

Background: Post-transcriptional modifications of rRNAs play important roles in biogenesis and function of ribosome.
Results: Identification of an essential RNA acetyltransferase Rra1p responsible for forming N4-acetylcytidine at position 1773 in 18 S rRNA.
Conclusion: Rra1p and ac4C1773 are required for pre-18 S rRNA processing.
Significance: Rra1p modulates 40 S subunit biogenesis through a single acetylation of 18 S rRNA by sensing nuclear acetyl-CoA concentration.
Keywords: Acetyl Coenzyme A (Acetyl-CoA), Acetyltransferase, Ribosomal RNA Processing (rRNA Processing), Ribosome Assembly, RNA Modification
Abstract
Biogenesis of eukaryotic ribosome is a complex event involving a number of non-ribosomal factors. During assembly of the ribosome, rRNAs are post-transcriptionally modified by 2′-O-methylation, pseudouridylation, and several base-specific modifications, which are collectively involved in fine-tuning translational fidelity and/or modulating ribosome assembly. By mass-spectrometric analysis, we demonstrated that N4-acetylcytidine (ac4C) is present at position 1773 in the 18 S rRNA of Saccharomyces cerevisiae. In addition, we found an essential gene, KRE33 (human homolog, NAT10), that we renamed RRA1 (ribosomal RNA cytidine acetyltransferase 1) encoding an RNA acetyltransferase responsible for ac4C1773 formation. Using recombinant Rra1p, we could successfully reconstitute ac4C1773 in a model rRNA fragment in the presence of both acetyl-CoA and ATP as substrates. Upon depletion of Rra1p, the 23 S precursor of 18 S rRNA was accumulated significantly, which resulted in complete loss of 18 S rRNA and small ribosomal subunit (40 S), suggesting that ac4C1773 formation catalyzed by Rra1p plays a critical role in processing of the 23 S precursor to yield 18 S rRNA. When nuclear acetyl-CoA was depleted by inactivation of acetyl-CoA synthetase 2 (ACS2), we observed temporal accumulation of the 23 S precursor, indicating that Rra1p modulates biogenesis of 40 S subunit by sensing nuclear acetyl-CoA concentration.
Introduction
The ribosome, a large ribonucleoprotein complex composed of two subunits, translates the genetic information in mRNA into protein sequence. In Saccharomyces cerevisiae the small 40 S subunit contains 18 S rRNA (see Fig. 1A) and 33 species of ribosomal proteins, whereas the large 60 S subunit consists of three rRNAs (25 S, 5.8 S, and 5 S) and 46 species of ribosomal proteins. The 40 S subunit serves as the decoding center where the codon-anticodon interaction between mRNA and aminoacyl-tRNA takes place, whereas the 60 S subunit has the peptidyltransferase center that catalyzes peptide-bond formation. Over the course of a single yeast cell cycle, 200,000 ribosomes are produced; thus, >2000 ribosomes are generated every minute in a rapidly growing cell (1). To sustain such high rates of synthesis, biogenesis of ribosomes must be very efficient. Ribosome biogenesis is a complicated, energy-intensive process that requires dozens of different small nucleolar RNAs (snoRNAs)3 as well as >200 transacting assembly factors, including RNA-binding proteins, RNA helicases, GTPases, rRNA-modifying enzymes, and endo/exonucleases (2–5). These factors interact transiently with pre-ribosomal particles at different times to facilitate rRNA processing, assembly of ribosomal proteins, and transport/localization of pre-ribosomal particles to the appropriate subcellular compartments.
FIGURE 1.

N4-acetylcytidine is present at position 1773 in S. cerevisiae 18 S rRNA. A, the secondary structure of S. cerevisiae 18 S rRNA (left), with a detailed description of helix 45 and the surrounding region (right box), including modified nucleosides: N4-acetylcytidine (ac4C) at position 1773, N6,N6-dimethyladenosine (m62A) at positions 1782 and 1783, and 2′-O-methylcytidine (Cm) at position 1639. The RNA segment G1745-A1800 transcribed in vitro, used for ac4C formation, is shown in blue. The ac4C-containing hexamer fragment generated by RNase T1 digestion is colored green. Watson-Crick base pairs and wobble pairs are shown as bars and dots, respectively. B, capillary LC/ESI-MS analysis of RNA fragments of S. cerevisiae rRNAs digested with RNase T1. The upper panel shows a base-peak chromatogram (BPC), and the lower panel represents mass chromatogram for detecting the double-charged ion of the ac4C-containing hexamer fragment (UUUCac4CGp, m/z 965.6). C, collision-induced dissociation spectrum of the ac4C-containing hexamer fragment. The double-charged ion (m/z 965.6) was used as the parent for collision-induced dissociation. The sequence was confirmed by assignment of the product ions. Nomenclature for the product ions is as suggested in a previous report (54).
In S. cerevisiae the 35 S pre-rRNAs are transcribed by RNA polymerase I from the rDNA repeat region on chromosome XII and eventually processed into 18 S, 5.8 S, and 25 S rRNAs through multistep processing events. The 35 S pre-rRNA contains four spacers (5′-ETS, ITS1, ITS2, and 3′-ETS) that are removed during rRNA processing (6) (see Fig. 3A). Once transcribed, the 35 S pre-rRNA is cleaved endonucleolytically and then trimmed exonucleolytically in a processing complex called processome (7). During rRNA processing, rRNAs are modified post-transcriptionally. These modifications are involved in fine-tuning translational fidelity and/or modulating ribosome biogenesis. In eukaryotic ribosomes the most prevalent rRNA modifications are 2′-O-methylation and pseudouridylation (Ψ), which are introduced by box C/D and box H/ACA snoRNA-guided enzymes, respectively (8). These snoRNA-dependent modifications are considered to occur co-transcriptionally (9). On the other hand, the 18 S and 25 S rRNAs are also subjected to several snoRNA-independent modifications, which are introduced by specific RNA-modifying enzymes. In S. cerevisiae 18 S rRNA, three species of base-specific modifications have been found at four positions (10); 1-methyl-3-(3-amino-3-carboxypropyl)pseudouridine (m1acp3Ψ) at position 1191, 7-methylguanosine at position 1575, and N6,N6-dimethyladenosine at positions 1781 and 1782. During biogenesis of m1acp3Ψ1191, the box H/ACA snoRNA snR35 is responsible for Ψ1191 formation. Next, Emg1p, an essential rRNA methyltransferase, methylates Ψ1191 to form m1Ψ1191 in nucleolus (11, 12). EMG1 is an essential gene in S. cerevisiae and is well conserved in archaea and eukaryote. Inactivation of Emg1p showed impaired formation of 18 S rRNA with a decreased 40 S subunit. In the last step the 3-amino-3-carboxypropyl group is formed on m1acp3Ψ1191; this reaction is known to occur in cytoplasm (13), but the responsible modifying enzyme remains unknown. Missense mutations found in human EMG1 are associated with a genetic disorder Bowen-Conradi syndrome characterized by severe developmental growth delays (14). It implies the importance of m1acp3Ψ1191 modification in 40 S subunit formation. The methyltransferase responsible for 7-methylguanosine (m7G) 1575 formation is Bud23p (15). Deletion of BUD23 leads to severe growth defects and reduction of 40 S biogenesis with impaired processing of the 20 S precursor to 18 S rRNA. Dim1p, another essential methyltransferase, is responsible for dimethylation of adenine bases at positions 1781 and 1782 in the 3′-terminal region of 18 S rRNA (16). These dimethylations mediated by Dim1p are required for processing at the 5′ terminus (A1 site) of 18 S rRNA.
