

Background: Isoprenylcysteine carboxyl methyltransferase (ICMT) is an integral membrane enzyme that modifies CAAX proteins.
Results: Mutational analysis has identified residues important for activity and substrate recognition.
Conclusion: Crucial residues are found in cytosolic and membrane-embedded regions, consistent with the ability of ICMT to bind both water-soluble and lipophilic substrates.
Significance: Insights into the enzymatic mechanism of ICMT may assist with inhibitor development.
Keywords: Enzyme Mechanism, Membrane Enzyme, Post-translational Modification (PTM), Protein Methylation, Ras Protein, Site-directed mutagenesis, Isoprenylcysteine Carboxyl Methyltransferase (ICMT), Substrate Recognition
Abstract
The eukaryotic integral membrane enzyme isoprenylcysteine carboxyl methyltransferase (ICMT) methylates the carboxylate of a lipid-modified cysteine at the C terminus of its protein substrates. This is the final post-translational modification of proteins containing a CAAX motif, including the oncoprotein Ras, and therefore, ICMT may serve as a therapeutic target in cancer development. ICMT has no discernible sequence homology with soluble methyltransferases, and aspects of its catalytic mechanism are unknown. For example, how both the methyl donor S-adenosyl-l-methionine (AdoMet), which is water-soluble, and the methyl acceptor isoprenylcysteine, which is lipophilic, are recognized within the same active site is not clear. To identify regions of ICMT critical for activity, we combined scanning mutagenesis with methyltransferase assays. We mutated nearly half of the residues of the ortholog of human ICMT from Anopheles gambiae and observed reduced or undetectable catalytic activity for 62 of the mutants. The crystal structure of a distantly related prokaryotic methyltransferase (Ma Mtase), which has sequence similarity with ICMT in its AdoMet binding site but methylates different substrates, provides context for the mutational analysis. The data suggest that ICMT and Ma MTase bind AdoMet in a similar manner. With regard to residues potentially involved in isoprenylcysteine binding, we identified numerous amino acids within transmembrane regions of ICMT that dramatically reduced catalytic activity when mutated. Certain substitutions of these caused substrate inhibition by isoprenylcysteine, suggesting that they contribute to the isoprenylcysteine binding site. The data provide evidence that the active site of ICMT spans both cytosolic and membrane-embedded regions of the protein.
Introduction
Members of the Ras family of GTPases and many other eukaryotic proteins critical to oncogenesis and tumor progression undergo a series of post-translational modifications signaled by a C-terminal amino acid CAAX motif (where C is a cysteine, A is an aliphatic amino acid, and X is one of several potential amino acids) (1). The processing of proteins with a CAAX motif (CAAX proteins) includes isoprenylation of the cysteine by either a farnesyl or geranylgeranyl lipid, endoproteolytic cleavage of the AAX residues, and carboxyl methylation of the resulting isoprenylcysteine. The enzyme responsible for the methylation step is isoprenylcysteine carboxyl methyltransferase (ICMT)2 (2) (Fig. 1). ICMT is a multi-spanning endoplasmic reticulum integral membrane protein conserved among eukaryotic organisms. Methylation by ICMT is important for partitioning of CAAX proteins into lipid membranes as this modification neutralizes the negative charge on the carboxylate and increases the hydrophobicity of the modified protein (3). Without carboxyl methylation, K-Ras is mislocalized away from the plasma membrane and diminished in its ability to regulate cell growth and proliferation (4, 5). Thus, inhibiting ICMT is a possible anti-cancer strategy (6).
FIGURE 1.
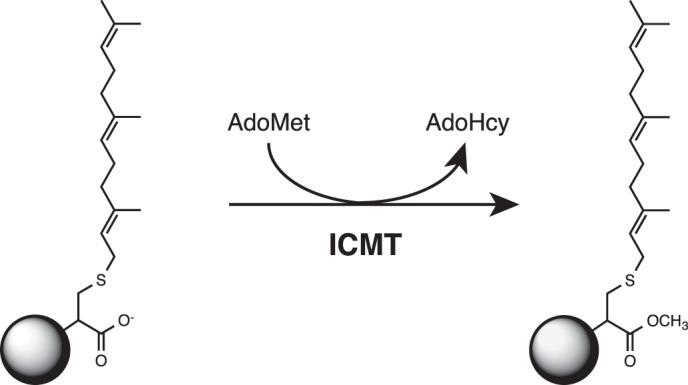
Reaction catalyzed by ICMT. ICMT methylates the carboxylate of a farnesylated (15 carbons) cysteine residue (as depicted here) or a geranylgeranylated (20 carbons) cysteine residue at the C terminus of its protein substrates. AdoMet is the methyl donor in the reaction. Additional elements of the protein substrate that are not required for substrate recognition by ICMT are depicted as a circle.
A number of farnesyltransferase inhibitors that target this pathway of post-translational modifications were tested in the clinic; however, these drugs had limited utility and efficacy (7) perhaps due to the alternative prenylation of Ras by a geranylgeranyl lipid (8) or due to changes in the behavior of other CAAX proteins relevant to cancer progression (9). Despite the complexities encountered with farnesyltransferase inhibitors, inactivation of ICMT has been shown to decrease cellular transformation by oncogenic K-Ras in rodent and human cell-based cancer models, which include colon, prostate, breast, and lung (5, 10–14). A recent report that knocking out ICMT in a mouse model of pancreatic cancer exacerbated the disease (15) underscores the need for further studies exploring the potential utility of ICMT inhibitors. To date, only a few inhibitors of ICMT have been identified, and no clinical trials using these inhibitors have been conducted (13, 16, 17). The development of effective ICMT inhibitors will benefit from a detailed understanding of the active site of the enzyme.
The mechanism by which ICMT methylates its substrates is not fully understood. ICMT has no sequence homology with soluble methyltransferases, and the three-dimensional structure of a eukaryotic ICMT family member has yet to be determined. Studies addressing the topology of the enzyme have suggested that Homo sapiens ICMT (Hs ICMT) contains eight transmembrane helices (TMs) and Saccharomyces cerevisiae ICMT (Sc ICMT) has six TMs (Fig. 2) (18, 19). Plant orthologs (e.g. Arabidopsis thaliana ICMT) may contain only five TMs (Fig. 2). Sequence conservation among ICMT family members is highest for the region of the protein corresponding to the six C-terminal TMs of Hs ICMT (M3-M8). Although limited mutational analysis has identified a few amino acids implicated in ICMT function (18–20), the binding site for the isoprenylcysteine substrate has not been identified.
FIGURE 2.

Sequence alignment of ICMT orthologs and Ma MTase. Sequence alignment of Ag ICMT (283 amino acids (aa)), Hs ICMT (284 aa), Sc ICMT (239 aa), At ICMT (197 aa), and a prokaryotic integral membrane methyltransferase of known structure, Ma MTase (194 aa). Helices are indicated by horizontal rectangles below the aligned sequences. Helical regions of ICMT (black rectangles) were predicted using Jpred (43) and are also based on hydropathy plots and experimental mapping of Hs ICMT (19), where solid black rectangles represent regions predicted to reside in the membrane and open rectangles indicate helical regions in the cytosol. The Ma MTase helices (gray rectangles) and transmembrane helices (solid gray) were determined from the crystal structure (20). ICMT residues identified in this study as important for substrate binding are indicated: stars mark residues involved in AdoMet binding, and hexagons mark residues for which mutants have altered isoprenylcysteine substrate binding properties. All ICMT residues whose mutants were inactive are colored red; a red square indicates a residue that severely impaired activity when mutated but whose particular role has not been assigned. The UniProt accession numbers for the sequences in the alignment are: O60725, Q7PXA7 (version 2), P32584, Q93W54, and Q8TMG0. Alignment was made with ClustalW with manual adjustments. Conservation of the residues at the N terminus of Ag ICMT and Hs ICMT (M1 and M2) is poor, and as such, the alignment is less certain in this region.
