

Abstract
Cisplatin is a cytostatic agent used in the treatment of many types of cancer, but its use is associated with increased incidences of secondary leukemia. We evaluated cisplatin's in vivo genotoxic potential by analyzing peripheral blood for Pig-a mutant phenotype erythrocytes and for chromosomal damage in the form of micronuclei. Mutant phenotype reticuloyte and erythrocyte frequencies, based on anti-CD59 antibody labeling and flow cytometric analysis, were determined in male Sprague Dawley rats treated for 28 consecutive days (days 1–28) with up to 0.4 mg cisplatin/kg/day, and sampled on days −4, 15, 29, and 56. Vehicle and highest dose groups were evaluated at additional time points post-treatment up to 6 months. Day 4 and 29 blood samples were also analyzed for micronucleated reticulocyte frequency using flow cytometry and anti-CD71-based labeling. Mutant phenotype reticulocytes were significantly elevated at doses ≥0.1 mg/kg/day, and mutant phenotype erythrocytes were elevated at doses ≥0.05 mg/kg/day. In the 0.4 mg/kg/day group, these effects persisted for the 6 month observation period. Cisplatin also induced a modest but statistically significant increase in micronucleus frequency at the highest dose tested. The prolonged persistence in the production of mutant erythrocytes following cisplatin exposure suggests that this drug mutates hematopoietic stem cells and that this damage may ultimately contribute to the increased incidence of secondary leukemias seen in patients cured of primary malignancies with platinum-based regimens.
Keywords: cisplatin, Pig-a gene, mutation, flow cytometry, micronuclei, genotoxicity, stem cells
Cisplatin and other platinum drugs are cytostatic agents used widely in the treatment of malignancies, including testicular, ovarian, lung, and breast cancer (MedLine Plus, 2014). The use of platinum-based drugs in the treatment of testicular cancer is an example of a remarkably effective therapy, as the current 5-year survival rate is greater than 95% (Travis et al., 2000, 2010; Verdecchia et al., 2007). However, secondary effects of platinum therapy include cardiovascular disease, neurotoxicity, nephrotoxicity, pulmonary toxicity, and secondary malignant neoplasms (Travis et al., 2010). Given the relatively young age of testicular cancer patients and the high cure rate of this disease, it is important to understand cisplatin's deleterious side effects, including those that take years to manifest.
Given our laboratory's focus on biomarkers related to DNA damage and carcinogenesis, we were interested in cisplatin's association with the development of secondary leukemia, an outcome that occurs in excess following treatment for testicular and ovarian cancers (Howard et al., 2008; Kaldor et al., 1990; Pedersen-Bjergaard et al., 1991; Travis et al., 1996, 1997). In their analysis of 18,567 testicular cancer patients, Travis et al. (2000) estimated that 650 mg cisplatin/m2 cumulative exposure carries an increased relative risk of 3.2-fold, with larger doses (1000 mg/m2) further increasing the risk to 6-fold. While the dose-related association between cisplatin and leukemia is strong, it must be qualified. As described by the National Toxicology Program's Report on Carcinogens (2011) and a review by the International Agency for Research on Cancer (1981), cisplatin is used in combination with other potentially carcinogenic agents that include, but are not limited to, radiation, doxorubicin, and etoposide. The lack of epidemiological studies for cisplatin as a monotherapy has resulted in a class 2A IARC designation; that is, probably carcinogenic to humans.
There is considerable evidence in animals for the carcinogenicity of cisplatin (IARC, 1981, 1987). Cisplatin is a metallic coordination agent that forms DNA adducts, causing DNA-intrastrand and interstrand cross-links (Fichtinger-Schepman et al., 1985). Genotoxic activity is observed in a variety of pre-clinical models, including bacteria, mammalian cells cultured in vitro, and rodents. These effects include gene mutation and also cytogenetic damage in the form of double-strand breaks (Bhalli et al., 2013; Brower et al., 1981; de Boer et al., 1989; Louro et al., 2002; MacGregor et al., 2006). Leukemia has been observed in rats following repeated intraperitoneal injections of cisplatin (IARC, 1981, 1987), and tissues including liver, lung, and skin have exhibited increased incidence of benign or malignant tumors in a variety of mouse and rat models (Diwan et al., 1993, 1995; Satoh et al., 1993; Waalkes et al., 2006).
Given this profile, cisplatin represented an interesting agent to evaluate for in vivo mutagenicity using a relatively new assay based on the X-linked phosphatidylinositol glycan-class A (Pig-a) gene. In this system, reticulocytes and erythrocytes that lack cell surface CD59 (RETCD59- and RBCCD59-) serve as phenotypic reporters of Pig-a gene mutation, and their frequency is determined via flow cytometric analysis. To date, rodent studies have mainly focused on circulating erythrocytes, as these cells are easily obtained in sufficient quantity with low volume blood draws (Dobrovolsky et al., 2010). In a study with male F344 rats, Bhalli and colleagues reported that cumulative doses of 3 and 6 mg cisplatin/kg given over 3 days significantly induced RETCD59- and RBCCD59- (Bhalli et al., 2013). This group also reported that cisplatin-exposed cancer patients (in combination with etoposide or gemcitabine) exhibited elevated frequencies of mutant phenotype erythrocytes (Dobrovolsky et al., 2011).
