


Abstract
Short-wave ultraviolet light induces both mildly helix-distorting cyclobutane pyrimidine dimers (CPDs) and severely distorting (6–4) pyrimidine pyrimidone photoproducts ((6–4)PPs). The only DNA polymerase (Pol) that is known to replicate efficiently across CPDs is Polη, a member of the Y family of translesion synthesis (TLS) DNA polymerases. Phenotypes of Polη deficiency are transient, suggesting redundancy with other DNA damage tolerance pathways. Here we performed a comprehensive analysis of the temporal requirements of Y-family Pols ι and κ as backups for Polη in (i) bypassing genomic CPD and (6–4)PP lesions in vivo, (ii) suppressing DNA damage signaling, (iii) maintaining cell cycle progression and (iv) promoting cell survival, by using mouse embryonic fibroblast lines with single and combined disruptions in these Pols. The contribution of Polι is restricted to TLS at a subset of the photolesions. Polκ plays a dominant role in rescuing stalled replication forks in Polη-deficient mouse embryonic fibroblasts, both at CPDs and (6–4)PPs. This dampens DNA damage signaling and cell cycle arrest, and results in increased survival. The role of relatively error-prone Pols ι and κ as backups for Polη contributes to the understanding of the mutator phenotype of xeroderma pigmentosum variant, a syndrome caused by Polη defects.
INTRODUCTION
Exposure to ultraviolet (UV) light induces both mildly helix-distorting cis-syn cyclobutane pyrimidine dimers (CPDs) and strongly helix-distorting (6–4) pyrimidine pyrimidone photoproducts ((6–4)PPs) in the genome (1). Both CPDs and (6–4)PPs form blocks for replicative DNA polymerases (Pols), since their active sites are unable to accommodate these photolesions. The only DNA polymerase known to efficiently replicate across CPDs both in vitro and in vivo is Polη, a member of the Y family of DNA polymerases which, in mammalian cells, also includes Pols ι, κ and Rev1 (2,3). Polη is capable of containing a thymine-thymine CPD, the most frequent photolesion, in its enlarged active site (4). In contrast to CPDs, (6–4)TT lesions form a poor substrate for Polη in vitro, as Polη frequently inserts a G opposite the 3′ T but is unable to carry out the subsequent extension step (5). In vivo, TLS at (6–4)PP may involve either Polη or Polι, followed by extension by another polymerase, likely the B family polymerase Polζ (6).
The importance of Polη in DNA damage responses is stressed by patients suffering from the xeroderma pigmentosum-variant syndrome (XP-V), a rare autosomal recessive human disorder, caused by mutations in the gene that encodes Polη (7,8). Clinically, XP-V is characterized by photosensitivity of the skin and high susceptibility to develop cancer of sunlight-exposed areas of the skin. After exposure to ultraviolet C (UVC) light, the conversion of low molecular weight to high molecular weight nascent DNA is much slower in XP-V cells than in normal cells (9). The TLS defect results in the accumulation of ssDNA regions that activate the ataxia-telangiectasia-mutated and Rad3-related (Atr) kinase (10,11). Activated Atr phosphorylates multiple proteins, including Checkpoint kinase 1 (Chk1) that controls S-phase progression by inhibiting origin firing, slowing down replication fork progression, stabilizing stalled replication forks and delaying cell cycle progression (12,13). Nevertheless, XP-V cells are only mildly sensitive to the cytotoxic effects of UVC light and the defect in the progression of replication at damaged DNA is only transient, suggesting that most lesions are ultimately bypassed in these cells. Since XP-V cells display increased mutagenesis upon exposure to UVC light (14), an alternative TLS process presumably operates as a backup to convert ssDNA regions into dsDNA in XP-V cells.
Recently, we have analyzed the in vivo roles of individual TLS Pols, including Polη and other Y family Pols, in the suppression of DNA damage signaling and genome instability in immortalized mouse embryonic fibroblast (MEF) lines upon exposure to UVC light (15). We found that Polι and Polκ-deficient MEFs only displayed minor phenotypes in response to UVC light, whereas Rev1 appears to be mainly involved in the bypass of (6–4)PP (15,16). In addition, we observed that, similar to XP-V cells, Polη-deficient MEFs display a transient defect in TLS, resulting in the accumulation of cells in mid-S phase and activation of DNA damage signaling (15). Mainly TLS across genomic CPDs is affected in these cells (15). Possibly, this transient TLS defect in the absence of Polη might be due to the Y-family Pols ι and κ that act as backup Pols in bypassing UVC lesions at the genome, and in the suppression of DNA damage responses.
To address this question, here we have used MEF lines with well-defined single, double and triple deficiencies in Pols η, ι and κ. To provide quantitative data on the UV damage responses in these cell lines, the same UVC dose was applied in most experiments. We report that in Polη-deficient MEFs exposed to UVC light, Polκ is the predominant TLS polymerase to bypass both genomic CPDs and (6–4)PPs, contributing to (i) alleviating cell cycle arrest, (ii) quenching DNA damage signaling and (iii) promoting cell survival. Pol ι may play a role in TLS of a subset of (6–4)PP. Our results support and greatly extend previous studies of cells in which the expression of multiple TLS polymerases was reduced using siRNA knock-down strategies (6,17–20).
MATERIALS & METHODS
Cell culture
MEFs lacking Polη, Polι or Polκ were isolated from day 13.5 embryos of Polη, Polι or Polκ-deficient mice (21,22); Aoufouchi et al., in preparation). Crossings between Polη, Polι and Polκ-deficient mice produced 13.5-day embryo that were doubly-deficient for Polη and Polι, for Polη and Polκ or for all three Pols. From these embryos, MEFs were isolated and immortalized following transfection of SV40 large T antigen. Immortalized MEFs homozygous for a targeted disruption of Rev1 were described previously (16). All MEF lines were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 4,5 g/l glucose, Glutamax and pyruvate (Invitrogen) supplemented with 10% fetal calf serum, penicillin (100 U/ml), and streptomycin (100 μg/ml; MEF medium) at 37°C in a humidified atmosphere containing 5% CO2.
DNA fiber analysis
Per well of a 6-well plate, 7.5 × 104 MEFs were seeded and cultured overnight in an MEF medium. Prior to UVC exposure (13 J/m2), MEFs were incubated in a medium containing 25 μM 5-Chloro-2′-deoxyuridine (CldU) for 20 min at 37°C. After UVC exposure, a medium containing 500 μM 5-Iodo-2′-deoxyuridine (IdU) was added, resulting in a final concentration of 250 μM IdU and 12.5 μM CldU. After 20 min at 37°C, cells were trypsinized, 2 μl of a suspension of 3 × 105 MEFs/ml were spotted onto a microscope slide, incubated for 5 min and lysed with 7 μl lysis buffer (200 mM Tris-HCl pH7.4, 50 mM EDTA, 0.5% SDS) for 3 min. Slides were tilted to 15°C to allow the DNA to run down the slide. Next, slides were air dried and subsequently fixed in methanol-acetic acid (3:1). After rehydration, fixed DNA fibers were denatured in 2.5 M HCl for 75 min. Incorporation of CldU was detected using rat-α-BrdU antibodies (1:500; BU1/75, AbD Serotec) and Alexafluor-555-labeled goat-α-rat antibodies (1:500; Molecular Probes, Life Technologies Europe BV, Bleiswijk, The Netherlands), whereas incorporated IdU was detected using mouse-α-BrdU antibodies (1:750; Clone B44, BD) and Alexafluor-488-labeled goat-α-mouse antibodies (1:500; Molecular Probes, Life Technologies Europe BV, Bleiswijk, The Netherlands). Finally, slides were mounted in Fluoro-Gel (Electron Microscopy Sciences, Hatfield, USA). Microscopy was performed using a fluorescent microscope (Carl Zeiss BV, Sliedrecht, The Netherlands).
