


Abstract
4-Hydroxy-2-nonenal (HNE) is a reactive α,β-unsaturated aldehyde generated during oxidative stress and subsequent peroxidation of polyunsaturated fatty acids. Here, Werner protein (WRN) was identified as a novel target for modification by HNE. Werner syndrome arises through mutations in the WRN gene that encodes the RecQ DNA helicase which is critical for maintaining genomic stability. This hereditary disease is associated with chromosomal instability, premature aging and cancer predisposition. WRN appears to participate in the cellular response to oxidative stress and cells devoid of WRN display elevated levels of oxidative DNA damage. We demonstrated that helicase/ATPase and exonuclease activities of HNE-modified WRN protein were inhibited both in vitro and in immunocomplexes purified from the cell extracts. Sites of HNE adduction in human WRN were identified at Lys577, Cys727, His1290, Cys1367, Lys1371 and Lys1389. We applied in silico modeling of the helicase and RQC domains of WRN protein with HNE adducted to Lys577 and Cys727 and provided a potential mechanism of the observed deregulation of the protein catalytic activities. In light of the obtained results, we postulate that HNE adduction to WRN is a post-translational modification, which may affect WRN conformational stability and function, contributing to features and diseases associated with premature senescence.
INTRODUCTION
Werner syndrome (WS) is a rare human autosomal, recessive disease that results in premature aging. WS cases are characterized by numerous progeroid pathologies including osteoporosis, atherosclerosis, type II diabetes, cataracts and a marked increase in the incidence of cancer, particularly sarcomas (1). The leading cause of death is myocardial infarction. WS cells display genomic instability with chromosomal deletions, elevated rates of homologous recombination, prolonged S-phase DNA synthesis and defective telomere maintenance (2). WS is caused by the loss of function of a single gene (3) that encodes a 1432-residue protein, Werner protein (WRN) (3,4). WRN is a member of the RecQ helicase family. RecQ helicases play important roles in the maintenance of genomic stability. They act in many DNA metabolic processes, including DNA replication, recombination, base excision repair (BER) and transcription (5). DNA helicases of the RecQ family have a broad amino acid sequence homology to the Escherichia coli RecQ helicase. These 3′–5′ DNA helicases unwind a wide variety of potentially recombinogenic DNA structures, including four-way junctions, D-loops and G-quadruplex DNA. Humans have five RecQ homologs, RECQL/RECQL1, BLM/RECQL2, WRN/RECQL3, RECQL4 and RECQL5 (6). Mutations in Bloom (BLM) and RECQLQ4 cause Bloom syndrome and Rothmund–Thomson syndrome, respectively, and similarly to the defect in WRN predispose to cancer. WRN is unique among all RecQ helicases in it displaying both 3′–5′ helicase/adenosine triphosphatase (ATPase) and 3′–5′ exonuclease activities (7).
WRN contains four domains comprising an exonuclease (8), a helicase/ATPase, a RecQ C-terminal (RQC) (9), a helicase-and-ribonuclease D/C-terminal (10) and a nuclear localization signal (NLS, Supplementary Figure S1) (11). The helicase and RQC domains combine to form the catalytic core of the protein. The helicase domain has the RecQ consensus helicase motifs, I, Ia, Ib and II–VI (3). Motif I, motif II and motif VI are all needed for adenosine triphosphate (ATP) binding and hydrolysis. A part of motif I known as the Walker A motif is crucial for ATP binding. The residues of the motif interact with ATP phosphate groups and with magnesium ions. Mutations found in the WRN gene are stop codons, splice site variants and insertions/deletions that generate frameshift mutations (12). Most mutations lead to the synthesis of C-terminal truncated protein without the NLS, which cannot localize to the nucleus (13). Biochemical characterization of the human WRN has shown its preferential activity on various branched DNAs, including bubbles, stem-loops, forks and Holliday junctions, as well as on RNA-DNA duplexes, triplexes and tetraplexes, implying for WRN roles in DNA replication, recombination and repair (14,15). Post-translational modifications of WRN modulate its enzymatic activity, and thereby could be an important mechanism for the regulation of WRN functions in response to DNA damage (16,17).
Since mutations in the WRN gene result in so many aging-like phenotypic changes, it has been reasoned that polymorphisms in the WRN gene may also contribute to age-related pathologies in subjects not affected by the syndrome, thus influencing aging in the population at large. One of the most common and intensively studied is the polymorphism at amino acid 1367 Cys (TTG)/Arg (CTG). The 1367Arg variant of WRN has been associated with protection from age-related diseases (18–22), suggesting it has a functional role.
Lipid peroxidation (LPO), which is significantly increased during inflammation, is implicated in aging as well as in the pathogenesis of numerous human diseases, including atherosclerosis, cancer, diabetes and arthritis (23,24). One of the major LPO products is 4-hydroxy-2-nonenal (HNE), a highly reactive α,β-unsaturated hydroxyalkenal, produced by the oxidative degradation of ω-6 polyunsaturated fatty acids of phospholipids, such as linoleic and arachidonic acids (25). HNE is considered a biomarker of oxidative stress in pathophysiological processes since elevated levels of HNE were detected in vivo in several pathological conditions, including inflammation, and various pathologies like liver fibrosis, Alzheimer's diseases, amyotrophic lateral sclerosis, Parkinson's diseases and Huntington diseases (4,26–28). Increased levels of HNE-protein adducts have also been detected in blood plasma from patients with acute myocardial infarction undergoing coronary angioplasty, a finding indicating a consistent association of oxidative stress with acute myocardial infarction (29). LPO may also occur under physiological conditions. HNE level in mammals is tissue dependent and increases with age. HNE concentration in plasma ranges from 0.2 to 2.8 μM; however, under conditions of oxidative stress, including inflammation, HNE may accumulate in plasma and tissues (especially locally in membranes) at concentrations from 10 to 100 μM, or even to milimolar (30–36).
HNE is highly reactive and readily reacts with cellular macromolecules, such as proteins and DNA. This may lead to protein carbonylation and formation of exocyclic DNA adducts, most of which are anticipated to be highly mutagenic. However, HNE reacts much better with proteins than DNA. Thus, protein modification most likely represents one of the main mechanisms by which HNE influences physiological as well as pathological processes (37). HNE is an electrophile due to its three functional groups that react with nucleophilic amino acid residues of proteins forming covalent adducts with protein nucleophilic side chains. HNE reaction with the thiol group of cysteine, the primary amino-group of lysine or the imidazole group of histidine leads to formation of stable Michael addition adducts with a hemiacetal structure (38). The nucleophilic attack by thiol or amino groups occurs primarily at carbon 3 and, second, at the carbonyl carbon 1. In most cases adduction of HNE to proteins leads to the inhibition of protein biological activity. For example, we showed previously that the ATPase activity of Cockayne syndrome group B protein was abolished after pre-incubation with 200 μM HNE (39). Accordingly, Schwarzer et al. found that formation of HNE–protein kinase C (PKC) adduct after treatment of human monocytes with 10–100 μM HNE totally inhibited PKC activity (40). Higher doses of HNE (150–300 μM) were needed to inactivate 50% activity of microsomal glucose-6-phosphatase (34). In the case of Hsp72 or Hsp90, the modification of very sensitive critical thiols by physiologically relevant concentrations of HNE (10 and 45 μM, respectively) impaired protein binding and ATPase activity (41,42). The exposure of purified human SIRT3 to 100 μM HNE resulted in about 75% inhibition (43). Also treatment of recombinant human Akt2 with 20 or 40 μM HNE inhibited Akt2 activity by 30% or 85%, respectively (44). Furthermore, it has been demonstrated that HNE-induced cellular senescence in leukemic cell lines by inhibiting telomerase activity (45). Moreover, adduction by HNE made protein more susceptible to proteolysis by the proteasomal pathway (46). However, despite overwhelming data showing impairment of catalytic activity of HNE modified proteins, in some cases formation of HNE-protein adducts can have an activating effect. Low concentrations of HNE (1–10 μM) stimulated the activity of phosphoinositide-specific phospholipase C (47).
We report here that the WRN protein undergoes HNE adduction both in vitro and in vivo. The results show that such post-translational modification of WRN by HNE affects its catalytic activities. Mass spectrometry and in silico modeling enabled us to identify HNE-modified amino acids and determine how they affect WRN conformation and function. We speculate that accumulation of HNE-WRN adducts may contribute to the premature aging resembling the WS phenotype.
MATERIALS AND METHODS
Materials
4-Hydroxy-2-nonenal was synthesized as described (48) with some modifications done by Marek Komisarski and Dr. Anna Biela. Monoclonal mouse HNE antibody was from R&D Systems (Minneapolis, MN, USA), polyclonal rabbit WRN (H-300), monoclonal mouse WRN (D-6), normal rabbit and mouse immunoglobulin Gs (IgGs) and Protein A/G PLUS-Agarose beads were from Santa Cruz Biotechnology (Dallas, TX, USA). Recombinant histidine-tagged human WRN protein was purified as described previously (49).
DNA substrates
DNA oligodeoxynucleotides were synthesized and purified by Metabion International AG (Martinsried, Germany). The sequences of the oligodeoxynucleotides are shown in Table 1. Template 22–15B or TelY or 43–5over together with respective top 22–15A or TelX or 32–5over primers were designed to form 37 or 49 nt fork or 32/43 nt 5′-overhang partial duplex substrates. Top primers were labeled at the 5′-end with T4 polynucleotide kinase (Optikinase, Santa Clara, CA, USA) and [γ-32P]ATP (GE Health Sciences) followed by annealing as described previously (50).
Table 1. Oligonucleotide sequences for DNA substrates (5′ to 3′).
| Oligonucleotide, size | Sequence |
|---|---|
| Helicase primers | |
| 22-15A, 37 nt | TTTTTTTTTTTTTTTTTAGGGTTAGGGCATGCACTAC |
| 22-15B, 37 nt | GTAGTGCATGCCCTAACCCTAATTTTTTTTTTTTTTT |
| Exonuclease primers | |
| TelX, 49 nt | TTTTTTTTTTTTTTTGGTGATGGTGTATTGAGTGGGATGCATGCACTAC |
| TelY, 49 nt | GTAGTGCATGCATCCCACTCAATACACCATCACCTTTTTTTTTTTTTTT |
| 32-5over, 32 nt | TGACGTGACGACGATCAGGGTACGTTCAGCAG |
| 43-5over, 43 nt | AGTGCAGACTGCTGCTGAACGTACCCTGATCGTCGTCACGTCA |
Partial duplex DNA substrates were constructed by annealing appropriate top primer to template oligonucleotide. 22-15A and 22-15B were used to construct the 37 nt fork, TelX and TelY – the 49 nt fork and 32-5over and 43-5over—the 32/43 nt 5′-overhang.