We undertook the work described herein to extend these studies, with an emphasis on (1) studying rats treated with cisplatin using a highly fractionated treatment schedule, a scenario that more closely mimics both typical cancer patient exposures and pivotal repeat-dose general toxicity study designs, (2) evaluating the dose-response relationship over a wider range of exposure levels, and (3) determining the kinetics of appearance and disappearance of cisplatin-induced RETCD59- and RBCCD59- over a relatively long timeframe. We considered the third objective to be particularly important, as we hypothesize that short-term (weeks) and long-term (4–6 month) persistence of mutant phenotype cells is indicative of mutations occurring in lineage-specific progenitors and long-term hematopoietic stem cells (HSCs), respectively. The results of these experiments are discussed in terms of the utility of the Pig-a assay to evaluate drugs for leukemogenic potential.
MATERIALS AND METHODS
Reagents
Cisplatin (CAS no. 15663-27-1) was purchased from R&D Systems, Minneapolis, MN. Reagents used for flow cytometric micronucleus scoring (e.g., Propidium Iodide Solution, Anti-CD71-FITC and Anti-CD61-PE Solutions, RNase, and Malaria Biostandards) were from In Vivo Rat MicroFlow Kits, Litron Laboratories, Rochester, NY. Reagents used for flow cytometric RBCCD59- and RETCD59- scoring (e.g., Nucleic Acid Dye Solution, Anti-CD59-PE, and Anti-CD61-PE) were from Rat MutaFlow Kits, Litron Laboratories. Additional supplies included Lympholyte-Mammal cell separation reagent from CedarLane, Burlington, NC; Anti-PE MicroBeads, LS Columns, and a QuadroMACS Separator from Miltenyi Biotec, Bergisch Gladbach, Germany; and CountBright Absolute Count Beads and fetal bovine serum from Invitrogen, Carlsbad, CA.
Animals, treatments, blood harvests
Experiments were conducted with the approval of the University of Rochester's Institutional Animal Care and Use Committee. Male Sprague Dawley rats were purchased from Charles River Laboratories, Wilmington, MA. Rodents were allowed to acclimate for approximately one week, and their age at the start of treatment was 7 weeks. Water and food were available ad libitum throughout the acclimation and experimental periods. Cisplatin was prepared in 0.9% saline solution. For each of 28 consecutive days of administration, treatment occurred by intraperitoneal injection at 5 ml/kg body weight/day for final dose levels of 0, 0.05, 0.1, 0.2, and 0.4 mg cisplatin/kg/day (n = 6/group). The 28 day treatment schedule was recently recommended by the IWGT Pig-a Workgroup (manuscript in preparation), and as noted above, relative to acute studies, it better approximates, but does not exactly correspond to, schedules that cancer therapy patients experience when undergoing platinum-based therapies (Platinol, U.S. FDA-approved drug label).
Start and end of treatment were designated days 1 and 28, respectively. Blood samples were collected for micronucleus analyses on days 4 and 29. Blood samples were collected for Pig-a mutant phenotype analyses before treatment (day −4), and again on days 15, 29, 56, 84, 112, 139, 168, and 196.
Peripheral blood was obtained by nicking a lateral tail vein with a surgical blade after animals were warmed briefly under a heat lamp. Approximately 200 μl of free-flowing blood was collected directly into heparinized capillary tubes (Fisher Scientific, cat. no. 22-260-950). For Pig-a analyses, 80 μl of each blood sample was transferred to tubes containing 100 μl kit-supplied heparin solution where it remained at room temperature for less than 2 h until leukodepletion as described previously (Dertinger et al., 2012). For the micronucleated reticulocyte (MN-RET) endpoint, 30 μl of each whole blood sample was transferred to tubes containing 100 μl kit-supplied heparin solution where it remained at room temperature for less than 2 h, after which it was fixed with ultracold methanol (Torous et al., 2003).
As described previously, pre-treatment Pig-a mutant phenotype cell frequencies were used as study inclusion criteria (Dertinger et al., 2012). Pre-treatment frequencies were compared to the distribution of 586 historical control animals, and rats that exhibited RBCCD59- > 4.2 × 10−6 and/or RETCD59− > 5.1 × 10−6 were considered outliers and excluded from the study (i.e., 1 out of 34 rats was excluded based on these criteria). Animals that did not exceed these threshold values were randomized across treatment groups.
Micronucleated reticulocytes: Sample preparation, data acquisition
MN-RET and reticulocyte (RET) frequencies were scored via flow cytometry according to the In Vivo Rat MicroFlow Kit manual and described in detail elsewhere (Dertinger et al., 2004; Torous et al., 2003). The frequency of MN-RET was based on the acquisition of approximately 20,000 CD71-positive RET/blood sample. Instrument setup and calibration was performed using kit-supplied biological standards (Plasmodiumberghei-infected blood cells; Tometsko et al., 1993). A BD FACSCalibur flow cytometer running CellQuest Pro v5.2 software was used for data acquisition and analysis.
Pig-a mutation: Sample preparation, data acquisition
As described previously, determinations of RETCD59- and RBCCD59- frequencies employed immunomagnetic depletion of wild-type erythrocytes prior to flow cytometric analysis (Dertinger et al., 2011a,b, 2012). One variation on the published labeling and analysis method was used for the current studies, and involved the use of deep-well 96 well plates (Axygen Scientific, cat. no. P-DW-20-C) for sample processing. Flow cytometric analyses were also conducted using 96 well plates (U-bottom, Corning, cat. no. 3799) in conjunction with a BD High Throughput Sampler (HTS).
An Instrument Calibration Standard was generated on each day of data acquisition. These samples contained approximately 50% wild-type and 50% mutant-mimic erythrocytes and, as described previously, provided a means to rationally and consistently define the location of CD59-negative cells (Phonethepswath et al., 2010). A BD FACSCanto II flow cytometer running Diva v6.1.2 software was used for data acquisition and analysis.