Like most cellular methyltransferases, ICMT uses S-adenosyl-l-methionine (AdoMet) as the methyl donor in the reaction. Kinetic analysis suggests that the enzymatic reaction proceeds in an ordered manner, with AdoMet binding first followed by the isoprenylcysteine substrate (21, 22). Product release is also ordered with the methylated isoprenylcysteine dissociating first followed by S-adenosyl-l-homocysteine (AdoHcy) (21, 22).
AdoMet-dependent methyltransferases can be classified based on a consensus sequence and the structural fold of their AdoMet binding site (23). Five methyltransferase classes have been identified for water-soluble methyltransferase enzymes (classes I-V). A prokaryotic integral membrane methyltransferase from Methanosarcina acetivorans (Ma MTase), for which a crystal structure has been determined (20), is the founding member of an integral membrane class of methyltransferase enzymes (class VI). The portion of ICMT containing its two C-terminal TMs (M7 and M8 of Hs ICMT) has sequence homology (28.4% identity) with the corresponding portion of Ma MTase and contains the region known to bind AdoHcy from the crystal structure (Fig. 2) (20). This sequence similarity suggests that ICMT is a member of the class VI methyltransferase enzymes and that Ma MTase and ICMT bind AdoMet similarly.
In ICMT, recognition of the lipid-modified protein substrate is governed by the carboxylate of the isoprenylated cysteine residue at the C terminus of the protein substrate and by the attached isoprenoid lipid, but recognition does not require additional elements of the protein substrate (24, 25). Minimal isoprenylcysteine substrates of ICMT include N-acetyl farnesyl cysteine, N-acetyl geranylgeranyl cysteine, and S-farnesyl-3-thiopropionic acid, which have similar Km values to a full-length isoprenylated protein substrate (25).
The biological methyl acceptor substrate(s) of the prokaryotic Ma MTase are not known, and Ma MTase does not efficiently methylate substrates of ICMT such as N-acetyl farnesyl cysteine (20). Aside from the residues observed to bind AdoHcy in the crystal structure of Ma MTase, the sequence similarity with ICMT is low (∼15% overall identity), suggesting that the regions of their active sites that bind methyl acceptor substrates differ between the enzymes.
Mutational analysis of ICMT would help identify residues that are important for enzymatic activity and those that are involved in substrate binding. Here we present a mutagenesis study encompassing approximately half of the amino acids of the ortholog of Hs ICMT from Anopheles gambiae (Ag ICMT). We find that many conserved residues are important for ICMT function, and we identify residues that likely contribute to the binding sites for its substrates. The amino acids involved in binding AdoMet are conserved with Ma MTase. Amino acids implicated in binding of the isoprenylcysteine substrate are conserved only among eukaryotic ICMT family members.
EXPERIMENTAL PROCEDURES
Cloning and Expression
Using complementary DNA (cDNA) as a template, PCR products were obtained for Hs ICMT and Ag ICMT. The products were ligated into the expression vector pCGFP-EU (26) to create ICMT constructs with green fluorescent protein (GFP) fused to the C terminus, and the coding regions were sequenced in their entirety. Point mutations were introduced using standard site-directed mutagenesis PCR techniques. Total cell lysate containing overexpressed GFP-tagged Hs ICMT or Ag ICMT was prepared as follows. HEK293 cells were transfected with the plasmids using Lipofectamine 2000 (Invitrogen) and cultured for 48 h after transfection. Cells were harvested, divided into two aliquots, and each aliquot was resuspended in 150 μl of buffer consisting of 50 mm NaH2PO4, pH 7.5, 190 mm NaCl, 10 mm KCl, 20 mm DTT, and 1:500 dilution of Protease Inhibitor Mixture Set III, EDTA-free (Calbiochem). One aliquot was used for analysis by fluorescence-detection size-exclusion chromatography (FSEC) (26). For this aliquot, the buffer was supplemented with 200 μm AdoHcy (Sigma) and 10 mm of the detergent lauryl maltoside neopentyl glycol (Anatrace). Samples were rotated for 1 h at 4 °C and then centrifuged at 20,800 × g for 1 h at 4 °C before FSEC analysis. The cells in the second aliquot were lysed by sonication (10 s) in a bath sonicator and stored at 4 °C for no more than a few hours before use in the enzymatic assay.
Enzymatic Assay
A radioactivity assay involving the quantification of [3H]methyl incorporation into biotin-S-farnesyl-l-cysteine (BFC) was used to detect ICMT activity. This assay is based on a previously described protocol with additions and modifications (21). The cell lysate was diluted into reaction buffer containing 100 mm HEPES, pH 7.4, 150 mm NaCl, 5 mm MgCl2, and 1 mm DTT to yield an ICMT protein concentration of ∼10 nm (determined from the fluorescence of the GFP fusion protein relative to a purified GFP standard, which was quantified using FSEC (26) as the area under the curve for the corresponding peaks). The concentration of substrates in the reaction was 4 μm BFC (Duke University Small Molecule Synthesis Facility) and 5 μm [3H]AdoMet (1.3 Ci/mmol) (PerkinElmer Life Sciences) under standard reaction conditions. For further analyses the concentrations of BFC and AdoMet were varied as detailed in the figures legends.
The reaction was carried out for 20 min at 37 °C and was stopped by the addition of 500 μl of ice-cold quench buffer containing 20 mm NaH2PO4, pH 7.4, 150 mm NaCl, and 0.2% Tween 20. 10 μl of streptavidin beads (25 μl for reactions using >4 μm BFC) (Streptavidin Sepharose High Performance (GE Healthcare) slurry in 20% ethanol) were added, and the samples were rotated for 1 h at 4 °C. The beads were collected by centrifugation and washed 3 times with wash buffer containing 20 mm NaH2PO4, pH 7.4, and 150 mm NaCl. The beads were then resuspended in 100 μl of wash buffer, transferred to scintillation vials, and 5 ml of scintillation fluid (Ultima Gold, PerkinElmer Life Sciences) was added. [3H]Methyl incorporation was determined by scintillation counting. Background activity due to endogenous Hs ICMT (obtained using lysate from HEK293 cells transfected with GFP alone) was subtracted for all experiments and represented ∼1% of the total activity obtained from cells transfected with GFP-tagged Hs ICMT or Ag ICMT. To account for differences in the expression levels of the various ICMT constructs, the measured activity was divided by the expression level of the GFP-tagged construct, as quantified by FSEC. When comparing the activity of the mutants, the measured activity was normalized using measurements for wild-type enzyme conducted on the same day.
We found that a small amount of BFC bound to the plastic tubes and/or tips. To account for this, we determined the approximate concentration of available BFC in the reactions (using the method described below). For the initial velocity versus BFC experiments, we plotted available, not total, BFC. Using the total BFC value (the theoretical concentration of BFC assuming that none of it is lost due to binding to plastic) does not change the shape of the curves (they are still sigmoidal) but overestimates the Khalf by ∼1 μm. To approximate the amount of available BFC, a batch of radiolabeled BFC-[3H]methyl was enzymatically produced using Ag ICMT (from a reaction containing 4 nm ICMT, 4 μm BFC, and 5 μm [3H]AdoMet (1.3 Ci/mmol), incubated for 1 h at 37 °C). This reaction mixture containing BFC-[3H]methyl was then used to dope reaction mixtures that were prepared and processed in an identical manner to our initial velocity experiments, except that no enzyme was added and the incubation step at 37 °C was omitted. After pulldown by streptavidin, the available concentration of BFC was calculated based on scintillation counting, plotted in comparison to the total BFC value, and fitted to the polynomial equation of Fig. 3D. The correction is fairly minor (for example, a theoretical total BFC value of 5 μm corresponds to a 3.8 μm concentration of available BFC), and it relies on the assumption that BFC and BFC-[3H]methyl bind to plastic to a similar degree.
FIGURE 3.

