute window. n=5 movies for each genotype.
(D) ORN axotomy assays in WldS backgrounds and miro mutants. A single antennal lobe where ORN axons were severed is shown.
(E) Quantification of data from (D). Age-matched uninjured controls at the same time points are shown at right. n≥15 samples for each genotype and time point. **, p<0.01; ***, p<0.001.
Axon injury in mammals leads to extracellular Ca2+ entry, which is necessary and sufficient for Wallerian degeneration [10]. Mitochondrial motility is known to be potently modulated by Ca2+ [11, 12]. We therefore sought to determine whether Drosophila axons showed axotomy-induced changes in axonal Ca2+, and whether axonal Ca2+ signaling was modulated by WldS. We drove the expression of the genetically-encoded Ca2+ indicator GCaMP3 in axons and measured changes in GCaMP3 signals in distal axon segments after laser-induced axotomy. In control animals we found a rapid increase in Ca2+ levels within seconds after axotomy, with Ca2+ levels peaking within 1 minute after cut, and then returning towards baseline levels over the next hour (Figure 2A,B,C; Supplementary Movie 3). However, >1 hour after axotomy axonal Ca2+ levels remained significantly elevated above baseline (~20% increase). Strikingly, while baseline Ca2+ levels in WldS+ axons were indistinguishable from those in controls (Supplementary Figure 3C), in Wlds+ axons injury-induced Ca2+ bursts were almost completely eliminated: Ca2+ levels only rose to ~15% control levels and returned to baseline within 1.5 minutes (Figure 2C; Supplementary Movie 4).
Figure 2. In vivo laser axotomy induces a dramatic rise in axonal Ca2+ that is suppressed by WldS.

(A) tdc2-Gal4 labels 3 axons in each peripheral nerve, only one segment is illustrated.
(B) Axons were labeled with mCD8::mCherry, axonal Ca2+ was monitored by co-expressing GCaMP3 in the tdc2-Gal4+ subset of neurons. Note the breakage of the axon after laser axotomy (red, mCherry). Axonal Ca2+ levels one minute after axotomy (green, GCaMP3).
(C,D) Representative traces showing Ca2+ responses in axon fragments distal to the injury site over time. Genotypes as indicated.
(E) Quantification of peak Ca2+ intensities and time to ½ recovery from average peak intensity for each genotype listed. n≥5 live samples for each genotype and time point versus control. *, p<0.05; **, p<0.01; ***, p<0.001.
In previous work we generated a collection of UAS-regulated Wlds-derived molecules that suppress Wallerian degeneration to varying degrees [8] (Supplementary Figure 1C). We assayed axotomy-induced changes in GCaMP3 fluorescence in axons expressing each of these molecules in live larval preparations. As with Wlds, we found no evidence for changes in axonal mitochondrial number, morphology, or size in these backgrounds (Supplementary Figure 2A,B). However, we found a striking correlation between axon protective function and suppression of axotomy-induced increases in axonal Ca2+: Wlds and Nmnat3 strongly suppressed Wallerian degeneration and post-injury axonal Ca2+ increases; Nmnat1, ΔN16::Wlds, and Wlds-dead partially suppressed Wallerian degeneration and post-injury axonal Ca2+ increases; and Nmant1dead, which lacks NAD+ biosynthetic activity and provided no protection from Wallerian degeneration [8], did not affect post-injury axonal Ca2+ increases. These changes were evident in both the distal and proximal axon segment, and affected both peak axonal Ca2+ intensities and recovery times to baseline Ca2+ levels (Figure 2C,D,E; Supplementary Figures 3A,B).
Wlds enhances mitochondrial movement, which is essential for maximal axonal protection after injury
Since mitochondrial motility is Ca2+-modulated we reasoned that changes in axonal Ca2+ buffering might affect axonal mitochondrial motility. We therefore assayed mitochondrial flux in axons expressing WldS-derived neuroprotective molecules. Surprisingly, we found a significant change in basal mitochondrial motility: in control animals ~35% of total axonal mitochondria were motile, however ~65% of mitochondria were motile in Wlds+ axons. Moreover, we found that molecules that provide partial suppression of Wallerian degeneration (Nmnat1, ΔN16::WldS, and WldS-dead) led to a modest, but significant, increase in the number of motile mitochondria, while molecules that maximally suppress Wallerian degeneration (WldS, N16::Nmnat1, and Nmnat3) led to a robust increase in the number of motile mitochondria (Figure 3B). This change in mitochondrial flux in WldS axons appears to represent a decrease in docked mitochondria, an increase in motile mitochondria, but no significant change in pause/release rates for individual mitochondria (Figure 3C).
