

Background: Angiotensin II induces skeletal muscle atrophy, but the function of other renin-angiotensin system components in skeletal muscle physiology is understood poorly.
Results: Angiotensin II type 2 receptor (AT2R) blockade inhibits skeletal muscle regeneration and myoblast differentiation.
Conclusion: AT2R positively regulates skeletal muscle regeneration.
Significance: Stimulation of AT2R signaling may be beneficial in muscle wasting conditions.
Keywords: Angiotensin II, Heart Failure, Regeneration, Skeletal Muscle, Stem Cells, AT2 Receptor
Abstract
Patients with advanced congestive heart failure (CHF) or chronic kidney disease (CKD) often have increased angiotensin II (Ang II) levels and cachexia. Ang II infusion in rodents causes sustained skeletal muscle wasting and decreases muscle regenerative potential through Ang II type 1 receptor (AT1R)-mediated signaling, likely contributing to the development of cachexia in CHF and CKD. However, the potential role of Ang II type 2 receptor (AT2R) signaling in skeletal muscle physiology is unknown. We found that AT2R expression was increased robustly in regenerating skeletal muscle after cardiotoxin (CTX)-induced muscle injury in vivo and differentiating myoblasts in vitro, suggesting that the increase in AT2R played an important role in regulating myoblast differentiation and muscle regeneration. To determine the potential role of AT2R in muscle regeneration, we infused C57BL/6 mice with the AT2R antagonist PD123319 during CTX-induced muscle regeneration. PD123319 reduced the size of regenerating myofibers and expression of the myoblast differentiation markers myogenin and embryonic myosin heavy chain. On the other hand, AT2R agonist CGP42112 infusion potentiated CTX injury-induced myogenin and embryonic myosin heavy chain expression and increased the size of regenerating myofibers. In cultured myoblasts, AT2R knockdown by siRNA suppressed myoblast differentiation marker expression and myoblast differentiation via up-regulation of phospho-ERK1/2, and ERK inhibitor treatment completely blocked the effect of AT2R knockdown. These data indicate that AT2R signaling positively regulates myoblast differentiation and potentiates skeletal muscle regenerative potential, providing a new therapeutic target in wasting disorders such as CHF and CKD.
Introduction
Angiotensin II (Ang II),2 the main effector molecule of the renin-angiotensin system (RAS), has multiple physiological actions, including regulation of blood pressure and the salt/water balance, through a variety of effects on the central nervous system, the adrenal gland, the vasculature, and the kidneys. Furthermore, the RAS plays an important role in the pathogenesis of cardiovascular diseases ranging from early endothelial dysfunction to congestive heart failure (CHF) and renal or cerebrovascular disease (1). Although most physiological effects of Ang II are mediated via the AT1 receptor, Ang II also signals via the AT2 receptor, whose function is incompletely understood.
There is increasing interest in the potential effects of Ang II on skeletal muscle, not previously considered a major target organ for Ang II effects. Therefore, we (2–9) and others (10–12) have shown that Ang II causes skeletal muscle wasting via increased protein breakdown and apoptosis and decreased protein synthesis. These findings have important clinical relevance because patients with chronic diseases such as advanced CHF or chronic kidney disease often have increased Ang II levels (13–15) and cachexia, which independently worsens the outcome (16). Angiotensin-converting enzyme inhibitor treatment improves weight loss in CHF patients (14), and Ang II type 1 receptor (AT1R) blockade prevents skeletal muscle atrophy in a rat model of CHF (17), suggesting that Ang II could play an important role in the development of cachexia and that the RAS could represent a common mechanism regulating skeletal muscle function in chronic diseases.
We have shown that most of the atrophic effects of Ang II on skeletal muscle are indirect, i.e. mediated by intermediate molecules, including cytokines and glucocorticoids (4, 18), which is consistent with the fact that adult muscle fibers express little to no Ang II receptors (18, 19). However, we demonstrated recently that Ang II also acts directly on skeletal muscle via its effects on satellite cells (19). These mitotically quiescent mononuclear muscle stem cells are the key orchestrators of the regenerative response to injury. They become activated, proliferate, form myoblasts, and then terminally differentiate and fuse to form multinucleated myotubes, thereby repairing damaged myofibers (20). We found that satellite cells express high levels of AT1R and that Ang II inhibits satellite cell proliferation via AT1R signaling, leading to depletion of the satellite cell pool and reduced muscle regenerative capacity in a cardiotoxin injury-induced model (19). We also found that differentiated satellite cells expressed the AT2R. Our findings were consistent with reports that AT1R blockade or AT1R deficiency was beneficial in mouse models of myopathy and injury (21, 22), although there are contradictory reports that an angiotensin-converting enzyme inhibitor or AT1R ablation significantly impaired skeletal muscle regeneration (23, 24).
The focus of this study was to determine the potential functional role of the AT2R in skeletal muscle. Our findings indicate that AT2R expression is increased dramatically in satellite cells during differentiation and that the AT2R is a critical positive regulator of myoblast differentiation and skeletal muscle regenerative processes.
EXPERIMENTAL PROCEDURES
Animals
Animal protocols were approved by the Institutional Animal Care and Use Committee at Tulane University. 8- to 10-week-old male C57BL/6 mice (Charles River Laboratories) were used in this study. Ang II (Phoenix Pharmaceuticals, 1.5 μg/kg/min), PD123319 (Pfizer, 30 mg/kg/day), and CGP42112 (Sigma, 10 μg/kg/min) were administered via osmotic minipump (Alzet model 1007D). CTX was injected as previously described (25).
Antibodies and Reagents
Antibodies used for immunoblotting included anti-AT2R (Millipore, 1:500), anti-myogenin (Abcam, 1:500), anti-desmin (Cell Signaling Technology, 1:1000), anti-ERK1/2 (Cell Signaling Technology, 1:1000), anti-phospho-ERK1/2 (Cell Signaling Technology, 1:2000), anti-p38MAPK (Cell Signaling Technology, 1:1000), anti-phospho-p38MAPK (Cell Signaling Technology, 1:1000), anti-MEK1/2 (Santa Cruz Biotechnology, 1:200), anti-phospho-MEK1/2 (Santa Cruz Biotechnology, 1:200), anti-MKP-1 (Santa Cruz Biotechnology, 1:100), anti-JNK (Cell Signaling Technology, 1:1000), anti-phospho-JNK (Cell Signaling Technology, 1:2000), and anti-mouse β-actin (Sigma, 1:10,000). Antibodies used for immunohistochemical staining included anti-eMyHC (Developmental Studies Hybridoma Bank, 1:100), anti-fast skeletal myosin (Sigma, 1:100), anti-slow skeletal myosin (Sigma, 1:2000), anti-laminin (Sigma, 1:50), anti-mouse IgG-Alexa Fluor 488 (Invitrogen, 1:500), and anti-rabbit IgG-Alexa Fluor 594 (Invitrogen, 1:500). The ERK inhibitors U0126 and PD98059 were purchased from Cell Signaling Technology and used at final concentrations of 10 and 25 μm, respectively.
Immunohistochemical Staining
Skeletal muscle slow and fast myosin were stained on paraffin (6 μm) sections using a Mouse on Mouse immunodetection kit (Vector Laboratories) according to the instructions of the manufacturer. Sections were costained with laminin to identify each myofiber.