Calculations, statistical analyses
The incidences of RET and MN-RET are expressed as frequency percent. The formulas used to calculate RBCCD59- and RETCD59- frequencies based on data from pre- and post-immunomagnetic separation specimens have been described previously (Dertinger et al., 2012). RBCCD59- and RETCD59- frequencies are expressed as number per one million total RBC or RET, respectively. All %MN-RET, %RET, mutant cell frequencies, averages, and standard error calculations were performed with Excel Office X for Mac (Microsoft, Seattle, WA).
For statistical evaluations, proportions of MN-RET among RETs were transformed in Excel as follows: transformed data = asin(sqrt(proportion of MN-RET)). RBCCD59- and RETCD59- frequencies were log(10) transformed (note that since zero RETCD59- readings were occasionally observed, a 0.1 offset was added to each RETCD59− frequency prior to log transformation). Each time point was studied separately, where the effect of treatment on these transformed MN-RET, RBCCD59-, and RETCD59- data was compared to concurrent vehicle control using Dunnett's multiple comparison t-tests in the context of a one-way analysis of variance (ANOVA) model (JMP, v8.0.1, SAS Institute Inc., Cary, NC). Significance was evaluated at the 5% level using a one-tailed test for increases relative to vehicle control. These same analyses were performed for untransformed RET frequencies, however, in these cases the tests were two-tailed.
As recommended by the 2013 IWGT Pig-a working group (manuscript in preparation), statistical significance was not the sole determinant of a positive Pig-a result. For a statistically significant increase in Pig-a mutant phenotype cells to be considered biologically relevant, mean RBCCD59- and mean RETCD59- frequencies also needed to exceed a lab-specific historical negative control distribution (Dertinger et al., 2014).
RESULTS
While cisplatin was well tolerated over the dose levels studied, it was observed to affect weight gain (Fig. 1). Also, the two highest dose levels caused reductions to %RET at early time point(s)—day 15 and 29 (Fig. 2). Later, at days 56 and 139, mean %RET for the 0.4 mg/kg/day treatment group were significantly elevated over vehicle control, suggestive of stimulated erythropoiesis (Fig. 2).
FIG. 1.

Mean body weight for each of five cisplatin treatment groups. Weights were determined each day dosing occurred.
FIG. 2.
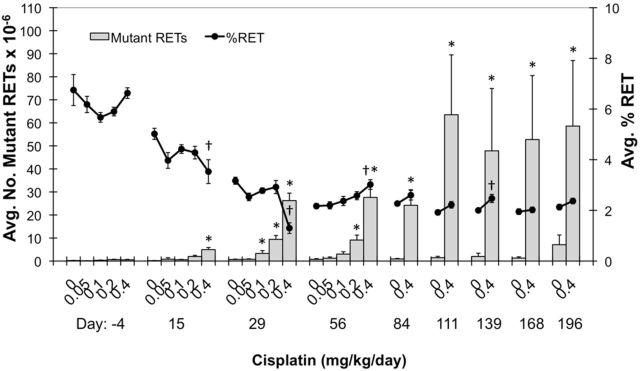
Mean mutant phenotype reticulocyte (RETCD59-) frequencies are graphed for each of nine time points (y-axis). Percent reticulocytes (%RET) are also graphed on the yy-axis (black line). Each treatment group consisted of six rats, and all error bars are SEM. Asterisks associated with mean RETCD59− values indicate significance compared to same-day vehicle control values (based on a pair-wise test, p < 0.05; mean also needed to exceed historical control 90% tolerance interval, upper limit, alpha 0.1). Daggers indicate a significant difference in mean %RET compared to same-day vehicle control values (i.e., two-sided Dunnett's t-test, p < 0.05).
Cisplatin induced significant increases in RETCD59- frequency as early as day 15 (Fig. 2). RETCD59- frequencies continued to rise, generally stabilizing over days 29 through 84. At day 84, high dose mean RETCD59- frequencies (24.2 × 10−6) were 26-fold greater than mean vehicle control (0.92 × 10−6). From day 111 and beyond, markedly more variation in the cisplatin-induced RETCD59- values was observed. Higher variability was also noted for the vehicle control group at the last blood sampling time. Figure 3 shows individual rat RETCD59- frequencies for the vehicle control and 0.4 mg/kg/day groups over the entire 6 month study period.
FIG. 3.
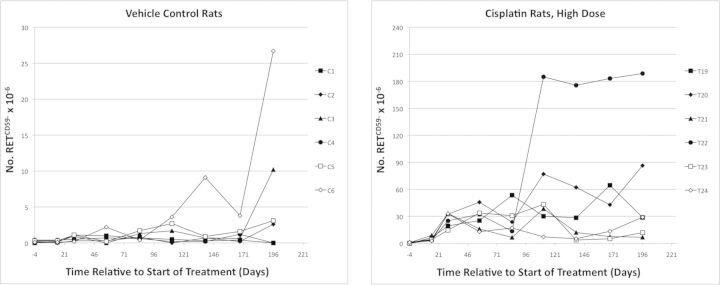
Mutant phenotype reticulocyte (RETCD59-) frequencies are graphed over time for each vehicle control rat (left panel), and each individual high dose rat (0.4 mg cisplatin/kg/day). Note that different scales were used for the y-axis.
As expected, the lagging indicator of mutation, RBCCD59- frequency, increased more slowly than RETCD59−. The first significantly elevated frequencies were evident at day 29 (Fig. 4). As with RETCD59-, RBCCD59- frequencies were relatively stable through day 84. At day 84, high dose mean RBCCD59- frequencies (29 × 10−6) were 51-fold greater than mean vehicle control frequencies (0.57 × 10−6). Greater variability became evident as time progressed. Figure 5 illustrates individual rat RBCCD59- frequencies for the vehicle control and 0.4 mg/kg/day groups over the entire 6 month study period.