Is increased mitochondrial flux critical for WldS-mediated axon protection? The adapter protein Miro functions to tether mitochondria to cytoskeletal motor proteins and modulate mitochondrial movement in a Ca2+-dependent fashion [12]. Impressively, mutations in miro dominantly decrease mitochondrial motility [13]. We therefore crossed strong alleles of miro (miroSD32 and miroSD26) and a UAS-regulated version of Miro (UAS-myc::Miro), which when overexpressed acts as a dominant negative [13] into the WldS background and assayed mitochondrial flux. We found that miro mutants or expression of Myc::Miro dominantly suppressed mitochondrial movement in controls. In addition, we found that loss of Miro function also decreased mitochondrial movement in the presence of WldS to levels found in control animals (Supplementary Figure 2C,D). Remarkably, reduced miro function also dominantly suppressed the neuroprotective effects of WldS in ORN axotomy assays. In animals with reduced miro function, axon loss was evident by 5 days after axotomy, with synaptic regions showing significant degeneration, and by 30 days after axotomy the protection afforded by WldS is almost completely blocked (Figure 3D,E).
Mitochondria from Wlds-expressing axons show enhanced Ca2+ buffering capacity and resistance to formation of the permeability transition pore
Mitochondria are major sinks for cellular Ca2+ in both axons and synapses [14]. A powerful mechanism by which WldS could exert all of these effects would be by altering mitochondrial Ca2+ buffering capacity. We therefore assessed mitochondrial Ca2+ cycling/capacity in cortical mitochondria isolated from young (~p25) wild type (NJ) and WldS mice. Mitochondrial isolations [15, 16] from both control and WldS animals yielded healthy, well-coupled mitochondria (Figure 4A,B). No apparent difference was observed in the Ca2+ uptake rates in mitochondria isolated from WldS versus control mice (Figure 4A,B). In contrast, the threshold for mitochondrial permeability, indicated by the loss of membrane potential (Figure 4A) and mitochondrial release of Ca2+ (Figure 4B), was significantly greater in mitochondria from WldS mice (Figure 4C). Thus, following increases in cytoplasmic Ca2+, WldS mitochondria isolated from mouse brain buffer higher loads of Ca2+ before releasing it back into the cytoplasmic compartment via the mitochondria permeability transition pore (PTP).
Figure 4. WldS brain mitochondria display higher Ca2+ load capacity than age matched wild type (NJ) controls.

(A,B) TMRE (membrane potential indicator) and CaG5N (extra-mitochondrial Ca2+ indicator) fluorescence were monitored over time simultaneously for each sample of non-synaptic mitochondria. As illustrated in TMRE traces for the first three minutes, the addition of pyruvate and malate (PM) an oxidative substrate, causes a marked downward deflection at 1 minute due to increased mitochondrial membrane potential (Δψm). Following ADP (A) addition, the loss of Δψm is indicated by upward deflection at 2 mins as Δψm is utilized to phosphorylate ADP to ATP via proton flow thru the ATP synthase. The ATP synthase inhibitor, oligomycin (O) addition at 3 min results in maximum Δψm as proton flow is inhibited. The Ca2+ infusion began at 5 min (infusion rate 160 nmol of Ca2+/mg protein/min) and was monitored by CaG5N fluorescence, and is illustrated by the initial upward deflection followed by constant signal due to mitochondrial Ca2+ uptake into the matrix. The subsequent rise in CaG5N fluorescence accompanied by a loss of membrane potential signifies mitochondrial permeability transition and subsequent release of mitochondrial Ca2+.
(C) Quantification of mitochondrial Ca2+ buffering capacity (nmols/mg protein) indicates that WldS non-synaptic mitochondria sequestered significantly higher amounts of Ca2+ compared to control group (n=6/group, * p<0.05, unpaired t-test).