Primary Satellite Cell and C2C12 Cell Culture
Satellite cells were collected as described previously (26, 27). Briefly, hind limb muscles (gastrocnemius, soleus, tibialis anterior, and extensor digitorum longus) were incubated with 0.2% (w/v) collagenase type II in DMEM and 10% horse serum for 2 h at 37 °C. Digested muscles were dissociated by triturating with a Pasteur pipette, washed, and digested further with 1 unit/ml dispase for 1 h at 37 °C. Cells were filtered, washed, and incubated in proliferation medium (DMEM supplemented with 20% FBS (Mediatech), 10% HS (horse serum), 2% chicken embryo extract (US Biological), 1 mm sodium pyruvate, 2 mm l-glutamine, and penicillin/streptomycin) for 40 min to let the fibroblasts attach onto the plate, and unattached cells were plated onto Matrigel-coated plates in the proliferation medium at a density of 1 × 104 cells/cm2. C2C12 myoblasts (ATCC) were cultured in C2C12 growth medium (DMEM supplemented with 20% FBS, 1 mm sodium pyruvate, and 2 mm l-glutamine). After satellite cells and C2C12 cells reached confluence, cells were switched to differentiation medium (DMEM supplemented with 2% horse serum, 1 mm sodium pyruvate, and 2 mm l-glutamine) to induce differentiation. The myoblast fusion index was calculated as the percentage of nuclei in eMyHC-positive myotubes divided by the total number of nuclei.
Quantitative RT-PCR
Quantitative RT-PCR was performed as described previously (5), and the PCR primers used in this study were obtained commercially (SABiosciences; PPM04482A for myogenin, PPM41610F for eMyHC, PPM04811A for AT2R, and PPM03559F for Hprt).
Measurement of Cell Proliferation, Viability, and Apoptosis
Cells were plated on 96-well plates in proliferation medium, and cell proliferation, cell viability, and caspase 3/7 activity was measured when cells reached confluence (day 0) and 24 h after cells were switched to differentiation medium (differentiation day 1). For cell proliferation analysis, anti-Notch antibody (Millipore, 1:300) was added to the culture together with BrdU (10 μm) and incubated for 24 h. BrdU incorporation was measured using a BrdU cell proliferation ELISA kit (Roche) according to the instructions of the manufacturer. Cell viability and caspase 3/7 were measured using a CellTiter-Glo luminescent cell viability assay (Promega) and Caspase-Glo 3/7 assay (Promega), respectively, according to the instructions of the manufacturer. For the apoptosis and cell viability assay, staurosporine (1 mm, Cell Signaling Technology) and mitomycin C (0.4 mg/ml, Sigma), respectively, were added as positive controls.
siRNA Electroporation in Vivo and Transfection in Vitro
The siRNAs used in this study were obtained from Invitrogen (Silencer Select siRNA; AT2R siRNA-a, catalog no. s201014; AT2R siRNA-b, catalog no. s62151; and control siRNA, catalog no. 4390843). For in vivo siRNA electroporation, mice were anesthetized by ketamine/xylazine, the hind limbs were shaved, and 35 μl of siRNA (20 μm) was injected into five to six different locations of the gastrocnemius muscle with a 22-gauge needle and syringe (Hamilton model 705). Transcutaneous pulses were applied by two stainless steel plate electrodes (Caliper Electrode model 384, BTX) with a distance between the two plates of 0.5 cm, and conductive gel was used to ensure the electrical contact. Electric pulses with a standard square wave were delivered by an electroporator (ECM830 Electro Square Porator, BTX). Eight pulses of 100 V/cm were administered to the muscle with a delivery rate of 1 pulse/s, with each pulse 20 ms in duration. In vitro siRNA transfection was performed using Lipofectamine RNAiMAX reagent (Invitrogen) according to the instructions of the manufacturer. Cells were transfected when they reached ∼40% confluence, and the second transfection was performed on the following day to ensure a high efficiency of transfection. C2C12 myoblasts and primary satellite cells were induced to differentiate 2 days after the initial transfection.
RESULTS
AT2R Expression Increases during Muscle Regeneration
Consistent with previous reports (25), a number of myogenic transcription factors were up-regulated in the hind limb muscle after CTX injury. Myogenin mRNA expression reached a maximal level on day 3, which preceded maximal induction of eMyHC expression, a marker of differentiating myotubes. Although there was no detectable AT2R expression in skeletal muscle before the CTX injury, the AT2R mRNA level was increased robustly during regeneration and reached a maximal level on day 5, coinciding with the expression of eMyHC (Fig. 1A). These data suggest that AT2R expression is induced during the process of satellite cell differentiation. Consistent with these data, AT2R mRNA and protein expression was highly up-regulated in primary satellite cells and C2C12 myoblasts after cells were induced to differentiate (Fig. 1, B and C). These data strongly suggest that up-regulation of AT2R expression during satellite cell differentiation may play an important role in muscle regenerative processes.
FIGURE 1.
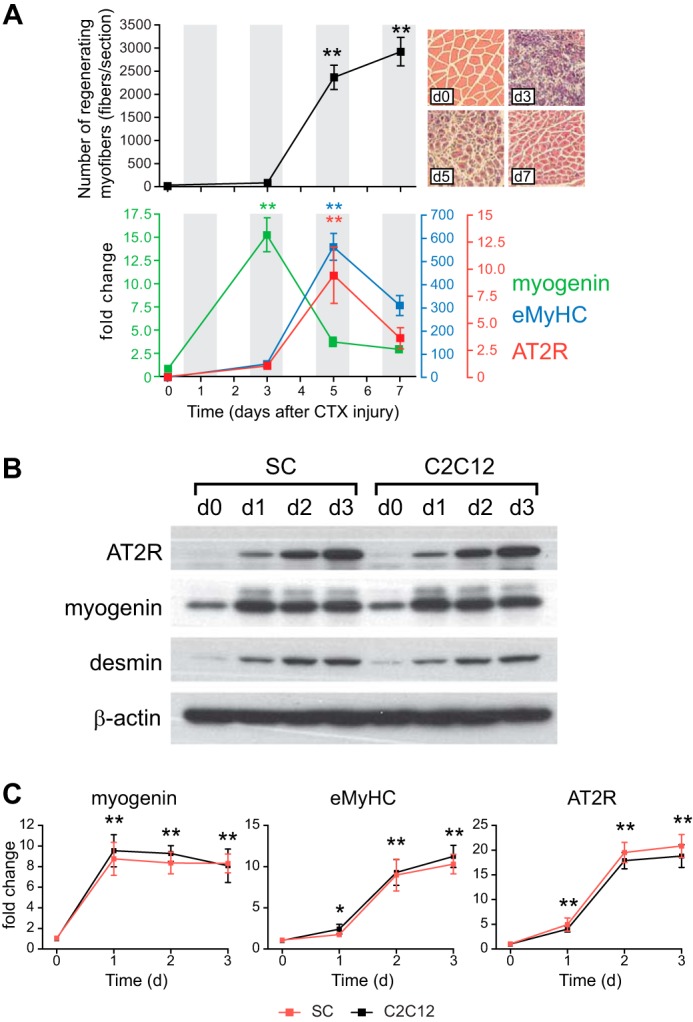
AT2R expression is increased during skeletal muscle regeneration and satellite cell differentiation. A, the number of regenerating myofibers (myofibers with centralized nuclei) after CTX injection (top panel with representative pictures). Gastrocnemius muscle was injected with CTX on day (d) 0, and muscles were harvested on days 3, 5, and 7. Paraffin sections were prepared from the middle of the gastrocnemius muscle, and the regenerating myofiber number was quantified per section. For qRT-PCR analysis (bottom panel), RNA was extracted from whole gastrocnemius muscles. B, expression of AT2R during primary satellite cell (SC) and C2C12 myoblast differentiation. Primary satellite cells were collected from C57BL/6 mouse hind limb muscles and cultured in proliferation medium. After cells reached confluence (day 0), the medium was switched to differentiation medium, and cells were harvested after 1, 2, and 3 days. C2C12 myoblasts were cultured and harvested under the same conditions as primary satellite cells. AT2R, myogenin, and desmin expression was analyzed by immunoblotting. C, qRT-PCR analysis of AT2R, myogenin, and eMyHC under the same conditions as in B. Data are mean ± S.E. (n = 5). *, p < 0.05; **, p < 0.01 compared with day 0.