FIG. 4.
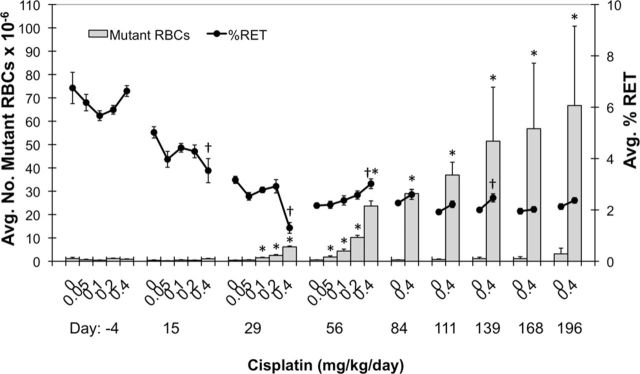
Mean mutant phenotype erythrocyte (RBCCD59-) frequencies are graphed for each of nine time points (y-axis). Percent reticulocytes (%RET) are also graphed on the yy-axis (black line). Each treatment group consisted of six rats, and all error bars are SEM. Asterisks associated with mean RBCCD59- values indicate significance compared to same-day vehicle control values (based on a one-sided pair-wise test, p < 0.05; mean also needed to exceed historical control 90% tolerance interval, upper limit, alpha 0.1). Daggers indicate a significant difference in mean %RET compared to same-day vehicle control values (i.e., based on a two-sided pair-wise test, p < 0.05).
FIG. 5.
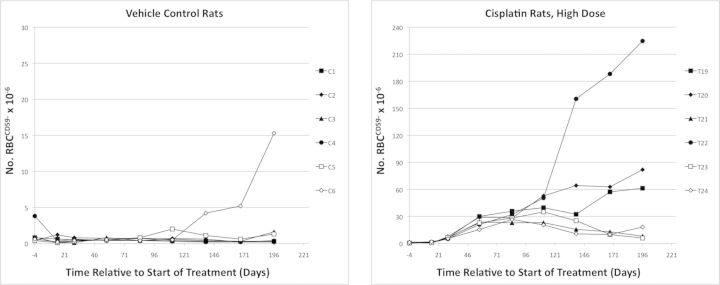
Mutant phenotype erythrocyte (RBCCD59-) frequencies are graphed over time for each individual vehicle control rat (left panel) and each individual high dose rat (0.4 mg cisplatin/kg/day). Note that different scales were used for the y-axis.
Cisplatin was also observed to induce %MN-RET, an endpoint indicative of cytogenetic damage to erythroid cells (Fig. 6). The only significant responses were associated with the highest dose level at the day 4 and 29 time points. The maximal mean MN-RET frequency caused by cisplatin was observed on day 29, and the 0.28% value corresponds to a 2.8-fold increase over the concurrent vehicle control.
FIG. 6.
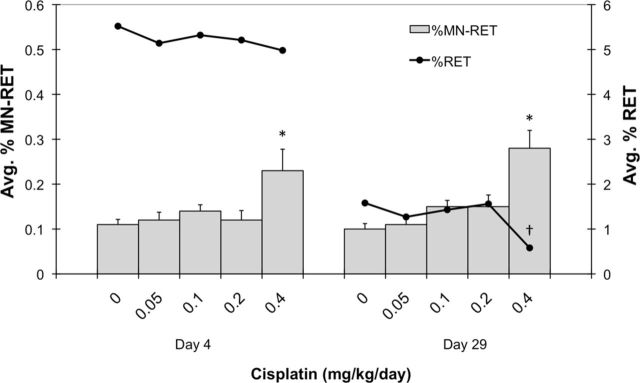
Mean micronucleated reticulocyte (MN-RET) frequencies are graphed for each of two time points (y-axis). Percent reticulocytes (%RET) are also graphed on the yy-axis (black line). Each treatment group consisted of six rats, and all error bars are SEM. Asterisks associated with mean MN-RET values indicate significance compared to same-day vehicle control values (based on a one-sided pair-wise test, p < 0.05). Daggers indicate a significant difference in mean %RET compared to same-day vehicle control values (i.e., based on a two-sided pair-wise test, p < 0.05).
DISCUSSION
Significant induction of the RBCCD59- frequency was observed for each of the dose levels of cisplatin tested. While mean RETCD59− frequencies were slightly elevated at the lowest dose studied (0.05 mg/kg/day, or 1.4 mg/kg cumulative), these results did not attain statistical significance. The higher sensitivity for detecting modest changes to baseline frequency in RBC compared to RET populations is not unexpected, and has been observed for other agents studied by this and other laboratories (Dertinger et al., 2014). We attribute this to the greater numbers of erythrocytes per specimen that are evaluated for the mutant phenotype compared to the immature subpopulation. For instance, in the present study, we were able to consistently analyze more than 150 × 106 erythrocytes for the rare RBCCD59- phenotype, whereas many fewer reticulocytes were evaluated for RETCD59− (typically on the order of 5 × 106 per specimen). This impacts the power of the assay to detect modest treatment-related changes to baseline values, and explains why we continue to study both cohorts, despite the fact that RBCCD59- take longer to fully manifest compared to the more rapidly responding RETCD59− endpoint (Phonethepswath et al., 2010).
The genotoxicity of cisplatin reported herein is in agreement with previous rodent-based studies. In a mouse study by Louro et al. (2002), a single administration of 6 mg/kg caused a significant increase in mean mutation frequency in liver tissue (lacZ transgene). More recently, Bhalli et al. (2013) studied F344 rats and reported that 3 days of treatment with 3 and 6 mg/kg (cumulative) caused significant Pig-a responses in both erythrocytes and reticulocytes. In the same study, the highest dose level of 6 mg/kg also significantly increased the frequency of 6-thioguanine-resistent lymphocytes, a reporter of Hprt mutation.
