

Abstract
The 6,7-indolyne shows remarkable regioselectivity in its cycloaddition with 2-substituted furans. Electron-donating groups give predominantly the more sterically crowded product, while electron-withdrawing groups display the opposite regioselectivity. By contrast, 4,5- and 5,6-indolynes show no regioselectivity. Optimized electronic structure calculations using the M06-2X density functional and 6-311+G(2df,p) basis set revealed that the 6,7-indolynes are highly polar structures and that their cycloadditions have substantial electrophilic substitution character that leads to the observed preference for contrasteric products.
Benzynes and other common arynes (e.g., naphthalynes and pyridynes) have fascinated chemists for decades. Surprisingly, arynes occurring at all three benzenoid positions of the ubiquitous indole nucleus (indole arynes or indolynes 1–3, Figure 1) were, until recently, unknown. These newest members of the aryne family represent, inter alia, potentially attractive and powerful new tools for the total synthesis of complex natural products such as the nodulisporic acids,1 trikentrins,2 herbindoles,2a,c and the teleocidins.3 (Figure 2). We recently discovered the first successful and general route to these reactive intermediates via metal–halogen exchange of o-dihalides4 and later via fluoride-induced decomposition of o-silyltriflates.5 We subsequently examined the regioselectivity of indolynes in Diels–Alder reactions with 2-substituted furans.5a These studies quickly led to the first application of indolynes in the construction of natural products and culminated in a concise and efficient total synthesis of (±)-cis-trikentrin A and the (±)-herbindoles A and B.6 This work and recently that of Garg clearly validated the utility of indolynes to gain quick access to architecturally challenging structures.7
Figure 1.
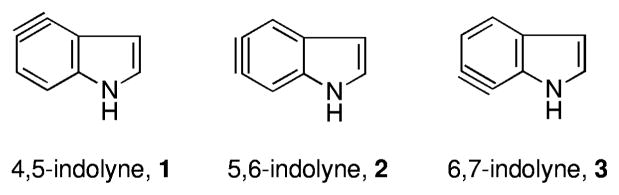
Benzenoid indole aryne (indolyne) systems.
Figure 2.

Examples of complex indole natural products.
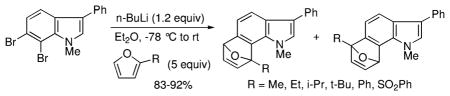
During the course of these investigations, we were surprised to find that 2-substituted furans in particular displayed a striking regioselectivity when paired with the 6,7-indolynes (Scheme 1).5a
Scheme 1.
Regioselective 6,7-Indolyne Cycloadditions with 2-Substituted Furans
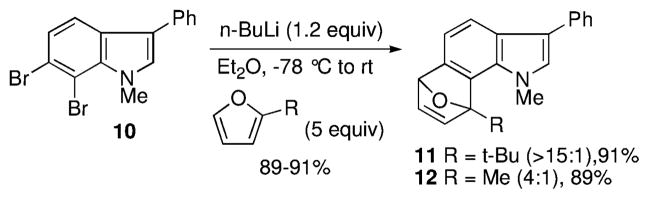
Using 2-t-butylfuran, for example, the contrasteric product 11 was formed in at least a 15:1 preference over the alternate regioisomer. Even with the less sterically demanding 2-methylfuran, there was a 4:1 preference for the indicated isomer. Analysis of the products by various 2D NMR methods (COSY, NOESY, HMBC, and HSQC) allowed for the unequivocal assignment of the structures.
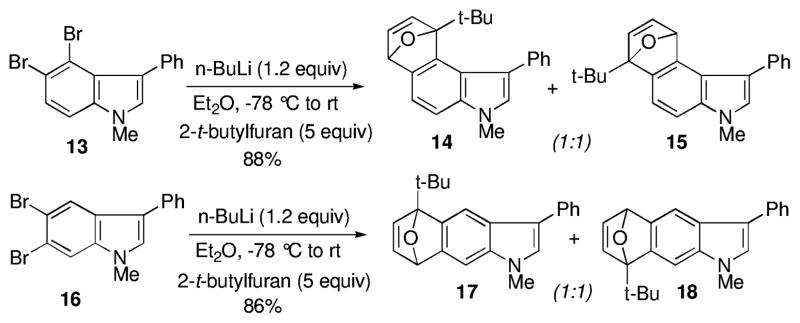
By contrast, there was no preference for either regioisomer in the 4,5- and 5,6-indolyne cycloaddition examples involving 2-t-butylfuran. In each case, a nearly 1:1 ratio of cycloadducts was formed in excellent yield (Scheme 2).5a
Scheme 2.