The AT2R Antagonist Reduces and the AT2R Agonist Potentiates Muscle Regeneration in Vivo
The robust increase of AT2R expression during muscle regeneration in vivo and during satellite cell/C2C12 myoblast differentiation in vitro (Fig. 1) suggested that the AT2R plays an important role in the regulation of satellite cell differentiation and muscle regeneration. To investigate the role of the AT2R during muscle regeneration in vivo, we infused mice with the AT2R antagonist PD123319 (28) in a setting of CTX-induced injury (Fig. 2A). We found that PD123319 infusion significantly reduced the size of regenerating myofibers, whereas the number of regenerating myofibers was not altered (Fig. 2, B and C). To gain more insight into the AT2R-mediated regulation of regenerating myofiber number and size, we analyzed the slow and fast fiber sizes in regenerating areas. As shown in Fig. 2B, the sizes of regenerating myofibers positive for slow and fast myosin were both reduced to the same extent, suggesting that the effect of AT2R suppression is not fiber type-specific. We also analyzed the number of regenerating myofibers on the basis of the number of centralized nuclei in each myofiber (Fig. 2E). There was a significant reduction in the number of regenerating myofibers with two or more centralized nuclei, suggesting that AT2R suppression prevents myoblast fusion. Furthermore, these alterations of skeletal muscle regeneration were associated with a decline in the expression of myogenin and eMyHC (Fig. 2, F and G). Next we infused the AT2R agonist CGP42112 into C57BL/6 mice after CTX injury to determine the effect of AT2R activation on muscle regeneration (Fig. 3A). In contrast to the effects of AT2R antagonism and AT2R knockdown (Fig. 2), the infusion of the AT2R agonist in the setting of CTX-induced injury increased the size of regenerating myofibers without altering the total number of regenerating myofibers (Fig. 3, B and C). The increase of regenerating myofiber size was not fiber type-specific (Fig. 3, D and E) as in the case of AT2R antagonist infusion. We also found that there was an increase in the number of centralized nuclei in regenerating myofibers. These changes in regenerating myofibers after AT2R agonist infusion were associated with increased expression of myogenin (Fig. 3D) and eMyHC (Fig. 3E). These data strongly suggest that AT2R activation positively regulates satellite cell differentiation and potentiates muscle regeneration.
FIGURE 2.
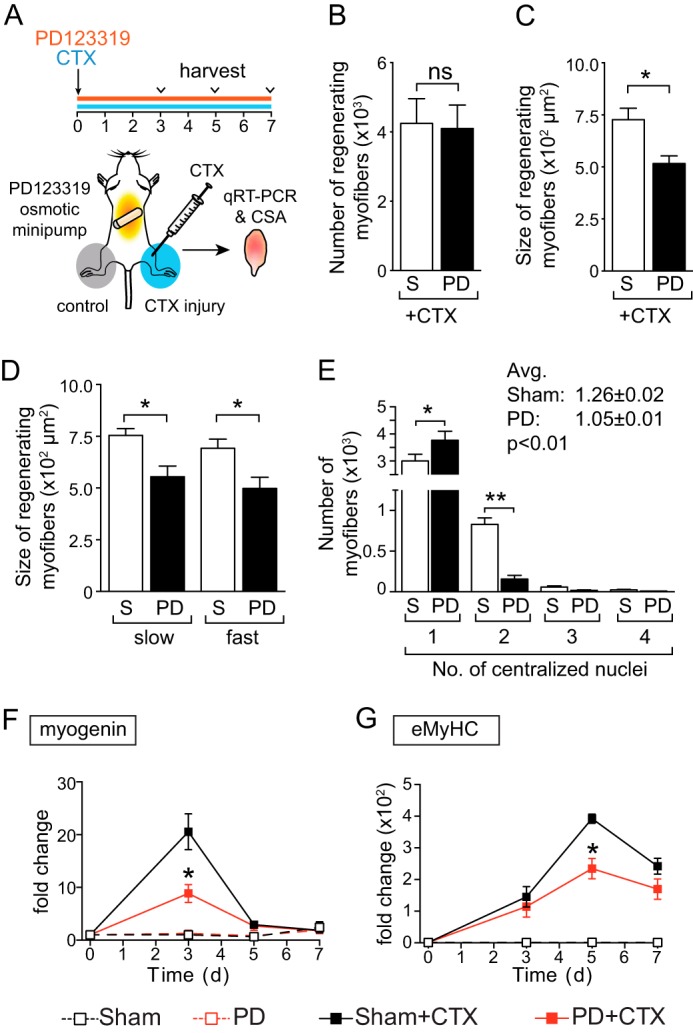
AT2R antagonist inhibits skeletal muscle regeneration in vivo. A, experimental model. CTX was injected, and AT2R antagonist PD123319 infusion (30 mg/kg/day) was started on day 0. Gastrocnemius muscles were collected on days 3, 5, and 7. For H&E staining, paraffin sections were prepared from the middle of the gastrocnemius muscles. For qRT-PCR analysis, RNA was extracted from whole gastrocnemius muscle. B, number of regenerating myofibers (myofibers with centralized nuclei) after CTX injection and sham (S) or PD123319 (PD) infusion on day 5. The number of regenerating myofibers is shown per section. C, cross-sectional area (CSA) of regenerating myofibers after CTX injection and PD infusion on day 5. D, the same sections as in C were stained for skeletal muscle slow and fast myosin, and the size of the regenerating myofibers was quantified. E, the number of regenerating myofibers was analyzed on the same sections as in B and categorized on the basis of the number of centralized nuclei in each myofiber. Avg, average. F and G, qRT-PCR analysis of myogenin (F) and eMyHC (G) in the gastrocnemius muscle after CTX injection and PD infusion on days 0, 3, 5, and 7. Data are mean ± S.E. (n = 5–6). *, p < 0.05; ns, not significant.
FIGURE 3.

The AT2R agonist potentiates skeletal muscle regeneration in vivo. A, experimental model. CTX was injected, and AT2R antagonist CGP42112 infusion (10 μg/kg/min) was started on day 0. Gastrocnemius muscles were collected on days 3, 5, and 7. For H&E staining, paraffin sections were prepared from the middle of the gastrocnemius muscle. For qRT-PCR analysis, RNA was extracted from whole gastrocnemius muscle. B, number of regenerating myofibers (myofibers with centralized nuclei) after CTX injection and sham (S) or CGP42112 (CG) infusion on day 5. The number of regenerating myofibers is shown per section. C, Cross sectional area (CSA) of regenerating myofibers after CTX injection and CGP infusion on day 5. D, the same sections as in C were stained for skeletal muscle slow and fast myosin, and the size of the regenerating myofibers was quantified. E, the number of regenerating myofibers was analyzed on the same sections as in B and categorized on the basis of the number of centralized nuclei in each myofiber. F and G, qRT-PCR analysis of myogenin (F) and eMyHC (G) in the gastrocnemius muscle after CTX injection and CGP infusion on days 0, 3, 5, and 7. Data are mean ± S.E. (n = 5–6). *, p < 0.05; ns, not significant.