FIGURE 3.

Rra1p plays a critical role in pre-18 S rRNA processing. A, schematic depiction of the rRNA processing pathways in S. cerevisiae. In the canonical pathway 35 S pre-rRNA is processed by endonucleolytic cleavages at the A0, A1, and A2 sites to yield 20 S pre-rRNA. In an alternative pathway A3 site cleavage in ITS1 takes place before cleavages at the A0, A1, and A2 sites to yield the 23 S and 27SA3 pre-rRNAs (27SA3 is not shown is this scheme). Upon depletion of Rra1p, endonucleolytic cleavages at the A0, A1, and A2 sites are inhibited. The position of ac4C1773 is indicated by circle flag. B, high level accumulation of 23 S pre-rRNA with severely reduced 18 S rRNA upon Rra1p depletion. Precursors of 18 S rRNA were detected by northern blotting at the indicated times after heat treatment of the rra1ts (right panels) and its parental (WT/YKL200) (left panels) strains. Steady-state levels of 25 S (top panels) and 18 S (second panels) rRNAs were visualized by ethidium bromide (EtBr) staining. The 23 S (third panels) and 20 S (bottom panels) pre-rRNAs are detected by northern blotting with the ITS1 probe. C, relative 18 S/25 S ratio of WT (open circles) and rra1ts (closed squares) strains after culture temperature was raised to 37 °C. The data were calculated from the band intensities on the gel stained by EtBr as shown in B. D, sucrose density gradient profiling of ribosomal subunits in cell lysates of the WT (YKL200) (left) and the rra1ts strain (right). E, capillary LC/ESI-MS analyses of RNase T1-digested RNA fragments of 18 S rRNAs from the WT (YKL200) (upper panels) and rra1ts strain (middle panels) and of 23 S pre-rRNA from the rra1ts strain (lower panels). Mass chromatograms for detecting the double-charged ions of the ac4C-containing hexamers (UUUCac4CGp, m/z 965.6) and the control fragments A973-G976 (AAmCGp, m/z 669.1) are shown in the left and right panels, respectively. The intensities of the ac4C-containing hexamers in the mass chromatograms were normalized to those of the control fragments. F, 23 S pre-rRNA accumulated in the rra1ts strain resides in the nucleus. Shown are mass chromatograms detecting the m1acp3Ψ1191-containing fragment (ACm1acp3ΨCAACACGp, m/z 1105.8) (left panels) and the m1Ψ1191-containing fragment (ACm1ΨCAACACGp, m/z 1072.2)(right panels) from 18 S rRNA (WT, YKL200 strain) (upper panels) and 23 S pre-rRNA (rra1ts strain) (lower panels), respectively. Each peak is indicated by an arrowhead.
In addition to these modifications, N4-acetylcytidine (ac4C) is present in 18 S rRNA from rat liver, chicken liver, and S. cerevisiae, although the exact locations of ac4C are yet to be determined (17). In Dictyostelium discoideum, ac4C has been detected specifically at position 1844 in the 3′-terminal region of 18 S rRNA (18). Together these previous reports indicate that ac4C is highly conserved among eukaryotic 18 S rRNAs. However, the biogenesis and function of this modification remain to be elucidated.
In this study we used mass spectrometric analysis to show that ac4C is present at position 1773 in 18 S rRNA from S. cerevisiae (see Fig. 1A). In addition, we found that the essential gene RRA1 (ribosomal RNA cytidine acetyltransferase 1) encodes an RNA acetyltransferase responsible for ac4C1773 formation using ATP and acetyl-CoA as substrates. We demonstrate that ac4C1773 plays a critical role in processing of 23 S precursor to 18 S rRNA during biogenesis of the 40 S subunit. Moreover, we provide evidence that 18 S rRNA processing is modulated in response to the nuclear concentration of acetyl-CoA.
EXPERIMENTAL PROCEDURES
Yeast Strains, Media, and Growth Conditions
The S. cerevisiae wild-type strain (BY4741: MATa; his3-1; leu2-0; met15-0; ura3-0), the degron strain rra1ts [Y40097: MATa; ade2-1; ura3-1; his3-11,15; trp1-1; leu2–3,112; can1-100; UBR1::GAL-HA-UBR1 (HIS3); YNL132W::CUP1-UBI4-myc-YNL132w (kanMX)], and the parental strain [YKL200: MATa; ade2-1; ura3-1; his3-11,15; trp1-1; leu2-3,112; can1-100; UBR1::GAL-HA-UBR1 (HIS3)] were obtained from EUROSCARF (19). The WT strain was grown in YPD medium (1% yeast extract, 2% peptone, and 2% glucose) at 30 °C. For rapid depletion of Rra1p, the rra1ts strain (Y40097) and its parental strain (YKL200) were first grown in YPDCu medium (YPD medium with 0.1 mm CuSO4) at 24 °C until the cell density reached 0.7 A600, at which time they were harvested and resuspended in YPG medium (1% yeast extract, 2% peptone, and 2% galactose). After incubation at 24 °C for 35 min, an aliquot of the culture was harvested as the 0-h time point, and the remaining culture was transferred to fresh YPG medium prewarmed to 37 °C to initiate depletion of Rra1p. Aliquots were harvested at the indicated time points (see Fig. 3B). The strain acs2ts (YHT652) [MATa; his3-1; leu2-0; met15-0; ura3-0; acs2::HygMX (pHT215, acs2-Ts1-CEN-URA3)] and its control strain, YHT651 [MATa; his3-1; leu2-0; met15-0; ura3-0; acs2::HygMX (pHT215, ACS2-CEN-URA3)] were kindly provided by Prof. Jef D. Boeke (20). These strains were grown in YPD medium at 25 °C until the cell density reached 1.0 A600 and then mixed with an equal amount of fresh YPD medium prewarmed at 49 °C to immediately adjust the culture temperature to 37 °C. Aliquots were harvested at 0, 1, and 2 h after cultivation.
Preparation of Total RNA and 18 S rRNA
For small-scale preparation, the harvested cells were frozen in liquid nitrogen and crushed in an SK-Mill (FUNAKOSHI). Total RNA was extracted from the crushed cells using TriPure Isolation Reagent (Roche Applied Science). For large scale preparations, the harvested cells were lysed using EmulsiFlex C-3 (AVESTIN) followed by RNA extraction by the acid guanidinium thiocyanate-phenol-chloroform extraction (AGPC) method (21). For mass spectrometric analysis, total RNA was resolved on a 4% PAGE in a gel containing 7 m urea and then stained with ethidium bromide. The 18 S rRNA was excised from the gel, eluted by shaking in elution buffer (0.3 m NaOAc, 1 mm EDTA, 0.1% SDS) at 37 °C for 8 h, and precipitated with ethanol.