Furan and Azide Cycloadditions with 4,5-, and 5,6-Indolynes
Intrigued by these observations, we wished to identify the fundamental electronic and steric parameters that account for these fascinating reaction profiles. The information obtained from this study will provide a better understanding of the structure and reactivity of the indole aryne system. This knowledge in turn should result in more versatile strategies and benefits for complex molecule total synthesis.
We began our investigation by examining a series of 2-substituted furans featuring electron-donating straight chain alkyl, branched alkyl, and aryl groups and electron-withdrawing groups (Table 1).8 The electron-donating groups (entries 1–5) showed a remarkable preference for the more crowded cycloadduct 19, while the opposite regiochemistry was found for an electron-withdrawing case (entry 6).
Table 1.
Regioselective 6,7-Indolyne Cycloadditions with 2-Substituted Furans
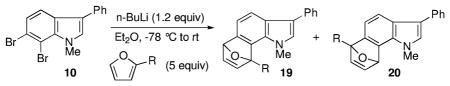
| ||||
|---|---|---|---|---|
| entry | furan, R | 19 | 20 | yield, % |
| 1 | Me | 80 | 20 | 89 |
| 2 | Et | 84 | 16 | 90 |
| 3 | i-Pr | 94 | 6 | 88 |
| 4 | t-Bu | 98 | 2 | 91 |
| 5 | Ph | >99 | <1 | 92 |
| 6 | SO2Ph | <1 | >99 | 83 |
In earlier studies involving the reaction of monosubstituted 3-benzynes with 2-substituted furans (Me, Et, i-Pr, t-Bu),9 regioselectivity correlated well with the strength of inductively electron-withdrawing groups such as F and OMe on the benzyne to favor contrasteric products, while it was reversed with inductively electron-donating groups such as Me.
To characterize this phenomenon in additional detail, electronic structure calculations were undertaken to predict the structures and reactivities of the N-methyl-4,5-, 5,6-, and 6,7-indolynes with furan and 2-alkylfurans. Similar calculations were also undertaken for the benzofuran and benzothiophene analogues of the indolynes as these heterocycles may prove useful for the construction of other interesting natural (or unnatural) products. With respect to methodology, all structures were optimized using the M06-2X density functional10 and 6-311+G(2df,p) basis set11 as implemented in MN-GFM,12 a locally modified version of the Gaussian03 software package.13 Analytical frequency calculations were employed to characterize the nature of all gas-phase structures as minima or transition states. In select instances, the effects of diethyl ether solvation were taken into account using the SMD14 implicit solvation model.
Table 2 lists the activation free energies computed for the reaction of the N-methylindolynes with the furans. With unsubstituted furan, ΔG‡ is similar for the 4,5- and 5,6-indolyne isomers, and slightly smaller for the 6,7-isomer. Considering 2-t-butylfuran, the predicted free energies of activation are reduced by 3–4 kcal/mol compared to reaction with furan itself in the 4,5- and 5,6-indolyne cases. Regioselection becomes possible upon 2-substitution of the furan, and the differential free energies of activation in these two instances are predicted to be 1 kcal/mol or less. In the 6,7-indolyne case, by contrast, a free energy of activation similar to those for 4,5 and 5,6 is predicted for the formation of 26a (which is equivalent to 19 but lacks the 3-phenyl substituent), but no transition-state (TS) structure for the formation of 26b (corresponding to desphenyl 20) could be located—the approach of the furan to the indolyne led smoothly and without barrier to the final tetracyclic product in every instance.
Table 2.