In the present experiments, the treatment followed for the longest period of time, 0.4 mg/kg/day, represents a cumulative exposure of 11.2 mg/kg, which is a human equivalent dose of 67.2 mg/m2 (U.S. Food and Drug Administration, Guidance for Industry, 2005). This cumulative dose is below those used in typical cisplatin-based testicular cancer therapies, which generally involve treatment with 20 mg cisplatin/m2/day on five successive days, and a repeat of this cycle on several occasions (Platinol, U.S. FDA-approved drug label).
The kinetics by which RETCD59- and RBCCD59- induced by prototypical mutagens enter the peripheral blood stream has been studied extensively, as manifestation time is a key parameter in the conduct of any phenotype-based genotoxicity assay (Kimoto et al., 2012; Phonethepswath et al., 2010). Less attention has been given to the rate and degree to which Pig-a mutant phenotype cell frequencies change over protracted periods of time. Phonethepswath et al. studied male rats for 3 months following acute exposures to N-ethyl-N-nitrourea (ENU), N-methyl-N-nitrourea, 7,12-dimethylbenz[a]anthracene, 4-nitroquinoline 1-oxide, and benzo[a]pyrene (Phonethepswath et al., 2010). Pig-a responses were generally still evident at 3 months, but in most cases with some reductions relative to maximal values that occurred at earlier time points. A clear exception was ENU, where maximal frequencies were stable for the 3 month observation period. A report by Miura and colleagues (Miura et al., 2009) showed a similar pattern for ENU-treated F344 rats, with essentially maximal mean responses maintained for 6 months post-treatment. These authors noted that at late time points the frequencies observed in the individual ENU-treated rats were more variable, an observation that is consistent with our current study's day 111+ results. Bhalli et al. (2011) also studied Pig-a mutant phenotype responses for 6 months following acute exposure of C57Bl/6 mice to ENU. In the case of this mouse model, responses were maintained throughout the study period, but appreciable reductions to mean mutant cell frequencies were observed.
We designed the current study with a lengthy follow-up period in order to better understand two important questions: (1) Whether the remarkable persistence noted for ENU applies to other agents, including the non-classical alkylator cisplatin, and (2) whether this animal model and resulting Pig-a data support translational work aimed at evaluating cisplatin-based therapies’ leukemogenic potential as a consequence of stem cell mutagenesis. We hypothesized that if cisplatin is playing a key role in secondary leukemogenesis, then Pig-a mutation would not be restricted to erythroid precursors that undergo terminal maturation to erythrocytes. Rather, for cisplatin to be a causative factor, acting at least in part through a mutagenic mode of action, then HSCs should also be targets of treatment-induced mutation.
The 6 month period of time following cessation of treatment was chosen to ensure that elevated mutant frequencies, if observed, were related to mutations in HSC. Given that the lifespan of mouse and rat red blood cells is 45 and 56 days, respectively, rodents replace approximately 2% of their red cell mass every day (Tarbutt, 1967). This prodigious cellular output of reticulocytes is derived from morphologically recognizable erythroid precursors in the marrow that in rats undergo seven cell divisions as they accumulate hemoglobin and ultimately enucleate (Tarbutt, 1967). This precursor compartment is supported by erythroid progenitors, defined by their capacity to form colonies of maturing red cells in semisolid media. The most immature erythroid progenitor, termed BFU-E, requires 7–10 days to fully mature in vitro (Heath et al., 1976; Kasai et al., 1980). Lineage-restricted hematopoietic progenitors are ultimately derived from HSCs, which contain the self-renewal capacity to sustain life-long hematopoiesis. HSC function has been most studied in rodents, particularly mice, where inbred strains facilitate their transplantation. Marking studies indicate that murine HSCs provide stable hematopoietic output within 4–6 months following transplantation (Jordan and Lemischka, 1990) and that individual HSCs have differing lifespans, indicative of self-renewal capacity that varies from months to several years (Sieburg et al., 2011). Thus, the persistence of mutant red cell production, as evidenced by the presence of RETCD59- and RBCCD59- cells 6 months following cessation of cisplatin treatment, suggests that the underlying mutation has occurred in HSCs. Furthermore, a recent analysis of whole genome sequencing of human hematopoietic cells reported by Welch et al. (2012) suggest that HSCs randomly accumulate mutations over time, a finding that may explain elevated frequencies of mutant phenotype cells in vehicle control animals at the last time point studied. Collectively, these data, taken together with our findings, implicate cisplatin-induced mutations in HSCs in the pathogenesis of secondary leukemias following platinum-based therapies.
Although our data support a genotoxic mode of action for secondary leukemias related to the use of cisplatin, the risk/benefit ratio remains decidedly in favor of the continued use of platinum agents due to their remarkable effectiveness in the treatment of primary malignancies, especially testicular cancer. Even so, it is important to investigate ways of maintaining the clinical efficacy of therapeutic regimens while minimizing undesirable side effects. This is especially the case for anti-neoplastic agents when the cure rate is high and when patients are of relatively young ages (Travis et al., 2010). Work in this area is particularly important when classical drugs are combined with new agents in an attempt to magnify their effectiveness. A recent example is the use of Poly (ADP-ribose) polymerase (PARP) inhibitors in combination with platinum and other drugs (Donawho et al., 2007). In these instances it is important to evaluate secondary effects, including late effects such as secondary malignant neoplasms, when new drugs are used to enhance platinum drugs’ efficacy (Kaufmann and Patel, 2010).