Isolation of 23 S pre-rRNA by Reciprocal Circulating Chromatography
Total RNA was extracted from a 1-liter culture of the rra1ts strain cultured at 37 °C for 24 h. 23 S pre-rRNA was isolated by reciprocal circulating chromatography (RCC) using an automated RCC device (22), essentially following a previously described method (23). We designed three 5′-terminal ethylcarbamate amino-modified DNA probes (listed in Table 1), each of which was covalently immobilized on N-hydroxysuccinimide (NHS)-activated Sepharose 4 Fast Flow (GE Healthcare). The DNA-immobilized resins were packed into custom-made tips attached to a multichannel head on the reciprocal circulating chromatography device. The operation temperatures for hybridization step, washing step, and elution step were set to 66 °C, 50 °C, and 72 °C, respectively. The 23 S pre-rRNA isolated using each of three probes was combined, resolved by 4% PAGE in a gel containing 7 m urea, and stained with ethidium bromide. The 23 S pre-rRNA was excised from the gel, eluted by shaking in elution buffer (0.3 m NaOAc, 1 mm EDTA, 0.1% SDS) at 37 °C for 8 h, and precipitated with ethanol.
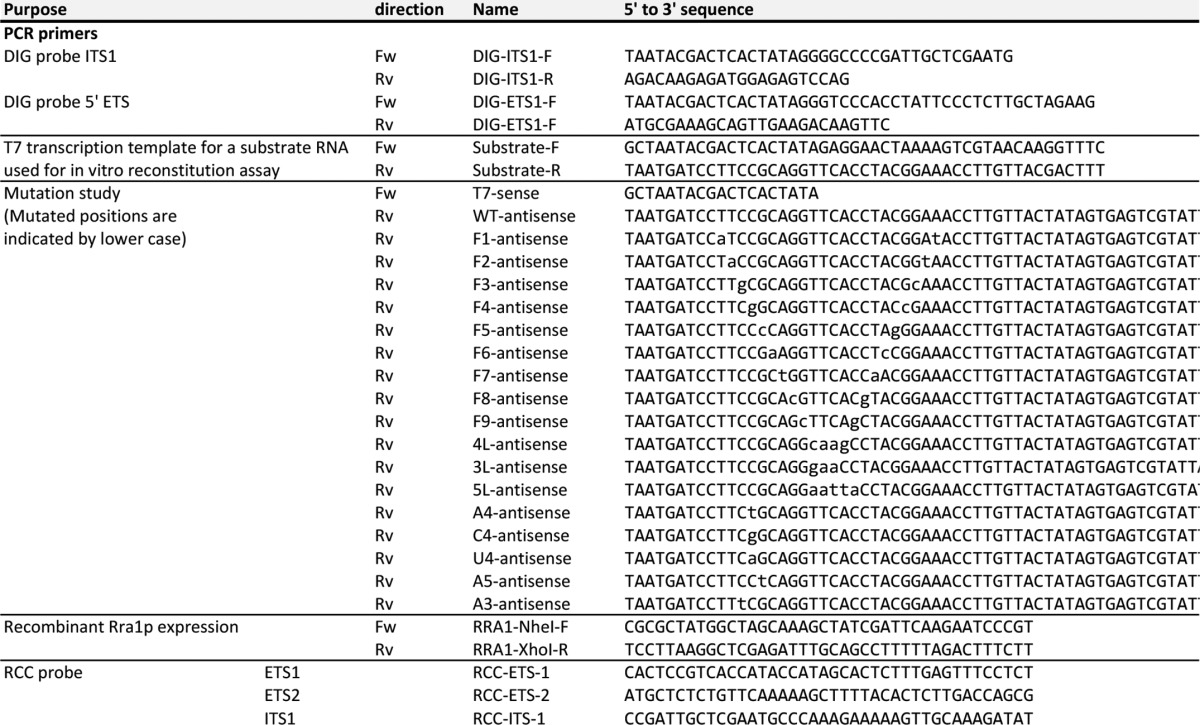
TABLE 1.
List of primers and probes used in this study
RCC, reciprocal circulating chromatography.
RNA Mass Spectrometry
Highly sensitive analysis of RNA fragments by mass spectrometry was carried out essentially as described previously (23, 24). The isolated 18 S rRNA or 23 S pre-rRNA was digested with RNase T1 at 37 °C for 30 min in a 10-μl reaction mixture containing 10 mm ammonium acetate (pH 5.3) and 5 units/μl RNase T1 (Epicenter) followed by the addition of an equal volume of 0.1 m triethylamine acetate (pH 7.0). The digested samples were then subjected to capillary liquid chromatography (LC) coupled with nanoelectrospray ionization (ESI) mass spectrometry using a linear ion trap-orbitrap hybrid mass spectrometer (LTQ Orbitrap XL, Thermo Fisher Scientific).
Northern Blotting
Total RNA (2 μg) in loading solution (10 mm MOPS, 2.5 mm NaOAc (pH 5.0), 0.5 mm EDTA-NaOH (pH 8.0), 6.5% formaldehyde, and 50% deionized formamide) was denatured at 65 °C for 5 min and left on ice. After adding 2 μl of dye solution (0.25% bromphenol blue, 0.25% xylene cyanol, 50% glycerol, and 1 mm EDTA-NaOH (pH 8.0)), total RNA was electrophoresed on a 1% agarose gel (10 × 10 cm, SeaKem GTG agarose, TaKaRa) containing 0.6 m formaldehyde in MOPS buffer (20 mm MOPS, 5 mm NaOAc (pH 5.0), and 1 mm EDTA-NaOH (pH 8.0)) for 2.5 h at 100 V. The gel was stained with ethidium bromide and treated with alkaline solution (50 mm NaOH, and 1 μg/ml ethidium bromide) followed by neutralization twice in 200 mm NaOAc (pH 5.0) for 20 min each. 18 S and 25 S rRNAs in the gel were visualized using a FLA7000 imaging analyzer (Fujifilm). The gel was then soaked twice for 10 min each in 10 × SSC with gently shaking. RNAs in the gel were transferred to a Hybond-N+ nylon membrane (GE healthcare) in 10 × SSC using a Model 785 Vacuum Blotter (Bio-Rad).
Northern blotting was carried out using the DIG Northern Starter kit (Roche Applied Science). RNA probes (∼200-mers) complementary to 5′-ETS or ITS1 were transcribed by T7 RNA polymerase in the presence of digoxigenin-11-UTP. Primer sequences used to amplify the templates for the RNA probes are listed in Table 1. Hybridization was carried out at 68 °C overnight. The hybridized bands were visualized by chemiluminescence of CDP-Star (Roche Applied Science) using alkaline phosphatase-conjugated anti-digoxigenin-alkaline phosphatase and detected by LAS 4000 mini (GE healthcare).
Sucrose Density Gradient Centrifugation
The rra1ts strain (Y40097) and its parental strain (YKL200) were cultured in YPG medium at 37 °C and then harvested after 24 h of growth. Preparation of whole cell lysate was carried out basically as described (25). Cell pellets were ground in a mortar with liquid nitrogen and then resuspended in the lysis buffer (50 mm Hepes-KOH (pH 7.7), 100 mm KCl, 5 mm MgCl2, 1 mm DTT, 1 mm PMSF, protease inhibitor mixture (Roche Applied Science), and 0.02 units/μl SUPERase-In RNase Inhibitor (Invitrogen)). The suspension was centrifuged at 5000 g for 5 min at 4 °C to clear the lysate, which was then centrifuged at 10,000 × g for 10 min at 4 °C. Each supernatant (500 μl) was loaded onto a gradient of 10–40% (w/v) sucrose in the lysis buffer, and the gradients were ultracentrifuged in a Beckman SW-28 rotor at 20,000 rpm for 14 h at 4 °C. The gradients were fractionated into >70 fractions on a Piston Gradient Fractionator (BIOCOMP), and the absorbance at 254 nm of each fraction was measured using an ultraviolet monitor (ATTO AC-5200).