ΔG‡ Values in kcal/mol for the Reaction of Indolynes with Substituted Furans
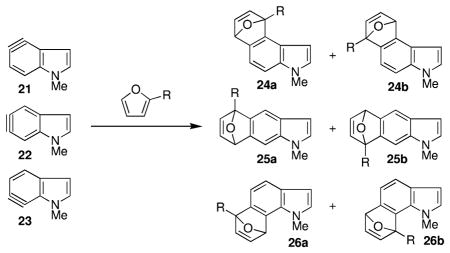
| ||||
|---|---|---|---|---|
| entry | aryne | furan, R | ΔG‡ (a) | ΔG‡ (b) |
| 1 | 21 | H | 10.5 | |
| 2 | 22 | H | 10.0 | |
| 3 | 23 | H | 8.7 (8.4)a | |
| 4 | 21 | t-Bu | 6.3 | 7.3 |
| 5 | 22 | t-Bu | 6.9 | 6.1 |
| 6 | 23 | t-Bu | 7.0 (8.0) | b |
| 7 | 23 | Me | 9.2 (9.4) | 7.5 (7.8) |
| 8 | 23 | Et | 9.0 (9.4) | 7.8 (8.2) |
| 9 | 23 | i-Pr | 9.7 (10.3) | 7.6 (8.2) |
Values in parentheses include continuum ethereal solvation effects.
No barrier to reaction is predicted in the gas phase or continuum solution.
To better understand this point, the reactions of other 2-alkylfurans with 23 were modeled. With R = Me, Et, or i-Pr, TS structures for formation of 26b could be found in every instance. Interestingly, the differential free energies of activation for formation of 26a vs 26b favored formation of the latter in each case, with the margin being largest (2.1 kcal/mol) for R = i-Pr. Ethereal solvation effects on the free energies of activation are predicted from the SMD continuum model to be 1 kcal/mol or smaller in every instance and to have no influence on regioselection.
Inspection of the TS structures for the formation of 26b with the smaller alkyl groups provides insight into the failure to find such a TS structure for t-Bu. There are significant steric interactions between the N-Me group of the indolyne and the 2-alkyl group on the furan so that the bond forming to the 6 position is substantially shorter than that to the 7 position. The difference in the two formation bond lengths becomes increasingly long as the substituent is varied from Me to Et to i-Pr. This increase, however, is probably not associated with larger steric bulk since each of these furan 2-alkyl substituents orients a C–H bond toward the N-methyl group to minimize steric clash. Instead, it becomes clear from consideration of the preference for 26b over 26a and inspection of partial atomic charges that the 6,7-“cycloaddition” has substantial electrophilic substitution character.
Thus, the electron-poor indolyne attacks the 2-substituted furan to generate the more stable 2-alkyldihydrofurylcarbe-nium ion. As the 2-substituent is varied from Me to Et to i-Pr, the increased stabilization provided by the larger alkyl groups increases this character and leads to increased regioselection and increased electrophilic substitution character so that bond formation becomes decreasingly synchronous. Thus, it appears that in the case of R = t-Bu the combination of unavoidably increased sterics (there is no longer a C–H bond that can be disposed toward the N-Me group) and enhanced carbenium ion stabilization switches the mechanisms for a highly asynchronous but still concerted cycloaddition to a “stepwise” electrophilic substitution and subsequent ring closure. We place “stepwise” in quotes here because no TS structures nor intermediates can be located in either the gas phase or ethereal continuum solution for this process. To the extent that the reaction has an actual free energy of activation, it is likely associated with the displacement of discrete solvent molecules between the reacting partners that is not included in the present computational models, which otherwise are in reasonable quantitative agreement for predicted regioselection in 26a and 26b compared to 19 and 20.