Comparison of the activities of Ag ICMT and Hs ICMT. A and B, activities of Ag ICMT and Hs ICMT shown as a plot of the formation of BFC-[3H]methyl ester as a function of BFC concentration (0.8–50 μm) (A) or AdoMet concentration (0.5–50 μm) (B). The data shown are the mean of duplicate measurements, with error bars representing ± 1 S.D. C, the Khalf, Vmax, kcat, and h values obtained for BFC, and the Km, Vmax, and kcat values obtained for AdoMet as determined from the initial velocity curves in A and B. Best-fit values calculated in GraphPad Prism 6 are reported with the S.E. of the fit. D, correlation between total and available BFC used to correct for BFC binding to plastic tubes and pipette tips in A. The data shown are the mean of triplicate measurements, with error bars representing ± 1 S.D. E, dose-response curves comparing the inhibitory effect of S-farnesylthioacetic acid (FTA) on Ag ICMT and Hs ICMT activity. The data shown are the mean of duplicate measurements, with error bars representing ± 1 S.D.
For initial velocity curves, the data were fit to the following models using least squares nonlinear regression with GraphPad Prism 6 software. 1) For the initial velocity versus AdoMet concentration curves, a Michaelis-Menten model was used.
2) For the initial velocity versus BFC concentration curves when substrate inhibition was not observed, an allosteric sigmoidal model was used.
3) For the initial velocity versus BFC concentration curves when substrate inhibition was observed, an allosteric sigmoidal model that takes into account substrate inhibition was used.
In these equations Y is the initial velocity, X is the substrate concentration, h is the Hill coefficient, Vmax is the maximum enzyme velocity, Khalf and Km are the concentrations of half-maximal velocity for sigmoidal and Michaelis-Menten models, respectively, and Ki is the inhibition constant. For experiments where the concentration of BFC was varied, 5 μm AdoMet or 20 μm AdoMet (for data in Fig. 4B) was used. For experiments where the concentration of AdoMet was varied, 4 μm BFC was used. To account for the background activity due to endogenous Hs ICMT in the HEK293 cell lysate, initial velocity curves were constructed using lysate from cells transfected with GFP alone (e.g. Fig. 4A) and subtracted for all analyses.
FIGURE 4.

Sigmoidal kinetics for BFC observed under different conditions. A, activity of endogenous Hs ICMT shown as a plot of the formation of BFC-[3H]methyl ester as a function of BFC concentration (0.8–50 μm). For comparison, the Vmax for Hs ICMT-GFP in terms of radioactive counts is 192,000 ± 5,000 cpm (see Fig. 3A). B, activity of Ag ICMT shown as a plot of the formation of BFC-[3H]methyl ester as a function of BFC concentration (1.3–50 μm) when AdoMet was saturating (20 μm). C, activity of Hs ICMT in the presence of 20-fold less membranes than used under standard reaction conditions shown as a plot of the formation of BFC-[3H]methyl ester as a function of BFC concentration (0.5–18.2 μm). For A–C, the data shown are the mean of duplicate determinations of a single experiment, with error bars representing ± 1 S.D. D, kinetic parameters derived from A–C. Values were determined by fitting the data to an allosteric sigmoidal model (details are indicated under “Experimental Procedures”). Values are reported with the S.E. of the fit.
RESULTS
Identification and Characterization of A. gambiae ICMT
To efficiently characterize various ICMT enzymes, we developed an assay that measures the catalytic activity of overexpressed ICMT in cell lysate and one in which the amount of ICMT enzyme in the assay can be quantified from the fluorescence of a GFP tag attached to ICMT. After transient transfection of GFP-tagged ICMT in HEK293 cells, enzymatic activity was detected from the transfer of [3H]methyl to the minimal methyl acceptor substrate BFC (21). The amount of ICMT enzyme present in each reaction was determined by solubilizing an identical sample in detergent, analyzing it using size-exclusion chromatography by monitoring the fluorescence of the GFP tag (FSEC) (26), and calculating the amount of enzyme from the area under the FSEC peak corresponding to ICMT-GFP in comparison to purified GFP standards (e.g. Fig. 5A). FSEC analysis also provided an indication of the overall structural integrity of the various ICMT constructs.
FIGURE 5.

Scanning mutagenesis of ICMT. A, representative FSEC traces for wild-type Ag ICMT, an inactive mutant (Y212A), and a biochemically unstable mutant (E141A) in the detergent lauryl maltoside neopentyl glycol. B, radioactive methyl transfer relative to wild-type Ag ICMT for the mutants studied. The substrate concentrations were 4 μm BFC and 5 μm AdoMet. The data shown are the mean of triplicate determinations from a single experiment, with error bars representing ± 1 S.D. 100% activity is equivalent to ∼55 pmol of methyl transfer in 20 min. Asterisks indicate the biochemically unstable mutants E141A and G162L as assessed by FSEC.
We selected Ag ICMT for mutational analysis from among 55 other eukaryotic ICMT orthologs that we evaluated using FSEC because it has superior biochemical stability in detergent and might be useful in future structural studies. Human ICMT (Hs ICMT) and Ag ICMT share 51% identity, are of similar overall length, and are both predicted to have eight TMs (Fig. 2).
Some ICMT family members from lower eukaryotes, such as S. cerevisiae ICMT (Sc ICMT), are significantly shorter at their N-terminal region than Hs ICMT and Ag ICMT (Fig. 2). To determine whether the N-terminal extension in Hs ICMT and Ag ICMT is essential for function, we compared the activities of full-length ICMT with truncated versions of the enzymes (Hs ICMT60–284 and Ag ICMT58–283, in which the N-terminal 59 or 57 amino acids, respectively, were replaced by methionine). Hs ICMT, Ag ICMT, Hs ICMT60–284, and Ag ICMT58–283 all had similar FSEC profiles, suggesting that the truncated enzymes were expressed and biochemically stable. However, Hs ICMT60–284 and Ag ICMT58–283 had no detectable activity, indicating that the N-terminal region of the protein is important for the activity of these enzymes despite its absence from other ICMT enzymes.
To evaluate whether Ag ICMT and Hs ICMT have comparable enzymatic properties, initial velocities were plotted as functions of BFC and AdoMet concentration, and kinetic parameters were obtained by fitting the data (Fig. 3, A–C). The enzymes exhibited similar kinetics for both substrates and had similar kcat values. The Km of AdoMet was 6.7 ± 0.6 and 8.4 ± 0.7 μm for Ag ICMT and Hs ICMT, respectively. This agrees well with the value determined previously for Hs ICMT (7.8 ± 1.2 μm) (21).
The curves of initial velocity versus BFC concentration were sigmoidal in shape with a Hill coefficient of 3.4 ± 0.5 for Ag ICMT and 3.9 ± 0.5 for Hs ICMT (Fig. 3, A and C). The concentration of BFC that produced a half-maximal enzyme velocity (Khalf) was 1.9 ± 0.1 and 2.4 ± 0.1 μm for Ag ICMT and Hs ICMT, respectively. We observed sigmoidal kinetics for BFC under all of the assay conditions that we tested. Analysis of the endogenous Hs ICMT that is present within HEK293 cells also revealed sigmoidal kinetics (Fig. 4, A and D). Overexpressed Hs ICMT in the absence of a tag exhibited comparable kinetic parameters, indicating that the effect of the GFP tag was minimal (data not shown). Sigmoidal kinetics were also observed for Ag ICMT when using 20 μm AdoMet, a concentration well above its Km (Fig. 4, B and D). Using 20-fold less membranes than the standard reaction also yielded indistinguishable sigmoidal curves, suggesting that the total quantity of BFC relative to the quantity of membranes was not the cause (Fig. 4, C and D). The reason that we observe a sigmoidal relationship between the initial velocity and the concentration of BFC, whereas previous characterization of Hs ICMT yielded Michaelis-Menten behavior (21), is not clear, but it might be due to differences in the source of the expressed protein (HEK293 cell membranes in the present study and insect cell membranes in Baron and Casey (21)). Nevertheless, the Km value for BFC determined previously (2.1 ± 0.4 μm) (21) is in accord with the Khalf reported here (2.4 ± 0.1 μm).
