

Abstract
Background
Because annulated indoles have almost no representation in the PubChem or MLSMR databases, an unprecedented class of an indole-based library was constructed, using the indole aryne methodology, and screened for antitumor activity. Sixty-six novel 6,7-annulated-4-substituted indole compounds were synthesized, using a strategic combination of 6,7-indolyne cycloaddition and cross-coupling reactions under both Suzuki-Miyaura and Buchwald-Hartwig conditions, and tested for their effectiveness against murine L1210 tumor cell proliferation in vitro.
Materials and Methods
Various markers of tumor cell metabolism, DNA degradation, mitotic disruption, cytokinesis and apoptosis were assayed in vitro to evaluate drug cytotoxicity.
Results
Most compounds inhibited the metabolic activity of leukemic cells in a time- and concentration-dependent manner but only 9 of them were sufficiently potent to inhibit L1210 tumor cell proliferation by 50% in the low-μM range after 2 (IC50: 4.5–20.4 μM) and 4 days (0.5–4.0 μM) in culture. However, the antiproliferative compounds that were the most effective at day 4 were not necessarily the most potent at day 2, suggesting different speeds of action. A 3-h treatment with antiproliferative annulated indole was sufficient to inhibit, in a concentration-dependent manner, the rate of DNA synthesis measured in L1210 cells over a 0.5-h period of pulse-labeling with 3H-thymidine. Four of the antiproliferative compounds had weak DNA-binding activities but one compound reduced the fluorescence of the ethidium bromide-DNA complex by up to 53%, suggesting that some annulated indoles might directly interact with double-stranded DNA to disrupt its integrity and prevent the dye from intercalating into DNA base pairs. However, all 9 antiproliferative compounds induced DNA cleavage at 24 h in L1210 cells, containing 3H-thymidine-prelabeled DNA, suggesting that these antitumor annulated indoles might trigger an apoptotic pathway of DNA fragmentation. Indeed the antiproliferative annulated indoles caused a time-dependent increase of caspase-3 activity with a peak at 6 h. Interestingly, the compounds with the most potent antiproliferative IC50 values at day 2 were consistently the most effective at inhibiting DNA synthesis at 3 h and inducing DNA fragmentation at 24 h. After 24–48 h, antiproliferative concentrations of annulated indoles increased the mitotic index of L1210 cells and stimulated the formation of many bi-nucleated cells, multi-nucleated cells, apoptotic cells and micronuclei, suggesting that these antitumor compounds might enhance mitotic abnormality, induce chromosomal damage or missegregation, and block cytokinesis to induce apoptosis.
Conclusion
Although annulated indoles may have interesting bioactivity, novel derivatives with different substitutions must be synthesized to elucidate structure-activity relationships, identify more potent antitumor lead compounds, and investigate their molecular targets and mechanisms of action.
Keywords: Annulated indoles, tumor cell proliferation, DNA synthesis, interaction and fragmentation, cells with mitotic figures, several nuclei and micronuclei, cytokinesis, apoptosis
Indole arynes or indolynes were discovered and reported as a completely new class of reactive aryne intermediate (1–5). An unprecedented 93-member library of 6,7-annulated-4-substituted indoles was constructed, using a strategic combination of 6,7-indolyne cycloaddition and cross-coupling reactions under both Suzuki-Miyaura and Buchwald-Hartwig conditions (6). This work may represent the first example of library development that employs the indole aryne methodology (6). The 4 steps of the synthetic process, showing cycloaddition at the 6,7 position, followed by cross-coupling at the C4 position, are summarized in Figure 1. 1) The main 4,6,7-tribromoindole scaffold was synthesized by the Bartoli reaction (7, 8). 2) The N-methyl-4,6,7-tribromoindole scaffold was treated with n-butyllithium to generate the 6,7-indole aryne which reacted with both cyclopentadiene (X=CH2) and furan (X=O) to give the respective Diels-Alder cycloadducts in high yields. 3) The olefin (alkene) in the cycloadduct had to be reduced quantitatively with diimide prior to cross-coupling in order to produce higher yields (9). 4) The 4-Br was reacted either with various boronic acids under a carefully prescribed set of conditions to give the Suzuki-Miyaura cross-coupled products or with various commercially-available secondary amines under a carefully prescribed set of conditions to give the Buchwald-Hartwig cross-coupled products (6). The reason such annulated indoles were synthesized is that they have almost no representation in the NIH PubChem and Molecular Library Small Molecule Repository (MLSMR) databases and their biological properties have not been fully evaluated and exploited. The only exceptions are a few biologically active natural products, such as trikentrins (10), which are antibacterial agents, herbindoles (11), which possess cytotoxic and anti-feedant properties, teleocidins (12), which are tumor promoters used to study multistage carcinogenesis, and nodulisporic acids (13), which have antiparasitic activity. Annulated indoles were therefore predicted to have unique chemical property space characteristics and a good probability of exhibiting interesting biological activity (6). Hence, a total of 66 newly-synthesized 6,7-annulated-4-substituted indoles were evaluated for their antiproliferative activity in rapidly growing suspension cultures of L1210 leukemia cells, using tests of metabolic activity, DNA degradation, mitotic disruption, cytokinesis and apoptosis in vitro (14, 15).
Figure 1.

First example of library development that employs the indole aryne methodology to construct 6,7-annulated-4-substituted indole compounds. Top: Bartoli route to prepare the N-methyl-4,6,7-tribromoindole scaffold. Bottom: Selective generation of 4-bromo-6,7-indole aryne, and strategic combination of 6,7-indolyne cycloaddition and cross-coupling reactions under both Suzuki-Miyaura and Buchwald-Hartwig conditions to construct polycyclic indole libraries.
Materials and Methods
Drug treatment, cell culture and proliferation assay
Solutions of synthetic 6,7-annulated-4-substituted indoles and known anticancer drugs used as positive controls (all from Sigma-Aldrich, St. Louis, MO, USA) were dissolved and serially-diluted in dimethyl sulfoxide (DMSO). Suspension cultures of murine L1210 lymphocytic leukemia cells (ATCC, Manassas, VA, USA) were incubated at 37°C in a humidified atmosphere containing 5% CO2 and maintained in continuous exponential growth by twice-a-week passage in RPMI 1640 medium supplemented with 10% fetal bovine calf serum (FCS; Atlanta Biologicals, Norcross, GA, USA) and penicillin (100 IU/ml)-streptomycin (100 μg/ml). L1210 cell suspensions were grown in triplicate in 48-well Costar cell culture plates for 2 and 4 days in the presence or absence (control) of serial concentrations of synthetic 6,7-annulated-4-substituted indoles to evaluate their antiproliferative activity. Since compounds were supplemented to the culture medium in 1-μl aliquots, the concentrations of vehicle in the final incubation volume (0.5 ml) never exceeded 0.2% and did not interfere with the data. Decreasing concentrations of cells, such as 45,000 and 2,500 L1210 cells/0.5 ml/well, were initially plated in triplicate at time 0 in order to collect control samples with approximately equal cell densities after 2 and 4 days in culture, respectively (14, 15). The proliferation of control and drug-treated tumor cells was assessed from their mitochondrial ability to bioreduce the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) reagent (Promega, Madison, WI, USA) in the presence of phenazine methosulfate (PMS; Sigma) into a water-soluble formazan product that absorbs at 490 nm (16). After 2 or 4 days in culture, control and drug-treated L1210 cell samples were further incubated at 37°C for 2 h in the dark in the presence of 0.1 ml of MTS:PMS (2:0.1) reagent and their relative metabolic activity was estimated by recording the absorbance at 490 nm, using a Cambridge model 750 automatic microplate reader (Packard, Downers Grove, IL, USA) (14, 15). Blank values for culture medium supplemented with MTS:PMS reagent in the absence of cells were subtracted from the results. Data of all biochemical experiments were analyzed using the Student’s t-test with the level of significance set at p<0.05.