Covalent modification of recombinant WRN with HNE
Covalent modification of human WRN was achieved by incubating purified protein (0.1 μM) in HW buffer (40 mM Tris, pH 8, 4 mM MgCl2, 5 mM DTT and 100 μg/ml bovine serum albumin (BSA)) with various concentrations of HNE. Treatment involved final concentrations of 10, 25, 50, 100, 500 and 1000 μM of HNE for 10 min at 30°C. These reaction mixtures were then used to measure WRN helicase, exonuclease and DNA binding activities or were boiled in sodium dodecyl sulphate (SDS) loading buffer and applied to 4–15% SDS-polyacrylamide gel electrophoresis(SDS-PAGE) followed by transfer to polyvinylidene difluoride (PVDF) membrane (BIO-RAD, Hercules, CA, USA). Visualization of adduct formation was monitored by western blot with polyclonal rabbit anti-HNE antibody. Membranes were then stripped and reprobed for monoclonal mouse anti-WRN antibody. Immunoblots were visualized by chemiluminescence using a MultiImage Light Cabinet and analyzed by associated FluorChem Q Software (Alpha Innotech Corporation, Santa Clara, CA, USA).
WRN helicase, exonuclease and ATPase activity assay
Helicase reactions were performed at 37°C in HW buffer. Reaction mixtures (10 μl) contained 1 nM 5′-[32P]-labeled oligonucleotide-based partial duplex (37 nt fork, 22-15A/22-15B), 2 mM ATP and 2.5 or 5 nM WRN modified with different concentrations of HNE. Reactions were started with the enzyme and terminated after 30 min by adding 0.5 reaction volumes of SDS/stop solution (2% SDS, 50 mM ethylenediaminetetraacetic acid (EDTA), 30% glycerol, 0.1% bromophenol blue and 0.1% xylene cyanol). The reaction products were separated on 8% native PAGE (acrylamide to bis-acrylamide, 19:1) and run in Tris-borate-EDTA buffer (90 mM Tris-borate, pH 8.3, 2 mM EDTA) at 150 V at room temperature. Radiolabeled DNA bands were visualized by autoradiography using a PhosphorImager (FLA-7000, FUJIFILM, Tokyo, Japan). Quantitative analyses of the band shifts were performed using Multi Gauge software (FUJIFILM). The relative amounts of displaced products were expressed as percentage of total DNA.
Exonuclease assays were carried out at 37°C in HW buffer. Reaction mixtures (10 μl) contained 1 nM 5′-[32P]-labeled oligonucleotide-based partial duplex (49 nt fork, TelX/TelY or 32/42 nt 5′-overhang, 32-5over/43-5over) and 5 or 10 nM WRN protein modified with different concentrations of HNE. When 32/42nt 5′-overhang was used as a substrate, mixtures were supplemented with 2 mM ATP. Reactions were started with the enzyme and terminated after 30 min by adding 0.5 reaction volumes of formamide/stop solution (95% formamide, 5 mM EDTA, 0.1% bromophenol blue and 0.1% xylene cyanol). The reactions were heat-denatured for 5 min at 90°C and reaction products were separated on 15% denaturing PAGE (acrylamide to bis-acrylamide, 19:1). Products were visualized using a PhosphorImager, and quantitifications were performed using the associated software (FUJIFILM). The relative amounts of cut products were expressed as percentage of total DNA.
ATPase assays were performed at 37°C in AW Buffer (20 mM HEPES–NaOH, pH 7.8, 0.05 mM ATP, 40 μg/ml BSA, 1 mM DTT). Reaction mixtures (10 μl) contained 1 μCi [γ-32P] ATP, 200 nM oligonucleotide-based partial duplex (37 nt fork, 22-15A/22-15B) and 10 nM WRN modified with different concentrations of HNE. Reactions were started with the enzyme and stopped after 1 h by the addition of 5 μl of 0.5 M EDTA. ATP hydrolysis was analyzed on a polyethylenimine-cellulose thin layer chromatography plate (PEI Cellulose F, Merck, Darmstadt, Germany) developed in 1 M formic acid and 0.8 M LiCl. Plates were visualized by PhosphorImaging and quantitification was performed using Multi Gauge software (FUJIFILM).
WRN catalytic activities on DNA templates pre-incubated with HNE
Radiolabeled oligonucleotide partial duplexes (1 nM 37 nt fork, 22-15A/22-15B and 1 nM 49 nt fork, TelX/TelY) were incubated with HNE (0.5 and 1 mM) in HW buffer for 30 min at 37°C. Templates were then purified using clean-up columns (Merk Millipore, Darmstadt, Germany) and were used as substrates for WRN helicase and exonuclease assays (5 or 20 nM of protein was used, respectively). The catalytic reactions were run and analyzed as described above.
Cell culture
Human normal (GM00637) fibroblasts and fibroblasts obtained from patients with WS (AG11395) were purchased from Coriell Cell Repositories (Camden, NJ, USA). Cells were maintained in Eagle's Minimum Essential Medium with Earle's salts supplemented with 10% fetal bovine serum and non-essential amino acids (Life Technologies, Carlsbad, CA, USA) in a humidified 5% CO2 atmosphere at 37°C.
Co-immunoprecipitation (co-IP) assay
Whole cell extracts were prepared from normal (WT) or WS fibroblasts treated in serum-free medium with 100 μM HNE for 2 h at 37°C. Cells were harvested and disrupted by re-suspending in 1 ml of cell extraction buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM NaF, 1 mM Na3VO4, 0.5% NP-40, 0.5% Triton X-100, 0.5 mM PMSF and 1 mM DTT) supplemented with a protease inhibitor cocktail (Complete, Roche, Basel, Switzerland) and by incubating for 30 min on ice followed by centrifugation at 16 000 × g for 15 min.
Cell extracts (1 mg each) were pre-cleared with Protein A/G PLUS-Agarose beads and incubated with 2 μg of polyclonal rabbit anti-WRN antibody or monoclonal mouse anti-HNE, or 2 μg of normal rabbit or mouse IgG as a negative controls in a total volume of 500 μl of cell extraction buffer for 16 h at 4°C. Each sample was then incubated with Protein A/G PLUS-Agarose beads (30 μl) for 4 h at 4°C. After extensive washing with cell extraction buffer, bound proteins were eluted by boiling beads in sample buffer for 5 min and analyzed by western blotting with mouse anti-HNE or rabbit anti-WRN or mouse anti-WRN followed by chemiluminescence analysis.
For helicase, exonuclease and ATPase assays with immunocomplexes, rabbit IgG or rabbit anti-WRN immunoprecipitates were washed three times with cell extraction buffer and two times with HW or AW buffer, followed by the addition of the radiolabeled DNA substrates. The catalytic reactions were run and analyzed as described above but incubation times were longer: 1 h for the helicase and exonuclease activity on fork substrates and 2 h for the ATPase and exonuclease on 5′-overhang.
Electrophoretic mobility shift assay
The binding reaction mixtures (10 μl) contained 10 mM Hepes-KOH, pH 7.8, 1 mM EDTA, 0.5 mM DTT, 50 ng/μl BSA, 1 nM of 37 nt 5′-[32P]-labeled 22-15A/22-15B or 1 nM of 49 nt 5′-[32P]-labeled TelX/TelY and 25 nM WRN. The mixtures were incubated for 30 min on ice, after which glycerol was added to final concentration of 25%. Aliquots were removed and analyzed by electrophoresis on 4% non-denaturing polyacrylamide gels (80:1 or 37.5:1 acrylamide:bisacrylamide) in buffer containing 20 mM Hepes-KOH pH 7.8, and 0.1 mM EDTA at 100 V for 2 or 3 h, respectively, at 4°C. The gels were visualized using a PhosphorImager.
Mass spectrometry
Recombinant human WRN was incubated with increasing concentrations of HNE followed by NaBH4 (5 mM) reduction at 25°C for 30 min to stabilize protein adducts. Samples were digested overnight with trypsin (sequencing Grade Modified Trypsin–Promega V5111). Peptide mixtures were analyzed by LC-MS-MS/MS (liquid chromatography coupled to tandem mass spectrometry) using Nano-Acquity (Waters) LC system and Orbitrap Velos mass spectrometer (Thermo Electron Corp., San Jose, CA, USA). Briefly, peptide mixture was applied to RP-18 pre-column (nanoACQUITY Symmetry® C18 – Waters 186003514) using water containing 0.1% trifluoroacetic acid (TFA) as mobile phase and then transferred to nano-HPLC RP-18 column (nanoACQUITY BEH C18 – Waters 186003545) using an acetonitrile gradient (0–30% AcN in 160 min) in the presence of 0.05% formic acid with the flow rate of 250 nl/min. Column outlet was directly coupled to the ion source of the spectrometer working in the regime of data dependent MS to MS/MS switch. A blank run ensuring lack of cross-contamination from previous samples preceded each analysis. Acquired raw data were processed by Mascot Distiller followed by Mascot Search (Matrix Science, London, UK, on-site license) against SwissSprot database (v. 10_2012) restricted to human entries. Search parameters for precursor and product ions mass tolerance were 20 ppm and 0.4 Da, respectively; enzyme specificity: trypsin, missed cleavage sites allowed: 1, fixed modification of cysteine by carbamidomethylation and variable modification of lysines carboxymethylation, methionine oxidation and HNE of cysteines, lysines and histidines. Peptides with Mascot Score exceeding the threshold value corresponding to <5% false positive rate, calculated by Mascot procedure, and with the Mascot score above 30 were considered to be positively identified. Peptides with indicated HNE modification identified by Mascot were manually validated. During fragmentation, the peptide dissociates into two fragments. Fragments containing the N-terminal part of the peptides are termed b series ions (singly ‘b’ and doubly ‘b++’ charged), and, respectively, fragments containing the C-terminal part are named y series ions (singly ‘y’ and doubly ‘y++’ charged) (51).