To further characterize Ag ICMT, the inhibitory effect of S-farensylthioacetic acid (25) was tested and compared with that for Hs ICMT. We found that S-farensylthioacetic acid inhibits both ICMT enzymes with similar IC50 values (∼25 μm for Ag ICMT and Hs ICMT) (Fig. 3E). Taken together with the kinetic analysis, the results suggest that the active sites of these enzymes are similar and that functional studies of Ag ICMT regarding the mechanisms of substrate recognition and catalysis will be relevant to the human enzyme.
Scanning Mutagenesis
To identify amino acid residues important for ICMT function, we mutated 137 of the 283 residues in Ag ICMT, and we analyzed the mutants for enzymatic activity and FSEC profile. We focused on regions of highest sequence conservation among eukaryotic ICMT enzymes (Fig. 2). In addition, all cysteine residues and numerous amino acids located in cytosolic loop regions were mutated. Residues that were not alanine or glycine in wild-type ICMT were mutated to alanine. Alanine residues were mutated to leucine, and additionally glycine in some positions, whereas glycine residues were changed to both alanine and leucine. The results from this analysis are summarized in Figs. 5 and 6. Most of the mutants gave FSEC profiles nearly identical to wild-type protein, indicating that these mutations did not cause structural changes substantial enough to cause protein aggregation or large scale misfolding (Fig. 5A). Two exceptions were the E141A and G162L mutants, which had significantly altered FSEC profiles (Fig. 5A). For initial screening, the amount of methyl transferred to BFC was measured for wild-type or mutant ICMT using the standard reaction conditions (substrate concentrations of 4 μm BFC and 5 μm AdoMet).
FIGURE 6.

Topology model of Ag ICMT. Putative TMs are illustrated as numbered cylinders (M1-M8), lines indicate predicted loop regions, and site-directed mutants are indicated by an oval containing the residue number (Ag ICMT numbering). Most of the residues studied were mutated to alanine. Glycine residues were mutated to alanine and leucine, and alanine residues were mutated to leucine and in some cases glycine. The residues are colored as indicated based on the percentage of radioactive methyl transfer relative to wild-type Ag ICMT as shown in Fig. 5B.
62 mutants were identified that had normal FSEC profiles but had activities of <50% relative to wild-type ICMT (Figs. 5B and 6). Of these mutants, 34 exhibited activities <5% in comparison to wild-type, 13 exhibited activities between 5 and 30%, and 15 exhibited activities between 30 and 50% (Fig. 6). The 62 residues are located in both the cytosolic and membrane-embedded regions of ICMT, and they can be grouped into two categories. One category contains residues that are conserved with Ma MTase and likely form the AdoMet binding site. Residues in the second category, which includes most of the catalytically compromising mutations, have high sequence conservation among eukaryotic ICMT orthologs but little or no sequence conservation with Ma MTase.
Mutation of the AdoMet Binding Site
The residues of Ma MTase that are located within 5 Å of AdoHcy in the structure are fully conserved between Ma MTase and ICMT (Fig. 7, A and C) (20). There are two sequence motifs that form the AdoMet/AdoHcy binding site: AdoMet binding motif I (HXLVXXXXYXXXRHPXY), located in the M4-M5 cytosolic loop of Ma MTase (corresponding to the M6-M7 loop of Ag ICMT), and AdoMet binding motif II (RXXXEEXXLXXXFXXXYXXXY), which contains amino acids in the cytosolic portion of an α-helix extending from M5 (termed the “M5 extension” in Ma MTase and the “M8 extension” in Ag ICMT) and a cytosolic C-terminal helix (Fig. 7A). We mutated all of these conserved amino acids (His-196, Leu-198, Val-199, Tyr-204, Arg-208, His-209, Pro-210, Tyr-212, Arg-246, Glu-250, Glu-251, Leu-254, Phe-258, Tyr-262, and Tyr-265) in Ag ICMT to alanine. Except for H196A, all of the mutants had reduced methyltransferase activity (<50%) (Fig. 7B).
FIGURE 7.

Mutation of the AdoMet binding motif. A, sequence alignment of the conserved AdoMet binding region in Ag ICMT, Hs ICMT, Hs PEMT, and Ma MTase. Residues that interact with AdoHcy in the Ma MTase structure are indicated by a star. The UniProt accession number for Hs PEMT is CAG33380. Alignment was made in ClustalW with manual adjustments. B, mutational analysis of the residues conserved between Ma MTase and ICMT that coordinate AdoHcy in the Ma MTase structure. All residues were mutated to alanine. For Glu-250 and Glu-251, conservative substitutions to aspartic acid and glutamine were also evaluated. The activity assay was conducted using 4 μm BFC and 5 or 50 μm AdoMet. The data shown are the mean of duplicate measurements with error bars representing ± 1 S.D. Ag ICMT numbering is shown with Hs ICMT numbering in parentheses. C, details of the AdoMet binding site from the crystal structure of Ma MTase (PDB code 4A2N). AdoHcy and residues involved in binding AdoHcy (Ma MTase numbering with Ag ICMT numbering in parentheses) are drawn as sticks, and hydrogen bonds are indicated as dashed lines. D, details of the interaction of Glu-167 and Glu-168 (Glu-250 and Glu-251 in Ag ICMT) with AdoHcy. C and D were generated in PyMOL.
To investigate whether activity of these mutants was impaired due to an increased Km for AdoMet, we assayed the enzymatic activity of the mutants using both 5 and 50 μm AdoMet. Using 50 μm AdoMet, an increase in methyl transfer was observed for a number of the mutants tested (L198A, V199A, P210A, L254A, F258A, Y262A, and Y265A), suggesting that these mutants have higher Km values for AdoMet in comparison to wild-type ICMT (Fig. 7B). For several of the mutants, increasing the AdoMet concentration to 50 μm did not increase the amount of methyl transferred to an appreciable degree, which is consistent with a larger increase in the Km or an effect on kcat for these mutants.
In Ma MTase, two glutamic acids, Glu-167 and Glu-168 (corresponding to Glu-250 and Glu-251 in Ag ICMT), participate in a network of hydrogen bonds that interact with the amino group of AdoHcy (Fig. 7D). Glu-167 makes a hydrogen bond directly with it, and Glu-168 participates in an indirect interaction involving His-126 (His-209 in Ag ICMT) (20). To investigate whether the presence or position of the negative charge on these residues is important in ICMT, we made conservative changes to Glu-250 and Glu-251 by mutating them to aspartic acid and glutamine. Of these, the only mutant that had detectable activity was E251D, which had ∼20% activity in comparison to wild-type ICMT when assayed using 5 μm AdoMet (Fig. 7B). The E251D mutant regained wild-type activity levels at 50 μm AdoMet, indicating an effect on Km for this mutant (Fig. 7B). In agreement with the Ma MTase structure (20) as well as a previous Sc ICMT site-directed mutagenesis study (18), our results suggest that the negative charge of Glu-251 plays an important role in stabilization of the imidazole ring of His209 that interacts with AdoMet (Fig. 7D). Our data suggest that Ma MTase and ICMT have very similar AdoMet binding sites and that the binding site for AdoMet in Ag ICMT consists of the following residues: 198LVXXXXYXXXRHPXY212 and 246RXXXEExxLXXXFXXXYXXXY265. The residues that interact with AdoMet in ICMT are most likely located in the cytosol and flanked by hydrophobic regions of the enzyme that form the binding site for the isoprenylcysteine substrate.