DNA synthesis
To estimate the rate of DNA synthesis, L1210 cells were re-suspended in fresh FCS-containing RPMI 1640 medium at a density of 0.5×105 cells/0.5 ml, incubated at 37°C for 2.5 h in the presence or absence (control) of drugs and then pulse-labeled for an additional 30 min with 1 μCi of [methyl-3H]thymidine (52 Ci/mmol; GE Healthcare-Amersham, Piscataway, NJ, USA) (14, 15). The incubations were terminated by the addition of 2 ml of 10% trichloroacetic acid (TCA). After holding on ice for 15 min, the acid-insoluble material was recovered over Whatman GF/A glass microfiber filters and washed thrice with 2 ml of 5% TCA and twice with 2 ml of 100% EtOH. After drying the filters, the radioactivity bound to the acid-precipitable material was determined by liquid scintillation counting (LSC) in 6 ml of Bio-Safe NA (Research Products International, Mount Prospect, IL, USA) (14, 15).
DNA binding and fragmentation assays
An ethidium bromide (EB) displacement assay was used to assess the eventual DNA-binding potencies of 6,7-annulated-4-substituted indoles, based on the IC50 concentrations of drugs that cause a 50% reduction in the fluorescence of the DNA-EB complex at 525 nm excitation/600 nm emission (17). The fluorescence of EB, which is extremely low when unbound in water, is boosted 25-fold after binding and intercalating in the hydrophobic environment of the intact double-stranded DNA. Since a highly fluorescent complex is formed within milliseconds between DNA and EB, any drug directly binding to and/or damaging DNA would disrupt EB intercalation and compromise the fluorescence of this EB-DNA complex. The reaction mixtures (200 μl), containing final concentrations of 20 μg/ml calf thymus (ct) DNA (Sigma) and 1 μg/ml EB (Sigma) in 5 mM Tris-HCl buffer, pH 7.6, with 0.5 mM EDTA, were pipetted into 96-well Costar white opaque polystyrene assay plates and, after incubation in the presence or absence (control) of drugs for 30 min at room temperature, the fluorescence of the EB-DNA complex was detected at 525 nm excitation and 600 nm emission, using a Cary Eclipse Fluorescence Spectrophotometer equipped with a microplate reader accessory (Varian, Walnut Creek, CA, USA) (14, 15). Drug-induced DNA cleavage was determined by intact chromatin precipitation, using L1210 cells which were pre-labeled with 1 μCi of [3H]thymidine for 2 h at 37°C, washed with 3×1 ml of ice-cold Ca2+/Mg2+-free Dulbecco’s phosphate-buffered saline (PBS), collected by centrifugation, resuspended in fresh medium at a density of 1.2×106 cells/ml, and then incubated at 37°C for 24 h in the presence or absence of drugs (14, 15). After centrifugation at 200 ×g for 10 min to discard the drugs and wash the cells, the cell pellets were lysed for 20 min in 0.5 ml of ice-cold hypotonic lysis buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA and 0.2% Triton X-100), and centrifuged at 12,000 ×g for 15 min to collect the supernatants. The radioactivity in the supernatants (detergent-soluble low molecular weight DNA fragments) and the pellets (intact chromatin DNA) was determined by LSC. Before being counted in 6 ml of Bio-Safe NA, the intact pelleted chromatin was incubated for 2 h at 60°C in the presence of 0.6 ml of NCS tissue solubilizer (Amersham) (14, 15).
Fluorogenic assay of caspase-3 activity
Control and drug-treated L1210 cells (1.2×106/ml) were incubated for various periods of time at 37°C, collected by centrifugation (200 g × 10 min), and washed with 1 ml of ice-cold PBS. The cell pellets were resuspended in 120 μl of chilled 10 mM Hepes buffer, pH 7.4, containing 100 mM NaCl, 100 mM KCl, 5 mM MgCl2, 1 mM EDTA, 10 mM EGTA, 10% sucrose, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM dithiothreitol (DTT), and 100 μM digitonin, and were lysed for 10 min on ice. The cell lysates were centrifuged (14,000 ×g for 20 min) at 4°C to precipitate cellular debris and 100-μl aliquots of the clear supernatants were stored at −70°C overnight (14, 18–20). The caspase-3-like activities of the lysates were determined in reaction mixtures that contained 50 μl of lysis buffer (blank) or supernatant (control or drug-treated samples) and 50 μl of reaction buffer (100 mM Hepes, pH 7.5, containing 1 mM EDTA, 10 mM EGTA, 10% sucrose, and 10 mM DTT) and that were initiated by the addition of 5-μl aliquots of the specific 1 mM benzyloxycarbonyl (z)-Asp-Glu-Val-Asp (DEVD)-7-amino-4-trifluoromethylcoumarin (AFC) stock of AFC-substrate conjugate (Calbiochem, EMD Biosciences, San Diego, CA, USA) (14, 18–20). After incubation for 1 h at 37°C in 96-well Costar white opaque polystyrene assay plates, the fluorescence of the free AFC released upon proteolytic cleavage of the substrate by caspase-3 was detected at 400-nm excitation and 505-nm emission, using a Cary Eclipse Fluorescence Spectrophotometer with a microplate reader. Arbitrary fluorescence units were quantified with reference to calibration curves ranging from 0.1 to 1.2 nmol of AFC (from Sigma), the protein concentrations of the supernatants were determined using the BCA Protein Assay Kit (Pierce, Rockford, IL, USA), and the DEVD-specific cleavage activities of the samples were expressed as nmol of AFC released/mg of protein (14, 18–20).
Mitotic index and abnormalities
To determine the mitotic index, L1210 cells (0.5×106/0.5 ml of FCS-containing RPMI 1640 medium) were incubated in triplicate for various periods of time at 37°C in the presence or absence (control) of serial concentrations of experimental drugs or known microtubule-disrupting agents, collected by centrifugation (200 ×g for 10 min), and resuspended in 1 ml of hypotonic 75 mM KCl for 20 min at 4°C. After fixation in 1 ml of MeOH:acetic acid (3:1), the final cell pellets were collected by centrifugation, resuspended in 75 μl of MeOH:acetic acid (3:1), dispensed onto glass slides, air dried, and stained by spreading 40 μl of 0.1% crystal violet under a coverslip (14, 15). Mitotic figures with condensed chromosomes were identified microscopically and published criteria were followed to score binucleated cells (BNCs), multinucleated cells (MULTI) and micronuclei (MNi) (21). Cytokinesis-blocked BNCs contained either 2 separate nuclei of equal size, 2 nuclei that touched or overlapped with distinct nuclear boundaries, or 2 nuclei that were linked by a small nucleoplasmic bridge (21). Viable cells with 3 (trinucleated) or 4 (quadrinucleated) distinct nuclei were scored as typical MULTI cells (21). Dot-like chromatin-containing structures in the cytoplasm, at least 1/3 smaller than the main nucleus, surrounded by a membrane either separated from or marginally overlapping the main nucleus, and having the same staining as the main nucleus were scored as MNi containing either a whole chromosome or an acentric chromosomal fragment (21). The frequencies of BNCs, MULTI cells and MNi were used to estimate the ability of cytotoxic or genotoxic (mutagenic or clastogenic) drugs to block cytokinesis and induce chromosomal damage or missegregation (21). The % of cells in mitosis or with 2 nuclei, 3–4 nuclei and MNi were determined microscopically by counting a total of at least 2,000 cells/slide and the mitotic, BNC and MULTI cell indexes were calculated as % of mitotic figures or BNC and MULTI cells in drug-treated cultures/% of mitotic figures or BNC and MULTI cells in vehicle-treated controls (14, 15, 21).