Molecular modeling
All simulations were performed using Yasara Structure package (version 13.4.21, Graz, Austria). Helicase and RECQ domains of the Werner protein (569 residues corresponding to fragment 521–1089) were modeled by homology, basing on four closest templates identified in RCSB Protein Data Bank (PDB) records, IDs: 1OYW, 2V1X, 1OYY, 2WWY. Three of them, for which re-refinement was found to improve the structure quality Z-score, were downloaded from a databank of updated and optimized X-ray structure models, PDB_REDO (www.cmbi.ru.nl/pdb_redo). According to overall quality Z-score, the best models were built on the basis of 2V1X and 1OYW PDB records. However, the final hybrid model, which combines the best parts of top models, was scored higher than the letters, and thus was further used in the analyses. ADP molecule was placed according to known structure of human RECQ-like DNA helicase (PDB ID: 2V1X). Model of single-strand DNA bound to WRN was adopted from human RECQ-like helicase in complex with a DNA substrate (PDB ID: 2WWY). HNE molecules were attached either to Lys577 or Cys727 of the WRN hybrid model with the YAMBER force field, and further subjected to minimization (1000 iteration) followed by simulated annealing procedure in the presence of explicit water molecules. In these steps coordinates of residues located outside 5Å sphere of HNE were preserved. Finally, the whole system was refined by 200 ps unrestrained molecular dynamics in NPT ensemble.
RESULTS
Human WRN is a target for covalent modification by HNE
To determine whether HNE covalently binds to WRN protein, we examined its in vitro adduction to WRN by incubating the mixture of purified recombinant human WRN (0.1 μM) and BSA (1.5 μM) with HNE in the concentration range of 10–1000 μM over a 10 min time course at 30°C. The presence of BSA in the reaction mixture assured WRN stability throughout the course of HNE modification and enzymatic activity assays. The molar ratio of HNE to WRN/BSA varied from 6- to 600-fold. The reaction products were analyzed by western blot with the use of anti-HNE antibody. As shown in Figure 1A, upper panel, the extent of HNE modification of WRN protein increased in a concentration-dependent manner. Quantification of the western blots indicated a 5- to 200-fold increase in the level of HNE-modified proteins compared to the mock-treated control (Figure 1B). Analysis of the same blot with anti-WRN antibody showed equal amounts of WRN in each reaction (Figure 1A, lower panel). These results indicate that HNE binds to WRN in vitro.
Figure 1.
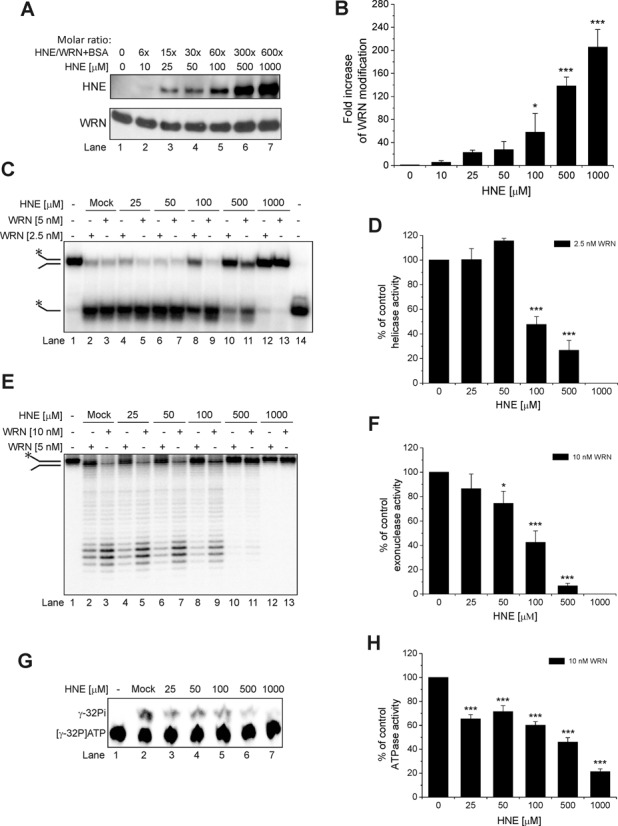
Effect of HNE on recombinant WRN. (A) HNE covalently modifies WRN in a concentration-dependent manner. Purified WRN in the presence of BSA was incubated with increasing molar ratios of HNE for 10 min at 30°C. Samples were boiled in SDS loading buffer, subjected to SDS-PAGE, blotted and probed for HNE followed by stripping and reprobing for anti-WRN antibodies. (B) Fold increase in WRN modification from the data shown in (A). (C) Effect of HNE modification on WRN helicase activity. Purified WRN was incubated with increasing molar ratios of HNE and enzymatic assays were performed as described in Materials and Methods. (D) Percent of control helicase activity from the data shown in (C). (E) Effect of HNE modification on WRN exonuclease activity. Purified WRN was incubated with increasing molar ratios of HNE, and enzymatic assays were performed. (F) Percent of control exonuclease activity from the data shown in (E). (G) Effect of HNE modification on WRN ATPase activity. (H) Percent of control ATPase activity from the data shown in (G). Mock, control enzymatic activity performed with WRN that has been treated as HNE modified WRN but without HNE. Lane 14, heat denatured helicase substrate. All data bars are the mean of at least three independent experiments with SDs indicated by error bars. Statistical analysis occurred via one-way analysis of variance with Tukey's multiple-comparison test (*P < 0.05; ***P <0.001).
HNE adduction alters the enzymatic activity of the WRN protein in vitro
It was previously shown that HNE directly affects the activity of some DNA repair proteins. Here, we investigated whether HNE could also affect the catalytic activities of WRN protein. First, we focused on the helicase activity. The DNA substrate used in this assay consisted of a 37 bp partial duplex with 15 unannealed nucleotides (37 nt fork). The fork substrate was susceptible to WRN unwinding and 37 nt single-stranded products were visible (Figure 1C, lanes 2 and 3). In the presence of 2.5 nM WRN, 79% of 1 nM substrate was unwound. Under these conditions, we analyzed WRN unwinding as a function of HNE concentration. Depending on the concentration used for the modification, HNE had different effects on WRN helicase activity (Figure 1C, lanes 4–13). At a concentration of 25 μM, HNE did not influence the enzyme (Figure 1C, lanes 4–5 and D). WRN modified with 50 μM HNE displayed slightly increased helicase activity, which was indicated by slight increase in the percent of unwound substrate (116% compared to the mock-treated WRN; Figure 1C, lanes 6–7 and D). When WRN was adducted with higher HNE concentrations (100–1000 μM), helicase activity decreased gradually. Only 48% and 27% of the substrate was unwound by 2.5 nM WRN, in comparison to control, in the reaction with WRN pre-treated with 100 and 500 μM HNE, respectively (Figure 1C, lanes 8 and 10 and D). WRN modified with 1000 μM HNE did not unwind the 37 nt fork at all (Figure 1C, lanes 12–13 and D).
The DNA substrate used in the exonuclease assay consisted of a 49 bp partial duplex with 15 unannealed nucleotides (49 nt fork). This fork substrate was susceptible to WRN degradation and several shorter products were generated (Figure 1E, lanes 2 and 3). Mock-treated WRN (10 nM) degraded 77% of the 1 nM substrate (Figure 1E, lane 2). Increasing HNE concentration (25–1000 μM) used for WRN modification yielded gradual inhibition of the DNA substrate cleavage (Figure 1E, lanes 4–13 and F). WRN (10 nM) modified with 25 μM HNE displayed a slight reduction in exonuclease activity (87% of the control activity) but after adduction with 50, 100 and 500 μM HNE only 75%, 42% and 7%, respectively, of 49 nt fork substrate was degraded compared to the mock-treated WRN (Figure 1E, lanes 7, 9 and 11 and F). There was no cleavage by WRN modified with 1000 μM HNE (Figure 1E, lanes 12 and 13 and F).
Next, we investigated whether HNE was able to inactivate the ATPase activity of the WRN protein in vitro. The DNA substrate used as cofactor in the ATPase assay was a 37 nt fork. Mock-treated WRN (10 nM) in the presence of 200 nM co-factor hydrolyzed 28% of the 1 μCi ATP (Figure 1G, lane 2). We observed gradual inhibition of ATP hydrolysis using WRN protein pre-treated with HNE, (Figure 1G, lanes 3–7 and H). At 25–100 μM HNE, compared to the mock-treated WRN, some reduction of ATPase activity was observed (65% to 60% of ATP underwent hydrolysis). At 500 μM HNE, 46% of ATP was hydrolyzed to inorganic phosphate and at 1000 μM HNE, 21% as compared to control.
BSA also reacts with HNE and it is possible that if BSA was not added to the reactions WRN would be even more sensitive to HNE modification than reported by above data. However, our experimental conditions with BSA resemble more precisely conditions in the cells and tissues (e.g. serum albumin in blood), where there is a lot of shielding proteins, which could sequester HNE away from WRN.
It is well established that HNE reacts with DNA much less efficiently than with proteins, however, in order to exclude the possibility that high concentrations of HNE could react with the DNA fork substrates and that potential HNE adduction to the DNA was responsible, at least partially, for the decreased WRN activities, we performed additional control experiments using the radiolabeled oligonucleotides (37 nt fork, 22-15A/22-15B and 49 nt fork, TelX/TelY) pre-incubated with high HNE concentrations (0.5 and 1 mM). Supplementary Figure S1 clearly shows that WRN helicase and exonuclease activities on untreated templates and on the templates incubated with HNE were very similar. Untreated 1 nM 37 nt fork in the presence of 5 nM WRN was unwound to 92% (Supplementary Figure S1A, lane 2 and B) and this substrate preincubated with 0.5 and 1 mM HNE was unwound to 92% and 91%, respectively (Supplementary Figure S1A, lanes 3 and 4 and B). Untreated 1 nM 49 nt fork in the presence of 20 nM WRN was cleaved to 87% (Supplementary Figure S1C, lane 2 and D) and this substrate pre-incubated with 0.5 and 1 mM HNE was degraded to 92% (Supplementary Figure S1A, lane 3 and 4 and B).