Expression and Purification of Recombinant Rra1p
The open reading frame of YNL132W was amplified by PCR from S. cerevisiae genomic DNA with primers listed in Table 1. The PCR product was cloned into the NheI and XhoI sites of pET-21b(+) (Novagen) to generate pET-21b-RRA1. Escherichia coli strain Rosetta (Merck Millipore) was used as a host strain for the expression of recombinant Rra1p. The C-terminal hexahistidine-tagged Rra1p protein was expressed in soluble form by mild induction with 2% lactose at 25 °C and then purified on an AKTA chromatography system using a His-trap column (GE Healthcare). Fractions containing the recombinant protein were pooled and dialyzed against a buffer consisting of 20 mm HEPES-KOH (pH 8.5), 300 mm KCl, 10 mm MgCl2, and 1 mm DTT. The protein concentration was determined using a Bradford protein assay kit (Bio-Rad) with bovine serum albumin as the standard. Glycerol was added to the protein solution to a final concentration of 33% (v/v).
In Vitro Reconstitution of ac4C1773 Formation
The DNA templates for in vitro transcription of RNA fragments containing helix 45 of 18 S rRNA and its variants were constructed by PCR using synthetic DNAs (Table 1). Substrate RNA was transcribed by T7 RNA polymerase basically as described (26). Transcripts were extracted from the reaction mixture with phenol-chloroform and then passed through NAP-5 column (GE Healthcare) to remove free NTPs followed by ethanol precipitation. Each transcript was further purified by 10% PAGE in a gel containing 7 m urea.
In vitro reconstitution of ac4C formation was carried out at 37 °C for 2 h in a reaction mixture (10 μl) consisting of 50 mm HEPES-KOH (pH 7.6), 150 mm KCl, 5 mm MgCl2, 1 mm DTT, 1 mm ATP, 1 mm acetyl-CoA, 1 μm substrate RNA, and 1.26 μm recombinant Rra1p. After the reaction the RNA fragment was extracted with phenol-chloroform followed by ethanol precipitation. The ac4C formation was analyzed by mass spectrometry as described above. For the mutation study of RNA substrates, ac4C formation was carried out at 37 °C for 2 h in a reaction mixture (100 μl) consisting of 50 mm HEPES-KOH (pH 7.6), 12 mm KCl, 10 mm MgCl2, 1 mm DTT, 0.1 mm ATP, 0.1 mm [1-14C] acetyl-CoA (American Radiolabeled Chemicals, 55 mCi/mmol), 0.02 units/μl SUPERase-In RNase Inhibitor (Invitrogen), 5 μm substrate RNA, and 0.1 μm recombinant Rra1p. After the reaction the substrate RNAs were extracted by phenol-chloroform, precipitated by ethanol, and analyzed by 10% PAGE in a gel containing 7 m urea. The substrate RNAs were stained by SYBR Safe (Invitrogen), visualized, and quantified by FLA-7000 imaging analyzer (Fujifilm). Then the gel was dried in vacuo, and the radioactivities of the acetylated RNA fragments were exposed on the imaging plate which was then visualized and quantified by FLA-7000 imaging analyzer. Relative activity of each variant was normalized by the band intensity on the gel stained by SYBR safe.
RESULTS
Determination of the Position of ac4C in S. cerevisiae 18 S rRNA
To determine the exact position of ac4C modification, we performed mass spectrometric analysis on 18 S rRNA from S. cerevisiae. To this end we digested total RNA from S. cerevisiae (BY4741) with RNase T1 and subjected the digests to the capillary LC coupled with ESI-MS to detect RNA fragments containing modified nucleotides. Most of the RNase T1-digested RNA fragments deduced from the 18 S rRNA sequence with modified nucleotides could be assigned in this analysis. We detected the double-charged negative ion of an ac4C-containing hexamer fragment (Fig. 1B) and analyzed it by collision-induced dissociation to sequence the fragment. Based on the assignment of product ions, the hexamer was found to be UUUCac4CGp (Fig. 1C), showing that ac4C was present at position 1773 in helix 45 in the 18 S rRNA (Fig. 1A). This position is equivalent to ac4C1844 in D. discoideum 18 S rRNA (18).
Identification of RRA1 and in Vitro Reconstitution of ac4C1773
ac4C is a modified nucleoside frequently detected in both tRNAs and rRNA from all domains of life (27). For instance, in E. coli, ac4C is present at the wobble position of elongator tRNAMet. We previously identified a bacterial tRNA acetyltransferase, TmcA (tRNAMet cytidine acetyltransferase), that catalyzes ac4C formation in the presence of acetyl-CoA and ATP (28). In bacteria, homologs of tmcA (COG1444) are limited to the γ-proteobacterial subphylum (28). On the other hand, tmcA homologs are widely distributed among archaea and eukaryotes; ac4C is found at the wobble positions of archaeal tRNAs and at position 12 in a subset of eukaryotic tRNAs (27). In addition, as above mentioned, ac4C can be found in the 3′ terminal region of 18 S rRNAs in eukaryotes (17, 18). Based on these observations, the eukaryotic homolog of TmcA was hypothesized to be an RNA acetyltransferase responsible for ac4C formation in tRNA and/or 18 S rRNA (28). The S. cerevisiae ortholog of bacterial TmcA, encoded by YNL132W/KRE33, is an essential protein initially identified as a killer toxin-resistance protein (29). Kre33p, a putative ATPase, is a component of the 90 S pre-ribosomal particle (30), which localizes to the nucleolus (31). A large scale genetic approach revealed that transcription inhibition of KRE33 results in defects in formation and nuclear export of the 40 S subunit (30, 32), suggesting that Kre33p acts as an assembly factor in ribosome biogenesis (33). These data prompted us to speculate that Kre33p is an acetyltransferase responsible for ac4C1773 formation in 18 S rRNA. To determine whether Kre33p has the appropriate enzymatic activity to catalyze formation of ac4C, Kre33p was recombinantly expressed in E. coli and purified homogenously (Fig. 2A). A 56-mer RNA segment, including helix 45 (G1745-A1800) (Fig. 1A), transcribed in vitro, was used as a substrate for ac4C formation mediated by recombinant Kre33p. After the reaction, the RNA segment was digested with RNaseT1 and then subjected to LC/ESI-MS to detect ac4C-modified fragment. As shown in Fig. 2B, an ac4C-containing hexamer was clearly detected only in the presence of both ATP and acetyl-CoA. Next, we used collision-induced dissociation analysis to determine that ac4C is present specifically at position 1773 (data not shown). In addition, we examined ac4C formation in a shorter substrate, a 26-mer derived from helix 45 (G1768-G1793) (Figs. 1A). Once again, we clearly detected an ac4C-containing hexamer (Fig. 2C), although the modification efficiency was only ∼4% in this condition.
FIGURE 2.