AT2R Regulates Myoblast Differentiation via ERK1/2 Signaling
To further investigate mechanisms whereby the AT2R regulates skeletal muscle regeneration, the AT2R was knocked down both in vivo and in vitro using siRNA. AT2R siRNA was delivered to mouse gastrocnemius muscle via electroporation-mediated siRNA transfer (29). Under this experimental condition, the electric pulse itself damages the skeletal muscle (5), which results in induction of myogenin, eMyHC, and AT2R in a manner similar to that of CTX-induced injury (Fig. 4, A–D). AT2R siRNA markedly blunted the injury-induced increase in AT2R expression (Fig. 4B) and significantly suppressed the increase of myogenin and eMyHC (Fig. 4, C and D), consistent with our AT2R antagonist infusion experiment (Fig. 2). These data are consistent with our hypothesis that the AT2R regulates satellite cell differentiation and muscle regeneration. To obtain further insights, we transfected AT2R siRNA to C2C12 cells to analyze the effect of AT2R knockdown on myoblast differentiation. As shown in Fig. 5A, AT2R siRNA (we used two independent AT2R siRNA, see “Experimental Procedures” for detailed information) blocked the induction of AT2R expression that occurred during normal C2C12 myoblast differentiation. AT2R knockdown did not alter apoptosis, viability, or proliferation of C2C12 cells either before or after induction of differentiation (Fig. 5C). Importantly, AT2R knockdown markedly reduced the induction of myogenin and desmin, consistent with our in vivo observations (Fig. 5A). It has been shown that MAPK signaling plays an important role in C2C12 myoblast differentiation (30). Also, it has been suggested that AT2R signaling attenuates ERK1/2 activity in vascular smooth muscle cells (31), whereas nothing is known about AT2R downstream signaling in satellite cells. We analyzed MAPK signaling pathways in C2C12 myoblasts and myotubes after AT2R siRNA transfection. There was no detectable difference in phosphorylation of p38MAPK and JNK in response to AT2R knockdown (Fig. 5, A and B). The phosphorylation of ERK1/2 was down-regulated at the initial stage of C2C12 cell differentiation, followed by a progressive increase up to day 3 of differentiation, consistent with a previous report (30). Importantly, we found that phosphorylation of ERK1/2 was increased after AT2R knockdown throughout the process of myoblast differentiation (Fig. 5, A and B). This increase of p-ERK1/2 was associated with increased phosphorylation of MEK, especially at the initial stages of differentiation, suggesting that ERK1/2 phosphorylation was regulated by MEK. To further explore the involvement of ERK1/2 in AT2R downstream signaling, ERK1/2 was inhibited by the ERK inhibitors U0126 and PD98059 together with AT2R knockdown (Fig. 6). As expected, AT2R knockdown markedly reduced myogenin and desmin expression in differentiating C2C12 cells. However, inhibition of ERK1/2 phosphorylation by U0126 and PD98059 largely restored the induction of myogenin and desmin expression in the setting of AT2R knockdown (Fig. 6A). The ability of ERK inhibition to rescue the AT2R siRNA-mediated reduction in C2C12 differentiation was further confirmed by immunohistochemical staining of eMyHC in differentiating myotubes (Fig. 6, B and C). The fusion index (calculated as the percentage of nuclei in eMyHC-positive myotubes divided by total number of nuclei) was decreased markedly by AT2R knockdown, and this decrease was completely abrogated by ERK inhibition. These data indicate that the AT2R positively regulates myoblast differentiation via down-regulation of ERK1/2 signaling.
FIGURE 4.
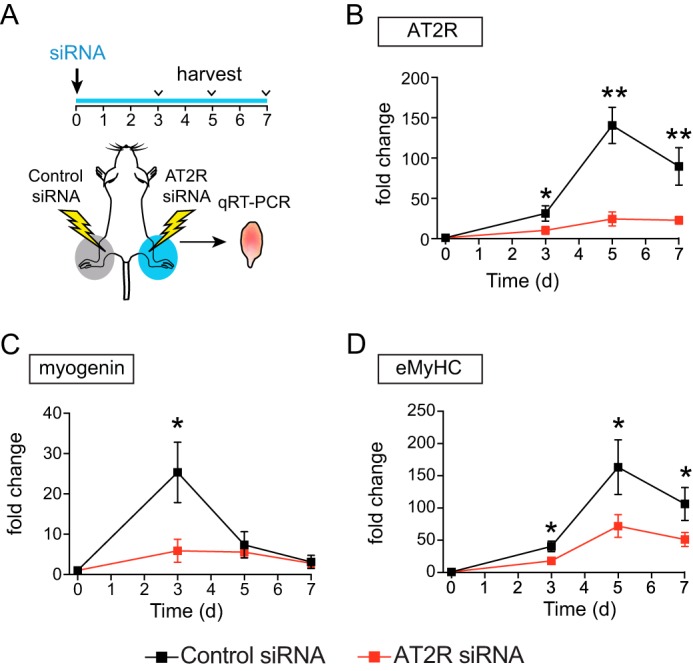
AT2R knockdown inhibits skeletal muscle regeneration in vivo. A, experimental model. siRNA electroporation was performed on day 0. Under this experimental condition, the electric pulse damages muscles and induces regeneration. Gastrocnemius muscles were collected on days 3, 5, and 7, and RNA was extracted. B–D, qRT-PCR analysis of AT2R (B), myogenin (C), and eMyHC (D) in the gastrocnemius muscle after siRNA electroporation on days 0, 3, 5, and 7. Data are mean ± S.E. (n = 5–6). *, p < 0.05; **, p < 0.01.
FIGURE 5.

AT2R knockdown inhibits myoblast differentiation. A, immunoblot of myoblast differentiation markers and MAPK signaling components in C2C12 cells after AT2R knockdown. C2C12 myoblasts were cultured in proliferation medium and transfected with AT2R or control siRNA. Two independent AT2R siRNAs were used, termed AT2R-a (Aa) and AT2R-b (Ab). After cells reached confluence (day 0), the medium was switched to differentiation medium, and cells were harvested after 1, 2, and 3 days. C, control. B, quantification of p-ERK1/2, p-p38MAPK, and p-JNK calculated from A. Data are presented as the ratio to total level of corresponding protein. C, cell proliferation, apoptosis, and viability in C2C12 cells. C2C12 myoblasts were transfected with control (C) or AT2R (A) siRNA, and cell proliferation, apoptosis, and viability were measured before and 1 day after induction of differentiation. Anti-notch antibody (N), staurosporine (S), and mitomycin C (M) were used as positive controls for cell proliferation, apoptosis, and cell viability, respectively. −, non-transfected control. Data are mean ± S.E. (n = 3 and 8 in B and C, respectively. *, p < 0.05; **, p < 0.01.
FIGURE 6.