Alkaline DNA unwinding (ADU)
This assay, which measures progression of replicons (23) was performed with minor modifications. The procedure is outlined in Figure 1C. Per well 5 × 104 MEFs were plated in a 24-well plate and cultured overnight in MEF medium. After pulse labeling with [3H]thymidine for 15 min, MEFs were washed once with PBS and subsequently exposed to 5 J/m2 UVC or mock treated. Then, at indicated times, DNA at replication forks was locally denatured upon incubation of MEFs with ice-cold denaturation solution (0.15 M NaCl and 0.03 M NaOH) for 30 min. The denaturation of DNA was terminated by adding ice-cold 0.02 M NaH2PO4. After sonication, SDS was added to a final concentration of 0.25% and the samples were stored at −20°C for at least 16 h. After thawing, the lysates were loaded onto hydroxyl apatite columns to elute ssDNA using 0.1 M K2HPO4 (pH6.8) and dsDNA using 0.3 M K2HPO4 (pH6.8), respectively. Radioactivity in each eluate was determined by liquid scintillation counting (PerkinElmer). Replication progression was calculated by determining the ratio of radioactivity in total DNA: ssDNA.
Figure 1.
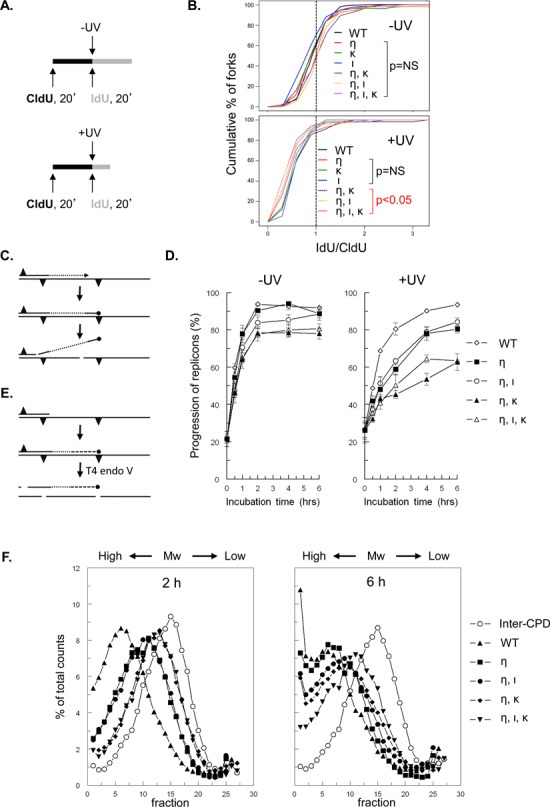
Both Pols ι and κ are required for replicon progression in Polη-deficient MEFs, late after UVC exposure. (A) Schematic representation of DNA fiber labeling with nucleotide analogs CldU and IdU in MEFs that were mock treated (-UV) or exposed to UVC (+UV). (B) Cumulative percentage of replication forks at ratios of lengths of ldU-labeled tracts to CldU-labeled tracts in wild-type MEFs (WT) or in MEFs with single, double or triple deficiencies in Polη (η), Polι (ι) and Polκ (κ) exposed to 13 J/m2 UVC (+UV). P values are shown of the two-sample Kolmogorov-Smirnov (K-S) test for the ratio distribution of each knock-out genotype compared to wild-type. (C) Scheme of the alkaline DNA unwinding assay. Nascent DNA is pulse labeled with [3H]thymidine (dotted line) immediately before the induction of photolesions (triangles; top). MEFs are then cultured in medium without label (middle). Stalling of a fork at a photolesion results in a DNA end containing [3H]thymidine that is locally denatured using alkaline, followed by sonication and isolation of [3H]thymidine-labeled ssDNA using hydroxyl apatite (bottom). (D) Replication fork progression in mock-treated MEFs (left panel) and in MEFs exposed to 5 J/m2 UVC (right panel; n = 4). Error bar, SEM. (E) Scheme of alkaline sucrose gradient sedimentation using T4 endonuclease V. Template DNA was uniformly labeled with [14C]thymidine (solid line) followed by exposure to UVC inducing CPD and (6–4)PP photolesions (triangles; top). Elongating daughter strands were pulse labeled with [3H]-thymidine for 30 min (dotted line) and cultured in a medium without label (dashed line; middle). At different times, cells were lysed and [14C]thymidine-containing DNA was cleaved by T4 endonuclease V at a CPD, followed by size fractionation using alkaline sucrose gradients (bottom). The [14C]thymidine-labeled inter-CPD size distribution serves as an internal standard, since CPDs are not removed in mouse cells. (F) Alkaline sucrose gradient profiles of [3H]thymidine-containing DNA of wild-type MEFs (WT, closed triangle), MEFs deficient in Polη (η; closed square) and of Polη-deficient MEFs containing an additional defect in Polι (ι, η; closed circle), Polκ (η, κ; closed diamond) or both TLS polymerases (η, ι, κ; closed inverted triangle) at 2 and 6 h after exposure to 5 J/m2 UVC. Also the profile of [14C]thymidine labeled, CPD-containing fragments is depicted (open circles). Mw, molecular weight.
Alkaline sucrose gradients
The replicative bypass of genomic CPDs and the increase in molecular mass of elongating nascent DNA molecules in MEFs exposed to 5 J/m2 UVC was determined by a sensitive variant of the alkaline sucrose sedimentation assay as described previously (24). The procedure is outlined in Figure 1E.
Immunostaining
MEFs were cultured overnight on coverslips, incubated with 10 μM 5-ethynyl-2′-deoxyuridine (EdU; Invitrogen) in an MEF medium for 30 min and subsequently exposed to UVC irradiation (5 J/m2). At indicated times after UVC treatment, cells were fixed and permeabilized as follows: for detection of Rpa, Chk1S345-P and Kap1S824-P, cells were preextracted and permeabilized by 0.3% Triton-X in CSK buffer pH 7.2–7.5 (10 mM HEPES pH7.4, 100 mM NaCl, 3 mM MgCl2, 0.3% Triton-X100, 300 mM sucrose) for 2 min on ice and immediately fixed with 3.7% paraformaldehyde for 20 min; for detection of AtmS1981-P, cells were fixed and permeabilized with ice-cold methanol:acetone (1:1) for 10 min at −20°C. Cells were blocked with 3% BSA+0.1% Tween-20 for at least 30 min to prevent nonspecific binding, and subsequently incubated overnight with antibodies against Rpa (Cell Signaling), Chk1S345-P (Cell Signaling), Kap1S824-P(Bethyl Laboratory) or AtmS1981-P (Rockland Immunochemicals) at 4°C. Then, appropriate fluorescent dye-conjugated secondary antibodies were applied and nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI). To visualize EdU-positive cells, which represent the S-phase cells at a time of UVC treatment, Alexafluor 647-conjuagated azide was used according to the manufacturer's recommendation (Click-iTTM Edu imaging kit, Invitrogen). Experiments were performed three times. Samples were mounted (Vectashield, Vector laboratories), and images were acquired by wide-field fluorescent microscopy (Axioplan M2, Carl Zeiss). Fluorescence intensity and numbers of foci were quantified using ImageJ software (National Institutes of Health) as described (25). Between 90 and 135 nuclei per cell line were analyzed for each time point. Detection of CPDs and (6–4)PPs in single-stranded DNA templates was essentially performed as described (16), except that to enable detection of (6–4)PPs the cells were fixed in 3.7% paraformaldehyde for 15 min after extraction with ice-cold 0.3% Triton-X100 in CSK buffer for 2 min.