These results clearly suggest that the decrease in helicase, exonuclease and ATPase activities was due to modification of the WRN protein by HNE. These observations demonstrate that adduction of WRN by pathologically relevant concentrations of HNE strongly affects WRN catalytic activities and point to the functional significance of this WRN modification.
Covalent modification of WRN by HNE in vivo
To determine whether WRN undergoes HNE adduction in cells, we performed co-IP experiments using cell extracts prepared from human fibroblasts obtained from either an unaffected (WT) or a WS individual. Cells were mock or HNE pre-treated. The incubation of the extracts with an antibody against WRN specifically immunoprecipitated endogenous WRN from WT but not WS extract, as expected (Figure 2A, middle panel, lanes 6–8), and also co-precipitated HNE adducted WRN from WT extract (Figure 2A, upper panel, lane 7). This result was confirmed by a reciprocal co-IP experiment, in which antibody to HNE co-immunoprecipitated endogenous HNE adducted WRN (Figure 2B, upper panel, lanes 6–7). As expected, there was no such signal in immunoprecipitate purified from WS cell extract (Figure 2B, upper panel, lane 8). Control experiments showed that pre-immune IgG did not immunoprecipitate WRN or HNE-WRN (Figure 2A, upper and middle panels, lanes 4–5 and B, upper panel, lanes 4–5). These results demonstrate that WRN forms a covalent complex with HNE in vivo.
Figure 2.
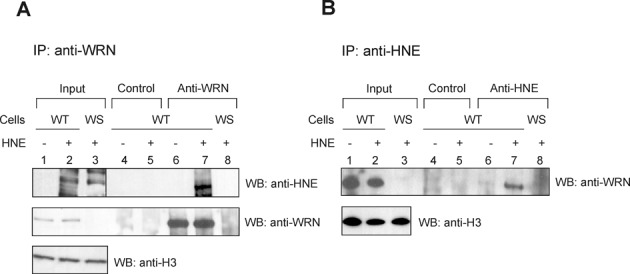
WRN is modified by HNE in vivo. Human normal (WT) or WS fibroblasts were pre-treated with 100 μM HNE for 2 h. Cell extracts were prepared and immunoprecipitated with rabbit anti-WRN or mouse anti-HNE antibodies (lanes 6–8, A and B, respectively) or control IgG (lanes 4 and 5, A and B). The immunoprecipitates were analyzed by SDS-PAGE and western blot with mouse anti-HNE or mouse anti-WRN (A), or rabbit anti-WRN antibodies (B). Input, 2.5% loaded. Lanes 1, input from WT cell extract. Lanes 2, the same as lanes 1, but cells were pre-treated with HNE. Lanes 3, Input from WS cell extract pre-treated with HNE.
HNE adduction inhibits helicase, exonuclease and ATPase activities of WRN in vivo
To investigate the potential consequence of HNE adducted WRN in cells, we measured the helicase and the exonuclease activities in WRN immunocomplexes purified from human WT and WRN null fibroblasts, mock or HNE pre-treated. The washed anti-IgG or anti-WRN immunoprecipitates were incubated with radiolabeled DNA substrates, 37 nt fork to assess the helicase activity and two substrates for the exonuclease activity, 49 nt forked and 32/43 nt 5′-overhang. The last substrate requires addition of ATP to be efficiently degraded by WRN. To assess the ATPase activity immunocomplexes were incubated with [γ-32P] ATP and 37 nt fork oligodeoxynucleotide as a cofactor. The results showed that the anti-WRN immunoprecipitates purified from untreated WT cells efficiently unwound the 37 nt forked duplex substrate (Figure 3A and E, lanes 3) and degraded both the 49 nt fork and the 32/43 nt 5′-overhang (Figure 3B, C and E, lanes 3). However, it is possible that other helicases which complex with WRN, e.g. BLM, could come down with the immunoprecipitation and contribute to the observed unwinding activity as well as to its inhibition. We also observed some degradation in immunocomplexes purified with control IgG and those obtained from WS cell extracts with the use of anti-WRN antibody (Figure 3B, C and E, lanes 1, 2 and 5). However, under these conditions, there was no detectable helicase or ATPase activity (Figure 3A–E, lanes 1, 2 and 5). The unwinding, degradation and ATP hydrolysis were strongly inhibited in immunocomplexes purified from WT fibroblasts pre-treated with HNE (Figure 3, lanes 4). The helicase activity was inhibited most efficiently (only 15% of the substrate was unwound in comparison to 51% of mock-treated control) (Figure 3A and E, lanes 4 and 3, respectively). The 49 nt fork and the 32/43 nt 5′-overhang were cleaved in 25% and 33%, respectively; whereas in reactions with untreated immunocomplexes they were cleaved in 64% and 75%, respectively (Figure 3B, C and E, lanes 4 and 3, respectively). In comparison with the very efficient inhibition of the helicase activity, the ATPase was much less inhibited, showing 33% hydrolysis in the presence of immunocomplexes treated with HNE. In the reaction with mock-treated ones, the hydrolysis was 50% (Figure 3D and E, lanes 4 and 3, respectively). These results suggest that WRN adduction by HNE inside the cell interferes with the catalytic activities of the enzyme.
Figure 3.
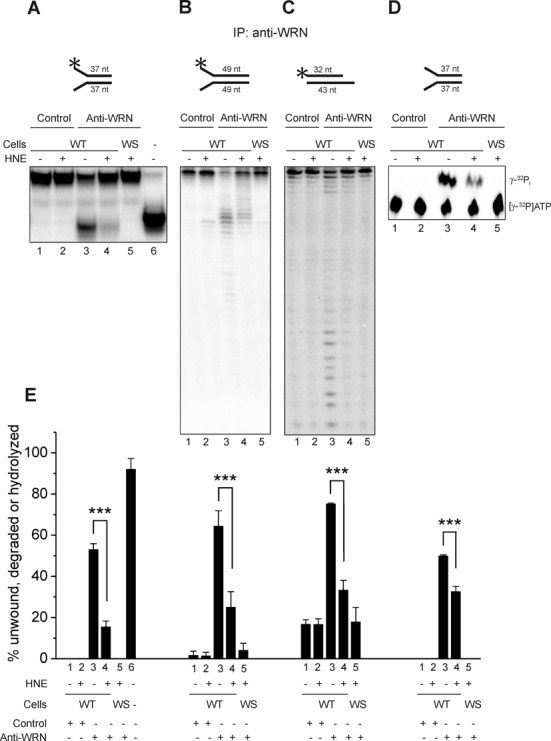
HNE adduction of WRN inhibits its helicase and exonuclease activities in vivo. Human normal (WT) or WS fibroblasts were pre-treated with HNE and the cell extracts were immunoprecipitated with rabbit anti-WRN antibody or control rabbit IgG. The immunoprecipitates were used in the measurements of WRN helicase (A), exonuclease (B and C) or ATPase activity (D) as described in Materials and Methods. Representative gels are shown. (E) Percent of helicase, exonuclease or ATPase activities from the data shown in A–D. Lanes 1, radiolabeled DNA substrate for helicase (A) or exonuclease (B and C) activity assays were reacted with control IgG immunocomplexes prepared from untreated WT fibroblasts. Lanes 2, the same as lanes 1, but cells were pre-treated with 100 μM HNE for 2 h. Lanes 3, the same as lanes 1, but substrates were reacted with immunocomplexes obtained against anti-WRN antibody. Lanes 4, the same as lanes 3, but cells were pre-treated with HNE. Lanes 5, the same as lane 4, but immunocomplexes were purified from WS fibroblasts. Lane 6, heat denatured helicase substrate. All data bars are the mean of three independent experiments with SDs indicated by error bars. Statistical analysis occurred via Student's t-test (***P < 0.001).
HNE modification does not alter the DNA binding affinity
To assess the mechanism by which aldehydic alkylation of WRN decreases its catalytic activities, a well established DNA binding assay with a 37 or 49 nt fork duplex substrate was used. As shown in Figure 4, the DNA binding of unmodified WRN was very slight and no reduction in the WRN affinity was observed following treatment with pathologically relevant concentrations of HNE (25–1000 μM). These data suggest that the functional effects of WRN modification by HNE are not related to decreased WRN DNA binding affinity.
Figure 4.
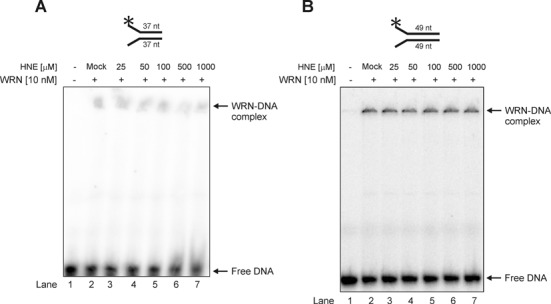
Modification of WRN by HNE does not change its DNA binding affinity on 22–15A/22–15B 37 nt (A) or TelX/TelY 49 nt partial DNA duplexes (B). Lane 1, control DNA substrate; lane 2, as 1 but DNA substrate was pre-incubated with mock-treated WRN; lanes 3–7, as 2 but WRN was treated with HNE at concentration of 25, 50, 100, 500 and 1000 μM, respectively. Mock, control enzymatic activity performed with WRN that has been treated as HNE modified WRN but without HNE.
Identification of HNE adducts on WRN protein
LC-MS/MS was applied for fine mapping of the HNE modification sites in human recombinant WRN. Analysis of trypsin digests of WRN resulted in MS peptide sequence coverage of 81% and revealed 6 HNE adducts (Supplementary Figure S2). Two of the identified adducts were localized within the ATPase domain, at Lys577 and Cys727. The remaining four modifications were in the C-terminal part of the protein, at His1290, Cys1367, Lys1371 and Lys1389 (Figures 5 and 6A and Supplementary Figure S3). Figure 5 displays the MS/MS spectrum of parent ions containing Lys577 and Cys727, respectively, where relative y and b ions confirm HNE adducts on WRN. The remaining MS/MS spectra of the HNE adducted peptides also confirmed modifications on His1290, Cys1367, Lys1371 and Lys1389 and are shown in Supplementary Figure S3.
Figure 5.