Results
Inhibition of tumor cell proliferation
All 66 synthetic 6,7-annulated-4-substituted indoles tested, inhibited the proliferation of suspension cultures of murine L1210 lymphocytic leukemia cells after 4 days in vitro (Table I). It would be unfeasible to show the structures and antitumor effects of all 66 compounds but, in summary Table I, groups of 6,7-annulated-4-substituted indoles are ranked according to the range of their concentrations required to inhibit the mitochondrial ability of L1210 cells to metabolize the MTS:PMS reagent by 50% after 2 days in culture. Only 3 compounds have no effect or inhibit tumor cell proliferation at day 2 by less than 50%, at the highest 156.25 μM concentration tested but the other groups of compounds have increasing antiproliferative activities with IC50 values in the 50–150, 40–50, 30–40, 20–30, 10–20 and less than 10 μM ranges (Table I). For individual compounds, the IC50 values at day 4 are always lower than values at day 2 because the magnitude of inhibition between the control cells that keep growing exponentially and the drug-treated cells that are inhibited keeps increasing over time. Hence, the 9 compounds that had the best combinations of antiproliferative IC50 values at day 2 and 4 were selected for further studies and their chemical structures and abilities to inhibit the metabolic activity of L1210 cells are detailed in Figure 2 and Table II, respectively. For simplification, the full KUC identification numbers of the compounds synthesized at the NIH Center of Excellence, in Chemical Methodologies and Library Development, at the University of Kansas (KU-CMLD) have been logically abbreviated in the present article: for example, KUC107069 is KU-69, KUC107072 is KU-72, etc… (Figure 2). Interestingly, among these 9 most potent antiproliferative compounds, 8 were from the Buchwald-Hartwig cross-coupled products (i.e. those that have an amine at the C-4 position of the indole) and just one (KU-191) came from the Suzuki-Miyaura series of library members (Figure 2). All these 9 antiproliferative 6,7-annulated-4-substituted indoles have promising antitumor effects in the low μM range but it should be noted that the compounds that seem to be the most effective at day 4 are not necessarily the ones the most potent at day 2 (Table II). Full concentration-response curves indicate that the inhibitions of tumor cell growth by antiproliferative compounds KU-69, KU-87 and KU-95, which are the most effective at day 4, begin around 102–256 nM and become maximal or near maximal around 10–25 μM (Figure 3). This is an example of 3 compounds that have excellent antiproliferative activities at day 4 but much weaker concentration-dependent inhibitory effects at day 2, so there is an unusually wide gap between their IC50 values at days 2 and 4 (Figure 3). In contrast, the concentration-dependent inhibitions of tumor cell growth by KU-70, KU-80 and KU-96 illustrate 3 compounds that may not be the most potent at day 4 but have already excellent antiproliferative activities at day 2 so the gap is much smaller between their antiproliferative IC50 values, which are almost similar at days 2 and 4 (Figure 4). Synthetic 6,7-annulated-4-substituted indoles, therefore, might differ in both antiproliferative potencies and speeds of action. However, when tested as positive controls in the same experiments, established anticancer drugs like daunorubicin (DAU) and mitoxantrone (Mitox) inhibited the growth of L1210 tumor cells at lower nM concentrations (data not shown).
Table I.
66 Newly-synthesized 6,7-annulated indole compounds screened for the inhibition of L1210 leukemic cell proliferation in vitro.
| Number of compounds tested | Range of IC50 values (μM) in L1210 cells at
|
|
|---|---|---|
| Day 2 | Day 4 | |
| 3 compounds | NAa | 18.7–137.8 |
| 5 compounds | 51.9–154.6 | 22.3–37.4 |
| 5 compounds | 42.0–49.6 | 24.9–29.4 |
| 8 compounds | 31.3–37.8 | 16.5–25.4 |
| 18 compounds | 20.0–29.5 | 1.4–20.4 |
| 18 compounds | 10.8–19.6 | 0.5–13.4 |
| 9 compounds | 4.9–9.3 | 2.0–6.9 |
Concentrations of novel synthetic indole-based compounds required to inhibit by 50% (IC50 values) the metabolic activity of L1210 leukemic cells, using the MTS:PMS assay after 2 or 4 days of culture in vitro. IC50 values (μM) were calculated from linear regression of the slopes of the log-transformed concentration-survival curves. Groups of compounds are ranked according to the range of their IC50 potencies at day 2.
Values are not available because the compounds have either no effect or decrease tumor cell proliferation by less than 50% at the highest 156.25 μM concentration tested.
Figure 2.
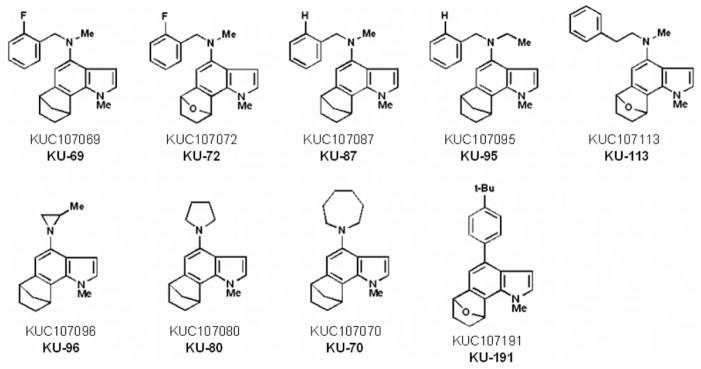
Chemical structures and identification numbers of the most effective antiproliferative 6,7-annulated-4-substituted indole compounds tested for their antitumor effects in L1210 cells in vitro.
Table II.
Antiproliferative activity of synthetic 6,7-annulated indole compounds in L1210 tumor cells in vitro.
| Compounds | IC50 values (μM)a in L1210 cells at
|
|
|---|---|---|
| Day 2 | Day 4 | |
| KU-69 | 13.2±0.5 | 0.5±0.02 |
| KU-70 | 5.8±0.2 | 2.8±0.2 |
| KU-72 | 7.8±0.6 | 2.0±0.2 |
| KU-80 | 7.0±0.4 | 2.4±0.2 |
| KU-87 | 16.1±0.6 | 1.1±0.1 |
| KU-95 | 20.4±0.7 | 1.4±0.1 |
| KU-96 | 4.9±0.2 | 2.3±0.1 |
| KU-113 | 6.1±0.2 | 4.0±0.1 |
| KU-191 | 4.5±0.1 | 2.7±0.1 |
Concentrations of novel synthetic indole-based compounds required to inhibit by 50% (IC50 values) the metabolic activity of L1210 leukemic cells, using the MTS:PMS assay after 2 or 4 days of culture in vitro. IC50 values (μM) were calculated from linear regression of the slopes of the log-transformed concentration-survival curves.
Means±SD (n=3).
Figure 3.

Comparison of the ability of serial concentrations (plotted on a logarithmic scale) of KU-69 (○, ●), KU-87 (□, ■) and KU-95 (△, ▲) to inhibit the metabolic activity of L1210 tumor cells at days 2 (open symbols) and 4 (solid symbols) in vitro. L1210 cell proliferation results were expressed as % of the net absorbance of MTS/formazan after bioreduction by vehicle-treated control cells after 2 (A490 nm=1.112±0.048) and 4 (A490 nm=1.177±0.051) days in culture (100±4.3%, striped area). The blank values (A490 nm=0.432 at day 2 and 0.434 at day 4) for cell-free culture medium supplemented with MTS:PMS reagent were subtracted from the results. Bars: means±SD (n=3). aNot different from respective controls; bp<0.05, cp<0.01 and dp<0.005, lower than respective controls.
Figure 4.

Comparison of the ability of serial concentrations (plotted on a logarithmic scale) of KU-70 (○, ●), KU-80 (□, ■) and KU-96 (△, ▲) to inhibit the metabolic activity of L1210 tumor cells at days 2 (open symbols) and 4 (solid symbols) in vitro. The conditions of the experiment and the determination of the results were identical to those of Figure 3. Vehicle-treated control cells after 2 and 4 days: 100±4.3%, striped area. Bars: means±SD (n=3). aNot different from respective controls; bp<0.05, cp<0.025, dp<0.01 and ep<0.005, lower than respective controls.
Inhibition of DNA synthesis
With one exception, a 3-h period of incubation is sufficient for the antiproliferative 6,7-annulated-4-substituted indole compounds to inhibit the incorporation of [3H]thymidine into the DNA used to assess the rate of DNA synthesis over a 30-min period of pulse-labeling in L1210 tumor cells in vitro (Figure 5). Interestingly, KU-70, KU-80 and KU-96, the compounds with some of the best antiproliferative IC50 values at day 2 (Table II) are also the best inhibitors of DNA synthesis at 3 h (Figure 5), whereas KU-69, KU-87 and KU-95, the compounds with the worst antiproliferative IC50 values at day 2 (Table II) have no or very little inhibitory effects on DNA synthesis at 3h (Figure 5). At 25 μM, KU-70, KU-80 and KU-96 can match the ability of 256 nM Mitox to inhibit DNA synthesis by more than 80%, but are still 100-times weaker than this known anti-cancer drug used as a positive control in the experiments (Figure 5). Increasing concentrations of KU-70 and KU-80, which have good antiproliferative IC50 values at 2 days (Table II), totally inhibit the ability of tumor cells to synthesize DNA after 3 h (Figure 6). These concentration-dependent inhibitions of DNA synthesis by KU-70 and KU-80 are maximal around 25–62.5 μM and characterized by IC50 values of 6.4 and 13.0 μM, respectively (Figure 6). However, under similar experimental conditions, Mitox inhibits DNA synthesis in L1210 cells with an IC50 value of 142 nM (data not shown).