Mutations of Conserved Residues That Perturb the Isoprenylcysteine Binding Site
Based on a proposed lipid binding tunnel in the Ma MTase structure (20) as well as proximity to the AdoMet binding site in ICMT, a number of mutants from our alanine-scanning mutagenesis survey that exhibited reduced activity were further evaluated with respect to possible defects in BFC binding (Phe-102, His-103, Thr-105, Glu-106, Tyr-107, Phe-123, Ile-124, Leu-125, Phe-184, His-186, Val-188, Tyr-212, Trp-215, Phe-216, Trp-218, Thr-222, Gln-223, Trp-240, Phe-242, and Phe-243) (Fig. 5B). The residues that were selected are located in M4, the M4-M5 loop, the M6-M7 loop, M7, and M8 of Ag ICMT (Fig. 6). We compared the activity of the mutants at 4 and 40 μm BFC to assess whether the diminished activity of these mutants was due to an increased Khalf for BFC (Fig. 8A). An increase in methyl transfer was not observed for any of the mutants when using 40 μm BFC. Rather, a decrease in activity was observed for several of the mutants at the elevated BFC concentration: T105A and Y107A from M4, F216A, T222A, and Q223A from M7, and W240A and F242A from M8 (Fig. 8A). In contrast to wild-type ICMT, these mutants were inhibited by higher BFC concentrations, consistent with the phenomenon of substrate inhibition, where an enzyme is inhibited by elevated concentrations of substrate (27). All of the residues that exhibited substrate inhibition when mutated to alanine are located in transmembrane regions. They are highly conserved among ICMT family members but are not conserved in Ma MTase (Fig. 2). With the exception of Trp-240 (Trp-241 in Hs ICMT) (19), these residues have not been previously implicated in ICMT function. These mutants were not inhibited by elevated concentrations of AdoMet (50 μm) (Fig. 8B). We evaluated the effect of the BFC concentration on the activity for one of the mutants (F216A) in the context of higher concentrations of AdoMet and observed substrate inhibition by BFC when using either 5 or 50 μm AdoMet (Fig. 8C). Our results indicate that the isoprenylcysteine substrate, but not AdoMet, inhibits these mutants and that this inhibition is independent of the AdoMet concentration, suggesting that binding of isoprenylcysteine is altered by these mutations.
FIGURE 8.

Identification of conserved residues that alter isoprenylcysteine substrate binding properties. A, radioactive methyl transfer relative to wild-type enzyme for select Ag ICMT mutants assayed in the presence of 5 μm AdoMet using either 4 or 40 μm BFC. B, relative methyl transfer using 5 and 50 μm AdoMet for Ag ICMT mutants shown to be inhibited by BFC in A. C, relative methyl transfer by wild-type and F216A mutant Ag ICMT measured using the indicated substrate concentrations. For all panels, the data shown are the mean of duplicate determinations from a single experiment, with error bars representing ± 1 S.D. The amino acid numbering corresponds to Ag ICMT with Hs ICMT numbering in parentheses.
We selected three alanine substitutions that exhibited the most dramatic substrate inhibition by BFC, Y107A, F216A, and W240A, and evaluated their kinetic behavior by plotting BFC and AdoMet initial velocity curves (Fig. 9, A and B). These residues are located in transmembrane helices M4, M7, and M8, respectively. The initial velocity curves for Y107A, F216A, and W240A indicated substrate inhibition by BFC, with Ki values of 37 ± 1, 40 ± 4, and 36 ± 1 μm, respectively (Fig. 9, A and C). Y107A and W240A also have slightly higher Khalf values for BFC (1.9 ± 0.1, 2.4 ± 0.1, and 3.1 ± 0.1 μm for wild-type, Y107A, and W240A, respectively) (Fig. 9, A and C). Interestingly, in addition to causing substrate inhibition by BFC, Y107A, F216A, and W240A have elevated Km values for AdoMet but are not inhibited by this substrate (Km values are 6.7 ± 0.6, 18 ± 2, 22 ± 3, and 26 ± 2 μm for wild type, Y107A, and W240A, respectively) (Fig. 9, B and C). The elevation of Khalf and Km for BFC and AdoMet in these mutants suggests that the binding of the BFC and AdoMet substrates may be energetically coupled.
FIGURE 9.

Comparison of the kinetic properties of Ag ICMT and mutants that have altered isoprenylcysteine substrate binding properties. A and B, activities of Ag ICMT and the Ag ICMT mutants Y107A, F216A, and W240A shown as a plot of the formation of BFC-[3H]methyl ester as a function of BFC concentration (0.8–50 μm) (A) or AdoMet concentration (0.5–50 μm) (B). The data shown are the means of duplicate determinations of a single experiment, with error bars representing ± 1 S.D. C, kinetic parameters derived from A and B. Values were determined by fitting the data to an allosteric sigmoidal model (for BFC with wild-type ICMT), an allosteric sigmoidal model that takes substrate inhibition into consideration (for BFC with Y107A, F216A, and W240A enzymes), or a classical Michaelis-Menten model (for AdoMet) (details are indicated under “Experimental Procedures”). Values are reported with the S.E. of the fit.
To further investigate the roles of Thr-105, Tyr-107, Phe-216, Thr-222, Gln-223, Trp-240, and Phe-242, we measured the activity of mutants with additional amino acid substitutions at these positions using 4 and 40 μm concentrations of BFC (Fig. 10A). The Y107F mutant had similar activity to wild-type ICMT and did not experience substrate inhibition. However, the Y107A, Y107W, and Y107M mutants showed substrate inhibition. Substrate inhibition was not observed for F216Y or F216W, whereas it occurred in the F216A, F216M, and F216R mutants. Phe-242 had a similar pattern, with the F242Y and F242W mutants behaving similarly to wild-type ICMT and the F242A and F242M mutants exhibiting substrate inhibition. For Trp-240, phenylalanine and tyrosine mutations did not show substrate inhibition, whereas methionine and alanine did.
FIGURE 10.

Extended mutational analysis of residues that alter isoprenylcysteine substrate binding properties. A, comparison of the methyl transfer activity of various amino acid substitutions in the presence of 5 μm AdoMet using either 4 or 40 μm BFC. The data shown are the means of duplicate determinations from a single experiment, with error bars representing ± 1 S.D., and are normalized to wild-type Ag ICMT using 4 μm BFC. Ag ICMT numbering with Hs ICMT numbering are in parentheses. B–D, kinetic analysis of Phe-216 mutants. B and C, activities of wild-type Ag ICMT and the F216A, F216Y, and F216R mutants shown as a plot of the formation of BFC-[3H]methyl ester as a function of BFC concentration (0.8–50 μm) (B) or AdoMet concentration (0.5–50 μm) (C). The data shown are means of duplicate determinations of a single experiment, with error bars representing ± 1 S.D. D, kinetic parameters derived from B and C. Values were determined by fitting the data to an allosteric sigmoidal model (for BFC with wild-type and F216Y enzymes), an allosteric sigmoidal model that takes substrate inhibition into consideration (for BFC with F216A and F216R enzymes), or a classical Michaelis-Menten model (for AdoMet) (details are indicated under “Experimental Procedures”). Values are reported with the S.E. of the fit.
Evaluating mutants of two threonines revealed that T105S is not affected by substrate inhibition, but there was an indication of substrate inhibition for T105L and T222S as well as the alanine mutants of these residues. These results indicate that the hydroxyl group is important in position 105, but a threonine to serine substitution is not tolerated in position 222. Neither Q223N nor Q223E had activity levels similar to wild-type ICMT, and both mutations caused substrate inhibition, indicating that conservative mutations of Gln-223 are not capable of recapitulating wild-type behavior. The differences in the behavior of these mutants provide initial insights into the chemical importance of the side chains of these residues in BFC binding.
We continued our characterization of the F216Y and F216R mutants by constructing full curves of initial velocity versus substrate concentration (Fig. 10, B–C). Similar to what was observed for the F216A mutant, the shape of the initial velocity curve for the F216R mutant with respect to BFC concentration was indicative of substrate inhibition by BFC (Ki = 47 ± 5 μm) (Fig. 10, B and D). In contrast, the F216Y mutant did not show substrate inhibition. Both the F216Y and the F216R mutants had slightly elevated Khalf values for BFC in comparison with wild-type ICMT (Fig. 10, B and D). As observed for the F216A mutant, the F216R mutant has a higher Km for AdoMet (9.4 ± 1.0 μm), but it does not exhibit substrate inhibition by AdoMet (Fig. 10, C and D). Thus, depending on the particular mutation, substitutions to Phe-216 result in changes in the Khalf for BFC, the Km for AdoMet, and the Vmax, and they can give rise to substrate inhibition by BFC. Taken together, it seems likely that Phe-216 is located within the active site. Its positioning within a hydrophobic region of M6 that does not interact with AdoHcy in the crystal structure of Ma MTase suggests that it forms part of the binding site for the isoprenylcysteine substrate.