In conclusion, the Pig-a erythrocyte-based assay represents an efficient method for evaluating the in vivo mutagenic potential of drugs and other chemicals. In addition, the kinetics of appearance and disappearance of mutant phenotype cells can provide information about the potential cellular targets. The prolonged persistence in the production of mutant erythrocytes following cisplatin exposure supports the hypothesis that this agent targets HSCs. It is this damage that may ultimately contribute to the increased incidence of secondary leukemias seen in patients cured of primary malignancies with platinum-based regimens. The assays employed in these rodent studies have been shown to be applicable to human studies (Abramsson-Zetterberg, 2000; Dertinger et al., 2004; Dobrovolsky et al., 2011; Witt et al., 2007), and it will be important to follow up these rodent findings with direct measurements in patients treated with cisplatin and related agents.
FUNDING
National Institute of Health/National Institute of Environmental Health Sciences (NIEHS; R44ES021973).
Acknowledgments
The authors would like to thank Drs Robert Heflich, Vasily Dobrovolsky, and Javid Bhalli for many helpful suggestions regarding study design and interpretation of results.
Disclosure: S.D.D., S.L.A., D.K.T., J.C.B., S.P., C.L., K.C., J.M., and J.C. are employees of Litron Laboratories, and J.T.M. serves as a consultant to Litron. Litron holds patents covering flow cytometric methods for scoring micronucleated reticulocytes and sells kits based on this technology (In Vivo MicroFlow). Litron holds patents covering flow cytometric methods for scoring GPI anchor-deficient erythrocytes and sells kits based on this technology (In Vivo MutaFlow).
REFERENCES
- Abramsson-Zetterberg L., Zetterberg G., Bergqvist M., Grawé J. Human cytogenetic biomonitoring using flow-cytometric analysis of micronuclei in transferring-positive immature peripheral blood reticulocytes. Environ. Mol. Mutagen. 2000;36:22–31. doi: 10.1002/1098-2280(2000)36:1<22::aid-em4>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Bhalli J. A., Pearce M. G., Dobrovolsky V. N., Heflich R. H. Manifestation and persistence of Pig-a mutant red blood cells in C57Bl/6 mice following single and split doses of N-ethyl-N-nitrosourea. Environ. Mol. Mutagen. 2011;52:766–773. doi: 10.1002/em.20682. [DOI] [PubMed] [Google Scholar]
- Bhalli B. A., Shaddock J. G., Pearce M. G., Dobrovolsky V. N. Sensitivity of the Pig-a assay for detecting gene mutation in rats exposed acutely to strong clastogens. Mutagenesis. 2013;28:447–455. doi: 10.1093/mutage/get022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer J. G., Glickman B. W. Sequence specificity of mutation induced by the anti-tumor drug cisplatin in the CHO aprt gene. Carcinogenesis. 1989;10:1363–1367. doi: 10.1093/carcin/10.8.1363. [DOI] [PubMed] [Google Scholar]
- Brower J., van de Putte P., Fichtinger-Schepman A. M. J., Reedijk J. Base-pair substitution hotspots in GAG and GGG nucleotide sequences in Escherichia coli K-12 induced by cis-diamminedichloroplatinum (II) Proc. Natl. Acad. Sci. U.S.A. 1981;78:7010–7014. doi: 10.1073/pnas.78.11.7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dertinger S. D., Bryce S. M., Phonethepswath S., Avlasevich S. L. When pigs fly: Immunomagnetic separation facilitates rapid determination of Pig-a mutant frequency by flow cytometric analysis. Mutat. Res. 2011a;721:163–170. doi: 10.1016/j.mrgentox.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dertinger S. D., Camphausen K., MacGregor J. T., Bishop M. E., Torous D. T., Avlasevich S., Cairns S., Tometsko C. R., Menard C., Muanza T., et al. Three-color labeling method for flow cytometric measurement of cytogenetic damage in rodent and human blood. Environ. Mol. Mutagen. 2004;44:427–435. doi: 10.1002/em.20075. [DOI] [PubMed] [Google Scholar]
- Dertinger S. D., Phonethepswath S., Avlasevich S. L., Torous D. K., Mereness J., Bryce S. M., Bemis J. C., Bell S., Weller P., MacGregor J. T. Efficient monitoring of in vivo Pig-a gene mutation and chromosomal damage: Summary of 7 published studies and results from 11 new reference compounds. Toxicol. Sci. 2012;130:328–348. doi: 10.1093/toxsci/kfs258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dertinger S. D., Phonethepswath S., Avlasevich S. L., Torous D. K., Mereness J., Cottom J., Bemis J. C., MacGregor J. T. Pig-a gene mutation and micronucleated reticulocyte induction in rats exposed to tumorigenic doses of the leukemogenic agents chlorambucil, thiotepa, melphalan, and 1,3-propane sultone. Environ. Mol. Mutagen. 2014;55:299–308. doi: 10.1002/em.21846. [DOI] [PubMed] [Google Scholar]