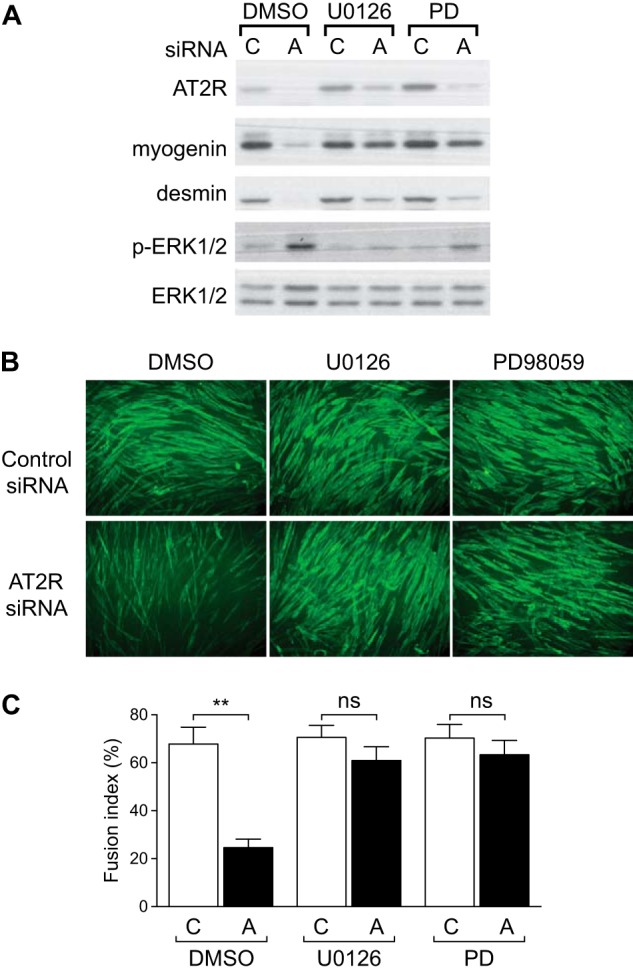
AT2R positively regulates myoblast differentiation via ERK1/2 signaling. A, immunoblot of myoblast differentiation markers of C2C12 cells. C2C12 myoblasts were cultured in proliferation medium, transfected with control (C) and AT2R (A) siRNA, and the medium switched to proliferation medium ± the ERK1/2 inhibitors U0126 and PD98059 (PD). After cells reached confluence, the medium was switched to differentiation medium, and cells were harvested after 24 h. DMSO, dimethyl sulfoxide. B, immunohistochemical staining of eMyHC in C2C12 myotubes. C2C12 myoblasts were cultured under the same condition as in A, and myotube formation was analyzed 2 days after differentiation. C, fusion index (calculated as the percentage of nuclei in eMyHC-positive myotubes divided by the total number of nuclei) of C2C12 myotubes calculated from B. Data are mean ± S.E. (n = 5). **, p < 0.01; ns, not significant.
DISCUSSION
Although RAS components such as angiotensinogen and angiotensin-converting enzyme are expressed in the skeletal muscle microcirculation (32) and in myoblasts (33), there have been limited data on the biological effects of Ang II or other components of the RAS on skeletal muscle. Ang (1–7), mainly produced from Ang II by ACE2 and signaling through the Mas receptor, has been reported to regulate skeletal muscle insulin sensitivity (34, 35), decrease fibrosis, and improve muscle function in a mouse model of muscle dystrophy (36). It has been shown by us (2–9) and by others (10–12) that Ang II causes skeletal muscle wasting via increased protein breakdown and apoptosis and decreased protein synthesis. Most of these effects of Ang II are indirect, i.e. mediated by intermediate molecules, including cytokines and glucocorticoids (4, 18), which is consistent with the fact that adult muscle fibers express little to no Ang II receptors (18, 19). However, recently, we discovered that satellite cells express the AT1R and that Ang II acts via these receptors to prevent satellite cell proliferation and suppress muscle regeneration (19). This mechanism could be an important contributory factor to the loss of skeletal muscle that occurs in many clinical conditions in which Ang II levels are elevated. A critical unanswered question is the potential role of the AT2R in skeletal muscle regeneration. Here we show that there is coordinated temporal expression of AT2R in differentiating muscle stem cells. AT2R expression is not detectable in primary satellite cells and in C2C12 myoblasts basally but is strongly induced during differentiation (Fig. 1). Inhibition of the AT2R by the AT2R antagonist PD123319 (Fig. 2) and by AT2R siRNA (Figs. 4–6) markedly inhibited myoblast differentiation and skeletal muscle regeneration, whereas the AT2R agonist CGP42112 potentiated muscle regeneration (Fig. 3). We found that the number of centralized nuclei in regenerating myofibers was reduced and increased by PD123319 and CGP42112, respectively, suggesting that AT2R regulates myoblast fusion processes (Figs. 2E and 3E). We also found that the effect of AT2R activation/inactivation on muscle regeneration is not fiber type-specific (Figs. 2D and 3D). During differentiation of myoblasts, phosphorylation of ERK1/2 was initially down-regulated, followed by an increase during myotube formation (Fig. 5). AT2R knockdown increased p-ERK1/2 in all stages of differentiation, indicating that the AT2R negatively regulated ERK1/2 signaling. Indeed, blockade of ERK1/2 signaling by the ERK inhibitors U0126 or PD98059 restored myoblast differentiation in the setting of AT2R knockdown (Fig. 6).
To our knowledge, this is the first report establishing a functional role of the AT2R in skeletal muscle regeneration. In fact, very little is known about the potential role of the AT2R in skeletal muscle biology, although a recent report has suggested that the AT2R may be involved in fibrotic responses in dystrophic skeletal muscle (37). Although AT2R expression is very low in rodent adult tissues, the AT2R is reported to be the dominant Ang II receptor expressed in the fetus. In many tissues, such as the adrenal gland, kidneys, heart, aorta, skin, and skeletal muscle, AT2R expression appears on embryonic days 11–13 and reaches a maximal level on embryonic day 19 (38). AT2R expression then rapidly declines in newborn animals to low or undetectable levels. Interestingly, AT2R expression in the adult reappears under pathophysiological conditions such as vascular injury and cardiac remodeling (39, 40). In some tissues, such as vascular smooth muscle, the AT2R is reported to mediate effects counterbalancing those mediated by the AT1R, e.g. vasodilatation, NO release, and inhibition of proliferation and growth (38, 41). Furthermore, it has been shown that the AT2R regulates neuronal cell differentiation and nerve regeneration following crush injury of the rat sciatic nerve (42). Therefore, the AT2R appears to play an important role in multiple organ development and neural regeneration. However, the molecular mechanisms whereby the AT2R may regulate tissue regeneration are understood poorly. Our data clearly show that the AT2R positively regulates satellite cell differentiation and skeletal muscle regeneration via down-regulation of ERK1/2 signaling. Considering the strong expression of AT2R during embryonic development and neuronal differentiation, one can speculate that a similar signaling mechanism is important for AT2R-mediated effects on cellular differentiation in other tissues.