Support for the polarization of the 6,7-indolyne that makes the 6 position substantially more electrophilic than the 7 position may be obtained from inspection of atomic polar tensor partial charges15 and the molecular geometry. Thus, C(6) is predicted to have a charge 0.26 au more positive than C(7), and the C(5)C(6)C(7) bond angle is predicted at the M06-2X level to be 135.3° while the C(6)C(7)C(7a) angle is predicted to be 117.2°. The former value is more consistent with carbocationic character, while the latter is more consistent with carbanionic character. For comparison, the corresponding bond angles in the analogous indyne (i.e., the hydrocarbon generated by replacing the N-methyl group with a methylene) are predicted at the DFT level to be 128.2 and 125.5°.16 Thus, the C(6)–C(7) bond is strongly polarized in the expected direction by the nearby C(7a)–N bond dipole.
At the M06-2X level, the C(7a)–N bond dipole is predicted to lead to some polarization of the formal triple bonds in the 4,5- and 5,6-indolynes as well, although to a substantially smaller degree than in the 6,7 case (cf. entries 4 and 5 in Table 2). The failure to observe any regioselection under experimental conditions (reactions of Scheme 2) may reflect reduced polarization associated with the 3-phenyl group in the experimental substrate, or some degree of solvent leveling of the gas-phase predictions, or simply overpolarization predicted at the DFT level, but the effect in any case is predicted to be only about 1 kcal/mol.
Given the substantial synthetic utility of the indolyne cycloadditions, we modeled the analogous cycloadditions of furan and 2-t-butylfuran with benzofuran and benzothiophene; results are presented in Table 3. Very similar reactivity profiles are predicted for all three heterocyclic systems, with the benzofuran case being predicted to have the lowest free energies of activation by a small margin, as would be expected given the greater electrophilicity of this heterocycle. This suggests that synthetic efforts based on these other heterocycles should be equally fruitful.
Table 3.
ΔG‡ Values in kcal/mol for the Reaction of Benzofurans and -Thiophenes with Substituted Furans
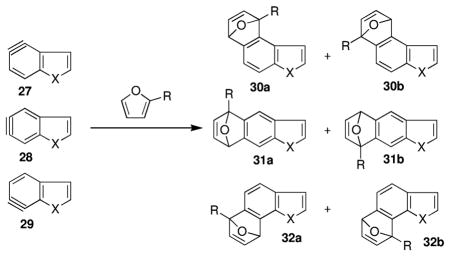
| ||||
|---|---|---|---|---|
| entry | aryne, X | furan, R | ΔG‡ (a) | ΔG‡ (b) |
| 1 | 27, O | H | 9.2 | |
| 2 | 28, O | H | 9.1 | |
| 3 | 29, O | H | 7.3 | |
| 4 | 27, O | t-Bu | 4.9 | 6.4 |
| 5 | 28, O | t-Bu | 5.8 | 4.9 |
| 6 | 29, O | t-Bu | 6.2 | a |
| 7 | 27, S | H | 9.6 | |
| 8 | 28, S | H | 8.9 | |
| 9 | 29, S | H | 8.4 | |
| 10 | 27, S | t-Bu | 5.4 | 6.5 |
| 11 | 28, S | t-Bu | 5.5 | 4.8 |
| 12 | 29, S | t-Bu | 5.8 | a |
No barrier to reaction is predicted in the gas phase or continuum solution.
Benzynes derived from benzannulated heterocycles are thus revealed to be powerful synthetic tools for the construction of highly functionalized tetracycles through [4 + 2] cycloaddition reactions. Dipolar effects can induce regioselection with asymmetrically substituted dienes when polarization of the benzyne triple bond imparts substantial asynchronous electrophilic substitution character to the initial bond forming step.
Supplementary Material
Acknowledgments
We acknowledge support of this work by the National Institutes of Health, NIGMS, Grant R01 GM069711 (KRB), and the National Science Foundation, CHE06-10183 (CJC). Additional support of this work was provided by the NIH, NIGMS, Grant P50 GM069663 via the University of Kansas Chemical Methodologies and Library Development Center of Excellence (KU-CMLD). We thank Dee Tanaka for synthetic technical assistance and Nalin Chandrasoma and Lincoln Maina for additional NMR acquisition.
Footnotes
Supporting Information Available: NMR and other spectroscopic data for compounds used in this study and experimental details for their preparation as well as optimized cartesian coordinates for all computed structures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Synthetic studies, see: Smith AB, III, Kuerti L, Davulcu AH, Cho YS, Ohmoto K. J Org Chem. 2007;72:4611–4620. doi: 10.1021/jo062423a.