Figure 5.

Comparison of the ability of 25 μM concentrations of the antiproliferative KU-69, KU-70, KU-72, KU-80, KU-87, KU-95, KU-96, KU-113 and KU-191 compounds to inhibit the rate of incorporation of [3H]thymidine into DNA measured in L1210 cells over 30 min following a 3-h period of incubation at 37°C in vitro. The magnitude of DNA synthesis inhibition caused by 0.256 μM mitoxantrone (Mitox) was used as a positive control. DNA synthesis in vehicle-treated control cells (C) at 37°C was 20,387±1,325 cpm (100±6.5%). The blank value (766±43 cpm) for control cells incubated and pulse-labeled at 2°C with 1.5 μCi of [3H]thymidine has been subtracted from the results. Bars: means±SD (n=3). aNot different from control; bp<0.025, less than control.
Figure 6.

Comparison of the ability of serial concentrations (logarithmic scale) of the antiproliferative KU-70 (●) and KU-80 (○) compounds to inhibit the rate of incorporation of [3H]thymidine into DNA measured in L1210 cells over 30 min following a 3-h period of incubation at 37°C in vitro. The conditions of the experiment and the determination of the results were identical to those of Figure 5. DNA synthesis in vehicle-treated control cells at 37°C: 100±6.5%, striped area. Bars: means±SD (n=3). aNot different from control; bp<0.05 and cp<0.025, less than control.
DNA binding and fragmentation
High concentrations of antiproliferative 6,7-annulated-4-substituted indoles and reference anticancer drugs were evaluated for their DNA- binding affinity in a cell-free system, using the classic EB displacement assay to identify intercalating or non-intercalating drugs that destabilize the DNA double helix. As expected, positive controls of 8 μM Mitox, 50 μM actinomycin D (Act-D) and 125 μM amsacrine (m-AMSA; 4′-[9-acridinylamino]methanesulfon-m-anisidide), which destabilize double-stranded ctDNA to prevent or disrupt EB intercalation, almost totally suppress the fluorescence of the EB-DNA complex (inhibition by 89.7, 84.2 and 73.8%, respectively) (Figure 7). Among our 9 antiproliferative annulated indoles, 4 compounds had no significant effect and 4 other compounds inhibited the fluorescence of the EB-DNA complex by only 12.4–14.9% when tested at 312.5–781.25 μM (data not shown). KU-80, which had a good antiproliferative IC50 value at day 2 (Table II and Figure 4) and was one of the best inhibitors of DNA synthesis at 3 h (Figures 5 and 6), was the only compound that inhibited the fluorescence of the EB-DNA complex in a concentration-dependent manner, with up to 52.6% inhibition at the highest 1,953.125 μM concentration tested (Figure 7). However, under similar experimental conditions, the concentration-dependent inhibitions of EB binding to DNA by Mitox, Act-D and m-AMSA have IC50 values of 1.2, 3.4 and 31.0 μM, respectively (data not shown). Hence, only a few of the antiproliferative annulated indoles might have weak direct interactions with double-stranded ctDNA to disrupt its structural and functional integrity and prevent the dye from intercalating into DNA base pairs. The abilities of our 9 antiproliferative annulated indoles to induce DNA fragmentation at 24 h were assessed and compared to those of DAU and Mitox, using L1210 cells containing 3H-thymidine-prelabeled DNA to detect low molecular weight DNA fragments after intact chromatin precipitation (Figure 8). As compared to untreated control tumor cells where there is only 3.8% of DNA fragmentation, anti-cancer treatments that induce DNA cleavage and apoptosis like 1.6 μM DAU and 0.256 μM Mitox respectively produce 11.6 and 24.0% of DNA fragmentation at 24 h and are used as positive controls in these experiments (Figure 8). When tested at 25 μM, all 9 of our antiproliferative annulated indoles substantially raised the levels of DNA fragmentation in L1210 cells at 24 h (Figure 8). Again, KU-80 (10.7% DNA fragmentation), KU-96 (21.7% DNA fragmentation) and KU-191 (14.6% DNA fragmentation), the compounds that are some of the most effective inhibitors of tumor cell proliferation at day 2 (Table II) and DNA synthesis at 3 h (Figure 5), clearly match the levels of DNA fragmentation caused by the lower concentrations of DAU and/or Mitox tested (Figure 8). Moreover, full concentration-response curves clearly show that, above 25 μM, KU-80, KU-96 and KU-191 are capable of producing huge 25–50% levels of DNA fragmentation (Figure 9). Even though they might be, on an equimolar concentration basis, less cytotoxic than known anti-cancer drugs, some of these antiproliferative annulated indoles are also likely to induce the internucleosomal fragmentation of DNA at 24 h, which is the ultimate marker of apoptosis in DAU- and Mitox-treated tumor cells.
Figure 7.

Comparison of the ability of 8 μM mitoxantrone (Mitox), 50 μM actinomycin D (Act-D), 125 μM m-amsacrine (Amsa) and serial concentrations of antiproliferative KU-80 compound to inhibit the binding of EB to double-stranded ctDNA. Results were expressed as % of the control (C) fluorescence of the EB-DNA complex in the absence of drug at 525 nm excitation and 600 nm emission (162.1±5.3 arbitrary units; 100±3.3 %). The background of EB fluorescence in the absence of DNA (26.3±1.0 arbitrary units) was subtracted from the results. Bars: means±SD (n=3). aNot different from control; bp<0.05, cp<0.025 and dp<0.005, smaller than control.
Figure 8.

Comparison of the ability of 25 μM concentrations of the antiproliferative KU-69, KU-70, KU-72, KU-80, KU-87, KU-95, KU-96, KU-113 and KU-191 compounds to induce DNA fragmentation at 24 h in L1210 cells containing 3H-prelabeled DNA in vitro. The levels of DNA fragmentation caused by 1.6 μM daunorubicin (DAU) and 0.256 μM mitoxantrone (Mitox) were used as positive controls. The results were expressed as [cpm in supernatant/(cpm in supernatant + pellet)] × 100 at 24 h. For vehicle-treated control (C) tumor cells (3.83±0.84% DNA fragmentation), the supernatant (DNA fragments) was 2,096±126 cpm and the pellet (intact DNA) was 52,628±3,368 cpm. Bars: means±SD (n=3). ap<0.05 and bp<0.025, greater than control.
Figure 9.

Comparison of the ability of serial concentrations (logarithmic scale) of the antiproliferative KU-80 (○), KU-96 (□) and KU-191 (△) compounds to induce DNA fragmentation at 24 h in L1210 cells, containing 3H-prelabeled DNA in vitro. The conditions of the experiment and the determination of the results were identical to those in Figure 8. DNA fragmentation in vehicle-treated control tumor cells: 3.83±0.84%, striped area. Bars: means±SD (n=3). aNot different from control; bp<0.05 and cp<0.025, greater than control.