In vitro reconstitution of ac4C1773 and substrate specificity of Rra1p. A, recombinant Rra1p (Kre33p) with C-terminal hexahistidine tag, analyzed by SDS-PAGE. B, in vitro reconstitution of ac4C1773 in the 56-mer RNA transcript, including helix 45 (Fig. 1A) in the presence or absence of recombinant Rra1p, ATP, and acetyl-CoA. The left and right panels show mass chromatograms detecting the hexamer fragment carrying ac4C1773 (UUUCac4CGp, m/z 965.6) or C1773 (UUUCCGp, m/z 944.6), respectively. C, ac4C1773 formation in the 26-mer RNA transcript in the presence (upper panels) or absence (lower panels) of recombinant Rra1p. The left and right panels show mass chromatograms detecting the hexamer fragment containing ac4C1773 (UUUCac4CGp, m/z 965.6) or C1773 (UUUCCGp, m/z 944.6), respectively. D, variants of the 41-mer transcript (G1760-A1800) used in this study. Variant names are the same as those in E and F. E, in vitro acetylation of the 41-mer variants by Rra1p. After the reaction, each variant was resolved by 10% denaturing PAGE and stained by SYBR Safe (upper panels). The radioactivity of the acetylated RNA fragment was visualized and quantified by FLA-7000 imaging analyzer (lower panels). M stands for a size marker. RI, radio isotope. F, relative acetylation activities of Rra1p on the 41-mer transcript variants, normalized by the activity on the wild-type transcript. Averaged values of three independent experiments with S.D. values are shown. Numerical values for activities are indicated at the bottom.
These data demonstrated that Kre33p is a bona fide acetyltransferase responsible for ac4C1773 in 18 S rRNA. Based on its enzymatic activity, we renamed YNL132W/KRE33 as RRA1 (ribosomal RNA cytidine acetyltransferase 1).
Substrate Specificity of Rra1p
To study the substrate specificity of Rra1p, we constructed a series of 41-mer transcripts (G1760-A1800) with various mutations (Fig. 2D). The relative activity of the enzyme on each of these variants was normalized to the activity on the wild-type transcript (WT) (Fig. 2, E and F). When each base pair of helix 45 was individually flipped (mutants F1-F9), ac4C formation was significantly reduced. In particular, no activity was detected on the mutants F3, F4, and F5, suggesting that the CCG sequence at positions 1772–1774 is essential for the ac4C formation. When the loop sequence of helix 45 was mutated (4L, 3L, and 5L), the activity persisted but at lower levels than on the WT transcript, indicating the bases in the loop sequence are involved in the activity. To investigate the functional significance of base-pairing at the target site (C1773-G1788), we replaced G1788 with A (A4), C (C4), and U (U4). Intriguingly, all of these mutants conferred higher activity than the WT transcript. In addition, when both sides of the target site were mutated, the two variants G1789A (A3) and C1787G (A5) also exhibited elevated activity. Taken together these data indicated that the CCG (1772–1774) sequence is critical for ac4C formation, but the Watson-Crick type base pairs of this sequence in helix 45 are not necessary for Rra1p activity. Indeed, to the contrary, disruption of these base pairs increased the activity.
Rra1p Plays a Critical Role in Pre-18 S rRNA Processing
Because Rra1p is involved in 40 S biogenesis (30, 32), we examined whether 18 S rRNA processing is affected by inactivation of Rra1p. To this end we used a degron construct of Rra1p (rra1ts) to rapidly deplete the protein upon heat induction (19) because RRA1 is an essential gene in yeast. When the rra1ts strain was cultured at 37 °C for 24 h, the steady-state level of 18 S rRNA was severely reduced relative to the parental strain (YKL200) (Fig. 3B), whereas the 25 S rRNA was unaffected (Fig. 3B). In fact, the 18 S/25 S ratio of the rra1ts strain dropped to only 16% that of the initial level (Fig. 3C), indicating that Rra1p is required for maturation of 18 S rRNA. In addition, in the rra1ts strain cultured at 37 °C for 24 h, the level of 80 S ribosome was markedly reduced, whereas free 60 S subunit accumulated to high levels (Fig. 3D), indicating that the steady-state level of the 40 S subunit was significantly lower than in the WT (YKL200) strain under the same conditions.
To determine which step in rRNA processing is affected by Rra1p depletion, we detected precursors of 18 S rRNAs by northern blotting after heat induction of both rra1ts and WT strains (Fig. 3B). In the rra1ts strain, we observed rapid accumulation of 23 S pre-rRNA (Fig. 3, A and B) immediately after raising the culture temperature to 37 °C. Concomitantly, the level of 20 S pre-rRNA (Fig. 3, A and B), the final precursor for 18 S rRNA, was reduced. After 24 h, we observed high levels of accumulation of 23 S pre-rRNA, which produced no 20 S pre-rRNA, eventually resulting in huge reduction of 18 S rRNA (Fig. 3B). This observation neatly explains why Rra1p is an essential protein. By contrast, in the WT strain, little accumulation of 23 S pre-rRNA was observed until 4 h after heat treatment (Fig. 3B). However, 24 h after heat treatment of the WT strain, the level of 23 S pre-rRNA increased (discussed below) and the level of 20 S pre-rRNA decreased, although the steady-state level of 18 S rRNA was not changed, unlike the case of rra1ts strain (Fig. 3B).
Under normal growth conditions, processing of 35 S pre-rRNA is initiated by endonucleolytic cleavage at A0 site (Fig. 3A). The 5′-ETS is removed by cleavages at A0 and A1, whereas cleavage at A2 in ITS1 generates 20 S and 27 S A2 pre-rRNAs, splitting pathways for small 40 S- and large 60 S-subunit maturation. In an alternative pathway, a separating cleavage at A3 site in ITS1, mediated by RNase MRP, produces the 23 S and 27 S A3 pre-rRNAs (Fig. 3A). As observed in the WT strain, 23 S pre-rRNA accumulated at high levels during stationary phase in both S. cerevisiae (34) and Candida albicans (35). Hence, 23 S pre-rRNA is a physiological precursor for 18 S rRNA formation in the WT strain. Therefore, rapid accumulation of 23 S pre-rRNA upon Rra1p depletion can be interpreted as the result of inhibition of cleavage at the A0, A1, and A2 sites. In other words either ac4C1773 formation or Rra1p binding to 90 S pre-ribosome is required for 23 S to be processed into 20 S pre-rRNA.
No ac4C1773 Is Formed in 23 S pre-rRNA Accumulated in the rra1ts Strain
We next compared levels of ac4C1773 in 18 S rRNAs between the WT and rra1ts strains. To this end we cultured both strains at 37 °C overnight and analyzed their 18 S rRNAs by capillary LC/ESI-MS (Fig. 3E). We detected the ac4C1773-containing fragment (UUUCac4CGp) in both strains. Unexpectedly, after normalization to the levels of the control fragment A973-G976 (AAmCGp), there was no reduction in ac4C1773 level in the rra1ts strain, showing that 18 S rRNA from the rra1ts strain was fully modified with ac4C1773. Next, we analyzed the 23 S pre-rRNA accumulated in the rra1ts strain. For this experiment, we isolated 23 S pre-rRNA by the reciprocal circulating chromatography (22) from the rra1ts strain cultured at 37 °C. The isolated 23 S pre-rRNA was further purified by PAGE and then subjected to RNaseT1 digestion followed by capillary LC/ESI-MS analysis. Several fragments derived from 5′-ETS and ITS1 were detected (data not shown), confirming the successful isolation of 23 S pre-rRNA. To confirm whether the 23 S pre-rRNA accumulated in the rra1ts strain resides in the nucleus, we searched for a fragment containing m1acp3Ψ1191, because 3-amino-3-carboxypropyl formation of m1acp3Ψ1191 takes place in the cytoplasm (13). When we analyzed 18 S rRNA as a control, we clearly detected the m1acp3Ψ1191-containing fragment (ACm1acp3ΨCAACACGp) but no hypomodified species, such as an m1Ψ1191-containing fragment (ACm1ΨCAACACGp) (Fig. 3F). By contrast, in our analysis of the accumulated 23 S pre-rRNA, we did not detect an m1acp3Ψ1191-containing fragment but clearly detected an m1Ψ1191-containing fragment (Fig. 3F). These results strongly suggested that 23 S pre-rRNA resides in the nucleus of the rra1ts strain. Regarding ac4C1773 formation, we detected no ac4C1773-containing fragment (UUUCac4CGp) in the isolated 23 S pre-rRNA (Fig. 3E), strongly suggesting that 23 S pre-rRNA without ac4C1773 is not further processed into 20 S pre-rRNA. Therefore, it is likely that in the rra1ts strain, the 18 S rRNA fully modified with ac4C1773 originated from a residual pool of 40 S subunit that accumulated in the cytoplasm before heat treatment.