Bivariate cell cycle analysis
Cell cycle progression of MEFs, exposed to 5 J/m2 UVC or mock-treated and pulse-labeled with BrdU, 30 min prior to fixing the cells, was determined by bivariate cell cycle analysis essentially as described previously (15).
Cell proliferation assay
Proliferation of MEFs was determined 3 days after mock treatment or exposure to various doses of UVC light (Philips T UVC lamp, predominantly 254 mm) as described previously (15).
RESULTS
An early role of Pols ι and κ in photolesion bypass
Recently, we have shown that replicative bypass of photolesions was delayed rather than abolished in Polη-deficient MEFs, suggesting the existence of a backup mechanism that almost completely rescues the Polη defect (15). Here we tested whether two other Y-family TLS polymerases, i.e. Pols ι and κ, are involved in this backup pathway. To this aim we compared the responses of Polη-deficient MEF lines with additional deficiencies in Polι, Polκ or both TLS Pols with wild-type and single-mutant MEF lines. We first determined the progression of replicons in the different MEF lines using DNA fiber labeling. This sensitive assay allows the analysis of replicon progression on single DNA molecules, shortly after exposure to UVC. Thus, cells were incubated with chlorodeoxyuridine (CldU) for 20 min to label replicating DNA, exposed to 0 or to 13 J/m2 UVC, and subsequently incubated with Iododeoxyuridine (IdU) for another 20 min. Fibers were generated and stained with specific antibodies for CldU and IdU, visualized by fluorescent microscopy and the lengths of CldU- and IdU-containing tracts were quantified to determine the replication speed and replication fork stalling (Figure 1A). When undamaged templates are replicated, no obvious differences in the replication speed among the cell lines were found (Supplementary Figure S1B), indicating that TLS Pols η, ι and κ are dispensable for replication of undamaged DNA templates. Following UVC exposure, the ratio of IdU to CldU decreased from 1 to approximately 0.5 in wild-type and the single mutant Polη, Polι or Polκ-deficient MEFs (Figure 1B). This result indicates that (i) UVC-induced DNA damage results in reduced replicon progression and (ii), since the differences in IdU to CldU ratios were statistically not significant, TLS Pols η, ι and κ are seemingly not essential for photolesion bypass at a very early stage after an UVC dose of 13 J/m2 (Figure 1B). Nevertheless, it is possible that the DNA fiber analysis is not sensitive enough to visualize discontinuous DNA synthesis by skipping the lesion on UV-damaged DNA as proposed for Polη-deficient cells (11). At this stage, however, compared with wild-type and single Y Pol mutant MEFs, a significant decrease of the IdU-to-CldU ratio was observed in the double- and triple-mutant MEFs (Kolmogorov-Smirnov test: P < 0.01). These results suggest that both Pols ι and κ play a role in alleviating replication fork stalling in Polη-deficient cells, early after UV exposure, indicating partial redundancy between Pols η, ι and κ (Figure 1B).
A late role of Pols ι and κ in photolesion bypass
To investigate the redundancy between Pols η, ι and κ in TLS at photolesions also at later times after UVC exposure, we employed an alkaline DNA unwinding assay. In this assay, the progression of replicons is determined by measuring the persistence of radioactively labeled ssDNA ends in proliferating cells, pulse-treated with [3H]thymidine immediately prior to mock-treatment or UVC exposure (Figure 1C). Normal replicon progression will result in the relocation of radioactive label from ssDNA ends to dsDNA. In contrast to the DNA fiber assay, the alkaline DNA unwinding assay can be used to detect discontinuous DNA synthesis in TLS-defective cell lines (11,16,26). Previously, we have used this assay to show that MEFs with single defects in Pols η, ι and κ are not measurably defective in replication of undamaged DNA templates (15). Interestingly, Polη-deficient MEF lines with additional defects in Polι, Polκ or both TLS Pols replicate undamaged DNA templates somewhat less efficiently compared to the Polη single-mutant cells, indicating a defect in TLS of endogenous DNA lesions or hard-to-replicate DNA sequences (27, 28; Figure 1D, left panel). Upon exposure to 5 J/m2 UVC, Polη single-mutant cells displayed a delay in replication fork progression compared to wild-type cells. This delay was not aggravated by an additional deficiency for Polι (Figure 1D, right panel), which at first sight contrasts with the DNA fiber analysis. However, it should be stressed that the UV dose used in the fiber assay is more than 2-fold higher (13 J/m2 versus 5 J/m2) and this may increase the frequency of substrates for Polι in the absence of Polη. Thus, Polι acts as a backup Pol in Polη-deficient MEF mainly at early times after UV exposure (Figure 1B). Conversely, as compared with Polη single-mutant MEFs, the MEF line deficient for both Pols η and κ displayed strongly reduced fork progression following exposure to UVC (Figure 1D, right panel). This defective fork progression was not exacerbated by an additional deficiency for Polι in these cells. Together, these data indicate that Polκ, but not Polι, can complement the Polη defect at later time points after UVC exposure. Nevertheless, in Polη, Polκ doubly-deficient MEFs replicons continue to progress, albeit slowly, revealing that Polκ is not essential for replicative bypass of photolesions in the absence of Polη.
We wanted to provide an independent approach to study the possible roles of Pols ι and κ as backup polymerases in Polη-deficient MEFs at later time points after exposure. To this aim, we utilized a sensitive alkaline sucrose gradient-based assay that measures the length of newly synthesized daughter strands, specifically beyond the most prevalent genomic CPD lesions, of which the density is represented by the internal ([14C]thymidine-labeled) standard (Figure 1E). Consequently, this assay is indicative for both replicon progression and maturation of DNA replication, whereas the alkaline unwinding assay measures only replicon progression. As expected, the generation of nascent DNA molecules was delayed in Polη-deficient MEFs compared with wild-type cells, especially at 2 h after exposure (Figure 1F). At this time point, maturation of nascent DNA in MEFs deficient for both Pols η and ι was indistinguishable from the single Polη-deficient MEFs, consistent with the alkaline DNA unwinding assay. The defect of the Polη-deficient MEFs, however, was aggravated when these cells are also deficient for Polκ (Figure 1F, left panel). Compared with the MEF line deficient for both Pols η and κ, the triple-mutant MEF line shows a similar deficiency in generating nascent DNA molecules at 2 h after UVC exposure (Figure 1F, left panel). We conclude that, at this time point, Polκ, but not Polι, can complement for the Polη deficiency. At 6-h post exposure, the maturation of nascent DNA molecules was delayed not only in the MEFs deficient for both Polη and Polκ, but also in the MEFs deficient for both Polη and Polι, compared with the Polη single mutant. In the triple mutant, daughter strand maturation was reduced to an even greater extent (Figure 1F, right panel). The specific defect in the cell lines with a Polι defect at 6 h after treatment suggests that, at least in the absence of Polη, Polι is required for TLS at a non-abundant lesion type, whereas Polκ can complement for the Polη deficiency in TLS at most photolesions.