Conclusions
The mechanistic action of Wlds has remained controversial, but recent work has established a non-nuclear role for Wlds [2] after injury [17]. In this study we show that WldS is localized to mitochondria in vivo. It is important to note that protein localization studies with Wlds must be interpreted cautiously—the primarily nuclear localization of Wlds suggested a nuclear role for Wlds and initially misled the field [2]. However, we also find that Wlds increases mitochondrial Ca2+ buffering capacity, and results in maintained mitochondrial motility after axotomy. Taken together, these data argue strongly that the mitochondrial compartment is a key site of action for WldS in vivo.
We have shown that axonal injury in live Drosophila preparations leads to a dramatic and transient rise in axonal Ca2+. Increased axonal Ca2+ has been observed in mammals after acute nerve crush [18] and entry of extracellular Ca2+ is necessary and sufficient for Wallerian degeneration [10]. Impressively, Wlds expression resulted in a striking suppression of this axotomy-induced rise in axonal Ca2+. The most plausible explanation for this enhanced buffering is that increased ATP/energy production observed in Wlds+ mitochondria [9]—presumably via increased mitochondrial NAD+ production, though we cannot formally exclude essential roles for other substrates of Nmnat—is linked to increased mitochondrial membrane potential (Δψm), and thereby increased Ca2+ entry through the Δψm–regulated mitochondrial Ca2+ uniporter [19]. This model is supported by our observation that Wlds-expressing mitochondria isolated from mouse brain exhibit an enhanced ability to maintain their membrane potential and avoid PTP formation in the face of increasing extramitochondrial Ca2+. In the future it will be important to confirm that such changes are also observed in Drosophila axonal mitochondrial physiology in vivo in Wlds-expressing neurons.
Axonal Ca2+ spikes could result solely from entry of extracellular Ca2+ into the axon after injury. This would be consistent with the observation that blocking Ca2+ channels inhibits Wallerian degeneration [10, 18]. Mitochondria are a well-established sink for Ca2+ in axons [14] and here we show that Wlds+ mitochondria exhibit enhanced Ca2+ buffering capacity and resistance to Ca2+-induced formation of the permeability transition pore (PTP). Indeed PTP formation appears to be a key final execution step in Wallerian degeneration [20–22]. We therefore favor a model whereby extracellular Ca2+ enters the axon after axotomy and normally acts as a switch to activate Wallerian degeneration. In Wlds axons this Ca2+ is instead rapidly buffered by mitochondria, thereby blocking induction of axonal destruction. Consistent with this model, uncoupling mitochondria, which suppresses mitochondrial Ca2+ uptake [23, 24], completely abrogates the protective effect of WldS in vitro [25].
WldS-expressing neurons exhibit a roughly 2-fold increase in the number of motile versus stationary mitochondria compared to wild type controls, which could result from changes in mitochondrial Ca2+ buffering. Notably, genetic suppression of enhanced mitochondrial flux using mutations in miro also resulted in a remarkable suppression of Wlds-mediated axonal protection in vivo. However, since this suppression was only partial, additional factors beyond increases in mitochondrial motility must also contribute to Wlds-mediated axonal protection. For example, axonal energy supplies are likely closely intertwined with mitochondrial transport and bioenergetics. Wlds+ mitochondria are known to exhibit an enhanced ability to generate ATP [9]. This change in bioenergetics, coupled with increased mitochondrial motility in Wlds+ axons might enhance distribution of ATP or other mitochondrially-derived metabolites. At the same time, enhanced mitochondrial motility could also speed the removal of metabolic byproducts normally processed by mitochondria. Similarly, increased mitochondrial motility in axons could further enhance mitochondrial Ca2+ buffering in Wlds+ axons since motile mitochondria would be predicted to traverse more — axonal space, and perhaps be exposed to more Ca2+ than stationary mitochondria. Together these could have the combined effect of increasing energy delivery, removing harmful byproducts, and increased buffering of Ca2+, a signal that can potently activate axonal degeneration.