Accumulation of 23 S Pre-rRNA upon Depletion of Nuclear Acetyl-CoA
The findings described above clearly demonstrate that ac4C1773 formation mediated by Rra1p is required for processing 23 S pre-rRNA during the biogenesis of the 40 S subunit. On the basis of these observations, we hypothesized that rRNA processing and ribosome biogenesis could be controlled by nuclear acetyl-CoA concentration.
Acetyl-CoA is produced in the mitochondrial matrix by the pyruvate dehydrogenase complex and then used for acetylation of oxaloacetate to generate citrate in the tricarboxylic acid (TCA) cycle for energy production (Fig. 4A). In mammals, mitochondrial citrate can be exported to cytoplasm, where acetyl-CoA is produced from citrate and ATP in a reaction catalyzed by ATP-citrate lyase (36). However, no enzyme corresponding to ATP-citrate lyase is present in S. cerevisiae (37). Hence, nuclear and cytoplasmic acetyl-CoA cannot be supplied from the mitochondria in S. cerevisiae. Instead, nuclear acetyl-CoA is synthesized by acetyl-CoA synthetase 2 (Acs2p), which catalyzes the ligation of acetate and CoA (Fig. 4A) (38). Acs1p, another paralog of acetyl-CoA synthetase, is transcriptionally repressed at high concentrations of glucose (39). Thus, nuclear acetyl-CoA can be depleted in the temperature-sensitive strain, acs2ts (20), cultured at non-permissive temperature in the presence of glucose. Judging from the deacetylation kinetics of acetylated histones, rapid depletion of nuclear acetyl-CoA is achieved within 1 h after heat treatment (20). We cultured the acs2ts strain at 37 °C and monitored rRNA precursors by northern blotting at early times (Fig. 4B); longer term cultivation of this strain at non-permissive temperature resulted in global repression of transcription and extensive lethality (20). As shown in Fig. 4B, 23 S pre-rRNA clearly appeared 1–2 h after heat treatment in the acs2ts strain, whereas no accumulation of 23 S pre-rRNA was observed in the control strain YHT651. Transcription of rRNA precursors was not repressed over the time scale of this experiment, judging from the intensity of the 35 S pre-rRNA band 2 h after heat treatment (Fig. 4B). These data indicate that Rra1p modulates biogenesis of 40 S subunit by sensing the nuclear concentration of acetyl-CoA, thereby helping to coordinate ribosome synthesis with the cell's energy budget.
FIGURE 4.

The 23 S pre-rRNA accumulates upon Acs2p inactivation. A, schematic depiction of acetyl-CoA metabolism in S. cerevisiae. Acetyl-CoA produced in mitochondria is used in TCA cycle for energy production. Because no ATP-citrate lyase is present in S. cerevisiae, cytoplasmic acetyl-CoA is not produced from mitochondrial citrate. Nuclear acetyl-CoA, which is synthesized only by Acs2p, is used for histone acetylation by histone acetyltransferases (HAT) in nucleus and for formation of ac4C1773 in pre-rRNA in the nucleolus. B, accumulation of 23 S pre-rRNA after depletion of Acs2p. Precursors of 18 S rRNA were detected by northern blotting (NB) with the 5′-ETS probe at the indicated times after heat treatment of the acs2ts strain (YHT652) (right panels) and the control strain (YHT651) (left panels). Total RNA from rra1ts strain cultured at non-permissive condition was used as a marker for 23 S pre-rRNA (right-most panel). Steady-state levels of 25 S and 18 S rRNAs were visualized by ethidium bromide (EtBr) staining (lower panels).
DISCUSSION
In this study we detected ac4C at position 1773 in 18 S rRNA of S. cerevisiae and showed that an essential acetyltransferase, Rra1p, is responsible for ac4C1773 formation. Previous reports have demonstrated that this modified base is present in 18 S rRNAs from rat, chicken, D. discoideum; thus, ac4C in the terminal helix of the small 40 S subunit rRNA is likely to be a highly conserved modification among eukaryotes. Homologs of RRA1/tmcA (COG1444) are widely distributed among all domains of life. Bacterial homologs are only found in the γ-proteobacterial subphylum (28), suggesting that bacterial TmcA is responsible for ac4C formation at the wobble position of tRNAMet in these bacteria. Among the archaea, both Euryarchaeota and Crenarchaeota have RRA1 homologs. The RNA substrates for archaeal RRA1 remain to be elucidated, as ac4C is found in both tRNAs and rRNAs of some archaeal species (40–42). Among eukaryotes, RRA1 homologs are widely distributed in organisms from fungi to mammals. RNAi-mediated knockdown of Caenorhabditis elegans homolog nath-10 resulted in embryonic lethality (43), and disruption of the Drosophila melanogaster homolog CG1994 by P element insertion caused increased mortality during the early development (44). These observations support the functional and physiological importance of RRA1 in other eukaryotes.
The mammalian homolog of RRA1 is NAT10. Human NAT10 (also known as hALP) was initially identified as a transcriptional factor that interacts with the promoter region of hTERT (telomerase reverse transcriptase) (45). Recombinant NAT10/hALP (amino acids 164–834) lacking the N-terminal domains had an ability to acetylate calf thymus histones in vitro in the presence of acetyl-CoA (without ATP), indicating that NAT10/hALP has a histone acetyltransferase activity that might promote hTERT transcription via decondensation of chromatin structure (45). However, based on the crystal structure of TmcA, the N-terminal domain of the recombinant NAT10/hALP that was missing from that construct contains a functionally important region (DUF1726) that constitutes the RNA helicase/ATPase domain required for the tRNA acetyltransferase activity (28, 46). Therefore, it remains to be conclusively determined whether full-length NAT10 has an efficient and specific histone acetyltransferase activity. NAT10 is predominantly localized in the nucleolus in interphase, but in the mitotic midbody during telophase (47). Knockdown of NAT10 caused defects in nucleolar assembly and cytokinesis and also reduced the level of acetylated α-tubulin, leading to G2/M arrest (47). NAT10 can acetylate porcine tubulin in vitro (48), although the precise positions that are acetylated have not yet been determined. These observations suggest that NAT10 plays an important role in cell division by facilitating re-formation of the nucleolus and midbody in the late phase of cell mitosis as well as stabilization of microtubules. Assuming that NAT10 is a functional homolog of RRA1, it may play multiple functions required for both ribosome biogenesis and cell division. Furthermore, this protein is associated with several human diseases; NAT10 was highly expressed specifically in malignant tumors (47), and it is essential for growth of a subtype of epithelial ovarian cancer with poor prognosis (49). More recent work showed that NAT10 is the molecular target of a chemical compound that treats laminopathies, including premature aging syndromes, by correcting nuclear architecture (48). Functional studies of NAT10 as an rRNA-acetyltransferase should facilitate the development of innovative strategies for treating cancer and premature aging.