Both AT1R and AT2R belong to the seven-transmembrane domain receptor superfamily. They have 31% amino acid identity and bind to Ang II with nanomolar affinity, but downstream signaling pathways are vastly different (38, 43). AT1R is a classical G protein-coupled receptor, signals through the Gαq subunit, and activates MAPKs. On the other hand, although the AT2R can couple to G proteins, in many cases it activates signaling cascades independent of G proteins. The major AT2R signaling transduction pathways include activation of protein phosphatases such as MAP kinase phosphatase 1 (MKP-1), activation of the NO/cGMP system, and stimulation of phospholipase A2 with subsequent release of arachidonic acid (38, 44). Activation of protein phosphatases is of particular interest because it has been shown that MAPK signaling may play an important role in satellite cell proliferation and differentiation (30, 45–47), although some studies are conflicting. Bennett et al. (30) showed that MKP-1 regulates ERK2 activity differently depending on the myoblast differentiation stages. ERK2 is inactivated upon mitogen withdrawal (differentiation cue), concomitant with up-regulation of muscle-specific genes, and this ERK2 inactivation is regulated by MKP-1. On the other hand, endogenous MKP-1 expression is decreased at a later stage of differentiation, and MKP-1 overexpression during differentiation prevented myotube formation. Contrary to these data, Jones et al. (46) reported that inactivation of the ERK1/2 pathway did not affect myoblast differentiation, whereas it prevented myoblast proliferation. Also, Jones et al. (47) showed that inhibition of p38MAPK promotes satellite cell quiescence and prevents differentiation. Although the basal activity of ERK1/2 changes dramatically during satellite cell differentiation, our data clearly show that the AT2R negatively regulates ERK1/2 signaling at all stages of differentiation (Fig. 5, A and B) and that ERK inhibitors prevented the reduction in regeneration caused by AT2R knockdown (Figs. 5 and 6). The mechanism whereby ERK1/2 may differentially regulate satellite cell differentiation, depending on the temporal stages of differentiation, remains to be determined. In our experimental setting, AT2R and ERK1/2 were inhibited throughout the differentiation processes, and future studies are required to precisely understand the function of the AT2R in satellite cells, especially in different stages of differentiation. In our experimental setting, we did not observe changes in p38MAPK and JNK signaling during differentiation, and AT2R knockdown did not affect these pathways. Although AT2R knockdown led to an increase in phospho-MEK and phospho-ERK1/2, we did not detect changes in MKP-1 levels during differentiation and after AT2R siRNA transfection, suggesting that the alteration of ERK1/2 activity was regulated independently of MKP-1. Interestingly, in ERK inhibitor-treated cells, AT2R expression was increased both basally (1.87 ± 0.20-fold increase in control siRNA + U0126 versus control siRNA, n = 3, p < 0.05) and in the presence of AT2R siRNA (3.87 ± 0.87-fold increase in AT2R siRNA + U0126 versus AT2R siRNA, n = 3, p < 0.05), and AT2R and myogenin/desmin expression was highly correlated, suggesting that there is a positive feedback loop whereby the AT2R and ERK1/2 interact to regulate myoblast differentiation. Namely, the AT2R inhibits ERK1/2 activity, which promotes differentiation, and the reduction in ERK1/2 activity increases AT2R expression, further sustaining myoblast differentiation (Fig. 7).
FIGURE 7.
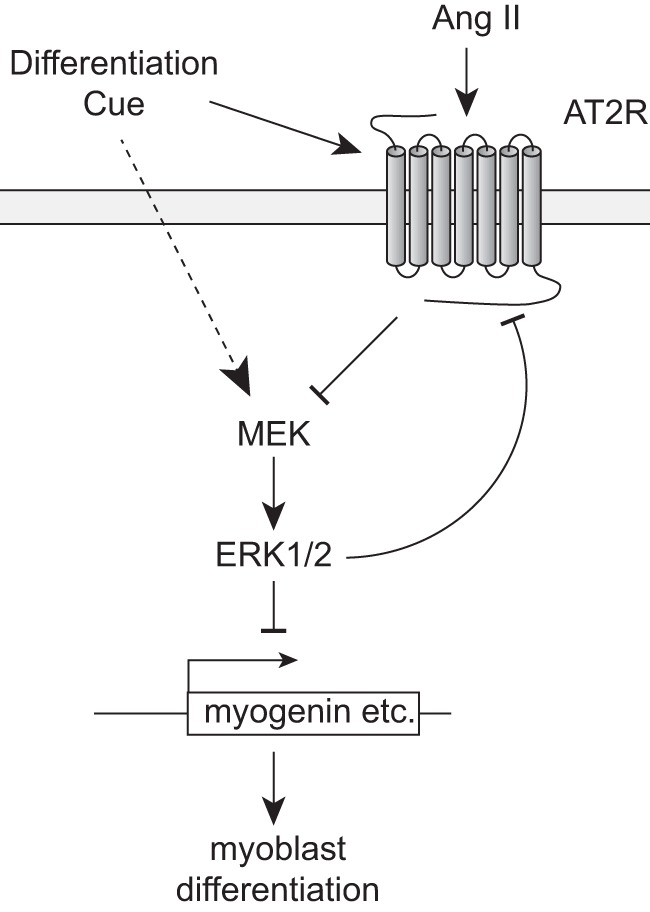
Proposed model for AT2R-mediated regulation of satellite cell differentiation. Initial cue(s) directing differentiation lead to rapid up-regulation of AT2R expression. During satellite cell differentiation, ERK1/2 phosphorylation is suppressed at the initial stage of differentiation, followed by an increase in the later stage (dotted line). AT2R inhibits ERK1/2 signaling at all stages of differentiation, resulting in activation of myogenic gene expression (myogenin and desmin) and myoblast differentiation. ERK1/2 inhibition increases AT2R expression, suggesting a positive feedback regulation. It is of note that it has been suggested that ERK1/2 positively regulates myotube formation in the later stage of differentiation, independently from the regulation of differentiation markers (data not shown; see “Discussion”).
Our findings raise the question of mechanisms involved in regulation of AT2R expression during myoblast differentiation. Our data indicate that the AT2R is strongly induced during myoblast differentiation, both transcriptionally and translationally. Little is known about AT2R transcriptional regulation. Ichiki et al. (48) showed that the DNA segment between −1497 and −874 upstream of the AT2R transcription start site was important for basal AT2R expression in mouse fibroblast R3T3 cells, whereas it was suppressive to basal AT2R expression in rat pheochromocytoma PC12W cells (49). These data suggest that there are different mechanisms regulating AT2R expression in different cell types, but, to date, no transcription factors have been identified that directly regulate AT2R expression in any cells/tissues. Future studies identifying cis- and trans-acting transcriptional pathways involved in the regulation of AT2R expression will be critical to further determine the mechanisms underlying AT2R-mediated satellite cell differentiation.
In summary, here we report, for the first time, that the AT2R plays a critical role in satellite cell differentiation and skeletal muscle regeneration. Expression of the AT2R is induced markedly in differentiating skeletal myoblasts, and the AT2R positively regulates myoblast differentiation via down-regulation of ERK1/2 activity. Taken together with our recent findings that Ang II AT1R signaling depletes the satellite cell pool and inhibits muscle regeneration, one can postulate that the balance between AT1R and AT2R signaling is critical for normal muscle regeneration. Because Ang II levels are elevated in chronic conditions that are accompanied by myopathy, such as CHF and chronic kidney disease, potential reductions in AT2R signaling could be particularly detrimental by resulting in preferential AT1R signaling. In fact, we have found recently that, in a mouse model of myocardial infarction, there is a markedly blunted increase in AT2R expression following injury and that this relative reduction in AT2R expression correlates with markedly reduced muscle regeneration.3 Skeletal muscle satellite cells could provide an excellent model system for the analysis of AT2R function in regulating tissue regeneration. Considering the function of the AT2R to potentiate myoblast differentiation, future studies assessing the mechanisms of AT2R regulation (e.g. identification of upstream transcription factors) have the potential to lead to the development of novel interventions to potentiate skeletal muscle regenerative capacity, particularly in chronic disease conditions.