- Dertinger S. D., Phonethepswath S., Weller P., Avlasevich S., Torous D. K., Mereness J. A., Bryce S. M., Bemis J. C., Bell S., Portugal S., et al. Interlaboratory Pig-a gene mutation assay trial: Studies of 1,3-propane sultone with immunomagnetic enrichment of mutant erythrocytes. Environ. Mol. Mutagen. 2011b;52:748–755. doi: 10.1002/em.20671. [DOI] [PubMed] [Google Scholar]
- Diwan B. A., Anderson L. M., Rehm S., Rice J. M. Transplacental carcinogenicity of cisplatin: Initiation of skin tumors and induction of other preneoplastic and neoplastic lesions in SENCAR mice. Cancer Res. 1993;53:3874–3876. [PubMed] [Google Scholar]
- Diwan B. A., Anderson L. M., Ward J. M., Henneman J. R., Rice J. M. Transplacental carcinogenesis by cisplatin in F344/NCr rats: Promotion of kidney tumors by postnatal administration of sodium barbital. Toxicol. Appl. Pharmacol. 1995;132:115–121. doi: 10.1006/taap.1995.1092. [DOI] [PubMed] [Google Scholar]
- Dobrovolsky V. N., Elespuru R. K., Bigger A. H., Robison T. W., Heflich R. H. Monitoring humans for somatic mutation in the endogenous PIG-A gene using red blood cells. Environ. Mol. Mutagen. 2011;52:784–794. doi: 10.1002/em.20667. [DOI] [PubMed] [Google Scholar]
- Dobrovolsky V. N., Miura D., Heflich R. H., Dertinger S. D. The in vivo Pig-a gene mutation assay, a potential tool for regulatory safety assessment. Environ. Mol. Mutagen. 2010;51:825–835. doi: 10.1002/em.20627. [DOI] [PubMed] [Google Scholar]
- Donawho C. K., Luo Y., Luo Y., Penning T.D., Bauch J.L., Bouska J.J., Bontcheva-Diaz V.D., Cox B.F., DeWeese T.L., Dillehay L.E. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin. Cancer Res. 2007;13:2728–2737. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- Fichtinger-Schepman A., van der Veer J. L., den Hartog J., Lohman P., Reedijk J. Adducts of the antitumor drug cis-diamminodichloroplatinum (II) with DNA: Formation, identification and quantitation. Biochemistry. 1985;24:707–713. doi: 10.1021/bi00324a025. [DOI] [PubMed] [Google Scholar]
- Heath D. S., Axelrad A. A., McLeod D. L., Shreeve M. M. Separation of the erythropoietin-responsive progenitors BFU-E and CFU-E in mouse bone marrow by unit gravity sedimentation. Blood. 1976;47:777–792. [PubMed] [Google Scholar]
- Howard R., Gilbert E., Lynch C. F., Hall P., Storm H., Holowaty E., Pukkala E., Langmark F., Kaijser M., Andersson M., et al. Risk of leukemia among survivors of testicular cancer: A population-based study of 42,722 patients. Ann. Epidemiol. 2008;18:416–421. doi: 10.1016/j.annepidem.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC. Cisplatin. IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Humans, Supplement 7. Lyon, France: International Agency for Research on Cancer; 1987. Overall Evaluations of Carcinogenicity; pp. 170–171. [Google Scholar]
- International Agency for Research on Cancer (IARC). Cisplatin. IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Humans. Vol. 26. Lyon, France: International Agency for Research on Cancer; 1981. Some antineoplastic and immunosuppressive agents; pp. 151–164. [Google Scholar]
- Jordan C. T., Lemischka I. R. Clonal and systemic analysis of long-term hematopoiesis in the mouse. Genes Dev. 1990;4:220–232. doi: 10.1101/gad.4.2.220. [DOI] [PubMed] [Google Scholar]
- Kaldor J. M., Day N. E., Pettersson F., Clarke A., Pedersen D., Mehnert W., Bell J., Høst H., Prior P., Karjalainen S, et al. Leukemia following chemotherapy for ovarian cancer. N. Engl. J. Med. 1990;322:1–6. doi: 10.1056/NEJM199001043220101. [DOI] [PubMed] [Google Scholar]
- Kasai S., Terasawa W., Kodama H., Terasawa T. Enhancement of erythroid colony formation in vitro by spleen extract from irradiated rats. Jpn. J. Physiol. 1980;30:767–774. doi: 10.2170/jjphysiol.30.767. [DOI] [PubMed] [Google Scholar]
- Kaufmann S. H., Patel A. Development of PARP inhibitors: An unfinished story. Oncology. 2010;24:66. [PubMed] [Google Scholar]
- Kimoto T., Chikura S., Suzuki-Okada K., Kobayashi X., Itano Y., Miura D., Kasahara Y. Effective use of the Pig-a gene mutation assay for mutagenicity screening: Measuring CD59-deficient red blood cells in rats treated with genotoxic chemicals. J. Toxicol. Sci. 2012;37:943–955. doi: 10.2131/jts.37.943. [DOI] [PubMed] [Google Scholar]
- Louro H., Silva M. J., Boavida M. G. Mutagenic activity of cisplatin in lacZ plasmid-based transgenic mouse model. Environ. Mol. Mutagen. 2002;40:283–291. doi: 10.1002/em.10118. [DOI] [PubMed] [Google Scholar]
- MacGregor J. T., Bishop M. E., McNamee J. P., Hayashi M., Asano N., Wakata A., Nakajima M., Saito J., Aidoo A., Moore M.M., et al. Flow cytometric analysis of micronuclei in peripheral blood reticulocytes: II. An efficient method of monitoring chromosomal damage in the rat. Toxicol. Sci. 2006;94:92–107. doi: 10.1093/toxsci/kfl076. [DOI] [PubMed] [Google Scholar]
- MedlinePlus. Cisplatin Injection. National Library of Medicine. Available at: http://www.nlm.nih.gov/medlineplus/druginfo/meds/a684036.html10.1093/toxsci/kfu078.html. Accessed January 24, 2014.