Activation of effector caspase-3
Since the activation of caspase-3 is required to trigger the internucleosomal fragmentation of DNA by endonucleases, the annulated indole treatments shown to induce DNA fragmentation at 24 h were tested for their ability to stimulate this key post-mitochondrial effector caspase in L1210 cells. The hypothesis that the caspase activation cascade is linked to apoptotic DNA fragmentation at 24 h is substantiated by the fact that 62.5 μM KU-80, 62.5 μM KU-96, 1.6 μM DAU and 0.64 μM Mitox all maximally induce, in a time-dependent manner, caspase-3 activity in L1210 cells (Figure 10). Interestingly, the activations of caspase-3 by KU-80 and KU-96 are noticeable as early as 3 h and respectively peak at 290.2 and 464.7% of the control after 6 h before slowly declining back towards basal levels between 12–24 h (Figure 10). That KU-80 induces caspase-3 activity to a lesser degree than KU-96 at 6 h (Figure 10) is consistent with the fact that KU-80 also produces less DNA fragmentation than KU-96 at 24 h (Figures 8 and 9). In contrast, the activations of caspase-3 by DAU and Mitox begin around 12 h and respectively peak at 605.9 and 379.2% of the control after 24 h (Figure 10), a finding that has already been observed in L1210 cells (14) and differ from the peak of DAU-induced caspase-3 activity reported at 6 h in human HL-60 promyelocytic leukemia cells (19). Different concentrations of drugs causing different ratios of cytostatic:cytotoxic effects might be responsible for such different sequences of apoptotic protease activations in different tumor cell lines.
Figure 10.
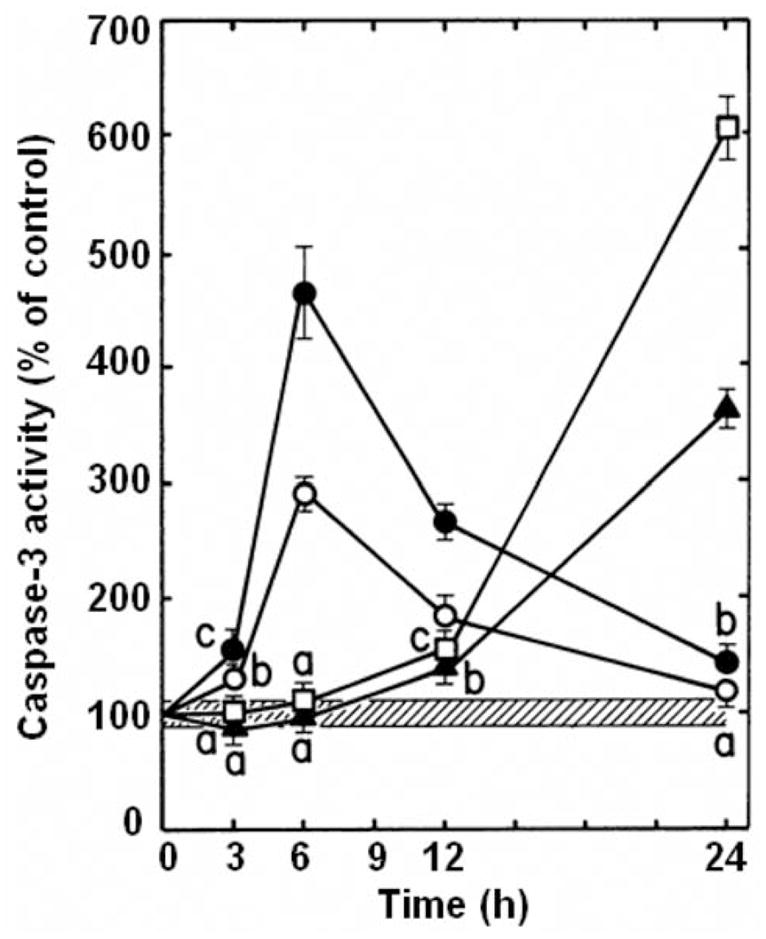
Fluorogenic assay of effector caspase activation in drug-treated L1210 cells. Comparison of the time-dependent inductions of caspase-3-like protease activity by 0.64 μM mitoxantrone (▲, Mitox), 1.6 μM daunorubicin (□, DAU), 62.5 μM KU-80 (○) and 62.5 μM KU-96 (●) in L1210 cells, incubated for 3–24 h in vitro. Results are expressed as percentage of DEVD cleavage activity in vehicle-treated control tumor cells (4.06±0.36 nmol AFC released/mg protein, 100±8.9%, striped area) at each time point tested. Bars: means±SD (n=3). aNot different from control; bp<0.025 and cp<0.005, greater than control.
Mitotic index and abnormalities
Microscopic studies were conducted to determine whether L1210 tumor cells treated for various periods of time with increasing concentrations of antiproliferative annulated indoles might undergo mitotic disruption. Control populations of L1210 tumor cells incubated in the absence of drugs contain only 0.42% of mitotic cells and 2.73% of BNCs. Known microtubule-disrupting agents like vincristine (VCR) and taxol were used as positive controls in these experiments. VCR blocks tubulin polymerization and microtubule assembly, whereas taxol lowers the critical concentration of free tubulin required to promote polymerization and blocks microtubule disassembly. Such antimitotic anticancer drugs, therefore, block cell-cycle progression in M-phase and dramatically increase the % of mitotic cells at 24 and 48 h: by 22.1- and 54.2-fold for 0.05 μM VCR and by 9.1- and 22.5-fold for 0.05 μM taxol (Figure 11A). Under similar conditions, increasingly antiproliferative concentrations of KU-69 also block mitosis, especially at 24 h in response to 1.6, 4 and 10 μM concentrations of KU-69, which respectively produce 9.1-, 11.1- and 8.3-fold increases in the % of mitotic cells (Figure 11A). This would suggest some microtubule disruption but the effect of KU-69 seems to decline at 48 h and for the highest 25 μM concentration tested (Figure 11A). VCR and taxol also increase by little the % of BNCs: by 2.8-fold for VCR at 48 h and by 6.9- and 3.9-fold for taxol at 24 and 48 h (Figure 11B). But increasing 4, 10 and 25 μM concentrations of KU-69 respectively produce huge 14.1-, 24.7- and 36.5-fold increases in the % of BNCs, uniquely at 24 h, indicating that they rapidly induce molecular events that block the process of cytokinesis in proliferating tumor cells (Figure 11B). VCR at 48 h and taxol at 24 and 48 h respectively increase by 15.2- and 10.6–29.4-fold the % of MULTI cells (Figure 12A) and respectively induce the formation of 0.49 and 7.56–0.70% of cells with MNi (Figure 12B), as compared to controls where those figures are extremely rare: 0.34% of MULTI cells and 0.06% of cells with MNi in vehicle-treated L1210 tumor cells. Under similar conditions, KU-69 also produces concentration-dependent increases in the % of MULTI cells and cells with MNi that match or surpass the effects of VCR and taxol, suggesting that these antiproliferative annulated indoles might enhance mitotic abnormalities, induce chromosomal damage or missegregation, and block cytokinesis. For instance, after 48 h, treatments with 10 and 25 μM KU-69 respectively cause 19.4- and 25.2-fold increases in the % of MULTI cells (Figure 12A) and induce the formation of 2.51 and 2.85% of cells that contain MNi (Figure 12B). It should be noted that the huge % of KU-69-induced BNCs observed at 24 h dramatically drops at 48 h (Figure 11B) while the % of KU-69-induced MULTI cells increases dramatically from 24 to 48 h (Figure 12A), an effect which is also apparent in response to taxol. This is consistent with the fact that drug-treated cells with disrupted microtubule kinetics escape from mitosis without cytokinesis and proceed as BNCs to the next cell cycle and round of DNA synthesis to form polynucleated and polyploid cells with 3–4 nuclei, which eventually die (21, 22). Hence, it is logical that the huge number of BNCs formed at 24 h drops at 48 h as these BNCs re-enter the cell cycle and are increasingly transformed into MULTI cells between 24 and 48 h. BNCs, therefore, do not disappear but simply evolve into MULTI cells. Finally, as compared to control L1210 tumor cells where the presence of nucleoplasmic bridges (NPBs) (0.002%) and nuclear buds (NBUDs) (0.006%) is extremely rare, treatments with 1.6, 4, 10 and 25 μM KU-69 respectively increased by 16.5-, 24.3-, 24.9- and 194.1-fold the % of tumor cells with NPBs and by 6.4-, 27.1-, 31.4- and 80.4-fold the % of cells with NBUDs (data not shown). Control populations of L1210 tumor cells incubated for 48 h in the absence of drugs contain only 0.046% of cells with 5 or more small nuclear structures, which is a microscopic evidence of nuclear fragmentation into smaller apoptotic chromatin bodies within an intact cytoplasm and cytoplasmic membrane (21). Using such criteria, treatments with 1.6, 4, 10 and 25 μM KU-69, respectively, increased by 3.5-, 6.9-, 9.5- and 45.9-fold the % of microscopically visible apoptotic tumor cells at 48 h (data not shown), confirming the results of our caspase-3 and DNA fragmentation studies.