This work was supported, in whole or in part, by NHLBI, National Institutes of Health Grants R01-HL070241 and R01-HL080682 and NIGMS, National Institutes of Health Grants P20-GM103629, P30-GM103337, and U54-GM104940.
T. Yoshida, unpublished observations.
- Ang II
- angiotensin II
- RAS
- renin-angiotensin system
- CHF
- congestive heart failure
- CTX
- cardiotoxin
- eMyHS
- embryonic myosin heavy chain
- qRT-PCR
- quantitative RT-PCR.
REFERENCES
- 1. Werner C., Pöss J., Böhm M. (2010) Optimal antagonism of the renin-angiotensin-aldosterone system: do we need dual or triple therapy? Drugs 70, 1215–1230 [DOI] [PubMed] [Google Scholar]
- 2. Brink M., Wellen J., Delafontaine P. (1996) Angiotensin II causes weight loss and decreases circulating insulin-like growth factor I in rats through a pressor-independent mechanism. J. Clin. Invest. 97, 2509–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brink M., Price S. R., Chrast J., Bailey J. L., Anwar A., Mitch W. E., Delafontaine P. (2001) Angiotensin II induces skeletal muscle wasting through enhanced protein degradation and down-regulates autocrine insulin-like growth factor I. Endocrinology 142, 1489–1496 [DOI] [PubMed] [Google Scholar]
- 4. Song Y.-H., Li Y., Du J., Mitch W. E., Rosenthal N., Delafontaine P. (2005) Muscle-specific expression of IGF-1 blocks angiotensin II-induced skeletal muscle wasting. J. Clin. Invest. 115, 451–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoshida T., Semprun-Prieto L., Sukhanov S., Delafontaine P. (2010) IGF-1 prevents ANG II-induced skeletal muscle atrophy via Akt- and Foxo-dependent inhibition of the ubiquitin ligase atrogin-1 expression. Am. J. Physiol. Heart Circ. Physiol. 298, H1565–H1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yoshida T., Semprun-Prieto L., Wainford R. D., Sukhanov S., Kapusta D. R., Delafontaine P. (2012) Angiotensin II reduces food intake by altering orexigenic neuropeptide expression in the mouse hypothalamus. Endocrinology 153, 1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Semprun-Prieto L. C., Sukhanov S., Yoshida T., Rezk B. M., Gonzalez-Villalobos R. A., Vaughn C., Michael Tabony A., Delafontaine P. (2011) Angiotensin II induced catabolic effect and muscle atrophy are redox dependent. Biochem. Biophys. Res. Comm. 409, 217–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tabony A. M., Yoshida T., Galvez S., Higashi Y., Sukhanov S., Chandrasekar B., Mitch W. E., Delafontaine P. (2011) Angiotensin II upregulates protein phosphatase 2Cα and inhibits AMP-activated protein kinase signaling and energy balance leading to skeletal muscle wasting. Hypertension 58, 643–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rezk B. M., Yoshida T., Semprun-Prieto L., Higashi Y., Sukhanov S., Delafontaine P. (2012) Angiotensin II infusion induces marked diaphragmatic skeletal muscle atrophy. PLoS ONE 7, e30276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sanders P. M., Russell S. T., Tisdale M. J. (2005) Angiotensin II directly induces muscle protein catabolism through the ubiquitin-proteasome proteolytic pathway and may play a role in cancer cachexia. Br. J. Cancer 93, 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Russell S. T., Sanders P. M., Tisdale M. J. (2006) Angiotensin II directly inhibits protein synthesis in murine myotubes. Cancer Lett. 231, 290–294 [DOI] [PubMed] [Google Scholar]
- 12. Morales M. G., Vazquez Y., Acuña M. J., Rivera J. C., Simon F., Salas J. D., Alvarez Ruf J., Brandan E., Cabello-Verrugio C. (2012) Angiotensin II-induced pro-fibrotic effects require p38MAPK activity and transforming growth factor β 1 expression in skeletal muscle cells. Int. J. Biochem. Cell Biol. 44, 1993–2002 [DOI] [PubMed] [Google Scholar]
- 13. Roig E., Perez-Villa F., Morales M., Jiménez W., Orús J., Heras M., Sanz G. (2000) Clinical implications of increased plasma angiotensin II despite ACE inhibitor therapy in patients with congestive heart failure. Eur. Heart J. 21, 53–57 [DOI] [PubMed] [Google Scholar]
- 14. Anker S. D., Negassa A., Coats A. J., Afzal R., Poole-Wilson P. A., Cohn J. N., Yusuf S. (2003) Prognostic importance of weight loss in chronic heart failure and the effect of treatment with angiotensin-converting-enzyme inhibitors: an observational study. Lancet 361, 1077–1083 [DOI] [PubMed] [Google Scholar]
- 15. Masson S., Latini R., Bevilacqua M., Vago T., Sessa F., Torri M., Anesini A., Salio M., Pasotti E., Agnello D., Santoro L., Catania A., Ghezzi P., Moccetti T., Maggioni A. P. (1998) Within-patient variability of hormone and cytokine concentrations in heart failure. Pharmacol. Res. 37, 213–217 [DOI] [PubMed] [Google Scholar]
- 16. Tan B. H., Fearon K. C. (2008) Cachexia: prevalence and impact in medicine. Curr. Opin. Clin. Nutr. Metab. Care 11, 400–407 [DOI] [PubMed] [Google Scholar]
- 17. Dalla Libera L., Ravara B., Angelini A., Rossini K., Sandri M., Thiene G., Battista Ambrosio G., Vescovo G. (2001) Beneficial effects on skeletal muscle of the angiotensin II type 1 receptor blocker irbesartan in experimental heart failure. Circulation 103, 2195–2200 [DOI] [PubMed] [Google Scholar]
- 18. Zhang L., Du J., Hu Z., Han G., Delafontaine P., Garcia G., Mitch W. E. (2009) IL-6 and serum amyloid A synergy mediates angiotensin II-induced muscle wasting. J. Am. Soc. Nephrol. 20, 604–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yoshida T., Galvez S., Tiwari S., Rezk B. M., Semprun-Prieto L., Higashi Y., Sukhanov S., Yablonka-Reuveni Z., Delafontaine P. (2013) Angiotensin II inhibits satellite cell proliferation and prevents skeletal muscle regeneration. J. Biol. Chem. 288, 23823–23832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yablonka-Reuveni Z., Day K. (2011) in Regenerating the Heart (Cohen I. S., Gaudette G. R., eds), pp. 173–200, Springer [Google Scholar]
- 21. Cohn R. D., van Erp C., Habashi J. P., Soleimani A. A., Klein E. C., Lisi M. T., Gamradt M., ap Rhys C. M., Holm T. M., Loeys B. L., Ramirez F., Judge D. P., Ward C. W., Dietz H. C. (2007) Angiotensin II type 1 receptor blockade attenuates TGF-β-induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 13, 204–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bedair H. S., Karthikeyan T., Quintero A., Li Y., Huard J. (2008) Angiotensin II receptor blockade administered after injury improves muscle regeneration and decreases fibrosis in normal skeletal muscle. Am. J. Sports Med. 36, 1548–1554 [DOI] [PubMed] [Google Scholar]