HNE modified WRN was characterized by LC-MS-MS/MS analysis. (A) Fragmentation spectrum of the DNVAVMATGYGK peptide—modified at Lys577. (B) Fragmentation spectrum of the NPQITCTGFDRPNLYLEVR peptide—modified at Cys727. During fragmentation, the peptide dissociates into two fragments. Fragments containing the N-terminal part of the peptides are termed b series ions (singly ‘b’ and doubly ‘b++’ charged), and, respectively, fragments containing the C-terminal part are named y series ions (singly ‘y’ and doubly ‘y++’ charged) (51). Tables represent a list of theoretical fragment ions calculated by the Mascot program, which served to identify modified peptide (fragment ions found in the spectra are shadowed). Assignment of dominating peaks from the spectra to the theoretical fragmentation ions definitely confirms the modification.
Figure 6.
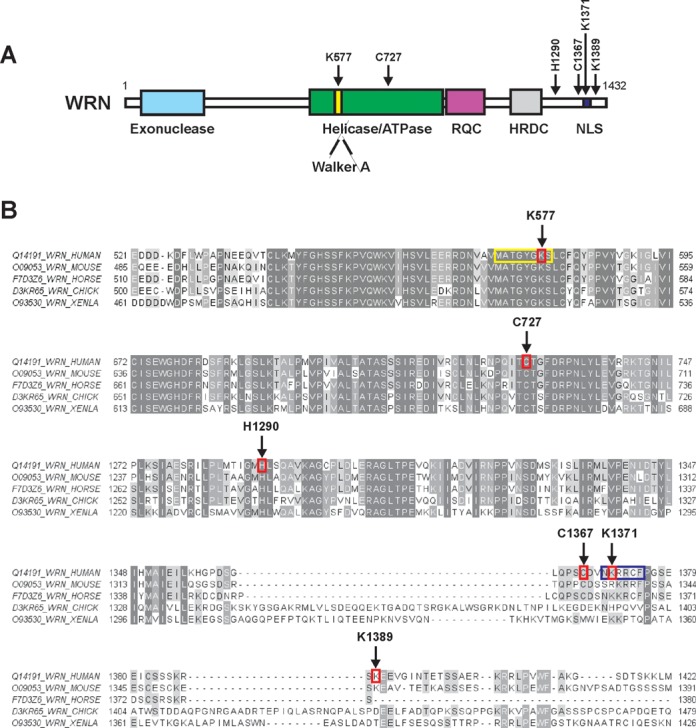
HNE modified amino acids identified in WRN. (A) Domain structure of the WRN protein. Positions of the HNE adducted amino acids are indicated by arrows. Functional domains are indicated below the structure. (B) Sequence alignment of WRN protein fragments (spanning part of the helicase and the C-terminus) of human, murine, horse, chicken (CHICK) and X. laevis (XENLA) origin. HNE modified amino acids, Lys577, Cys727, His1290, Cys1367, Lys1371 and Lys1389, are surrounded by red rectangles and indicated by arrows. Walker A motif (571–578) and nuclear localization sequence (1370–1375) are highlighted in yellow and dark blue rectangles, respectively. The peptides surrounding Lys577 are identically matched for seven residues toward the N-terminus and three residues toward the C-terminus. Human Lys577, Cys727, His1290, Cys1367, Lys1371 and Lys1389 correspond, respectively, to murine Lys541, Cys691, His1255, Cys1332, Arg1336 and Lys1354; horse Lys566, Cys716, His1280, Cys1359, Lys1363; chicken Lys556, Cys706, His1270, Cys1391, His1395 and Asp1438; and X. laevis Lys518, Cys668, His1238, Cys1348, Lys1352 and Thr1387. The picture was prepared with ClustalX2 (http://www.clustal.org) and visualized by Jalview (http://www.jalview.org).
Figure 6B provides a UniProt sequence comparison between forms of human, murine, horse, chicken and Xenopus laevis WRN (www.uniprot.org). This alignment shows strong conservation of Lys577, Cys727 and His1290, and suggests a shared susceptibility to alkylation of those specific amino acid residues for human, murine, horse, chicken and X. laevis forms of WRN.
Molecular modeling of HNE adducted WRN
In order to determine the structural consequences following HNE adduction of Lys577 and Cys727 and their impact on WRN function, we performed molecular modeling simulations. Based on the available PDB data, we constructed a model of the helicase and RQC domains of human WRN (521–1089 aa, Supplementary Figure S4A). The model was initially derived as a combination of 20 homology models built independently on the basis of E. coli RecQ catalytic core (PDB IDs: 1OYW and 1OYY) and human RECQL1 (PDB IDs: 2WWY and 2V1X; Supplementary Figure S4B). Further combination of energy minimization and restrained simulated annealing followed by 100 ps molecular dynamics was used to refine the protein structure with HNE modified Lys577 and Cys727. According to the model obtained, the addition of HNE to Lys577 precludes binding of ATP (ADP) to WRN since the locations of HNE and ATP overlap (Figure 7A and Supplementary Figure S4C). The HNE moiety that modifies Lys577 was found to occupy the ATP-binding site, thus interfering with normal ATPase and helicase functions. Figure 7B and Supplementary Figure S4C show peripheral location of HNE adducted to Cys727. Inspection of the modeled structures demonstrates that these modifications (i.e. Lys577 and Cys727) are not expected to influence the binding of DNA, which definitively interacts with distant regions of the WRN protein (Supplementary Figure S4D). Figure 7C and D illustrate the differences in the conformation of Lys577 and Cys727 as well as in several surrounding amino acid residues (including those of the Walker A motif) between native and aldehyde adducted WRN (amino acids residues are denoted as blue and green sticks, respectively). HNE addition results in minor structural differences, with root mean square deviation (RMSD) for side-chain heavy atoms of 1.55 Å. The RMSD for Lys577 was 1.37 Å, and for Cys727 equaled 1.24 Å. For the residues of the Walker A motif RMSD was 1.1 Å.
Figure 7.
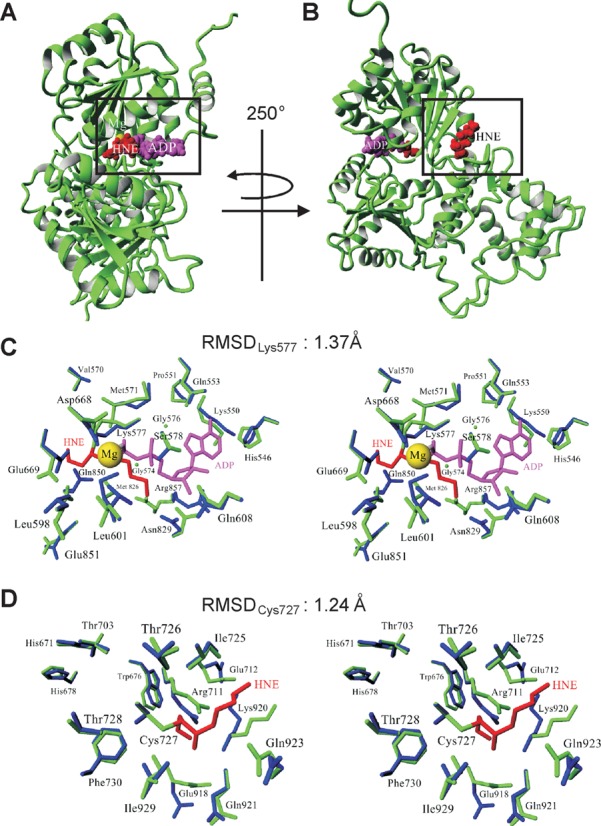
Structural changes in the helicase and RQC domains of the WRN protein upon HNE adduction. (A and B) Modeled structure of the WRN helicase and RQC domains with HNE modifications. Side chains of Lys577 (A) and Cys727 (B), which undergo HNE addition, are denoted in red using a space-filling model while the ADP residue is shown in magenta. Magnesium ion is marked as a yellow ball. (C and D) Stereo views demonstrating the changes in local architecture around Lys577 (C) and Cys727 (D) after HNE addition. The unmodified and modified proteins are denoted in blue and green, respectively. RMSDLys577 and RMSDCys727 denote the RMSD between unmodified and HNE adducted Lys577 and Cys727, respectively.
To further examine the effects of HNE modification, molecular surface maps with electrostatic potential were applied. Figure 8 and Supplementary Figure S5 clearly show differences in molecular surface and electrostatic potentials of the entire WRN model upon alkylation of Lys577 and Cys727, which may also affect the function of the enzyme. Moreover, an entrance site and internal cavity around the ATP binding pocket seen in native state clearly expanded upon HNE addition (Figure 8C and D, and Supplementary Figure S5C and D). Taken together, these data demonstrate the mechanism of HNE mediated inhibition of WRN helicase activity whereby the binding of ATP to the Walker motif is precluded by steric hindrances with HNE.
Figure 8.