Figure 11.

Comparison of the effects of the antiproliferative KU-69 compound, vincristine (VCR) and taxol on the frequency of mitotic figures (A) and binucleated cells (BNCs) (B) in L1210 tumor cells in vitro. L1210 cells were incubated in triplicate for 24 (open columns) and 48 h (closed columns) at 37°C in the presence, or absence (C: control) of the indicated concentrations of drugs. The percentage of cells in each category was determined by morphologic analysis, scoring at least 2,000 cells/slide to identify those containing mitotic figures or 2 nuclei. Results were expressed as % of mitotic (A) or BNCs (B) in drug-treated cultures divided by the % of mitotic (C: 0.42±0.08%) or BNCs (C: 2.73±0.57%) in vehicle-treated controls. Bars: means±SD (n=3). ap<0.05, smaller than control; bp<0.01 and cp<0.025, greater than respective controls.
Figure 12.

Comparison of the effects of the antiproliferative KU-69 compound, vincristine (VCR) and taxol on the frequency of multinucleated (MULTI) cells (A) and micronuclei (MNi) (B) in L1210 tumor cells in vitro. L1210 cells were incubated in triplicate for 24 (open columns) and 48 h (closed columns) at 37°C in the presence or absence (C: control) of the indicated concentrations of drugs. The percentage of cells in each category was determined by morphological analysis, scoring at least 2,000 cells/slide to identify those containing 3–4 nuclei or MNi. A: Results were expressed as % of MULTI cells in drug-treated cultures divided by the % of MULTI cells in vehicle-treated controls (C: 0.34±0.07%). B: Results were expressed as % of vehicle- (C: 0.06±0.01%) or drug-treated cells with MNi. Bars: means±SD (n=3). aNot different from control; bp<0.005, greater than respective controls.
Discussion
Since annulated indole structures are virtually unknown, we have exploited our ability to effect a tandem indole aryne cycloaddition/cross-coupling sequence of reactions with polybromoindole scaffolds to address this important gap in the literature. Using this tactic with the 4,6,7-tribromoindole platform (readily-obtained in scale via the Bartoli indole synthesis), for example, resulted in the formation of a 93-member polycyclic annulated indole library (1–9). This is the first library of annulated indoles reported in the literature to date and it is the first to employ indole arynes as a strategic device. The present study is the first to report that, out of the 66 newly-synthesized 6,7-annulated-4-substituted indoles tested for their bioactivity, 8 compounds from the Buchwald-Hartwig cross-coupled products and 1 compound from the Suzuki-Miyaura series exhibited interesting antitumor effects in L1210 cells with antiproliferative IC50 activities ranging between 0.5–4 μM after 4 days in culture. The results are encouraging, given the fact that strong anti-cancer compounds were discovered among a relatively small library of annulated indoles synthesized. The antitumor activities of such 6,7-annulated-4-substituted indoles open the possibility of designing new anticancer drugs based on this framework. An interesting structure-activity relationship (SAR) emerges with these entities. It appears that in annulated bicyclic structures, those with a –CH2 bridge generally showed higher activity than those with an oxygen bridge, and that fluorinated benzylic amines gave the highest antiproliferative activity. For example, substituting an oxygen bridge for a methylene bridge (KU-72 vs. KU-69), reduced the antiproliferative activity at day 4 by a factor of 4 (Figure 2). Removing the ortho-fluoro substituent in KU-69 reduced the antiproliferative activity only by a factor of about 2 (KU-69 vs. KU-87) (Figure 2). Also, changing the methyl-amine to an ethyl-amine (KU-87 vs. KU-95) only slightly decreased the antiproliferative activity (Figure 2). However, changing a benzyl-amine to a phenethylamine (KU113) reduced the antiproliferative activity from KU-72 by a factor of 2 and from KU-69 by a factor of 4 (Figure 2). Within the series of 3 cyclic amino products (KU-96, KU-80 and KU-70), marginally increased antiproliferative activity seems to correlate with smaller ring size (Figure 2). Since KU-69 has the best antiproliferative IC50 value at day 4, a quantitative SAR study is planned for this compound based, in part, on Topliss considerations. The Topliss correlation predicts the effect of having lipophilic/hydrophilic groups and electron-donating/electron-withdrawing groups at various positions (ortho, meta and para) on an isolated benzene ring. For example, it is predicted that by having a 3,4-dichloro substitution pattern on the benzylic amine of KU-69, the antiproliferative of this compound should increase. Unfortunately, the magnitude of such increased bioactivity cannot be predicted. In addition, to investigate the effect of N-alkyl substitution, we plan of replacing the N-methyl group with other alkyl groups of various lengths and branching patterns.
The concentration-dependent inhibitions of L1210 tumor cell proliferation by the annulated indoles are always more pronounced at 4 rather than 2 days, suggesting that the effectiveness of these bioactive compounds is a combination of drug concentration and duration of action. Although it may be somewhat misleading to compare biological responses measured at very different times, the concentration-dependent inhibitions of DNA synthesis by KU-70 and KU-80 (Figure 6) suggest that the ability of these compounds to prevent tumor cells from synthesizing macromolecules at 3 h may play a role in their antiproliferative activity at days 2 and 4 (Figure 4). In accordance with the effects of established anticancer drugs used as positive controls, concentrations of KU-80, higher than those sufficient to inhibit tumor cell proliferation must be used to inhibit DNA synthesis or maximally induce DNA fragmentation. Such apparent discrepancy may simply be due to different experimental conditions and cellular responses to various periods of drug exposure: the rate of DNA synthesis over 30 min is inhibited in cells treated for only 3 h with KU-80 or Mitox and DNA fragmentation occurs 24 h after treatment with KU-80, Mitox or DAU, whereas the more spectacular inhibitions of L1210 tumor cell proliferations are the result of 2- and 4-day long drug treatments. However, in spite of their excellent antiproliferative IC50 values at day 4, KU-69, KU-87 and KU-95 have the worst antiproliferative activities of the series at day 2 in relation with their weak abilities to inhibit DNA synthesis at 3 h and induce DNA fragmentation at 24 h. Conversely, the annulated indoles that have the most effective antitumor effects over shorter periods of time, such as KU-80 and KU-96, are some of the most potent inhibitors of DNA synthesis and inducers of DNA fragmentation within 3–24 h in relation with the fact that they already have some of the best antiproliferative activities at day 2. In contrast to slow-acting compounds like KU-69, KU-87 and KU-95 that might need more than 2 days to fully express their superior antiproliferative activities, therefore, it is postulated that the annulated indoles that are the most effective in short-term antitumor assays, such as KU-70, KU-80, KU-96 and KU-191, might have a more rapid mechanism of action and need less time to interact with and be incorporated into cells, undergo eventual metabolic activation, reach crucial cellular targets, and trigger various inhibitory pathways that finally damage and kill tumor cells.
FDA-approved Mitox is a synthetic anthracenedione related to DAU but less cardiotoxic. Besides targeting the DNA topoisomerase II enzyme to induce DNA damage, the mechanism of action of anthracycline-type antitumor drugs involves intercalation of the planar aglycone moiety into DNA to disrupt DNA replication and transcription (23). The 9-anilinoacridine drug m-AMSA is known to quench EB-DNA fluorescence and its anti-leukemic activity requires intercalative DNA binding (24, 25). Although alternative binding modes that include single-stranded DNA have also been proposed, the antitumor antibiotic Act-D blocks RNA polymerase and transcription by binding to double-stranded DNA. Act-D complexes with duplex DNA by intercalation of its planar aromatic rings between adjacent base pairs of the double helix, the dinucleotide site GpC exhibiting an especially high binding affinity for Act-D (26). In addition, m-AMSA preferentially targets and inhibits DNA topoisomerase II activity, whereas Act-D is likely a dual inhibitor of DNA topoisomerase I and II enzymes (27–29). Hence, the present EB results are consistent with the fact that Mitox (8 μM), Act-D (50 μM) and m-Amsa (125 μM) are DNA-reactive agents that intercalate into DNA and cause crosslinks and strand breaks. However, the observation that a single KU-80 compound is capable of directly binds to purified DNA to inhibit by 11.4–52.6% the fluorescence of the EB-DNA complex in a concentration-dependent manner over the 50–1,953 μM range (Figure 7), while the 8 other antiproliferative annulated indoles have no, or much weaker inhibitory effects in such DNA binding cell-free assay, suggests that most of the antiproliferative 6,7-annulated-4-substituted indoles synthesized are unlikely to directly target cellular DNA to exert their antitumor activities.