Polκ affects cell cycle progression of Polη-deficient MEFs
To investigate redundancy in the roles of Polη, Polι and Polκ in cell cycle progression upon UVC exposure we determined the incorporation of the nucleotide analog bromodeoxyuridine (BrdU) in different cell cycle stages of asynchronously growing MEFs. Thus, at different times after mock treatment or after exposure to 5 J/m2 UVC, MEFs were pulse-labeled with BrdU, immediately preceding their fixation. Subsequently, BrdU contents were analyzed by bivariate flow cytometry. Since MEFs deficient for Polι or κ display cell cycle progression similar to wild-type MEFs (15), we focused on wild-type MEFs and Polη-deficient MEFs with and without additional deficiencies for Pols ι and/or κ. No major differences were found between the cell lines after mock treatment, indicating that all tested MEF lines proliferate in a similar fashion in the absence of photolesions (Supplementary Figure S2). Exposure of cells to UVC, however, revealed marked differences in cell cycle distribution between the MEF lines (Figure 2A and B). More specifically, compared with UVC-exposed wild-type MEFs, all mutant MEFs displayed significantly increased levels of early S phase cells and decreased levels of late S phase cells, 12 h after UVC treatment (P < 0.05; Student's t-test). This suggests an S phase delay in the mutant MEFs. Moreover, at 16 h this phenotype persisted in Polη-deficient MEFs (P < 0.05; Student's t-test) and remained unaltered when also Polι was disrupted, indicating that Polι does not serve as a quantitatively important backup to Polη (Figure 2A and B). In contrast, the additional deficiency for Polκ in both Polη-deficient MEFs and MEFs double deficient for Polη and Polι strongly affects cell cycle progression as the proportion of non-replicating G1 and G2/M phase cells is in the absence of Polκ significantly increased at later times (Figure 2A and B; P < 0.05; Student's t-test). Together, these results suggest that S phase progression is perturbed in the absence of Polη, both early (Figure 1) and late after UVC exposure (Figure 2A and B). The residual S phase progression in these cells strongly depends on Polκ, rather than on Polι.
Figure 2.
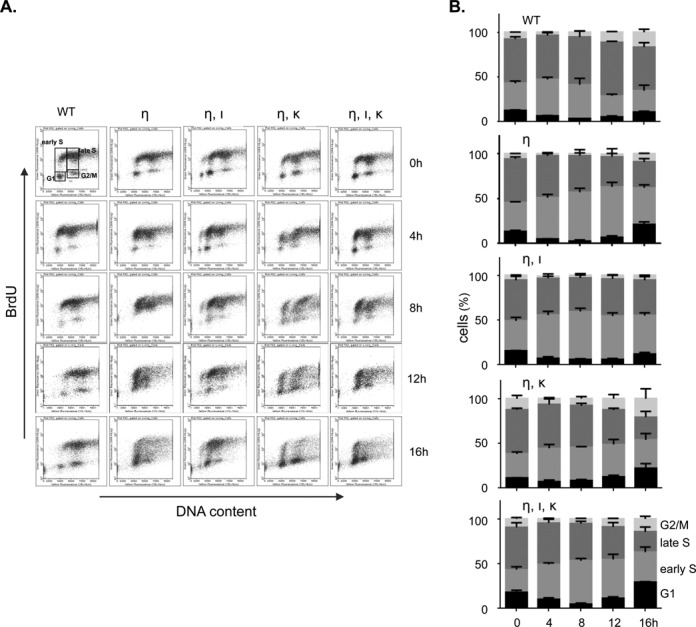
The replicative activity of Polη-deficient MEFs exposed to UVC relies mainly on Polκ. (A) FACS profiles showing BrdU content of wild-type MEFs (WT), MEFs deficient in Polη (η) and Polη-deficient MEFs containing an additional defect in Polι (η, ι), Polκ (η, κ) or both TLS polymerases (η, ι, κ) after exposure to 5 J/m2 UVC. Prior to fixation, MEFs were pulse labeled with BrdU for 30 min, immediately or at 4, 8, 12 and 16 h after UVC treatment. BrdU incorporation was determined by immunostaining and DNA content was measured using propidium iodide. (B) Quantification of MEFs in different cell cycle stages, up to 16 h after UVC exposure (n = 3; error bars: SD).
Low level of double strand DNA breaks in MEFs undergoing replication stress
Persistently stalled replication forks may collapse to double-strand DNA (dsDNA) breaks, underlying genome instability (29–31). To investigate the collapse of replication forks in the mutant cell lines, we assayed for phosphorylation of ataxia-telangiectasia-mutated (Atm) and of heterochromatic KRAB-ZFP-associated protein 1 (Kap1) in MEFs treated with UVC during S phase, as assessed by EdU incorporation, by immunostaining. Phosphorylation of Atm (AtmS1981-P) is an early step in the response to dsDNA breaks (32). Activation of Atm leads to phosphorylation of Kap1 at S824 (Kap1S824-P), although the formation of Kap1S824-P can also be mediated by other phosphatidylinositol-3 kinase-like kinases, including Atr (33). Except for the Rev1-deficient MEFs, we found only a minor UVC-dependent increase of Atm1981-P foci in nuclei of all other MEF lines tested, up to 8 h after UVC exposure (Figure 3A and B), suggesting that only few forks collapse at the UVC dose used (5 J/m2). Interestingly, strong induction of Kap1S824-P was found in all Polη-defective MEF lines and in MEFs deficient for Rev1, already 2 h after UVC exposure (see also below). Of note, with the exception of the Rev1-deficient MEFs, Kap1S824-P levels did not increase beyond 2 h after exposure, in agreement with the delay, rather than deficiency, of photolesion bypass in these cell lines (Figure 1).
Figure 3.
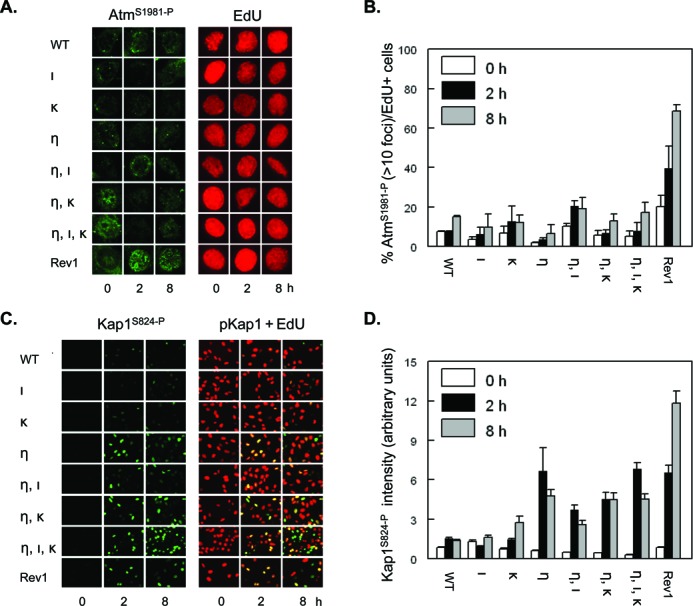
Few UVC-induced dsDNA breaks in Polη-deficient MEFs with or without additional deficiencies in Pols ι and κ. (A) Wild-type MEFs (WT), MEFs with single, double or triple deficiencies in Polη (η), Polι (ι) and Polκ (κ), or MEFs deficient in Rev1 (Rev1) were pulse labeled with EdU for 30 min, prior to exposure to 5 J/m2 UVC. Then, MEFs were fixed at 0, 2 and 8 h after treatment and immunostained for AtmS1981-P (left panel, online in green) in replicating, EdU-incorporating MEFs (online in red) at the time of UVC exposure. (B) Quantification of EdU-positive MEFs containing at least 10 AtmS1981-P foci. Error bar, SEM. (C) Similar experiment as in (A), except that MEFs were immunostained for Kap1S821-P (left panels, online in green) in replicating, EdU-incorporating MEFs (right panels, merge of staining for Kap1S821-P (online in green) + EdU (online in red)). (D) Quantification of the intensity of Kap1S821-P signal in EdU-positive MEFs. Error bars, SEM.