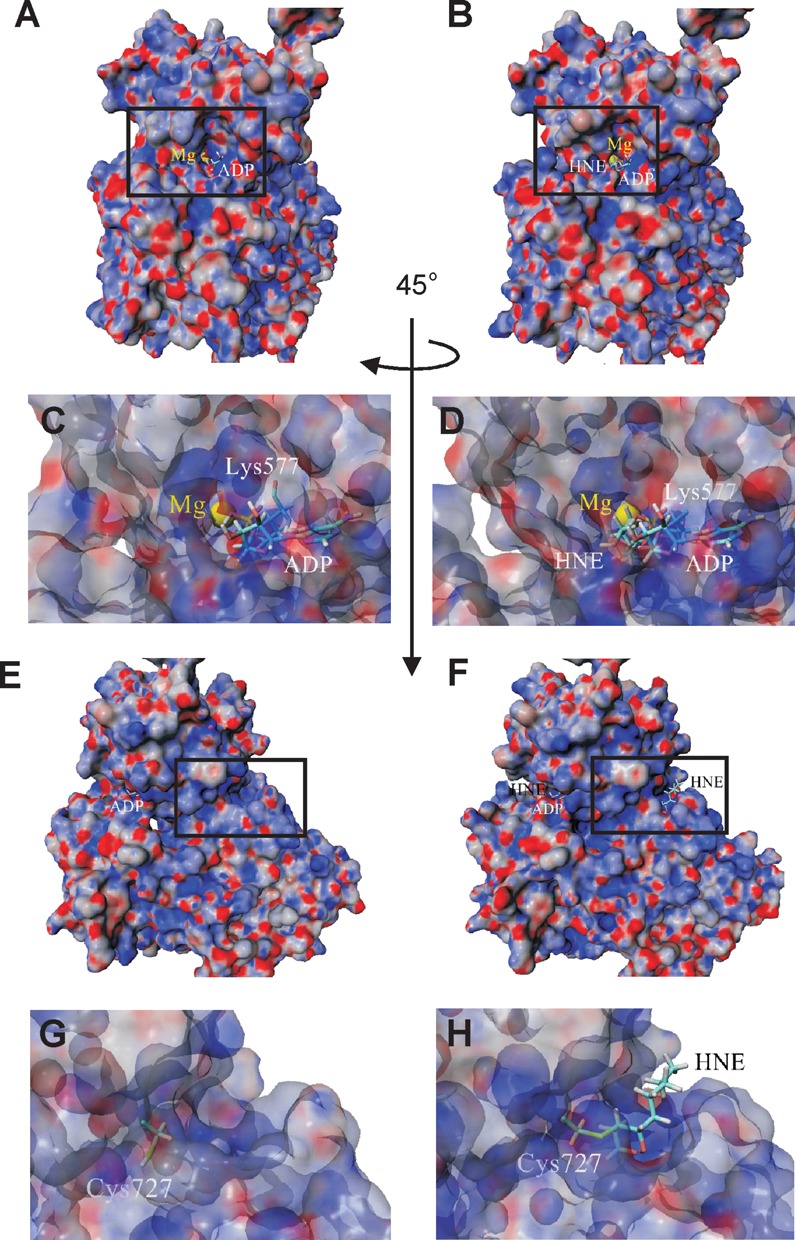
Changes in molecular surface of helicase and RQC domains of WRN protein upon HNE adduction. Surface representation of WRN electrostatic potential (±25 kT/e, red-negative, blue-positive, gray-neutral) before (A) and after HNE addition to Lys577 (B). A comparison of the vicinity of native (C) and HNE adducted Lys577 (D) show some changes in molecular surface in the proximity of the ATP (ADP) binding pocket. (E) Surface representation of helicase and RQC domains of the WRN protein after orthogonal rotation along the Y-axis of 45° in relation to structure shown in (A). (F) Surface representation of helicase and RQC domains of the WRN protein with HNE attached to Cys727. Detailed views of the vicinity of native (G) and modified (H) Lys727 illustrate changes in charge distribution and reveal surface location of HNE.
DISCUSSION
The maintenance of genome stability is critical for the suppression of cancer and premature aging. Mammalian cells are continuously exposed to reactive oxygen species, which are thought to be a major contributor to the aging process. Previous studies have indicated that the exposure of cells to even minimal transient stress causes substantial LPO (52). According to the oxidative theory of aging (53,54), oxidized lipoproteins may activate inflammatory events, which could lead to the generation of reactive oxygen species, DNA damage and mutagenesis. Genomic instability along with accumulation of DNA damage and cellular senescence is commonly seen in WS cells (14). Whether oxidative stress is a pathological factor in WS disease progression remains an issue of debate. There are many pieces of evidence suggesting such a link. WS cells are sensitive to some agents introducing oxidative damage to DNA and display a hyper-oxidation phenotype (55,56). Some features observed in WS patients may be directly related to a defect in repair of oxidative DNA lesions by BER. WRN is considered to be an important player in BER (57,58) and WRN deficient cells accumulate 8-oxoguanine, a marker of oxidative stress (59).
Our results show one of the possible molecular mechanisms contributing to the WS-like phenotype. We demonstrate that the WRN protein undergoes HNE adduction in vitro, but also in living cells (Figures 1A and B and 2). Such in vitro WRN modification was associated with modulation of ATP-dependant unwinding activity and abolishment of fork substrate degradation as well as ATP hydrolysis (Figure 1C–H). Significant inhibition occurred only at high HNE concentrations (100–1000 μM) that are rare in living organisms under physiological conditions. However, WRN protein was incubated with HNE only for 10 min. Experiments with the use of WRN immunocomplexes purified from HNE treated cells showed impairment of all catalytic activities with the strongest inhibition observed for the helicase (Figure 3). Since LPO products are quite stable in cells, inhibition of WRN protein and subsequently numerous DNA metabolic processes in aged individuals with enhanced LPO, might became appreciable. Current results are in line with previous data showing that iron-mediated oxidation of WRN protein led to inhibition of its catalytic activity (60). Moreover, exposure of cells to hydrogen peroxide resulted in oxidation of WRN in vivo (60). Taken together, these findings strongly suggest that oxidative stress/LPO via WRN inactivation results in premature aging features. Furthermore, our analysis of the functional effects of HNE adducts on WRN did not reveal decreased DNA binding (Figure 4). It was shown that WRN can unwind several BER intermediates, such as single-strand break intermediates, and stimulate polymerase β (pol β) strand displacement DNA synthesis via its helicase activity (57). The exonuclease domain of WRN has proofreading activity that can remove frequent errors made during nucleotide incorporation by pol β and thus potentially enhance the fidelity of BER (61). Significant HNE adduction to WRN might decrease the BER and contribute to lower fidelity of DNA repair synthesis.
Utilizing LC-MS/MS analysis, we have identified HNE adducts to Lys577 and Cys727 of the helicase domain, and to four amino acid residues in the terminal part of the protein (Figures 5 and 6 and Supplementary Figure S3). On the basis of the structural information obtained from the homology model of the WRN helicase and RQC domains and molecular modeling simulations, we showed that the Lys577-HNE adduct is located in the ATP binding pocket engaged in ATP binding and hydrolysis, necessary for catalytic activities, helicase/ATPase and for ATP-dependent exonuclease (Figure 7A and Supplementary Figure S4C). Moreover, we presented predicted changes in local conformation after HNE adduction to Lys577 (Figures 7C and 8A–D and Supplementary Figure S5A–D) and Cys727 (Figures 7D and 8E–H and Supplementary Figure S5E–H), which could further affect WRN catalytic activities. HNE adduction could thus prevent binding of ATP in the Walker motif within the binding pocket of WRN. It has been shown previously that mutants in the ATPase domain of WRN protein exhibited an almost complete inhibition of the ATPase activity and reduced ATP binding in vitro. Moreover, it has been reported that WRN polymorphism, R834C located in the helicase domain, significantly reduced not only WRN helicase but also exonuclease activity in immunoprecipitates from cell extracts of healthy individuals (62). Similar results were presented in assays with recombinant WRN protein containing the G574R missense mutation, which was recently identified in a WS patient. Both WRN helicase and exonuclease activities were reduced due to decreased ATP hydrolysis (63). Thus, prevention of ATP binding to the Walker motif and conformational change after HNE binding is proposed as a mechanism behind the disturbed unwinding and ATP-dependent degradation of DNA intermediates. However, stronger inhibition observed for the helicase (71%) than for ATPase (34%) could indicate a more complex mechanism of impaired unwinding than just due to blocked ATPase activity (Figure 3A, D and E). Molecular surface mapping around the ATP binding pocket revealed substantial changes in pocket structure and accessibility (Figure 8C and D, and Supplementary Figure S5C and D). These results suggest changed functional dynamics that could be attributed to WRN regulation. However, we did not determine the mechanism of the inhibited ATP-independent exonuclease activity on the 49 nt fork substrate (Figures 1E and F and 3B). The possible explanations are (i) that the region of exonuclease domain which lacked peptide coverage was modified with HNE (35% uncovered amino acids, Supplementary Figure S2) or (ii) that conformational shifts caused by HNE alkylation of the helicase domain and the C-terminal part of WRN protein were large enough to influence the conformation of the exonuclease domain and affect its function. The latter hypothesis seems to be supported by (i) structural differences within the entire helicase and RQC domains following binding of HNE to Lys577 and Cys727. HNE-WRN minimized structure revealed some differences in global conformation compared to the native protein, as shown by the RMSD value of 1.55 Å. It is also supported by (ii) global differences in surface electrostatics through the entire helicase and RQC domains between native and HNE modified WRN. Thus, one cannot rule out the possibility that even the more distant parts of the protein could also be affected.
Four of six identified HNE adducts were near the C-terminal part of WRN, at His1290, Cys1367, Lys1371 and Lys1389. Interestingly, among them Lys1371 is a part of NLS (Figure 6 and Supplementary Figure S2). The fact that the p53-binding domain of WRN resides near its C-terminus (64), a region eliminated by practically all the mutations found in Werner's syndrome patients, points to the importance of this region in preventing accumulation of genomic damage and suppressing tumors. According to the PolyPhenn database on single nucleotide polymorphisms (http://genetics.bwh.harvard.edu/pph2/), Cys1367 is vulnerable to damage. The Cys1367Arg variant is the most common WRN polymorphism, and comprehensive case–control study on German population showed that the Cys1367 variant allele was significantly associated with breast cancer risk. The risk was even higher in carriers of two risk alleles A of the p53 variant MspI 1798G>A, which is completely linked to the 16 bp insertion/duplication of p53PIN3, responsible for interaction with WRN (18). Our results are in line with these observations and suggest that accumulation of HNE modified amino acids at the C-terminus might interfere with the binding of p53 to WRN, leading to an attenuated apoptotic function of p53 and enhanced cellular senescence, which is characteristic for WS cells. Moreover, two studies have suggested that the wild-type Cys1367 allele variant of WRN protein was associated with myocardial infarction in the Japanese population (19,20); however, in Caucasians no associations with cardiovascular disease have been found (65). One association study has reported a protective effect for heterozygous carriers of the WRN Arg1367 variant allele against the development of soft tissue sarcomas (21) and type 2 diabetes mellitus (22). No significant association of WRN Cys1367Arg with age-related morbidity, cognitive function, longevity, coronary artery disease, Alzheimer and atherosclerosis was found in case–control studies (66–68). Although WRN Cys1367Arg is located adjacent to the NLS, a previous report has failed to detect any significant difference between Arg1367 and Cys1367 variants with respect to their nuclear localization or helicase and helicase-coupled exonuclease activities (65).