In contrast to the early cleavage of DNA into large 50–300 kbp fragments, an initial signaling event that may induce tumor cells treated with relatively low concentrations of DNA-damaging anticancer drugs to commit apoptosis, the secondary endonucleolytic degradation of DNA at internucleosomal linker sites to produce small 180–200 bp mono- and oligonucleosomal fragments at 24 h, is a late marker concurrent with morphological evidence of apoptosis (30). Upstream of internucleosomal DNA fragmentation, activation of the caspase cascade induces the proteolytic cleavage of a wide range of substrates. For instance, effector caspase-3 cleaves and inactivates the inhibitor of caspase-activated DNase, thus releasing active endonucleases that translocate into the nucleus to achieve internucleosomal DNA fragmentation (30). Antiproliferative concentrations of annulated indoles, especially KU-80, KU-96 and KU-191, all induce DNA fragmentation in L1210 cells at 24 h (Figures 8 and 9), suggesting that the ability of 6,7-annulated-4-substituted indoles to trigger apoptosis may play a significant role in their molecular mechanism of antitumor activity. This hypothesis is substantiated by the finding that KU-80 and KU-96 also induce a time-dependent increase of caspase-3 activity (Figure 10), which peaks at 6 h in L1210 cells and thus precedes the fragmentation of radiolabeled DNA and the appearance of apoptotic cells with 5 or more small nuclear structures detected at 24 h. Since apoptosis is an active ATP-driven and cell-cycle phase-specific process that requires the expression of specific genes, the synthesis of new RNA and proteins, and the activation of caspases, non-caspase proteases and nucleases, inhibition of such active mechanisms can prevent apoptosis (18–20). The discrepancy between the time-dependent activations of effector caspase-3 by annulated indoles, Mitox and DAU in L1210 cells might be due to differences in concentrations and mechanisms of action. Since KU-80 and KU-96 are tested at much higher concentrations than Mitox and DAU, it is possible that, even though they are increasingly cytostatic, the 62.5 μM concentrations of antiproliferative annulated indoles might inhibit DNA (Figures 5 and 6) and other macromolecule syntheses to such excessive degree and become so cytotoxic over time as to abolish their own ability to sustain the activation of caspase-3 (Figure 10) beyond 6–12 h (18–20). Even though most antiproliferative annulated indoles do not directly bind to DNA, the abilities of these antitumor compounds to interact with maintenance enzymes or regulatory proteins and indirectly cause high molecular weight DNA-strand breaks, crosslinks or chromosome aberrations and cytokinesis disruption remain to be determined. Therefore, it is rather premature to speculate on the nature of the primary molecular targets, massive damaging events and nuclear signals that induce tumor cells treated with antiproliferative annulated indoles to undergo mitotic disruption and apoptotic DNA degradation and nuclear fragmentation. The fact that 4 μM KU-69 is capable of causing 11.1- and 7.0-fold increases in the number of L1210 cells that display mitotic figures at 24 and 48 h (Figure 11) suggests an ability to disrupt microtubule kinetics. But 4 μM KU-69 cannot achieve the dramatic 54.1-and 22.5-fold increases in the mitotic index of L1210 cells caused by much smaller 0.05 μM concentrations of antimitotic VCR and taxol at 48 h, suggesting that this compound is not a potent mitotic spindle poison. Even though annulated indoles might weakly interact with tubulin and alter the polymerization/depolymerization of microtubules, these compounds are likely to target other molecular events in order to elicit their antiproliferative activity. For instance, increasingly antiproliferative 0.64–25 μM concentrations of KU-69 might trigger some genotoxic effects that induce chromosomal aberration, disrupt chromosomal segregation and block cytokinesis to substantially increase the levels of BNCs, MULTI cells and MNi in L1210 tumor cells at 24–48 h, the magnitudes of these responses often matching or even surpassing those induced by VCR and taxol (Figures 11 and 12). BNCs indicate that cytokinesis is inhibited following nuclear division (21). MULTI cells may follow if BNCs re-enter the cell cycle and escape further cytokinesis. The micronucleus test is an indicator of drug-induced chromosomal damage and aberration as such MNi structures generally form during the metaphase/anaphase transition of mitosis when a whole lagging chromosome or an acentric chromosome fragment resulting from a clastogenic or mutagenic event fail to integrate into the daughter nuclei (21). However, chromosome aberration and non-disjunction/missegregation might be the consequence of prolonged drug-induced mitotic disruption and might be responsible for cellular inability to undergo cytokinesis after regression of the cleavage furrow (31). If completion of cytokinesis requires accurate chromosome segregation, it is possible that KU-69 treatment induces chromosome aberration and missegregation to increase the frequency of BNCs, MULTI cells and MNi. NPBs between nuclei in BNCs originate from dicentric chromosomes in which the centromeres have been pulled to the opposite poles of the cell at anaphase and are indicative of DNA misrepair, chromosome re-arrangement or telomere end-fusions (21). NPBs may break to form MNi. NPBs, which are correlated with MNi frequency in BNCs, are thus biomarkers of dicentric chromosomes and might provide indirect evidence of KU-69-induced genome damage resulting from misrepaired DNA breaks. NBUDs occur during S-phase and form MNi that are still linked to the nucleus by a narrow or wide stalk of nucleoplasmic material depending on the stage of the budding process. Amplified and/or excess DNA is selectively localized to specific sites at the periphery of the nucleus and is eliminated via nuclear budding to form MNi during S phase. Hence, antiproliferative annulated indoles and drugs that inhibit DNA synthesis like hydroxyurea might increase the rate of elimination of amplified DNA and possibly DNA-repair complexes via nuclear budding (32, 33). Further SAR studies are required to identify more potent antitumor lead compounds and characterize their molecular targets and mechanisms of action. This series of synthetic 6,7-annulated-4-substituted indoles has also been submitted to the Molecular Library Screening Center Network (MLSCN) to assess their bioactivity in a wide range of cell-based and other biological assays.
Acknowledgments
This study was supported in part by grants from the National Institutes of Health (grant RO1 GM069711 to KRB and KU-CMLD grant NIGMS 5P50GM069663) and Kansas State University (Innovative Research Award from the Terry C. Johnson Center for Basic Cancer Research and Research Seed Grant Award from the Biology Research and Instruction Enhancement Fund Program).