Depletion of Rra1p resulted in high level accumulation of 23 S pre-rRNA, leading to a huge reduction of 18 S rRNA (Fig. 3B) and the 40 S subunit (Fig. 3D). No ac4C1773 was present in the 23 S pre-rRNA that accumulated in the rra1ts strain, whereas residual 18 S rRNA was fully modified with ac4C1773 (Fig. 3E). These results demonstrated that ac4C1773 formation is essential for processing of 23 S pre-rRNA at the A0, A1, and A2 sites to yield 20 S pre-rRNA (Fig. 3A). Cleavages at these sites in 35 S and 23 S pre-rRNAs are dependent on the presence of U3 snoRNA, which base pairs with pre-rRNA to assemble the small 40 S subunit processome, a complex that contains >70 proteins (3, 34). Given that Rra1p is a component of the 90 S pre-ribosomal particle, which contains U3 snoRNP and other components (50), it is likely that Rra1p itself or ac4C1773 in pre-rRNA plays a critical role in processing at the A0, A1, and A2 sites. For example, the RNA helicase-like activity of Rra1p might modulate the small 40 S subunit processome to facilitate the rRNA processing (33). Alternatively, a putative reader protein that specifically recognizes ac4C1773 might recruit unidentified endonucleases responsible for cleavage at these sites.
Based on the results of our in vitro ac4C1773 formation assay, Rra1p preferentially recognizes the conserved sequence and structure of helix 45 in 18 S rRNA (Fig. 2E). In particular, the CCG sequence at positions 1772–1774 (Fig. 1A) is essential for ac4C1773 formation. Intriguingly, we observed higher rates of ac4C1773 formation when the base pairs of the CCG sequence were disrupted. Considering that Rra1p has an RNA helicase domain that is essential for ac4C1773 formation, Rra1p might unwind the local duplex of helix 45 via its helicase activity to acetylate C1773. A similar mechanism was previously proposed based on a structural study of TmcA, which is responsible for ac4C formation in tRNA (46).
To investigate the physiological significance of ac4C modification in 18 S rRNA, we asked whether rRNA processing might be controlled by cellular energy status. Immediately after depletion of nuclear acetyl-CoA in the acs2ts strain, we clearly observed accumulation of the 23 S pre-rRNA over time (Fig. 4B), strongly indicating that low levels of nuclear acetyl-CoA result in hypomodification of ac4C1773, leading in turn to accumulation of 23 S rRNA. This finding suggests that 18 S rRNA processing can be attenuated by reduced acetyl-CoA concentration in the nucleus under some stress conditions, including nutrient starvation. Because ribosome biogenesis is a cellular process that consumes a lot of energy (5), it is physiologically reasonable that nutrient-starved cells would regulate ribosome assembly at the stage of rRNA processing. In particular, the 40 S subunit is responsible for translation initiation, a step targeted in numerous strategies for regulation of protein synthesis (51, 52). Regulation of biogenesis of the 40 S subunit is a more direct means to control overall protein synthesis than alteration of translational efficiency by regulation of the 60 S subunit. Upon acetyl-CoA depletion under certain stress conditions, shutting off the supply of 18 S rRNA by reducing the ac4C level in pre-rRNA would provide an effective means for translational control. The cellular concentration of acetyl-CoA is reduced upon entry into stationary phase from logarithmic phase (53), and rRNA transcription is down-regulated in response to such reduced levels of acetyl-CoA (20). Thus, regulation of 18 S rRNA processing might also occur in stationary phase.
In summary, we showed that ac4C1773 is present in S. cerevisiae 18 S rRNA and that Rra1p catalyzes ac4C1773 formation using ATP and acetyl-CoA as substrates. We also demonstrated that ac4C1773 is an essential modification required for processing of 23 S pre-rRNA to yield 20 S pre-rRNA. Together, our results reveal that biogenesis of the 40 S subunit can be regulated by sensing nuclear acetyl-CoA concentration.
Acknowledgments
We are grateful to the members of the Suzuki laboratory, in particular Yuriko Sakaguchi, Asuteka Nagao, and Takayuki Ohira for technical support and many insightful discussions. Special thanks are due to Shinichi Nakagawa (RIKEN), Hidekazu Takahashi (Nara Institute of Science and Technology), and Sarin Chimnaronk (Mahidol University) for productive advices and technical support. We also thank Jef D. Boeke (Johns Hopkins University) and Yoshikazu Ohya (University of Tokyo) for providing materials.
This work was supported by grants-in-aid for Scientific Research on Priority Areas from the Ministry of Education, Science, Sports, and Culture of Japan (to T. S.).
- snoRNA
- small nucleolar RNA
- ac4C
- N4-acetylcytidine
- RRA1
- ribosomal RNA cytidine acetyltransferase 1
- ACS2
- acetyl-CoA synthetase 2
- tmcA
- tRNAMet cytidine acetyltransferase
- m1acp3Ψ
- 1-methyl-3-(3-amino-3-carboxypropyl)pseudouridine
- ESI
- nanoelectrospray ionization.