Additional Regions That Affect Activity
Sequence analysis reveals a tandem GXXXG-like motif (68AXXXGXXXS76) in M3 of Ag ICMT (70AXXXGXXXG78 in Hs ICMT and 27SXXXGXXXGXXXG39 in Sc ICMT) (Fig. 2). GXXXG and GXXXG-like motifs (where G can be glycine, alanine or serine and X represents any amino acid) have been observed to facilitate oligomerization (e.g. dimerization) through interactions of helical transmembrane domains (28). Although the oligomeric state of ICMT has not been definitively resolved, it has been suggested that it may function as a dimer or higher order oligomer (29). To determine the importance of the residues in the GXXXG-like motif for ICMT activity, we mutated three residues of Ag ICMT in this motif (Ala-68, Gly-72, and Ser-76). Mutation to leucine in each position reduced the activity of Ag ICMT dramatically, whereas methyl transfer by conservative mutants A68G, G72A, and S76A was comparable to wild type (Figs. 5B and 6). These results are consistent with the GXXXG-like motif playing a role in ICMT function, perhaps by stabilization of an intermolecular interaction between ICMT monomers facilitating oligomerization or an intramolecular interaction between α-helices within a single ICMT monomer.
Our scanning mutagenesis also identified a patch of three hydrophobic residues (Phe-123, Ile-124, and Leu-125) within the cytosolic M4-M5 loop that dramatically reduce methyl transfer when mutated (Figs. 5B and 6). Mutants of amino acids flanking this hydrophobic patch were also found to reduce methyl transfer, although to a lesser extent (Figs. 5B and 6). Other studies have hinted at the importance of this region. Point mutants in ICMT orthologs, Hs ICMT F124A/R (Phe-123 in Ag ICMT), and Sc ICMT L81F (Ile-124 in Ag ICMT) were previously shown to have decreased methyltransferase activity (18, 19), offering further support that the M4-M5 cytosolic loop plays a role in ICMT function.
DISCUSSION
In comparison to water-soluble methyltransferases, relatively little is known about how enzymes like ICMT catalyze reactions within the context of lipid membranes. Our findings have revealed insights into the substrate binding sites and other regions important for catalytic activity of this eukaryotic integral membrane enzyme.
Our work confirms that the AdoMet binding site of ICMT is a tripartite motif that consists of two regions that interact with AdoMet (AdoMet binding motifs I and II) that are separated by a stretch of hydrophobic residues (M7 and M8 in Ag ICMT). The binding site is found within the C-terminal third of the amino acid sequence of ICMT and is formed by residues in the cytosolic M6-M7 loop, the M8 extension, and the C-terminal helix. These residues are conserved with the prokaryotic integral membrane methyltransferase Ma MTase and are analogous to the residues that were observed to bind AdoHcy in the Ma MTase crystal structure (Fig. 11). Our data support classification of ICMT together with Ma MTase as a member of a class of methyltransferases (class VI) that are integral membrane proteins residing within cellular membranes.
FIGURE 11.

Residues proposed to be involved in substrate binding in ICMT mapped on the structure of Ma MTase. The schematic representation of the Ma MTase structure (PDB code 4A2N) is colored according to amino acid sequence conservation with the ICMT family members (pink, high sequence conservation; green, low sequence conservation) in stereoview. The sequence alignment was made in ClustalW with manual adjustments according to Fig. 2, the conservation was analyzed and mapped on the structure using the ConSurf Server (44), and the figure was constructed with PyMOL. Amino acid numbering corresponds to Ma MTase, with Ag ICMT numbering in parentheses. Residues that contribute to AdoMet binding are drawn as sticks. The approximate locations of Ag ICMT residues in the transmembrane helices M4, M7, and M8 that our study suggests contribute to the isoprenylcysteine binding site (on the basis that mutants exhibit either substrate inhibition or are inactive) are indicated as spheres. AdoHcy is drawn as sticks and colored yellow.
Another eukaryotic integral membrane methyltransferase that uses AdoMet as a methyl donor is phosphatidylethanolamine N-methyltransferase (PEMT) (30). PEMT catalyzes the conversion of phosphatidylethanolamine to phosphatidylcholine through three consecutive methyl transfer reactions (31). Studies of Hs PEMT indicate that like ICMT, PEMT may bind AdoMet through a tripartite motif (32), and many of the residues that form the AdoMet binding site are conserved between ICMT, Ma MTase, and PEMT (Fig. 7A). Furthermore, mutants of Glu-180 and Glu-181 in Hs PEMT that align with ICMT residues that interact with AdoMet (Glu-250 and Glu-251 in Ag ICMT) were shown to be defective in AdoMet binding (32). Although ICMT, Ma MTase, and PEMT probably share structural similarities for AdoMet binding and can, therefore, be categorized within the same class of methyltransferases, the binding sites for their respective methyl acceptor substrates likely differ according to the chemical natures of these substrates.
Our study has identified several mutations in ICMT that cause substrate inhibition by the isoprenylcysteine substrate. It has been estimated that substrate inhibition occurs for ∼20% of all wild-type enzymes, and it typically is observed at substrate concentrations that are higher than the physiological levels (27). Although the molecular basis for substrate inhibition is not always apparent, one explanation for it is that the enzyme has two binding sites for its substrate: a productive site where binding produces product and a non-productive site where binding produces the product at a reduced rate (33). A plausible explanation for the substrate inhibition observed for the ICMT mutants is that these residues line the lipid binding pocket that recognizes the isoprenyl moiety of ICMT substrates and that mutating these residues alters the substrate binding pocket slightly such that the isoprenylcysteine substrate can bind in a non-productive conformation.
Single amino acid changes have been shown to cause substrate inhibition in other transferase enzymes, including N-acyltransferases, sulfotransferases, and uridine diphosphate (UDP)-glucuronosyltransferases (34–37). In AntD, an N-acyltransferase that is involved in the d-anthrose biosynthesis pathway in Bacillus anthracis, standard Michaelis-Menten kinetics were observed for the wild-type enzyme and a S84A mutant, whereas substrate inhibition kinetics were observed for S84T and S84C mutants (37). Crystal structures of AntD indicate that Ser-84 lines the substrate binding site and that the S84C mutant binds the substrate in an alternate conformation supporting the hypothesis that substrate inhibition arises for S84C due to altered substrate binding (37). Similarly, in the human sulfotransferase SULT1A1, substrate inhibition by dopamine was observed when Phe-247 was mutated to leucine and was interpreted to mean that this substitution created space for the binding of a second dopamine molecule (35). Finally, the UDP-glucuronosyltransferase human UGT1A10 has an amino acid motif (90FMVF93) that is postulated to be involved in binding of its phenolic substrates through ring stacking with the phenylalanine residues (38). Three mutants, V92A, F93G, and F93A, all show substrate inhibition kinetics (36). These examples are reminiscent of the behavior of the ICMT mutants that we observed.
ICMT, AntD, SULT1A1, and UGT1A10 are each transferase enzymes that transfer a specific functional group onto various substrates. Because of this plasticity for acceptor substrates, the substrate binding sites might be expected to be larger in size and more flexible than if they recognized only a single acceptor substrate. ICMT catalyzes the transfer of a methyl group to both farnesylcysteine (15-carbons) and geranylgeranylcysteine (20-carbons) substrates, and consequently the isoprenylcysteine substrate binding site needs to be able to accommodate substrates of different sizes. An active site that, depending on the substrate, is larger than it needs to be may be one reason that amino acid substitutions within it could give rise to non-productive binding conformations for the substrate.