Quenching of the UV-induced DNA damage response requires Pols η, ι and κ.
We stained the cell lines for phosphorylation of Chk1 (Chk1S345-P), a target for Atr-induced DNA damage signaling at ssDNA tracts (34). Thus, at different time points prior to staining, cells were exposed to 5 J/m2 UVC and immediately pulse-labeled with EdU. We included Rev1-deficient MEFs as a positive control, since these cells exhibit strong and persistent Atr/Chk1 signaling following UVC exposure (15,16). At 2h after exposure, all MEF lines displayed Chk1S345-P-positive cells among EdU-positive (replicating) cells. The intensity of Chk1S345-P staining in EdU-positive double deficient MEF lines, and to an even greater extent in the triple-deficient line, was higher than in wild type cells and cells deficient for Polι or Polκ (Figure 4A and B). Furthermore, it should be noted that the extent of Chk1S345-P correlated with that of Kap1S824-P in the different MEF lines, although Kap1S824-P is restricted to a subset of EdU-positive cells (compare Figure 4A, B with Figure 3C, D). This result suggests that the formation of Kap1S824-P rather is due to Atr signaling than to the formation of dsDNA breaks. To confirm the presence of ssDNA tracts we assessed the recruitment to chromatin of the heterotrimeric Replication Protein A (Rpa), which binds to ssDNA and recruits Atr. We observed a similar distinction between the MEF lines with respect to the level of Rpa as shown for Chk1S345-P (see Figure 4C and D). These results are in agreement with the pronounced replicon stalling in these MEF lines as observed in the replication assays (Figure 1). At 8 h after UVC exposure, the intensity of Rpa staining dropped considerably in all MEF lines, except in the Rev1-mutant and in the triple-mutant MEFs (Figure 4C and D). In conclusion, DNA damage signaling in these mutant cell lines qualitatively reflected their defect in TLS, suggesting that ssDNA at stalled replication forks is the primary determinant of DNA damage responses.
Figure 4.
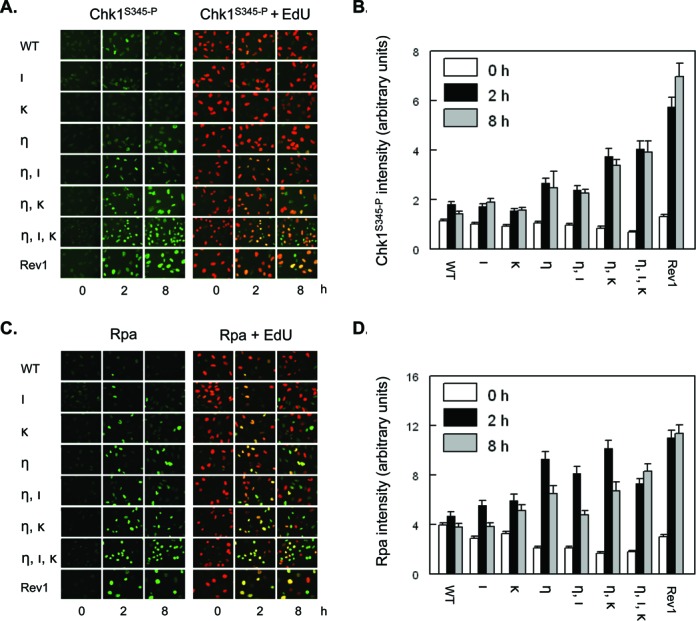
Quenching of DNA damage responses to photolesions requires Pols η, ι and κ. (A) Wild-type MEFs (WT), MEFs with single, double or triple deficiencies in Polη (η), Polι (ι) and Polκ (κ), or MEFs deficient in Rev1 (Rev1) were pulse labeled with EdU for 30 min, prior to exposure to 5 J/m2 UVC. Then, MEFs were fixed at 0, 2 and 8 h after treatment and immunostained for Chk1S345-P (left panels, online in green) in replicating, EdU-incorporating MEFs (right panels, merge of staining for Chk1S345-P (online in green) and EdU (online in red)) at the time of UVC exposure. (B) Quantification of the intensity of Chk1S345-P staining in EdU-positive MEFs. Error bar, SEM. (C) Similar experiment as in (A), except that MEFs were immunostained for Rpa (left panels, online in green). (D) Quantification of the intensity of Rpa staining in EdU-positive MEFs. Error bars, SEM.
Polκ protects Polη-deficient MEFs from UVC toxicity
To study the biological consequences of prolonged replication fork stalling, enhanced DNA damage signaling and impaired cell cycle progression, caused by defects in multiple Y family Pols, we analyzed cell proliferation at 3 days after exposure to various doses of UVC. Among the MEF lines tested, MEFs deficient for both Pols η and κ as well as the triple-mutant MEFs displayed the highest sensitivity to UVC light, whereas Polη-deficient MEFs and MEFs deficient for both Pols η and ι showed an intermediate UVC sensitivity (Figure 5). Confirming previous observations (15), the MEF line deficient for Polκ was slightly more sensitive to UVC light than wild-type MEFs, whereas Polι-deficient MEFs displayed no increased UVC sensitivity (Figure 5). These results are again consistent with an important role for Polκ as a backup TLS polymerase for Polη.
Figure 5.
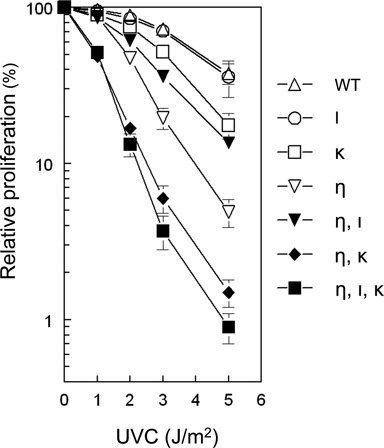
Effect of UVC on proliferation of MEFs with single, double or triple deficiencies in Pols η, ι and κ. MEFs were exposed to different doses of UVC and 3 days later the number of cells was assessed. The number of unexposed MEFs was set at 100%. Error bars, SEM.
Genomic CPDs are substrates for Pols ι and κ in Polη-deficient MEFs
By employing a novel immunostaining protocol using monoclonal antibodies that recognize CPDs or (6–4)PPs only when embedded in ssDNA we have previously observed that, in Polη-deficient MEFs, mainly CPDs cause stalling of replication forks (15). We applied this methodology to the current set of MEF lines to study which genomic photolesions are causing the phenotypes associated with MEFs deficient for both Pols η and κ and with the triple-mutant MEFs. Thus, cells were pulse-labeled with EdU, to identify the cells that were replicating during UVC exposure, and exposed to 5 J/m2 UVC. At 2 or 8 h after UVC exposure, cells were fixed and immunostained for CPDs, or for (6–4)PPs, embedded in ssDNA.