- 23. Johnston A. P., Baker J., Bellamy L. M., McKay B. R., De Lisio M., Parise G. (2010) Regulation of muscle satellite cell activation and chemotaxis by angiotensin II. PLoS ONE 5, e15212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murphy K. T., Allen A. M., Chee A., Naim T., Lynch G. S. (2012) Disruption of muscle renin-angiotensin system in AT1a−/− mice enhances muscle function despite reducing muscle mass but compromises repair after injury. Am. J. Physiol. Regul. Integr. Comp. Physiol. 303, R321–R331 [DOI] [PubMed] [Google Scholar]
- 25. Yan Z., Choi S., Liu X., Zhang M., Schageman J. J., Lee S. Y., Hart R., Lin L., Thurmond F. A., Williams R. S. (2003) Highly coordinated gene regulation in mouse skeletal muscle regeneration. J. Biol. Chem. 278, 8826–8836 [DOI] [PubMed] [Google Scholar]
- 26. Conboy I. M., Conboy M. J., Smythe G. M., Rando T. A. (2003) Notch-mediated restoration of regenerative potential to aged muscle. Science 302, 1575–1577 [DOI] [PubMed] [Google Scholar]
- 27. Conboy I. M., Rando T. A. (2002) The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev. Cell 3, 397–409 [DOI] [PubMed] [Google Scholar]
- 28. Blankley C. J., Hodges J. C., Klutchko S. R., Himmelsbach R. J., Chucholowski A., Connolly C. J., Neergaard S. J., Van Nieuwenhze M. S., Sebastian A., Quin J. (1991) Synthesis and structure-activity relationships of a novel series of non-peptide angiotensin II receptor binding inhibitors specific for the AT2 subtype. J. Med. Chem. 34, 3248–3260 [DOI] [PubMed] [Google Scholar]
- 29. Broderick K. E., Chan A., Lin F., Shen X., Kichaev G., Khan A. S., Aubin J., Zimmermann T. S., Sardesai N. Y. (2012) Optimized in vivo transfer of small interfering RNA targeting dermal tissue using in vivo surface electroporation. Mol. Ther. Nucleic Acids 1, e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bennett A. M., Tonks N. K. (1997) Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science 278, 1288–1291 [DOI] [PubMed] [Google Scholar]
- 31. Nakajima M., Hutchinson H. G., Fujinaga M., Hayashida W., Morishita R., Zhang L., Horiuchi M., Pratt R. E., Dzau V. J. (1995) The angiotensin II type 2 (AT2) receptor antagonizes the growth effects of the AT1 receptor: gain-of-function study using gene transfer. Proc. Natl. Acad. Sci. U.S.A. 92, 10663–10667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Agoudemos M. M., Greene A. S. (2005) Localization of the renin-angiotensin system components to the skeletal muscle microcirculation. Microcirculation 12, 627–636 [DOI] [PubMed] [Google Scholar]
- 33. Johnston A. P., Baker J., De Lisio M., Parise G. (2011) Skeletal muscle myoblasts possess a stretch-responsive local angiotensin signalling system. J. Renin Angiotensin Aldosterone Syst. 12, 75–84 [DOI] [PubMed] [Google Scholar]
- 34. Echeverría-Rodríguez O., Del Valle-Mondragón L., Hong E. (2014) Angiotensin 1–7 improves insulin sensitivity by increasing skeletal muscle glucose uptake in vivo. Peptides 51, 26–30 [DOI] [PubMed] [Google Scholar]
- 35. Prasannarong M., Santos F. R., Henriksen E. J. (2012) ANG-(1–7) reduces ANG II-induced insulin resistance by enhancing Akt phosphorylation via a Mas receptor-dependent mechanism in rat skeletal muscle. Biochem. Biophys. Res. Comm. 426, 369–373 [DOI] [PubMed] [Google Scholar]
- 36. Riquelme C., Acuña M. J., Torrejón J., Rebolledo D., Cabrera D., Santos R. A., Brandan E. (2014) ACE2 is augmented in dystrophic skeletal muscle and plays a role in decreasing associated fibrosis. PLoS ONE 9, e93449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Painemal P., Acuña M. J., Riquelme C., Brandan E., Cabello-Verrugio C. (2013) Transforming growth factor type β 1. BioFactors 39, 467–475 [DOI] [PubMed] [Google Scholar]
- 38. de Gasparo M., Catt K. J., Inagami T., Wright J. W., Unger T. (2000) International union of pharmacology: XXIII: The angiotensin II receptors. Pharmacol. Rev. 52, 415–472 [PubMed] [Google Scholar]
- 39. Akishita M., Horiuchi M., Yamada H., Zhang L., Shirakami G., Tamura K., Ouchi Y., Dzau V. J. (2000) Inflammation influences vascular remodeling through AT2 receptor expression and signaling. Physiol. Genomics 2, 13–20 [DOI] [PubMed] [Google Scholar]
- 40. Masaki H., Kurihara T., Yamaki A., Inomata N., Nozawa Y., Mori Y., Murasawa S., Kizima K., Maruyama K., Horiuchi M., Dzau V. J., Takahashi H., Iwasaka T., Inada M., Matsubara H. (1998) Cardiac-specific overexpression of angiotensin II AT2 receptor causes attenuated response to AT1 receptor-mediated pressor and chronotropic effects. J. Clin. Invest. 101, 527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Widdop R. E., Jones E. S., Hannan R. E., Gaspari T. A. (2003) Angiotensin AT2 receptors: cardiovascular hope or hype? Br. J. Pharmacol. 140, 809–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reinecke K., Lucius R., Reinecke A., Rickert U., Herdegen T., Unger T. (2003) Angiotensin II accelerates functional recovery in the rat sciatic nerve in vivo: role of the AT2 receptor and the transcription factor NF-κB. FASEB J. 17, 2094–2096 [DOI] [PubMed] [Google Scholar]
- 43. Berk B. C. (2003) Angiotensin type 2 receptor (AT2R): a challenging twin. Sci. STKE 2003, PE16. [DOI] [PubMed] [Google Scholar]
- 44. Steckelings U. M., Kaschina E., Unger T. (2005) The AT2 receptor: a matter of love and hate. Peptides 26, 1401–1409 [DOI] [PubMed] [Google Scholar]
- 45. Gredinger E., Gerber A. N., Tamir Y., Tapscott S. J., Bengal E. (1998) Mitogen-activated protein kinase pathway is involved in the differentiation of muscle cells. J. Biol. Chem. 273, 10436–10444 [DOI] [PubMed] [Google Scholar]
- 46. Jones N. C., Fedorov Y. V., Rosenthal R. S., Olwin B. B. (2001) ERK1/2 is required for myoblast proliferation but is dispensable for muscle gene expression and cell fusion. J. Cell Physiol. 186, 104–115 [DOI] [PubMed] [Google Scholar]
- 47. Jones N. C., Tyner K. J., Nibarger L., Stanley H. M., Cornelison D. D., Fedorov Y. V., Olwin B. B. (2005) The p38α/β MAPK functions as a molecular switch to activate the quiescent satellite cell. J. Cell Biol. 169, 105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ichiki T., Inagami T. (1995) Expression, genomic organization, and transcription of the mouse angiotensin II type 2 receptor gene. Circ. Res. 76, 693–700 [DOI] [PubMed] [Google Scholar]
- 49. Ichiki T., Inagami T. (1995) Transcriptional regulation of the mouse angiotensin II type 2 receptor gene. Hypertension 25, 720–725 [DOI] [PubMed] [Google Scholar]