In summary, this study demonstrates the dynamic processes associated with HNE adduction to WRN, pointing to perturbations in DNA metabolism observed in WS. Our results are in line with the oxidative theory of aging. We propose that elevated oxidative stress and subsequently WRN helicase/ATPase and exonuclease deficiency via HNE post-translational modification impairs DNA replication and repair, and that these functional defects are responsible for premature senescence associated features and diseases. Further studies on specific interactions of HNE-modified WRN with various target proteins may provide clues to the mechanisms responsible for such a global effect of HNE on various cellular processes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We would like to thank Dr. Artur Biela for help with HNE synthesis.
Footnotes
Present address: Zuzanna Bukowy, Institute of Human Genetics, Polish Academy of Sciences, Strzeszyńska 32, 60-479 Poznań, Poland.
FUNDING
National Science Center [UMO-2012/05/B/NZ5/01392]. Intramural Research Program of the National Institutes of Health, USA. Funding for open access charge: National Science Center [UMO-2012/05/B/NZ5/01392].
Conflict of interest statement. None declared.
REFERENCES
- 1.Matsumoto T., Imamura O., Yamabe Y., Kuromitsu J., Tokutake Y., Shimamoto A., Suzuki N., Satoh M., Kitao S., Ichikawa K., et al. Mutation and haplotype analyses of the Werner's syndrome gene based on its genomic structure: genetic epidemiology in the Japanese population. Hum. Genet. 1997;100:123–130. doi: 10.1007/s004390050477. [DOI] [PubMed] [Google Scholar]
- 2.Singh D.K., Ghosh A.K., Croteau D.L., Bohr V.A. RecQ helicases in DNA double strand break repair and telomere maintenance. Mutat. Res. 2012;736:15–24. doi: 10.1016/j.mrfmmm.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu C.E., Oshima J., Fu Y.H., Wijsman E.M., Hisama F., Alisch R., Matthews S., Nakura J., Miki T., Ouais S., et al. Positional cloning of the Werner's syndrome gene. Science. 1996;272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- 4.Zarkovic K. 4-hydroxynonenal and neurodegenerative diseases. Mol. Aspects Med. 2003;24:293–303. doi: 10.1016/s0098-2997(03)00024-4. [DOI] [PubMed] [Google Scholar]
- 5.Croteau D.L., Popuri V., Opresko P.L., Bohr V.A. Human RecQ helicases in DNA repair, recombination, and replication. Ann. Rev. Biochem. 2014;83:519–552. doi: 10.1146/annurev-biochem-060713-035428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hickson I.D. RecQ helicases: caretakers of the genome. Nat. Rev. Cancer. 2003;3:169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 7.Bohr V.A. Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance. Trends Biochem. Sci. 2008;33:609–620. doi: 10.1016/j.tibs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perry J.J., Yannone S.M., Holden L.G., Hitomi C., Asaithamby A., Han S., Cooper P.K., Chen D.J., Tainer J.A. WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat. Struct. Mol. Biol. 2006;13:414–422. doi: 10.1038/nsmb1088. [DOI] [PubMed] [Google Scholar]
- 9.Kitano K., Kim S.Y., Hakoshima T. Structural basis for DNA strand separation by the unconventional winged-helix domain of RecQ helicase WRN. Structure. 2010;18:177–187. doi: 10.1016/j.str.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 10.Kitano K., Yoshihara N., Hakoshima T. Crystal structure of the HRDC domain of human Werner syndrome protein, WRN. J. Biol. Chem. 2007;282:2717–2728. doi: 10.1074/jbc.M610142200. [DOI] [PubMed] [Google Scholar]
- 11.Matsumoto T., Imamura O., Goto M., Furuichi Y. Characterization of the nuclear localization signal in the DNA helicase involved in Werner's syndrome. Int. J. Mol. Med. 1998;1:71–76. doi: 10.3892/ijmm.1.1.71. [DOI] [PubMed] [Google Scholar]
- 12.Moser M.J., Oshima J., Monnat R.J., Jr WRN mutations in Werner syndrome. Human mutation. 1999;13:271–279. doi: 10.1002/(SICI)1098-1004(1999)13:4<271::AID-HUMU2>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 13.Matsumoto T., Shimamoto A., Goto M., Furuichi Y. Impaired nuclear localization of defective DNA helicases in Werner's syndrome. Nat. Genet. 1997;16:335–336. doi: 10.1038/ng0897-335. [DOI] [PubMed] [Google Scholar]
- 14.Rossi M.L., Ghosh A.K., Bohr V.A. Roles of Werner syndrome protein in protection of genome integrity. DNA Repair. 2010;9:331–344. doi: 10.1016/j.dnarep.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larsen N.B., Hickson I.D. RecQ helicases: conserved guardians of genomic integrity. Adv. Exp. Med. Biol. 2013;767:161–184. doi: 10.1007/978-1-4614-5037-5_8. [DOI] [PubMed] [Google Scholar]
- 16.Kusumoto-Matsuo R., Ghosh D., Karmakar P., May A., Ramsden D., Bohr V.A. Serines 440 and 467 in the Werner syndrome protein are phosphorylated by DNA-PK and affects its dynamics in response to DNA double strand breaks. Aging. 2014;6:70–81. doi: 10.18632/aging.100629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muftuoglu M., Kusumoto R., Speina E., Beck G., Cheng W.H., Bohr V.A. Acetylation regulates WRN catalytic activities and affects base excision DNA repair. PloS ONE. 2008;3:e1918. doi: 10.1371/journal.pone.0001918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wirtenberger M., Frank B., Hemminki K., Klaes R., Schmutzler R.K., Wappenschmidt B., Meindl A., Kiechle M., Arnold N., Weber B.H., et al. Interaction of Werner and Bloom syndrome genes with p53 in familial breast cancer. Carcinogenesis. 2006;27:1655–1660. doi: 10.1093/carcin/bgi374. [DOI] [PubMed] [Google Scholar]
- 19.Ye L., Miki T., Nakura J., Oshima J., Kamino K., Rakugi H., Ikegami H., Higaki J., Edland S.D., Martin G.M., et al. Association of a polymorphic variant of the Werner helicase gene with myocardial infarction in a Japanese population. Am. J. Med. Genet. 1997;68:494–498. doi: 10.1002/(sici)1096-8628(19970211)68:4<494::aid-ajmg30>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 20.Morita H., Kurihara H., Sugiyama T., Hamada C., Yazaki Y. A polymorphic variant C1367R of the Werner helicase gene and atherosclerotic diseases in the Japanese population. Thromb. Haemost. 1999;82:160–161. [PubMed] [Google Scholar]
- 21.Nakayama R., Sato Y., Masutani M., Ogino H., Nakatani F., Chuman H., Beppu Y., Morioka H., Yabe H., Hirose H., et al. Association of a missense single nucleotide polymorphism, Cys1367Arg of the WRN gene, with the risk of bone and soft tissue sarcomas in Japan. Cancer Sci. 2008;99:333–339. doi: 10.1111/j.1349-7006.2007.00692.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirai M., Suzuki S., Hinokio Y., Yamada T., Yoshizumi S., Suzuki C., Satoh J., Oka Y. WRN gene 1367 Arg allele protects against development of type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2005;69:287–292. doi: 10.1016/j.diabres.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 23.Pansarasa O., Bertorelli L., Vecchiet J., Felzani G., Marzatico F. Age-dependent changes of antioxidant activities and markers of free radical damage in human skeletal muscle. Free Radic. Biol. Med. 1999;27:617–622. doi: 10.1016/s0891-5849(99)00108-2. [DOI] [PubMed] [Google Scholar]
- 24.Halliwell B. Free radicals, reactive oxygen species and human disease: a critical evaluation with special reference to atherosclerosis. Brit. J. Exp. Pathol. 1989;70:737–757. [PMC free article] [PubMed] [Google Scholar]
- 25.Bartsch H., Nair J. Oxidative stress and lipid peroxidation-derived DNA-lesions in inflammation driven carcinogenesis. Cancer Detect. Prevent. 2004;28:385–391. doi: 10.1016/j.cdp.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 26.Sayre L.M., Zelasko D.A., Harris P.L., Perry G., Salomon R.G., Smith M.A. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J. Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 27.Selley M.L. (E)-4-hydroxy-2-nonenal may be involved in the pathogenesis of Parkinson's disease. Free Radic. Biol. Med. 1998;25:169–174. doi: 10.1016/s0891-5849(98)00021-5. [DOI] [PubMed] [Google Scholar]
- 28.Uchida K., Toyokuni S., Nishikawa K., Kawakishi S., Oda H., Hiai H., Stadtman E.R. Michael addition-type 4-hydroxy-2-nonenal adducts in modified low-density lipoproteins: markers for atherosclerosis. Biochemistry. 1994;33:12487–12494. doi: 10.1021/bi00207a016. [DOI] [PubMed] [Google Scholar]
- 29.Dominguez-Rodriguez A., Abreu-Gonzalez P., de la Rosa A., Vargas M., Ferrer J., Garcia M. Role of endogenous interleukin-10 production and lipid peroxidation in patients with acute myocardial infarction treated with primary percutaneous transluminal coronary angioplasty, interleukin-10 and primary angioplasty. Int. J. Cardiol. 2005;99:77–81. doi: 10.1016/j.ijcard.2003.12.027. [DOI] [PubMed] [Google Scholar]
- 30.Dianzani M.U., Barrera G., Parola M. 4-Hydroxy-2,3-nonenal as a signal for cell function and differentiation. Acta Biochimica Polonica. 1999;46:61–75. [PubMed] [Google Scholar]
- 31.Uchida K. Histidine and lysine as targets of oxidative modification. Amino Acids. 2003;25:249–257. doi: 10.1007/s00726-003-0015-y. [DOI] [PubMed] [Google Scholar]
- 32.Poli G., Schaur R.J., Siems W.G., Leonarduzzi G. 4-hydroxynonenal: a membrane lipid oxidation product of medicinal interest. Med. Res. Rev. 2008;28:569–631. doi: 10.1002/med.20117. [DOI] [PubMed] [Google Scholar]