A role for mitochondria in the Wlds neuroprotective mechanism is intriguing since defects in mitochondria respiration and dynamics are emerging as critical underlying factors in a number of neurological disorders [26]. For example, in mouse models of ALS (SOD1 transgenics) anterograde [27] and retrograde [28] mitochondrial transport is reduced; altered mitochondrial trafficking has been observed in models of Alzheimers disease [29]; and mutant, but not wild type Huntington protein blocks mitochondrial movement in cortical neurons [30]. However, in the majority of models, whether these mitochondrial alterations are a cause or consequence of disease remains an open question [26]. Our study shows, reciprocally, that enhanced mitochondrial flux is associated with, and is required for maximal axon protection by Wlds.
Supplementary Material
Highlights.
Axotomy induces dramatic Ca2+ bursts and termination of mitochondrial motility in axons
Wlds suppresses axotomy-induced Ca2+ bursts and axons maintain mitochondrial motility after axotomy
Wlds is localized to mitochondria and enhances mitochondrial Ca2+ buffering, resistance to mPTP, and mitochondrial motility in vivo
Enhanced mitochondrial motility is essential for Wlds neuroprotective function
Acknowledgments
This work was supported by NIH grant NS062993 (JWG and PGS), the Wellcome Trust (THG and MRF), NIH grant NS059991 (to MRF) and MRF is an Early Career Scientist with the Howard Hughes Medical Institute. TMR was supported by a grant from the DOD (BC093796). We thank Vimala Bondada and Dingyuan Lou for preliminary work on the mitochondrial Ca2+ dynamics, and all members of the laboratories for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Conforti L, Tarlton A, Mack TG, Mi W, Buckmaster EA, Wagner D, Perry VH, Coleman MP. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc Natl Acad Sci U S A. 2000;97:11377–11382. doi: 10.1073/pnas.97.21.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coleman MP, Freeman MR. Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci. 2010;33:245–267. doi: 10.1146/annurev-neuro-060909-153248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- 4.Perry VH, Brown MC, Lunn ER, Tree P, Gordon S. Evidence that Very Slow Wallerian Degeneration in C57BL/Ola Mice is an Intrinsic Property of the Peripheral Nerve. Eur J Neurosci. 1990;2:802–808. doi: 10.1111/j.1460-9568.1990.tb00472.x. [DOI] [PubMed] [Google Scholar]
- 5.Glass JD, Brushart TM, George EB, Griffin JW. Prolonged survival of transected nerve fibres in C57BL/Ola mice is an intrinsic characteristic of the axon. J Neurocytol. 1993;22:311–321. doi: 10.1007/BF01195555. [DOI] [PubMed] [Google Scholar]
- 6.Gillingwater TH, Ingham CA, Parry KE, Wright AK, Haley JE, Wishart TM, Arbuthnott GW, Ribchester RR. Delayed synaptic degeneration in the CNS of Wlds mice after cortical lesion. Brain. 2006;129:1546–1556. doi: 10.1093/brain/awl101. [DOI] [PubMed] [Google Scholar]
- 7.Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- 8.Avery MA, Sheehan AE, Kerr KS, Wang J, Freeman MR. Wld S requires Nmnat1 enzymatic activity and N16-VCP interactions to suppress Wallerian degeneration. J Cell Biol. 2009;184:501–513. doi: 10.1083/jcb.200808042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yahata N, Yuasa S, Araki T. Nicotinamide mononucleotide adenylyltransferase expression in mitochondrial matrix delays Wallerian degeneration. J Neurosci. 2009;29:6276–6284. doi: 10.1523/JNEUROSCI.4304-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.George EB, Glass JD, Griffin JW. Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J Neurosci. 1995;15:6445–6452. doi: 10.1523/JNEUROSCI.15-10-06445.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yi M, Weaver D, Hajnoczky G. Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J Cell Biol. 2004;167:661–672. doi: 10.1083/jcb.200406038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Schwarz TL. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Russo GJ, Louie K, Wellington A, Macleod GT, Hu F, Panchumarthi S, Zinsmaier KE. Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. J Neurosci. 2009;29:5443–5455. doi: 10.1523/JNEUROSCI.5417-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ganitkevich VY. The role of mitochondria in cytoplasmic Ca2+ cycling. Exp Physiol. 2003;88:91–97. doi: 10.1113/eph8802504. [DOI] [PubMed] [Google Scholar]