Almost no cells positive for CPDs were detected in wild-type MEFs and MEFs deficient for Polι or κ, indicating efficient bypass across genomic CPDs, independent of Pols ι and κ (Figure 6A). As expected, EdU-positive MEFs that are deficient for Polη displayed unreplicated CPDs at 2 h, and less at 8 h, after UVC exposure (Figure 6A and ref. 15), suggesting transient fork stalling at CPDs. Similar results were observed for Polη mutant MEFs with an additional deficiency for Polι (Figure 6A). MEFs deficient for both Pols η and κ displayed more EdU+CPD positive cells at 8 h upon UVC exposure, indicating that Polκ does perform TLS at CPDs in the absence of Polη. Nevertheless, EdU-positive triple-mutant MEFs exhibited the most pronounced staining for unreplicated CPDs at 8 h after UVC exposure (Figure 6A). This result suggests that, in the absence of Polη, Polι can perform TLS at CPDs, but only when also Polκ is inactive.
Figure 6.
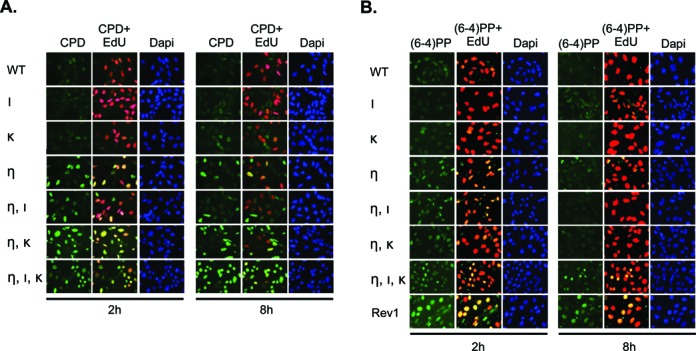
Pols ι and κ are required for efficient bypass of photoproducts in MEFs deficient in Polη. (A) Wild-type MEFs (WT) or MEFs with single, double or triple deficiencies in Polη (η), Polι (ι) and Polκ (κ) were pulse labeled with EdU for 30 min, prior to exposure to 5 J/m2 UVC, and 2 and 8 h later, MEFs were fixed and immunostained for CPD (online in green; upper panels) in ssDNA of nuclei (online in blue; lower panel) in replicating, EdU-incorporating (online in red, merged with CPD staining; middle panels) and non-replicating MEFs at the time of UVC exposure. (B) Similar as in (A) except that MEFs were immunostained for (6–4)PP (online in green; upper panel). Rev1-deficient MEFs (Rev1) were included as a positive control (15,16).
All Y family polymerases contribute to TLS of (6–4)PPs
In contrast to CPD lesions, (6–4)PPs impose a strong helical distortion to the DNA, and the 3′ pyrimidine base in the pyrimidine dimer is twisted outward and unable to engage in base-pairing (35). Thus far, it has been largely unclear what TLS polymerases are responsible for bypass of these ‘severe’ lesions at the genome of mammalian cells, although we have previously described a regulatory role for Rev1 (16). Immunostaining of unreplicated (6–4)PPs in EdU-positive cells revealed that TLS of (6–4)PPs is perturbed in MEF lines defective in Polη, as judged by the EdU-positive cells staining for single-stranded (6–4)PPs, at 2 h after UVC exposure of these cells (15). Nevertheless, the defect was less pronounced than in MEFs deficient for Rev1 (16 and Figure 6B). Staining for unreplicated (6–4)PPs was also found for double and, to a greater extent, triple-mutant MEFs, at 2 and 8 h after UVC exposure (Figure 6B). These results are the first to demonstrate the involvement of these Y-family polymerases in TLS of genomic (6–4)PP and, moreover, suggest that in the absence of Polη, both Pols ι and κ act as backup TLS polymerases to replicate across (6–4)PPs.
DISCUSSION
In this study, we have comprehensively analyzed the contributions of the three Y family Pols η, ι and κ in TLS, S phase progression, DNA damage signaling, checkpoint activation and survival in response to genomic CPD and (6–4)PP lesions, using single-, double-, and triple-deficient MEFs. Our results demonstrate that, in the absence of Polη, Polκ plays a more important role than Polι in responses to genomic photolesions, in agreement with previous studies of cells in which the expression of multiple TLS polymerases was reduced using siRNA knock-down strategies (6,17–20). In support, we and others have shown that Polκ (but not Polι)-deficient mammalian cells are slightly sensitive to UVC light (15,21,36). This sensitivity can be attributed to two not mutually exclusive functions of Polκ in response to UV light. First, some studies attribute this sensitivity to a defect in NER, at least outside of S phase (37,38). Second, using siRNA strategies, others provide evidence for a role of Polκ in TLS of a TT CPD on episomal substrates in vivo (19,20) while no effect was found on TLS across TT (6–4)PP (6). Indeed, Polκ can extend from a mismatched nucleotide inserted across 3′Ts of TT CPDs by another DNA polymerase (39). This TLS-related function of Polκ on abundantly-induced TT CPDs explains the strong defects in replication fork progression, increased staining for CPDs, enhanced DNA damage signaling, slow progression through S phase, and reduced cell proliferation observed in UVC-exposed MEFs deficient for both Pols η and κ. Since Polκ mainly acts as an extender DNA polymerase in TLS of CPDs, we postulate that in absence of both Pols η and ι another, yet unidentified, DNA polymerase functions as an inserter at these DNA lesions. The role of Polκ as backup for Polη is not restricted to UV lesions, since also in somatic hypermutation of immunoglobulin genes, Polκ acts as a backup in the absence of Polη (40).
Polι plays only a minor role in TLS at genomic photolesions in Polη-deficient cells, which is apparent from the delayed maturation of nascent strands upon UVC exposure. However, the triple-mutant MEFs displayed the most pronounced phenotypes of all cell lines tested in this study, suggesting that Polι is essential for TLS across some UVC-induced DNA lesions in the absence of both Polη and Polκ. In support, purified Polι can replicate TT (6–4)PPs (41,42) and Polι mediates part of the mutagenicity of (6–4)PP in vivo (6,43). In addition to a subset of (6–4)PPs, also some CPDs might be candidates for Polι-mediated TLS in MEFs deficient for both Pols η and κ. Indeed, Polι is capable of inserting nucleotides opposite TT CPDs in vitro (41,42,44) although CPDs are only poorly bypassed by Polι in human (Polη-deficient) XP-V cells (19,20). Of note, the efficiency by which UV photolesions are induced in the genome strongly depends on the dipyrimidine sequence. Thus, the order of preference for the formation of CPDs is TT > CT = TC > CC, whereas (6–4)PPs are mostly induced at TC and CC dipyrimidines, to a lesser extent at TT dimer sites and not at CT sites (45–49). Moreover, as these lesion types are structurally highly dissimilar (45), they may require different sets of TLS polymerases to allow efficient lesion bypass during DNA replication.
In triple-mutant MEFs, replication forks are permanently stalled only late after UVC exposure. This indicates that some photolesions can be bypassed independently from the three Y family polymerases. Thus, the B family TLS Pol ζ or the recently described archaeal-eukaryotic primase called Primase-Polymerase may play a role in an alternate backup TLS pathway (19,50,51). Nevertheless, as persistent CPDs and (6–4)PPs are observed in the triple-deficient cells, we infer that bypass of some lesions fully depends on the three Y family Pols.