- 33.Esterbauer H., Schaur R.J., Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 34.Koster J.F., Slee R.G., Montfoort A., Lang J., Esterbauer H. Comparison of the inactivation of microsomal glucose-6-phosphatase by in situ lipid peroxidation-derived 4-hydroxynonenal and exogenous 4-hydroxynonenal. Free Radic. Res. Commun. 1986;1:273–287. doi: 10.3109/10715768609051637. [DOI] [PubMed] [Google Scholar]
- 35.Tsukamoto H., Horne W., Kamimura S., Niemela O., Parkkila S., Yla-Herttuala S., Brittenham G.M. Experimental liver cirrhosis induced by alcohol and iron. J. Clin. Invest. 1995;96:620–630. doi: 10.1172/JCI118077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benedetti A., Esterbauer H., Ferrali M., Fulceri R., Comporti M. Evidence for aldehydes bound to liver microsomal protein following CCl4 or BrCCl3 poisoning. Biochimica et biophysica acta. 1982;711:345–356. doi: 10.1016/0005-2760(82)90044-3. [DOI] [PubMed] [Google Scholar]
- 37.Petersen D.R., Doorn J.A. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radic. Biol. Med. 2004;37:937–945. doi: 10.1016/j.freeradbiomed.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 38.Schaur R.J. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol. Aspects Med. 2003;24:149–159. doi: 10.1016/s0098-2997(03)00009-8. [DOI] [PubMed] [Google Scholar]
- 39.Maddukuri L., Speina E., Christiansen M., Dudzinska D., Zaim J., Obtulowicz T., Kabaczyk S., Komisarski M., Bukowy Z., Szczegielniak J., et al. Cockayne syndrome group B protein is engaged in processing of DNA adducts of lipid peroxidation product trans-4-hydroxy-2-nonenal. Mutat. Res. 2009;666:23–31. doi: 10.1016/j.mrfmmm.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwarzer E., Muller O., Arese P., Siems W.G., Grune T. Increased levels of 4-hydroxynonenal in human monocytes fed with malarial pigment hemozoin. A possible clue for hemozoin toxicity. FEBS Lett. 1996;388:119–122. doi: 10.1016/0014-5793(96)00523-6. [DOI] [PubMed] [Google Scholar]
- 41.Carbone D.L., Doorn J.A., Kiebler Z., Sampey B.P., Petersen D.R. Inhibition of Hsp72-mediated protein refolding by 4-hydroxy-2-nonenal. Chem. Res. Toxicol. 2004;17:1459–1467. doi: 10.1021/tx049838g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carbone D.L., Doorn J.A., Kiebler Z., Ickes B.R., Petersen D.R. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. J. Pharmacol. Exp. Therap. 2005;315:8–15. doi: 10.1124/jpet.105.088088. [DOI] [PubMed] [Google Scholar]
- 43.Fritz K.S., Galligan J.J., Smathers R.L., Roede J.R., Shearn C.T., Reigan P., Petersen D.R. 4-Hydroxynonenal inhibits SIRT3 via thiol-specific modification. Chem. Res. Toxicol. 2011;24:651–662. doi: 10.1021/tx100355a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shearn C.T., Fritz K.S., Reigan P., Petersen D.R. Modification of Akt2 by 4-hydroxynonenal inhibits insulin-dependent Akt signaling in HepG2 cells. Biochemistry. 2011;50:3984–3996. doi: 10.1021/bi200029w. [DOI] [PubMed] [Google Scholar]
- 45.Pizzimenti S., Briatore F., Laurora S., Toaldo C., Maggio M., De Grandi M., Meaglia L., Menegatti E., Giglioni B., Dianzani M.U., et al. 4-Hydroxynonenal inhibits telomerase activity and hTERT expression in human leukemic cell lines. Free Radic. Biol. Med. 2006;40:1578–1591. [Google Scholar]
- 46.Carbone D.L., Doorn J.A., Petersen D.R. 4-Hydroxynonenal regulates 26S proteasomal degradation of alcohol dehydrogenase. Free Radic. Biol. Med. 2004;37:1430–1439. doi: 10.1016/j.freeradbiomed.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 47.Rossi M.A., Di Mauro C., Esterbauer H., Fidale F., Dianzani M.U. Activation of phosphoinositide-specific phospholipase C of rat neutrophils by the chemotactic aldehydes 4-hydroxy-2,3-trans-nonenal and 4-hydroxy-2,3-trans-octenal. Cell Biochem. Function. 1994;12:275–280. doi: 10.1002/cbf.290120408. [DOI] [PubMed] [Google Scholar]
- 48.Komisarski M., Kaczmarska Z., Kusmierek J.T. Practical highly enantioselective synthesis of (R)- and (S)-(E)-4-hydroxynon-2-enal. Acta biochimica Polonica. 2009;56:189–193. [PubMed] [Google Scholar]
- 49.Tadokoro T., Kulikowicz T., Dawut L., Croteau D.L., Bohr V.A. DNA binding residues in the RQC domain of Werner protein are critical for its catalytic activities. Aging. 2012;4:417–429. doi: 10.18632/aging.100463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Speina E., Zielinska M., Barbin A., Gackowski D., Kowalewski J., Graziewicz M.A., Siedlecki J.A., Olinski R., Tudek B. Decreased repair activities of 1,N(6)-ethenoadenine and 3,N(4)-ethenocytosine in lung adenocarcinoma patients. Cancer Res. 2003;63:4351–4357. [PubMed] [Google Scholar]
- 51.Roepstorff P., Fohlman J. Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed. Mass Spectrom. 1984;11:601. doi: 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]
- 52.Yang Y., Sharma A., Sharma R., Patrick B., Singhal S.S., Zimniak P., Awasthi S., Awasthi Y.C. Cells preconditioned with mild, transient UVA irradiation acquire resistance to oxidative stress and UVA-induced apoptosis: role of 4-hydroxynonenal in UVA-mediated signaling for apoptosis. J. Biol. Chem. 2003;278:41380–41388. doi: 10.1074/jbc.M305766200. [DOI] [PubMed] [Google Scholar]
- 53.Harman D. Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 54.Martin G.M. Genetics and the pathobiology of ageing. Phil. Trans. R. Soc. Lond. B Biol. Sci. 1997;352:1773–1780. doi: 10.1098/rstb.1997.0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Imamura O., Fujita K., Itoh C., Takeda S., Furuichi Y., Matsumoto T. Werner and Bloom helicases are involved in DNA repair in a complementary fashion. Oncogene. 2002;21:954–963. doi: 10.1038/sj.onc.1205143. [DOI] [PubMed] [Google Scholar]
- 56.Szekely A.M., Bleichert F., Numann A., Van Komen S., Manasanch E., Ben Nasr A., Canaan A., Weissman S.M. Werner protein protects nonproliferating cells from oxidative DNA damage. Mol. Cell. Biol. 2005;25:10492–10506. doi: 10.1128/MCB.25.23.10492-10506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harrigan J.A., Wilson D.M., 3rd, Prasad R., Opresko P.L., Beck G., May A., Wilson S.H., Bohr V.A. The Werner syndrome protein operates in base excision repair and cooperates with DNA polymerase beta. Nucleic Acids Res. 2006;34:745–754. doi: 10.1093/nar/gkj475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahn B., Harrigan J.A., Indig F.E., Wilson D.M., 3rd, Bohr V.A. Regulation of WRN helicase activity in human base excision repair. J. Biol. Chem. 2004;279:53465–53474. doi: 10.1074/jbc.M409624200. [DOI] [PubMed] [Google Scholar]
- 59.Von Kobbe C., May A., Grandori C., Bohr V.A. Werner syndrome cells escape hydrogen peroxide-induced cell proliferation arrest. FASEB J. 2004;18:1970–1972. doi: 10.1096/fj.04-1895fje. [DOI] [PubMed] [Google Scholar]
- 60.Harrigan J.A., Piotrowski J., Di Noto L., Levine R.L., Bohr V.A. Metal-catalyzed oxidation of the Werner syndrome protein causes loss of catalytic activities and impaired protein-protein interactions. J. Biol. Chem. 2007;282:36403–36411. doi: 10.1074/jbc.M706107200. [DOI] [PubMed] [Google Scholar]
- 61.Huang S., Beresten S., Li B., Oshima J., Ellis N.A., Campisi J. Characterization of the human and mouse WRN 3′–>5′ exonuclease. Nucleic Acids Res. 2000;28:2396–2405. doi: 10.1093/nar/28.12.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kamath-Loeb A.S., Welcsh P., Waite M., Adman E.T., Loeb L.A. The enzymatic activities of the Werner syndrome protein are disabled by the amino acid polymorphism R834C. J. Biol. Chem. 2004;279:55499–55505. doi: 10.1074/jbc.M407128200. [DOI] [PubMed] [Google Scholar]
- 63.Tadokoro T., Rybanska-Spaeder I., Kulikowicz T., Dawut L., Oshima J., Croteau D.L., Bohr V.A. Functional deficit associated with a missense Werner syndrome mutation. DNA Repair. 2013;12:414–421. doi: 10.1016/j.dnarep.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blander G., Kipnis J., Leal J.F., Yu C.E., Schellenberg G.D., Oren M. Physical and functional interaction between p53 and the Werner's syndrome protein. J. Biol. Chem. 1999;274:29463–29469. doi: 10.1074/jbc.274.41.29463. [DOI] [PubMed] [Google Scholar]
- 65.Bohr V.A., Metter E.J., Harrigan J.A., von Kobbe C., Liu J.L., Gray M.D., Majumdar A., Wilson D.M., 3rd, Seidman M.M. Werner syndrome protein 1367 variants and disposition towards coronary artery disease in Caucasian patients. Mech. Ageing Dev. 2004;125:491–496. doi: 10.1016/j.mad.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 66.Kuningas M., Slagboom P.E., Westendorp R.G., van Heemst D. Impact of genetic variations in the WRN gene on age related pathologies and mortality. Mech. Ageing Dev. 2006;127:307–313. doi: 10.1016/j.mad.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 67.Castro E., Ogburn C.E., Hunt K.E., Tilvis R., Louhija J., Penttinen R., Erkkola R., Panduro A., Riestra R., Piussan C., et al. Polymorphisms at the Werner locus: I. Newly identified polymorphisms, ethnic variability of 1367Cys/Arg, and its stability in a population of Finnish centenarians. Am. J. Med. Genet. 1999;82:399–403. [PubMed] [Google Scholar]
- 68.Payao S.L., de Labio R.W., Gatti L.L., Rigolin V.O., Bertolucci P.H., Smith Mde A. Werner helicase polymorphism is not associated with Alzheimer's disease. J. Alzheimer's Dis. 2004;6:591–594. doi: 10.3233/jad-2004-6603. discussion 673–581. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.