The residues that altered isoprenylcysteine substrate binding properties when mutated are: Thr-105, Tyr-107, Phe-216, Thr-222, Gln-223, Trp-240, and Phe-242 (Fig. 8A). All of these residues are located in transmembrane-spanning regions where the side chains could potentially interact with the isoprenoid lipid moiety of the substrate (Fig. 11). As one might expect for a lipid binding pocket, most of these residues are aromatic amino acids. Aromatic residues have also been shown to interact with isoprenoid moieties in the CAAX protein prenyltransferases, farnesyltransferase, and geranylgeranyltransferase type-I (39, 40), and in farnesyl pyrophosphate synthase (41). These residues are strictly conserved among eukaryotic ICMT family membranes but are not conserved with PEMT or Ma MTase, offering support that the greatest degree of similarity between these enzymes is confined to the AdoMet binding site (Figs. 2 and 11). In contrast to the lipid access tunnel proposed for Ma MTase, which is situated between transmembrane helices M1 and M2 (roughly aligning to M4 and M5 in Ag ICMT), the ICMT residues that we have identified as capable of perturbing isoprenylcysteine binding are located in M4, M7, and M8. Although both ICMT and Ma MTase probably have somewhat analogous lipid-binding pockets, the locations of these and the specific interactions made between the protein and the methyl acceptor substrates probably differ between these two enzymes. In addition to the residues that cause substrate inhibition when mutated, we expect that other amino acids contribute to the binding site for the isoprenylcysteine substrate. For example, we have identified that Phe-102, His-103, Glu-106, Trp-215, Trp-218, Gly-221, and Phe-243 are key residues in hydrophobic portions of M4, M7, and M8 that severely impair catalytic activity when mutated (Figs. 2, 5B, 6, and 11).
Our kinetic analysis of Y107A, W240A, and the Phe-216 mutants F216A, F216Y, and F216R indicates that mutants with a defect in the binding of the isoprenylcysteine substrate may also have a defect in the binding of AdoMet. Mutation of Tyr-107 to alanine, Trp-240 to alanine, and Phe-216 to alanine and arginine affected both the enzyme kinetics with respect to BFC and the Km for AdoMet (Figs. 9 and 10, B–D). However, Tyr-107, Phe-216, and Trp-240 (Arg-21, Trp-133, and Ala-157 in Ma MTase) do not contact AdoHcy in the structure of MTase (Fig. 11). This suggests that the binding sites for AdoMet and the isoprenylcysteine substrate may be energetically coupled. There are several potential reasons as to why this could occur. In certain cases residues may span the binding sites for both substrates. It has been suggested that Arg-163 of Ma MTase (Arg-246 in Ag ICMT) may position the lipid substrate for catalysis through electrostatic interactions and also contribute to AdoMet binding (20). Alternatively, AdoMet, which is known from kinetic analysis to bind first to ICMT (21, 22), could be necessary to create an optimal binding site for the isoprenylcysteine substrate. This would be reminiscent of substrate recognition in farnesyltransferase where the farnesyl diphosphate substrate forms part of the binding surface for the CAAX peptide (39). Furthermore, it could be that ICMT undergoes a structural rearrangement after AdoMet binding and that this rearrangement is necessary to form the binding site for the isoprenylcysteine substrate. In this regard the hydrophobic patch that we identified in the M4-M5 loop of ICMT may be involved. Perhaps in a similar manner to the hydrophobic patch in sodium channels (42), the cytosolic M4-M5 loop of ICMT is capable of hinging toward the lipid membrane. Further investigation will be important to determine the specifics of substrate binding and any associated structural changes in ICMT.
Our study represents a comprehensive mutational analysis of the eukaryotic integral membrane methyltransferase ICMT. We offer support that the AdoMet binding site is contained within a tripartite motif formed by a cytosolic region of ICMT near its C terminus and provide evidence that certain residues in the M4, M7, and M8 transmembrane helices contribute to recognition of the isoprenylcysteine substrate. Knowledge regarding the substrate binding sites may be helpful for the design and optimization of ICMT inhibitors that could have utility for the treatment of cancer.
Acknowledgments
We thank C. D. Lima, S. Shuman, and members of the Long Laboratory for discussion and comments on the manuscript, E. Gouaux for the GFP fusion vector, and P. J. Casey for initial assistance with the ICMT activity assay protocol.
This work was supported in part by a Burroughs Wellcome Fund Career Award in the Biomedical Sciences (to S. B. L.) and an American Heart Association Pre-doctoral Fellowship (to M. M. D.).
- ICMT
- isoprenylcysteine carboxyl methyltransferase
- AdoMet
- S-adenosyl-l-methionine
- TM
- transmembrane helix
- AdoHcy
- S-adenosyl-l-homocysteine
- FSEC
- fluorescence-detection size-exclusion chromatography
- BFC
- biotin-S-farnesyl-l-cysteine
- PEMT
- phosphatidylethanolamine N-methyltransferase
- Hs
- H. sapiens
- Ag
- A. gambiae
- Sc
- S. cerevisiae
- Ma
- Methanosarcina acetivorans.
REFERENCES
- 1. Casey P. J., Seabra M. C. (1996) Protein prenyltransferases. J. Biol. Chem. 271, 5289–5292 [DOI] [PubMed] [Google Scholar]
- 2. Hrycyna C. A., Sapperstein S. K., Clarke S., Michaelis S. (1991) The Saccharomyces cerevisiae STE14 gene encodes a methyltransferase that mediates C-terminal methylation of a-factor and RAS proteins. EMBO J. 10, 1699–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Young S. G., Ambroziak P., Kim E., Clarke S. (2000) Post-isoprenylation protein processing: CXXX (CaaX) endoproteases and isoprenylcysteine carboxyl methyltranferase. in Protein Lipidation, 3rd Ed., pp. 155–213, Academic Press, Inc., San Diego, CA [Google Scholar]
- 4. Bergo M. O., Leung G. K., Ambroziak P., Otto J. C., Casey P. J., Young S. G. (2000) Targeted inactivation of the isoprenylcysteine carboxyl methyltransferase gene causes mislocalization of K-Ras in mammalian cells. J. Biol. Chem. 275, 17605–17610 [DOI] [PubMed] [Google Scholar]
- 5. Bergo M. O., Gavino B. J., Hong C., Beigneux A. P., McMahon M., Casey P. J., Young S. G. (2004) Inactivation of Icmt inhibits transformation by oncogenic K-Ras and B-Raf. J. Clin. Invest. 113, 539–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Winter-Vann A. M., Casey P. J. (2005) Post-prenylation-processing enzymes as new targets in oncogenesis. Nat. Rev. Cancer 5, 405–412 [DOI] [PubMed] [Google Scholar]
- 7. Cox A. D., Der C. J. (2002) Farnesyltransferase inhibitors: promises and realities. Curr. Opin. Pharmacol. 2, 388–393 [DOI] [PubMed] [Google Scholar]
- 8. Whyte D. B., Kirschmeier P., Hockenberry T. N., Nunez-Oliva I., James L., Catino J. J., Bishop W. R., Pai J. K. (1997) K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J. Biol. Chem. 272, 14459–14464 [DOI] [PubMed] [Google Scholar]
- 9. Cox A. D., Der C. J. (1997) Farnesyltransferase inhibitors and cancer treatment: targeting simply Ras? Biochim. Biophys. Acta 1333, F51–F71 [DOI] [PubMed] [Google Scholar]
- 10. Wang M., Hossain M. S., Tan W., Coolman B., Zhou J., Liu S., Casey P. J. (2010) Inhibition of isoprenylcysteine carboxylmethyltransferase induces autophagic-dependent apoptosis and impairs tumor growth. Oncogene 29, 4959–4970 [DOI] [PubMed] [Google Scholar]
- 11. Wang M., Tan W., Zhou J., Leow J., Go M., Lee H. S., Casey P. J. (2008) A small molecule inhibitor of isoprenylcysteine carboxymethyltransferase induces autophagic cell death in PC3 prostate cancer cells. J. Biol. Chem. 283, 18678–18684 [DOI] [PubMed] [Google Scholar]