- 2.(a) Jackson SK, Kerr MA. J Org Chem. 2007;72:1405–1411. doi: 10.1021/jo062350v. [DOI] [PubMed] [Google Scholar]; (b) Huntley RJ, Funk RL. Org Lett. 2006;8:3403–3406. doi: 10.1021/ol061259a. [DOI] [PubMed] [Google Scholar]; (c) Jackson SK, Banfield SC, Kerr MA. Org Lett. 2005;7:1215–1218. doi: 10.1021/ol047498k. [DOI] [PubMed] [Google Scholar]
- 3.(a) Dangel BD, Godula K, Youn SW, Sezen B, Sames D. J Am Chem Soc. 2002;124:11856–11857. doi: 10.1021/ja027311p. [DOI] [PubMed] [Google Scholar]; (b) Nakatsuka S, Matsuda T, Goto T. Tetrahedron Lett. 1987;38:3671–3674. [Google Scholar]
- 4.Buszek KR, Luo D, Kondrashov M, Brown N, VanderVelde D. Org Lett. 2007;9:4135–4137. doi: 10.1021/ol701595n. [DOI] [PubMed] [Google Scholar]
- 5.Brown N, Luo D, VanderVelde D, Yang S, Brassfield A, Buszek KR. Tetrahedron Lett. 2009;50:63–65. doi: 10.1016/j.tetlet.2008.10.086.Another o-silyltriflate method for 4,5-indolyne formation and further cycloaddition reactions have subsequently been reported by Garg: Bronner SM, Bahnck KB, Garg NK. Org Lett. 2009;11:1007–1010. doi: 10.1021/ol802958a.
- 6.Buszek KR, Brown N, Luo D. Org Lett. 2009;11:201–204. doi: 10.1021/ol802425m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tian X, Huters AD, Douglas CJ, Garg NK. Org Lett. 2009;11:2349–2351. doi: 10.1021/ol9007684. [DOI] [PubMed] [Google Scholar]
- 8.2-Methyl-, 2-ethyl-, and 2-t-butylfuran are readily available from commercial sources. The others were prepared as follows. (a) 2-i-Propylfuran: see ref 8c. 2-Phenylfuran: Negishi E, Takahashi T, King AO. Org Synth. 1988;66:67–72.2-Phenylsulfonylfuran: Malanga C, Aronica LA, Lardicci L. Synth Commun. 1996;26:2317–2327.
- 9.(a) Giles RGF, Sargent MV, Sianipar H. J Chem Soc, PT1. 1991:1571–1579. [Google Scholar]; (b) Newman MS, Kannan R. J Org Chem. 1976;41:3356–3359. [Google Scholar]; (c) Gribble GW, Keavy DJ, Branz SE, Kelly WJ, Pals MA. Tetrahedron Lett. 1988;29:6227–6230. [Google Scholar]
- 10.Zhao Y, Truhlar DG. Theor Chem Acc. 2008;120:215–241. [Google Scholar]
- 11.Hehre WJ, Radom L, Schleyer PvR, Pople JA. Ab Initio Molecular Orbital Theory. Wiley; New York: 1986. [Google Scholar]
- 12.Zhao Y, Truhlar DG. MN-GFM version 4.1. University of Minnesota; Minneapolis: 2008. [Google Scholar]
- 13.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, revision D.01. Gaussian, Inc; Wallingford, CT: 2004. [Google Scholar]
- 14.(a) Marenich AV, Cramer CJ, Truhlar DG. J Phys Chem B. 2009;113:6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]; (b) Marenich AV, Hawkins GD, Liotard DA, Cramer CJ, Truhlar DG. GESOL-version 2008. University of Minnesota; Minneapolis: 2008. [Google Scholar]
- 15.Cioslowski J. J Am Chem Soc. 1989;111:8333. [Google Scholar]
- 16.Cramer CJ, Thompson J. J Phys Chem A. 2001;105:2091–2098. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.