In conclusion, we have unveiled important but redundant roles for the three Y family of TLS polymerases in TLS of genomic CPD and (6–4)PP photolesions. Polκ appears the most important backup to Polη although, to a minor extent, Polι also functions as a backup. Nevertheless, also in the triple mutant most photolesions are ultimately bypassed, implicating the existence of yet other redundant pathways.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Disclaimer: The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Present address:
Piya Temviriyanukul, Institute of Nutrition, Mahidol University Salaya, 73170 Nakhon Pathom, Thailand
Niek Wit, Protein and Nucleic Acid Division, MRC laboratory of Molecular Biology, CB2 0QH Cambridge, United Kingdom
Frédéric Delbos, INSERM U1064, 44093 Nantes, France
FUNDING
Project Strategic Frontier Research (SFR-4) from Office of the Higher Education Commission, Royal Thai Government [Grant number MOE 0509(3)116715 to P.T.]; Dutch Cancer Foundation [Grant number NKI2012-5713 to H.J.]; Ligue Nationale contre le Cancer (Equipe labellisée) [to F.D. and C.-A.R.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Beukers R., Eker A.P.M., Lohman P.H.M. 50 years thymine dimer. DNA Repair. 2008;7:530–543. doi: 10.1016/j.dnarep.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 2.Sale J.E., Lehmann A.R., Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012;13:141–152. doi: 10.1038/nrm3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waters L.S., Minesinger B.K., Wiltrout M.E., D'Souza S., Woodruff R.V., Walker G.C. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 2009;73:134–154. doi: 10.1128/MMBR.00034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biertümpfel C., Zhao Y., Kondo Y., Ramón-Maiques S., Gregory M., Lee J.Y., Masutani C., Lehmann A.R., Hanaoka F., Yang W. Structure and mechanism of human DNA polymerase eta. Nature. 2010;465:1044–1048. doi: 10.1038/nature09196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson R.E., Haracska L., Prakash S., Prakash L. Role of DNA polymerase eta in the bypass of a (6–4) TT photoproduct. Mol. Cell. Biol. 2001;21:3558–3563. doi: 10.1128/MCB.21.10.3558-3563.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoon J.-H., Prakash L., Prakash S. Error-free replicative bypass of (6–4) photoproducts by DNA polymerase {zeta} in mouse and human cells. Genes Dev. 2010;24:123–128. doi: 10.1101/gad.1872810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson R.E., Kondratick C.M., Prakash S., Prakash L. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science. 1999;285:263–265. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- 8.Masutani C., Araki M., Yamada A., Kusumoto R., Nogimori T., Maekawa T., Iwai S., Hanaoka F. Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. EMBO J. 1999;18:3491–3501. doi: 10.1093/emboj/18.12.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lehmann A.R., Kirk-Bell S., Arlett C.F., Paterson M.C., Lohman P.H., de Weerd-Kastelein E.A., Bootsma D. Xeroderma pigmentosum cells with normal levels of excision repair have a defect in DNA synthesis after UV-irradiation. Proc. Natl. Acad. Sci. U.S.A. 1975;72:219–223. doi: 10.1073/pnas.72.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Despras E., Daboussi F., Hyrien O., Marheineke K., Kannouche P.L. ATR/Chk1 pathway is essential for resumption of DNA synthesis and cell survival in UV-irradiated XP variant cells. Hum. Mol. Genet. 2010;19:1690–1701. doi: 10.1093/hmg/ddq046. [DOI] [PubMed] [Google Scholar]
- 11.Elvers I., Johansson F., Groth P., Erixon K., Helleday T. UV stalled replication forks restart by re-priming in human fibroblasts. Nucleic Acids Res. 2011;39:7049–7057. doi: 10.1093/nar/gkr420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feijoo C., Hall-Jackson C., Wu R., Jenkins D., Leitch J., Gilbert D.M., Smythe C. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J. Cell Biol. 2001;154:913–923. doi: 10.1083/jcb.200104099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zachos G., Rainey M.D., Gillespie D.A.F. Chk1-dependent S-M checkpoint delay in vertebrate cells is linked to maintenance of viable replication structures. Mol. Cell. Biol. 2005;25:563–574. doi: 10.1128/MCB.25.2.563-574.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maher V.M., Ouellette L.M., Mittlestat M., McCormick J.J. Synergistic effect of caffeine on the cytotoxicity of ultraviolet irradiation and of hydrocarbon epoxides in strains of Xeroderma pigmentosum. Nature. 1975;258:760–763. doi: 10.1038/258760a0. [DOI] [PubMed] [Google Scholar]
- 15.Temviriyanukul P., van Hees-Stuivenberg S., Delbos F., Jacobs H., de Wind N., Jansen J.G. Temporally distinct translesion synthesis pathways for ultraviolet light-induced photoproducts in the mammalian genome. DNA Repair. 2012;11:550–558. doi: 10.1016/j.dnarep.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 16.Jansen J.G., Tsaalbi-Shtylik A., Hendriks G., Gali H., Hendel A., Johansson F., Erixon K., Livneh Z., Mullenders L.H.F., Haracska L., et al. Separate domains of Rev1 mediate two modes of DNA damage bypass in mammalian cells. Mol. Cell. Biol. 2009;29:3113–3123. doi: 10.1128/MCB.00071-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hendel A., Ziv O., Gueranger Q., Geacintov N., Livneh Z. Reduced efficiency and increased mutagenicity of translesion DNA synthesis across a TT cyclobutane pyrimidine dimer, but not a TT 6–4 photoproduct, in human cells lacking DNA polymerase eta. DNA Repair. 2008;7:1636–1646. doi: 10.1016/j.dnarep.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shachar S., Ziv O., Avkin S., Adar S., Wittschieben J., Reissner T., Chaney S., Friedberg E.C., Wang Z., Carell T., et al. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009;28:383–393. doi: 10.1038/emboj.2008.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoon J.-H., Prakash L., Prakash S. Highly error-free role of DNA polymerase eta in the replicative bypass of UV-induced pyrimidine dimers in mouse and human cells. Proc. Natl. Acad. Sci. U.S.A. 2009;106:18219–18224. doi: 10.1073/pnas.0910121106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ziv O., Geacintov N., Nakajima S., Yasui A., Livneh Z. DNA polymerase {zeta} cooperates with polymerases {kappa} and {iota} in translesion DNA synthesis across pyrimidine photodimers in cells from XPV patients. Proc. Natl. Acad. Sci. U.S.A. 2009 doi: 10.1073/pnas.0812548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schenten D., Gerlach V.L., Guo C., Velasco-Miguel S., Hladik C.L., White C.L., Friedberg E.C., Rajewsky K., Esposito G. DNA polymerase kappa deficiency does not affect somatic hypermutation in mice. Eur. J. Immunol. 2002;32:3152–3160. doi: 10.1002/1521-4141(200211)32:11<3152::AID-IMMU3152>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 22.Delbos F., de Smet A., Faili A., Aoufouchi S., Weill J.-C., Reynaud C.-A. Contribution of DNA polymerase eta to immunoglobulin gene hypermutation in the mouse. J. Exp. Med. 2005;201:1191–1196. doi: 10.1084/jem.20050292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johansson F., Lagerqvist A., Erixon K., Jenssen D. A method to monitor replication fork progression in mammalian cells: nucleotide excision repair enhances and homologous recombination delays elongation along damaged DNA. Nucleic Acids Res. 2004;32:e157. doi: 10.1093/nar/gnh154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Zeeland A.A., Smith C.A., Hanawalt P.C. Sensitive determination of pyrimidine dimers in DNA of UV-irradiated mammalian cells. Introduction of T4 endonuclease V into frozen and thawed cells. Mutat. Res. 1981;82:173–189. doi: 10.1016/0027-5107(81)90148-2. [DOI] [PubMed] [Google Scholar]