- 12. Wahlstrom A. M., Cutts B. A., Liu M., Lindskog A., Karlsson C., Sjogren A. K., Andersson K. M., Young S. G., Bergo M. O. (2008) Inactivating Icmt ameliorates K-RAS-induced myeloproliferative disease. Blood 112, 1357–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Winter-Vann A. M., Baron R. A., Wong W., dela Cruz J., York J. D., Gooden D. M., Bergo M. O., Young S. G., Toone E. J., Casey P. J. (2005) A small-molecule inhibitor of isoprenylcysteine carboxyl methyltransferase with antitumor activity in cancer cells. Proc. Natl. Acad. Sci. U.S.A. 102, 4336–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cushman I., Casey P. J. (2009) Role of isoprenylcysteine carboxylmethyltransferase-catalyzed methylation in Rho function and migration. J. Biol. Chem. 284, 27964–27973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Court H., Amoyel M., Hackman M., Lee K. E., Xu R., Miller G., Bar-Sagi D., Bach E. A., Bergö M. O., Philips M. R. (2013) Isoprenylcysteine carboxylmethyltransferase deficiency exacerbates KRAS-driven pancreatic neoplasia via Notch suppression. J. Clin. Invest. 123, 4681–4694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Judd W. R., Slattum P. M., Hoang K. C., Bhoite L., Valppu L., Alberts G., Brown B., Roth B., Ostanin K., Huang L., Wettstein D., Richards B., Willardsen J. A. (2011) Discovery and SAR of methylated tetrahydropyranyl derivatives as inhibitors of isoprenylcysteine carboxyl methyltransferase (ICMT). J. Med. Chem. 54, 5031–5047 [DOI] [PubMed] [Google Scholar]
- 17. Lau H. Y., Ramanujulu P. M., Guo D., Yang T., Wirawan M., Casey P. J., Go M. L., Wang M. (2014) An improved isoprenylcysteine carboxylmethyltransferase inhibitor induces cancer cell death and attenuates tumor growth in vivo. Cancer Biol. Ther. 10.4161/cbt.29692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Romano J. D., Michaelis S. (2001) Topological and mutational analysis of Saccharomyces cerevisiae Ste14p, founding member of the isoprenylcysteine carboxyl methyltransferase family. Mol. Biol. Cell 12, 1957–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wright L. P., Court H., Mor A., Ahearn I. M., Casey P. J., Philips M. R. (2009) Topology of mammalian isoprenylcysteine carboxyl methyltransferase determined in live cells with a fluorescent probe. Mol. Cell. Biol. 29, 1826–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang J., Kulkarni K., Manolaridis I., Zhang Z., Dodd R. B., Mas-Droux C., Barford D. (2011) Mechanism of isoprenylcysteine carboxyl methylation from the crystal structure of the integral membrane methyltransferase ICMT. Mol. Cell 44, 997–1004 [DOI] [PubMed] [Google Scholar]
- 21. Baron R. A., Casey P. J. (2004) Analysis of the kinetic mechanism of recombinant human isoprenylcysteine carboxylmethyltransferase (Icmt). BMC Biochem. 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shi Y. Q., Rando R. R. (1992) Kinetic mechanism of isoprenylated protein methyltransferase. J. Biol. Chem. 267, 9547–9551 [PubMed] [Google Scholar]
- 23. Schubert H. L., Blumenthal R. M., Cheng X. (2003) Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci. 28, 329–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Anderson J. L., Henriksen B. S., Gibbs R. A., Hrycyna C. A. (2005) The isoprenoid substrate specificity of isoprenylcysteine carboxylmethyltransferase: development of novel inhibitors. J. Biol. Chem. 280, 29454–29461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tan E. W., Pérez-Sala D., Cañada F. J., Rando R. R. (1991) Identifying the recognition unit for G protein methylation. J. Biol. Chem. 266, 10719–10722 [PubMed] [Google Scholar]
- 26. Kawate T., Gouaux E. (2006) Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure 14, 673–681 [DOI] [PubMed] [Google Scholar]
- 27. Chaplin M. F., Bucke C. (1990) Enzyme technology, 1st Ed., pp. 31–32, Cambridge University Press, Cambridge, UK [Google Scholar]
- 28. Lemmon M. A., Treutlein H. R., Adams P. D., Brünger A. T., Engelman D. M. (1994) A dimerization motif for transmembrane α-helices. Nat. Struct. Biol. 1, 157–163 [DOI] [PubMed] [Google Scholar]
- 29. Griggs A. M., Hahne K., Hrycyna C. A. (2010) Functional oligomerization of the Saccharomyces cerevisiae isoprenylcysteine carboxyl methyltransferase, Ste14p. J. Biol. Chem. 285, 13380–13387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shields D. J., Lehner R., Agellon L. B., Vance D. E. (2003) Membrane topography of human phosphatidylethanolamine N-methyltransferase. J. Biol. Chem. 278, 2956–2962 [DOI] [PubMed] [Google Scholar]
- 31. Ridgway N. D., Vance D. E. (1988) Kinetic mechanism of phosphatidylethanolamine N-methyltransferase. J. Biol. Chem. 263, 16864–16871 [PubMed] [Google Scholar]
- 32. Shields D. J., Altarejos J. Y., Wang X., Agellon L. B., Vance D. E. (2003) Molecular dissection of the S-adenosylmethionine-binding site of phosphatidylethanolamine N-methyltransferase. J. Biol. Chem. 278, 35826–35836 [DOI] [PubMed] [Google Scholar]
- 33. Reed M. C., Lieb A., Nijhout H. F. (2010) The biological significance of substrate inhibition: a mechanism with diverse functions. Bioessays 32, 422–429 [DOI] [PubMed] [Google Scholar]
- 34. Lu L. Y., Hsieh Y. C., Liu M. Y., Lin Y. H., Chen C. J., Yang Y. S. (2008) Identification and characterization of two amino acids critical for the substrate inhibition of human dehydroepiandrosterone sulfotransferase (SULT2A1). Mol. Pharmacol. 73, 660–668 [DOI] [PubMed] [Google Scholar]
- 35. Barnett A. C., Tsvetanov S., Gamage N., Martin J. L., Duggleby R. G., McManus M. E. (2004) Active site mutations and substrate inhibition in human sulfotransferase 1A1 and 1A3. J. Biol. Chem. 279, 18799–18805 [DOI] [PubMed] [Google Scholar]
- 36. Höglund C., Sneitz N., Radominska-Pandya A., Laakonen L., Finel M. (2011) Phenylalanine 93 of the human UGT1A10 plays a major role in the interactions of the enzyme with estrogens. Steroids 76, 1465–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kubiak R. L., Holden H. M. (2012) Structural studies of AntD: an N-acyltransferase involved in the biosynthesis of d-anthrose. Biochemistry 51, 867–878 [DOI] [PubMed] [Google Scholar]
- 38. Xiong Y., Bernardi D., Bratton S., Ward M. D., Battaglia E., Finel M., Drake R. R., Radominska-Pandya A. (2006) Phenylalanine 90 and 93 are localized within the phenol binding site of human UDP-glucuronosyltransferase 1A10 as determined by photoaffinity labeling, mass spectrometry, and site-directed mutagenesis. Biochemistry 45, 2322–2332 [DOI] [PubMed] [Google Scholar]
- 39. Long S. B., Casey P. J., Beese L. S. (2002) Reaction path of protein farnesyltransferase at atomic resolution. Nature 419, 645–650 [DOI] [PubMed] [Google Scholar]
- 40. Taylor J. S., Reid T. S., Terry K. L., Casey P. J., Beese L. S. (2003) Structure of mammalian protein geranylgeranyltransferase type-I. EMBO J. 22, 5963–5974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tarshis L. C., Proteau P. J., Kellogg B. A., Sacchettini J. C., Poulter C. D. (1996) Regulation of product chain length by isoprenyl diphosphate synthases. Proc. Natl. Acad. Sci. U.S.A. 93, 15018–15023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. West J. W., Patton D. E., Scheuer T., Wang Y., Goldin A. L., Catterall W. A. (1992) A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc. Natl. Acad. Sci. U.S.A. 89, 10910–10914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cole C., Barber J. D., Barton G. J. (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36, W197–W201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Glaser F., Pupko T., Paz I., Bell R. E., Bechor-Shental D., Martz E., Ben-Tal N. (2003) ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19, 163–164 [DOI] [PubMed] [Google Scholar]