- Miura D., Dobrovolsky V. N., Kimoto T., Kasahara Y., Heflich R. H. Accumulation and persistence of Pig-A mutant peripheral red blood cells following treatment of rats with single and split doses of N-Ethyl-N-nitrosourea. Mutat. Res. 2009;677:86–92. doi: 10.1016/j.mrgentox.2009.05.014. [DOI] [PubMed] [Google Scholar]
- National Toxicology Program. Report on Carcinogens, Twelfth Edition. Cisplatin. 2011.
- Pedersen-Bjergaard J., Daugaard G., Hansen S. W., Philip P., Larsen S. O., Rorth M. Increased risk of myelodysplasia and leukaemia after etoposide, cisplatin, and bleomycin for germ-cell tumours. Lancet. 1991;338:359–363. doi: 10.1016/0140-6736(91)90490-g. [DOI] [PubMed] [Google Scholar]
- Phonethepswath S., Franklin D., Torous D. K., Bryce S. M., Bemis J. C., Raja S., Avlasevich S., Weller P., Hyrien O., Palis J., et al. Pig-a mutation: Kinetics in rat erythrocytes following exposure to five prototypical mutagens. Toxicol. Sci. 2010;114:59–70. doi: 10.1093/toxsci/kfp289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platinol U.S. Food and Drug Administration-approved drug label. http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/018057s080lbl.pdf.
- Satoh M., Kondo Y., Mita M., Nakagawa I., Naganuma A., Imura N. Prevention of carcinogenicity of anticancer drugs by metallothionein induction. Cancer Res. 1993;53:4767–4768. [PubMed] [Google Scholar]
- Sieburg H. B., Rezner B. D., Muller-Sieburg C. E. Predicting clonal self-renewal and extinction of hematopoietic stem cells. Proc. Natl. Acad. Sci. U.S.A. 2011;108:4370–4375. doi: 10.1073/pnas.1011414108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarbutt R. G. A study of erythropoiesis in rats. Exp. Cell Res. 1967;48:473–483. doi: 10.1016/0014-4827(67)90370-9. [DOI] [PubMed] [Google Scholar]
- Tometsko A. M., Torous D. K., Dertinger S. D. Analysis of micronucleated cells by flow cytometry. 1. Achieving high resolution with a malaria model. Mutat. Res. 1993;292:129–135. doi: 10.1016/0165-1161(93)90140-u. [DOI] [PubMed] [Google Scholar]
- Torous D. K., Hall N. E., Murante F. G., Gleason S. E., Tometsko C. R., Dertinger S. D. Comparative scoring of micronucleated reticulocytes in rat peripheral blood by flow cytometry and microscopy. Toxicol. Sci. 2003;74:309–314. doi: 10.1093/toxsci/kfg143. [DOI] [PubMed] [Google Scholar]
- Travis L. B., Andersson M., Gospodarowicz M., van Leeuwen F.E., Bergfeldt K., Lynch C.F., Curtis R.E., Kohler B.A., Wiklund T., Storm H. Treatment-associated leukemia following testicular cancer. J. Natl. Cancer Inst. 2000;92:1165–1171. doi: 10.1093/jnci/92.14.1165. [DOI] [PubMed] [Google Scholar]
- Travis L. B., Beard C., Allan J. M., Dahl A.A., Feldman D.R., Oldenburg J., Daugaard G., Kelly J.L., Dolan M.E., Hannigan R. Testicular cancer survivorship: Research strategies and recommendations. J. Natl. Cancer Inst. 2010;102:1114–1130. doi: 10.1093/jnci/djq216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis L. B., Curtis R. E., Boice J. D., Jr, Platz C. E., Hankey B. F., Fraumeni J. F., Jr Second malignant neoplasms among long-term survivors of ovarian cancer. Cancer Res. 1996;56:1564–1570. [PubMed] [Google Scholar]
- Travis L. B., Curtis R. E., Storm H., Hall P., Holowaty E., Van Leeuwen F.E., Kohler B.A., Pukkala E., Lynch C.F., Andersson M., et al. Risk of second malignant neoplasms among long-term survivors of testicular cancer. J. Natl. Cancer Inst. 1997;89:1429–1439. doi: 10.1093/jnci/89.19.1429. [DOI] [PubMed] [Google Scholar]
- U.S. Food and Drug Administration. Guidance for Industry: Estimating the maximal safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. 2005. Available at: http://www.fda.gov/downloads/Drugs/Guidances/UCM078932.pdf. Accessed January 24, 2014.
- Verdecchia A., Francisci S., Brenner H., Gatta G., Micheli A., Mangone L., Kunkler I., Working Group EUROCARE-4. Recent cancer survival in Europe: A 2000–02 period analysis of EUROCARE-4 data. Lancet Oncol. 2007;8:784–796. doi: 10.1016/S1470-2045(07)70246-2. [DOI] [PubMed] [Google Scholar]
- Waalkes M. P., Liu J., Kasprzak K. S., Diwan B. A. Hypersusceptibility to cisplatin carcinogenicity in metallothionein-I/II double knockout mice: Production of hepatocellular carcinoma at clinically relevant doses. Int. J. Cancer. 2006;119:28–32. doi: 10.1002/ijc.21245. [DOI] [PubMed] [Google Scholar]
- Welch J. S., Ley T. J., Link D. C., Miller C. A., Larson D. E., Koboldt D. C., Wartman L. D., Lamprecht T. L., Liu F., Xia J., et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt K. L., Cunningham C. K., Patterson K. B., Kissling G. E., Dertinger S. D., Livingston E., Bishop J. B. Elevated frequencies of micronucleated erythrocytes in infants exposed to zidovudine in utero and postpartum to prevent mother-to-child transmission of HIV. Environ. Mol. Mutagen. 2007;48:322–329. doi: 10.1002/em.20266. [DOI] [PMC free article] [PubMed] [Google Scholar]