References
- 1.Buszek KR, Luo D, Kondrashov M, Brown N, VanderVelde D. Indole-derived arynes and their Diels-Alder reactivity with furans. Org Lett. 2007;9:4135–4137. doi: 10.1021/ol701595n. [DOI] [PubMed] [Google Scholar]
- 2.Buszek KR, Brown N, Luo D. Concise total synthesis of (±)-cis-trikentrin A and (±)-herbindol A via intermolecular indole aryne cycloaddition. Org Lett. 2009;11:201–204. doi: 10.1021/ol802425m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown N, Luo D, VanderVelde D, Yang S, Brassfield A, Buszek KR. Regioselective Diels- Alder cycloadditions and other reactions of 4,5-, 5,6-, and 6,7-indole arynes. Tetrahedron Lett. 2009;50:63–65. doi: 10.1016/j.tetlet.2008.10.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown N, Luo D, Decapo JA, Buszek KR. New synthesis of (±)-cis-trikentrin A via tandem indole aryne cycloaddition/Negishi reaction. Applications to library development. Tetrahedron Lett. 2009;50:7113–7115. doi: 10.1016/j.tetlet.2009.09.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garr AN, Luo D, Brown N, Cramer CJ, Buszek KR, VanderVelde D. Experimental and theoretical investigations into the unusual regioselectivity of 4,5-, 5,6-, and 6,7-indole aryne cycloadditions. Org Lett. 2010;12:96–99. doi: 10.1021/ol902415s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thornton PD, Brown N, Hill D, Neuenswander B, Lushington GH, Santini C, Buszek KR. Application of 6,7-indole aryne cycloaddition and Pd(0)-catalyzed Susuki-Miyaura and Buchwald- Hartwig cross-coupling reactions for the preparation of annulated indole libraries. ACS Comb Sci. 2011;13:443–448. doi: 10.1021/co2000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartoli G, Palmieri G, Bosco M, Dalpozzo R. The reaction of vinyl Grignard reagents with 2- substituted nitroarenes: a new approach to the synthesis of 7-substituted indoles. Tetrahedron Lett. 1989;30:2129–2132. [Google Scholar]
- 8.Dalpozzo R, Bartoli G. Bartoli indole synthesis. Curr Org Chem. 2005;9:163–178. [Google Scholar]
- 9.Buszek KR, Brown N. Improved method for the diimide reduction of multiple bonds on solid- supported substrates. J Org Chem. 2007;72:3125–3128. doi: 10.1021/jo0622173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson SK, Kerr MA. Total synthesis of (±)-herbindole A, (±)–herbindole B, and (±)cis- trikentrin A. J Org Chem. 2007;72:1405–1411. doi: 10.1021/jo062350v. [DOI] [PubMed] [Google Scholar]
- 11.Jackson SK, Banfield SC, Kerr MA. Total synthesis of (±)- herbindole B, and (±)-cis-trikentrin B. Org Lett. 2005;7:1215–1218. doi: 10.1021/ol047498k. [DOI] [PubMed] [Google Scholar]
- 12.Hitotsuyanagi Y, Fujiki H, Suganuma M, Aimi N, Sakai S-I, Endo Y, Shudo K, Sugimura T. Isolation and structure elucidation of teleocidin B-1, B-2 and B-4. Chem Pharm Bull. 1984;32:4233–4236. doi: 10.1248/cpb.32.4233. [DOI] [PubMed] [Google Scholar]
- 13.Singh SB, Ondeyka JG, Jayasuriya H, Zink DL, Ha SN, Dahl-Roshak A, Greene J, Kim JA, Smith MM, Shoop W, Tkacz JS. Nodulisporic acids D-F. structure, biological activities, and biogenetic relationships. J Nat Prod. 2004;67:1496–1506. doi: 10.1021/np0498455. [DOI] [PubMed] [Google Scholar]
- 14.Perchellet EM, Crow KR, Gakhar G, Nguyen TA, Shi A, Hua DH, Perchellet J-P. Bioactivity and molecular targets of novel substituted quinolines in murine and human tumor cell lines in vitro. Int J Oncology. 2010;36:673–688. doi: 10.3892/ijo_00000543. [DOI] [PubMed] [Google Scholar]
- 15.Perchellet J-P, Waters AM, Perchellet EM, Naganaboina VK, Chandra KL, Desper J, Rayat S. Bioactivity of synthetic 2-halo-3-aryl-4(3H)-quinazoliniminium halides in L1210 leukemia and SK- BR-3 mammary tumor cells in vitro. Anticancer Res. 2011;31:2083–2094. [PubMed] [Google Scholar]
- 16.Cory AH, Owen JC, Barltrop JA, Cory JG. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- 17.Morgan AR, Lee JS, Pulleyblank DS, Murray NL, Evans DH. Ethidium fluorescence assays. Part 1. Physico-chemical studies. Nucleic Acids Res. 1979;7:547–569. doi: 10.1093/nar/7.3.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perchellet EM, Wang Y, Weber RL, Sperfslage BJ, Lou K, Crossland J, Hua DH, Perchellet J-P. Synthetic 1,4-anthracenedione analogs induce cytochrome c release, caspase-9, -3, and -8 activities, poly(ADP-ribose) polymerase-1 cleavage and internucleosomal DNA fragmentation in HL-60 cells by a mechanism which involves caspase-2 activation but not Fas signaling. Biochem Pharmacol. 2004;67:523–537. doi: 10.1016/j.bcp.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 19.Perchellet EM, Wang Y, Weber RL, Lou K, Hua DH, Perchellet J-P. Antitumor triptycene bisquinones induce a caspase-independent release of mitochondrial cytochrome c and a caspase- 2-mediated activation of initiator caspase-8 and -9 in HL-60 cells by a mechanism which does not involve Fas signaling. Anti-Cancer Drugs. 2004;15:929–946. doi: 10.1097/00001813-200411000-00002. [DOI] [PubMed] [Google Scholar]
- 20.Perchellet EM, Ward MM, Skaltsounis A-L, Kostakis IK, Pouli N, Marakos P, Perchellet J-P. Antiproliferative and proapoptotic activities of pyranoxanthenones, pyranothio-xanthenones and their pyrazole-fused derivatives in HL-60 cells. Anticancer Res. 2006;26:2791–2804. [PubMed] [Google Scholar]
- 21.Fenech M, Chang WP, Kirsch-Volders M, Holland N, Bonassi S, Zeiger E. HUMN project: detailed description of the scoring criteria for the cytokinesis-block micronucleus assay using isolated human lymphocyte cultures. Mutat Res. 2003;534:65–75. doi: 10.1016/s1383-5718(02)00249-8. [DOI] [PubMed] [Google Scholar]
- 22.Lieu C-H, Chang Y-N, Lai Y-K. Dual cytotoxic mechanisms of submicromolar taxol on human leukemia HL-60 cells. Biochem Pharmacol. 1997;53:1587–1596. doi: 10.1016/s0006-2952(97)82450-1. [DOI] [PubMed] [Google Scholar]
- 23.Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol. 1999;57:727–741. doi: 10.1016/s0006-2952(98)00307-4. [DOI] [PubMed] [Google Scholar]
- 24.Baguley BC, Le Bret M. Quenching of DNA-ethidium fluorescence by amsacrine and other antitumor agents: a possible electron-transfer effect. Biochemistry. 1984;23:937–943. doi: 10.1021/bi00300a022. [DOI] [PubMed] [Google Scholar]
- 25.Denny WA, Twigden SJ, Baguley BC. Steric constraints for DNA binding and biological activity in the amsacrine series. Anticancer Drug Des. 1986;1:125–132. [PubMed] [Google Scholar]
- 26.Snyder JG, Hartman NG, Langlois D’Estantoit B, Kennard O, Remeta DP, Breslauer KJ. Binding of actinomycin D to DNA: evidence for a nonclassical high-affinity binding mode that does not require GpC sites. Proc Natl Acad Sci USA. 1989;86:3968–3972. doi: 10.1073/pnas.86.11.3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nelson EM, Tewey KM, Liu LF. Mechanism of antitumor drug action: poisoning of mammalian DNA topoisomerase II on DNA by 4′-(9-acridinylamino)-methanesulfon-m-anisidide. Proc Natl Acad Sci USA. 1984;81:1361–1365. doi: 10.1073/pnas.81.5.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu MH, Yung BY. Cell cycle phase-dependent cytotoxicity of actinomycin D in HeLa cells. Eur J Pharmacol. 1994;270:203–212. doi: 10.1016/0926-6917(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 29.Insaf SS, Danks MK, Witiak DT. A structure function analysis of DNA topoisomerase II inhibitors. Curr Med Chem. 1996;3:437–466. [Google Scholar]
- 30.Nagata S. Apoptotic DNA fragmentation. Exp Cell Res. 2000;256:12–18. doi: 10.1006/excr.2000.4834. [DOI] [PubMed] [Google Scholar]
- 31.Shi Q, King RW. Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature. 2005;437:1038–1042. doi: 10.1038/nature03958. [DOI] [PubMed] [Google Scholar]
- 32.Shimizu N, Itoh N, Utiyana H, Wahl GM. Selective entrapment of extrachromosomally amplified DNA by nuclear budding and micronucleation during S phase. J Cell Biol. 1998;140:1307–1320. doi: 10.1083/jcb.140.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shimizu N, Shimuara T, Tanaka T. Selective elimination of acentric double minutes from cancer cells through the extrusion of MNi. Mutat Res. 2000;448:81–90. doi: 10.1016/s0027-5107(00)00003-8. [DOI] [PubMed] [Google Scholar]