- 25.Gavet O., Pines J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell. 2010;18:533–543. doi: 10.1016/j.devcel.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jansen J.G., Tsaalbi-Shtylik A., Hendriks G., Verspuy J.W.A., Gali H., Haracska L., de Wind N. Mammalian polymerase zeta is essential for post-replication repair of UV-induced DNA lesions. DNA Repair. 2009;8:1444–1451. doi: 10.1016/j.dnarep.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 27.Bétous R., Rey L., Wang G., Pillaire M.-J., Puget N., Selves J., Biard D.S.F., Shin-ya K., Vasquez K.M., Cazaux C., et al. Role of TLS DNA polymerases eta and kappa in processing naturally occurring structured DNA in human cells. Mol. Carcinog. 2009;48:369–378. doi: 10.1002/mc.20509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baptiste B.A., Eckert K.A. DNA polymerase kappa microsatellite synthesis: two distinct mechanisms of slippage-mediated errors. Environ. Mol. Mutagen. 2012;53:787–796. doi: 10.1002/em.21721. [DOI] [PubMed] [Google Scholar]
- 29.Lundin C., Erixon K., Arnaudeau C., Schultz N., Jenssen D., Meuth M., Helleday T. Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol. Cell. Biol. 2002;22:5869–5878. doi: 10.1128/MCB.22.16.5869-5878.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petermann E., Helleday T. Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 2010;11:683–687. doi: 10.1038/nrm2974. [DOI] [PubMed] [Google Scholar]
- 31.Saintigny Y., Delacôte F., Varès G., Petitot F., Lambert S., Averbeck D., Lopez B.S. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001;20:3861–3870. doi: 10.1093/emboj/20.14.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiloh Y., Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013;14:197–210. [PubMed] [Google Scholar]
- 33.White D.E., Negorev D., Peng H., Ivanov A.V., Maul G.G., Rauscher F.J. KAP1, a novel substrate for PIKK family members, colocalizes with numerous damage response factors at DNA lesions. Cancer Res. 2006;66:11594–11599. doi: 10.1158/0008-5472.CAN-06-4138. [DOI] [PubMed] [Google Scholar]
- 34.Cimprich K.A., Cortez D. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim J.K., Choi B.S. The solution structure of DNA duplex-decamer containing the (6–4) photoproduct of thymidylyl(3'–>5')thymidine by NMR and relaxation matrix refinement. Eur. J. Biochem./FEBS. 1995;228:849–854. doi: 10.1111/j.1432-1033.1995.tb20331.x. [DOI] [PubMed] [Google Scholar]
- 36.Ogi T., Shinkai Y., Tanaka K., Ohmori H. Polkappa protects mammalian cells against the lethal and mutagenic effects of benzo[a]pyrene. Proc. Natl. Acad. Sci. U.S.A. 2002;99:15548–15553. doi: 10.1073/pnas.222377899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogi T., Lehmann A.R. The Y-family DNA polymerase kappa (pol kappa) functions in mammalian nucleotide-excision repair. Nat. Cell Biol. 2006;8:640–642. doi: 10.1038/ncb1417. [DOI] [PubMed] [Google Scholar]
- 38.Ogi T., Limsirichaikul S., Overmeer R.M., Volker M., Takenaka K., Cloney R., Nakazawa Y., Niimi A., Miki Y., Jaspers N.G., et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell. 2010;37:714–727. doi: 10.1016/j.molcel.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 39.Washington M.T., Johnson R.E., Prakash L., Prakash S. Human DINB1-encoded DNA polymerase kappa is a promiscuous extender of mispaired primer termini. Proc. Natl. Acad. Sci. U.S.A. 2002;99:1910–1914. doi: 10.1073/pnas.032594399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Faili A., Stary A., Delbos F., Weller S., Aoufouchi S., Sarasin A., Weill J.-C., Reynaud C.-A. A backup role of DNA polymerase kappa in Ig gene hypermutation only takes place in the complete absence of DNA polymerase eta. J. Immunol. 2009;182:6353–6359. doi: 10.4049/jimmunol.0900177. [DOI] [PubMed] [Google Scholar]
- 41.Haracska L., Johnson R.E., Unk I., Phillips B.B., Hurwitz J., Prakash L., Prakash S. Targeting of human DNA polymerase iota to the replication machinery via interaction with PCNA. Proc. Natl. Acad. Sci. U.S.A. 2001;98:14256–14261. doi: 10.1073/pnas.261560798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y., Yuan F., Wu X., Taylor J.S., Wang Z. Response of human DNA polymerase iota to DNA lesions. Nucleic Acids Res. 2001;29:928–935. doi: 10.1093/nar/29.4.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dumstorf C.A., Cleaver J.E., Lin Q., Kissling G.E., Yuan T., Kucherlapati G.C., McGregor W.G., Kunkel T.A. Participation of mouse DNA polymerase iota in strand-biased mutagenic bypass of UV photoproducts and suppression of skin cancer. Proc. Natl. Acad. Sci. U.S.A. 2006;103:18083–18088. doi: 10.1073/pnas.0605247103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaisman A., Frank E.G., Iwai S., Ohashi E., Ohmori H., Hanaoka F., Woodgate R. Sequence context-dependent replication of DNA templates containing UV-induced lesions by human DNA polymerase iota. DNA Repair. 2003;2:991–1006. doi: 10.1016/s1568-7864(03)00094-6. [DOI] [PubMed] [Google Scholar]
- 45.Rastogi R.P., Richa A., Tyagi M.B., Sinha R.P. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J. Nucleic Acids. 2010;2010:592980. doi: 10.4061/2010/592980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brash D.E. UV mutagenic photoproducts in Escherichia coli and human cells: a molecular genetics perspective on human skin cancer. Photochem. Photobiol. 1988;48:59–66. doi: 10.1111/j.1751-1097.1988.tb02786.x. [DOI] [PubMed] [Google Scholar]
- 47.Lippke J.A., Gordon L.K., Brash D.E., Haseltine W.A. Distribution of UV light-induced damage in a defined sequence of human DNA: detection of alkaline-sensitive lesions at pyrimidine nucleoside-cytidine sequences. Proc. Natl. Acad. Sci. U.S.A. 1981;78:3388–3392. doi: 10.1073/pnas.78.6.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mitchell D.L., Jen J., Cleaver J.E. Sequence specificity of cyclobutane pyrimidine dimers in DNA treated with solar (ultraviolet B) radiation. Nucleic Acids Res. 1992;20:225–229. doi: 10.1093/nar/20.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tornaletti S., Rozek D., Pfeifer G.P. The distribution of UV photoproducts along the human p53 gene and its relation to mutations in skin cancer. Oncogene. 1993;8:2051–2057. [PubMed] [Google Scholar]
- 50.Bianchi J., Rudd S.G., Jozwiakowski S.K., Bailey L.J., Soura V., Taylor E., Stevanovic I., Green A.J., Stracker T.H., Lindsay H.D., et al. PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol. Cell. 2013;52:566–573. doi: 10.1016/j.molcel.2013.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mourón S., Rodriguez-Acebes S., Martínez-Jiménez M.I., García-Gómez S., Chocrón S., Blanco L., Méndez J. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat. Struct. Mol. Biol. 2013;20:1383–1389. doi: 10.1038/nsmb.2719. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.