- 15.Brown MR, Sullivan PG, Dorenbos KA, Modafferi EA, Geddes JW, Steward O. Nitrogen disruption of synaptoneurosomes: an alternative method to isolate brain mitochondria. J Neurosci Methods. 2004;137:299–303. doi: 10.1016/j.jneumeth.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 16.Naga KK, Sullivan PG, Geddes JW. High cyclophilin D content of synaptic mitochondria results in increased vulnerability to permeability transition. J Neurosci. 2007;27:7469–7475. doi: 10.1523/JNEUROSCI.0646-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sasaki Y, Milbrandt J. Axonal degeneration is blocked by nicotinamide mononucleotide adenylyltransferase (Nmnat) protein transduction into transected axons. J Biol Chem. 2010;285:41211–41215. doi: 10.1074/jbc.C110.193904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knoferle J, Koch JC, Ostendorf T, Michel U, Planchamp V, Vutova P, Tonges L, Stadelmann C, Bruck W, Bahr M, et al. Mechanisms of acute axonal degeneration in the optic nerve in vivo. Proc Natl Acad Sci U S A. 2010;107:6064–6069. doi: 10.1073/pnas.0909794107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santo-Domingo J, Demaurex N. Calcium uptake mechanisms of mitochondria. Biochim Biophys Acta. 2010;1797:907–912. doi: 10.1016/j.bbabio.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 20.Sievers C, Platt N, Perry VH, Coleman MP, Conforti L. Neurites undergoing Wallerian degeneration show an apoptotic-like process with Annexin V positive staining and loss of mitochondrial membrane potential. Neurosci Res. 2003;46:161–169. doi: 10.1016/s0168-0102(03)00039-7. [DOI] [PubMed] [Google Scholar]
- 21.Sunio A, Bittner GD. Cyclosporin A retards the wallerian degeneration of peripheral mammalian axons. Exp Neurol. 1997;146:46–56. doi: 10.1006/exnr.1997.6484. [DOI] [PubMed] [Google Scholar]
- 22.Barrientos SA, Martinez NW, Yoo S, Jara JS, Zamorano S, Hetz C, Twiss JL, Alvarez J, Court FA. Axonal degeneration is mediated by the mitochondrial permeability transition pore. J Neurosci. 2011;31:966–978. doi: 10.1523/JNEUROSCI.4065-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pandya JD, Pauly JR, Sullivan PG. The optimal dosage and window of opportunity to maintain mitochondrial homeostasis following traumatic brain injury using the uncoupler FCCP. Exp Neurol. 2009;218:381–389. doi: 10.1016/j.expneurol.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 24.Pandya JD, Pauly JR, Nukala VN, Sebastian AH, Day KM, Korde AS, Maragos WF, Hall ED, Sullivan PG. Post-Injury Administration of Mitochondrial Uncouplers Increases Tissue Sparing and Improves Behavioral Outcome following Traumatic Brain Injury in Rodents. J Neurotrauma. 2007;24:798–811. doi: 10.1089/neu.2006.3673. [DOI] [PubMed] [Google Scholar]
- 25.Ikegami K, Koike T. Non-apoptotic neurite degeneration in apoptotic neuronal death: pivotal role of mitochondrial function in neurites. Neuroscience. 2003;122:617–626. doi: 10.1016/j.neuroscience.2003.08.057. [DOI] [PubMed] [Google Scholar]
- 26.Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Brownlees J, Ackerley S, Shaw PJ, McLoughlin DM, et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet. 2007;16:2720–2728. doi: 10.1093/hmg/ddm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi P, Strom AL, Gal J, Zhu H. Effects of ALS-related SOD1 mutants on dynein- and KIF5-mediated retrograde and anterograde axonal transport. Biochim Biophys Acta. 2010;1802:707–716. doi: 10.1016/j.bbadis.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Su B, Zheng L, Perry G, Smith MA, Zhu X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J Neurochem. 2009;109(Suppl 1):153–159. doi: 10.1111/j.1471-4159.2009.05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang DT, Rintoul GL, Pandipati S, Reynolds IJ. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol Dis. 2006;22:388–400. doi: 10.1016/j.nbd.2005.12.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.