REFERENCES
- 1. Warner J. R. (1999) The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 24, 437–440 [DOI] [PubMed] [Google Scholar]
- 2. Venema J., Tollervey D. (1999) Ribosome synthesis in Saccharomyces cerevisiae. Annu. Rev. Genet. 33, 261–311 [DOI] [PubMed] [Google Scholar]
- 3. Henras A. K., Soudet J., Gérus M., Lebaron S., Caizergues-Ferrer M., Mougin A., Henry Y. (2008) The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell. Mol. Life Sci. 65, 2334–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fromont-Racine M., Senger B., Saveanu C., Fasiolo F. (2003) Ribosome assembly in eukaryotes. Gene 313, 17–42 [DOI] [PubMed] [Google Scholar]
- 5. Woolford J. L., Jr., Baserga S. J. (2013) Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 195, 643–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mullineux S. T., Lafontaine D. L. (2012) Mapping the cleavage sites on mammalian pre-rRNAs: where do we stand? Biochimie 94, 1521–1532 [DOI] [PubMed] [Google Scholar]
- 7. Granneman S., Baserga S. J. (2004) Ribosome biogenesis: of knobs and RNA processing. Exp. Cell Res. 296, 43–50 [DOI] [PubMed] [Google Scholar]
- 8. Reichow S. L., Hamma T., Ferré-D'Amaré A. R., Varani G. (2007) The structure and function of small nucleolar ribonucleoproteins. Nucleic Acids Res. 35, 1452–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kos M., Tollervey D. (2010) Yeast pre-rRNA processing and modification occur cotranscriptionally. Mol. Cell 37, 809–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Piekna-Przybylska D., Decatur W. A., Fournier M. J. (2008) The 3D rRNA modification maps database: with interactive tools for ribosome analysis. Nucleic Acids Res. 36, D178–D183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eschrich D., Buchhaupt M., Kötter P., Entian K. D. (2002) Nep1p (Emg1p), a novel protein conserved in eukaryotes and archaea, is involved in ribosome biogenesis. Curr. Genet. 40, 326–338 [DOI] [PubMed] [Google Scholar]
- 12. Wurm J. P., Meyer B., Bahr U., Held M., Frolow O., Kötter P., Engels J. W., Heckel A., Karas M., Entian K. D., Wöhnert J. (2010) The ribosome assembly factor Nep1 responsible for Bowen-Conradi syndrome is a pseudouridine-N1-specific methyltransferase. Nucleic Acids Res. 38, 2387–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brand R. C., Klootwijk J., Planta R. J., Maden B. E. (1978) Biosynthesis of a hypermodified nucleotide in Saccharomyces carlsbergensis 17 S and HeLa-cell 18 S ribosomal ribonucleic acid. Biochem. J. 169, 71–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Armistead J., Khatkar S., Meyer B., Mark B. L., Patel N., Coghlan G., Lamont R. E., Liu S., Wiechert J., Cattini P. A., Koetter P., Wrogemann K., Greenberg C. R., Entian K. D., Zelinski T., Triggs-Raine B. (2009) Mutation of a gene essential for ribosome biogenesis, EMG1, causes Bowen-Conradi syndrome. Am. J. Hum. Genet. 84, 728–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. White J., Li Z., Sardana R., Bujnicki J. M., Marcotte E. M., Johnson A. W. (2008) Bud23 methylates G1575 of 18 S rRNA and is required for efficient nuclear export of pre-40 S subunits. Mol. Cell. Biol. 28, 3151–3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lafontaine D., Vandenhaute J., Tollervey D. (1995) The 18 S rRNA dimethylase Dim1p is required for pre-ribosomal RNA processing in yeast. Genes Dev. 9, 2470–2481 [DOI] [PubMed] [Google Scholar]
- 17. Thomas G., Gordon J., Rogg H. (1978) N4-acetylcytidine: a previously unidentified labile component of the small subunit of eukaryotic ribosomes. J. Biol. Chem. 253, 1101–1105 [PubMed] [Google Scholar]
- 18. McCarroll R., Olsen G. J., Stahl Y. D., Woese C. R., Sogin M. L. (1983) Nucleotide sequence of the Dictyostelium discoideum small-subunit ribosomal ribonucleic acid inferred from the gene sequence: evolutionary Implication. Biochemistry 22, 5858–5868 [Google Scholar]
- 19. Sanchez-Diaz A., Kanemaki M., Marchesi V., Labib K. (2004) Rapid depletion of budding yeast proteins by fusion to a heat-inducible degron. Sci. STKE 2004, PL8 [DOI] [PubMed] [Google Scholar]
- 20. Takahashi H., McCaffery J. M., Irizarry R. A., Boeke J. D. (2006) Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol. Cell 23, 207–217 [DOI] [PubMed] [Google Scholar]
- 21. Chomczynski P., Sacchi N. (2006) The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat. Protoc. 1, 581–585 [DOI] [PubMed] [Google Scholar]
- 22. Miyauchi K., Ohara T., Suzuki T. (2007) Automated parallel isolation of multiple species of non-coding RNAs by the reciprocal circulating chromatography method. Nucleic Acids Res. 35, e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miyauchi K., Kimura S., Suzuki T. (2013) A cyclic form of N6-threonylcarbamoyladenosine as a widely distributed tRNA hypermodification. Nat. Chem. Biol. 9, 105–111 [DOI] [PubMed] [Google Scholar]
- 24. Suzuki T., Ikeuchi Y., Noma A., Suzuki T., Sakaguchi Y. (2007) Mass spectrometric identification and characterization of RNA-modifying enzymes. Methods Enzymol. 425, 211–229 [DOI] [PubMed] [Google Scholar]
- 25. Inada T., Winstall E., Tarun S. Z., Jr., Yates J. R., 3rd, Schieltz D., Sachs A. B. (2002) One-step affinity purification of the yeast ribosome and its associated proteins and mRNAs. RNA 8, 948–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sampson J. R., Uhlenbeck O. C. (1988) Biochemical and physical characterization of an unmodified yeast phenylalanine transfer RNA transcribed in vitro. Proc. Natl. Acad. Sci. U.S.A. 85, 1033–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Machnicka M. A., Milanowska K., Osman Oglou O., Purta E., Kurkowska M., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K. M., Helm M., Bujnicki J. M., Grosjean H. (2013) MODOMICS: a database of RNA modification pathways: 2013 update. Nucleic Acids Res. 41, D262–D267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ikeuchi Y., Kitahara K., Suzuki T. (2008) The RNA acetyltransferase driven by ATP hydrolysis synthesizes N4-acetylcytidine of tRNA anticodon. EMBO J. 27, 2194–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pagé N., Gérard-Vincent M., Ménard P., Beaulieu M., Azuma M., Dijkgraaf G. J., Li H., Marcoux J., Nguyen T., Dowse T., Sdicu A. M., Bussey H. (2003) A Saccharomyces cerevisiae genome-wide mutant screen for altered sensitivity to K1 killer toxin. Genetics 163, 875–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grandi P., Rybin V., Bassler J., Petfalski E., Strauss D., Marzioch M., Schäfer T., Kuster B., Tschochner H., Tollervey D., Gavin A. C., Hurt E. (2002) 90 S pre-ribosomes include the 35 S pre-rRNA, the U3 snoRNP, and 40 S subunit processing factors but predominantly lack 60 S synthesis factors. Mol. Cell 10, 105–115 [DOI] [PubMed] [Google Scholar]
- 31. Huh W. K., Falvo J. V., Gerke L. C., Carroll A. S., Howson R. W., Weissman J. S., O'Shea E. K. (2003) Global analysis of protein localization in budding yeast. Nature 425, 686–691 [DOI] [PubMed] [Google Scholar]
- 32. Li Z., Lee I., Moradi E., Hung N. J., Johnson A. W., Marcotte E. M. (2009) Rational extension of the ribosome biogenesis pathway using network-guided genetics. PLoS Biol. 7, e1000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Strunk B. S., Karbstein K. (2009) Powering through ribosome assembly. RNA 15, 2083–2104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hughes J. M., Ares M., Jr. (1991) Depletion of U3 small nucleolar RNA inhibits cleavage in the 5′ external transcribed spacer of yeast pre-ribosomal RNA and impairs formation of 18 S ribosomal RNA. EMBO J. 10, 4231–4239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pendrak M. L., Roberts D. D. (2011) Ribosomal RNA processing in Candida albicans. RNA 17, 2235–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chypre M., Zaidi N., Smans K. (2012) ATP-citrate lyase: a mini-review. Biochem. Biophys. Res. Commun. 422, 1–4 [DOI] [PubMed] [Google Scholar]
- 37. Pronk J. T., Yde Steensma H., Van Dijken J. P. (1996) Pyruvate metabolism in Saccharomyces cerevisiae. Yeast 12, 1607–1633 [DOI] [PubMed] [Google Scholar]
- 38. Starai V. J., Escalante-Semerena J. C. (2004) Acetyl-coenzyme A synthetase (AMP forming). Cell. Mol. Life Sci. 61, 2020–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kratzer S., Schüller H. J. (1995) Carbon source-dependent regulation of the acetyl-coenzyme A synthetase-encoding gene ACS1 from Saccharomyces cerevisiae. Gene 161, 75–79 [DOI] [PubMed] [Google Scholar]
- 40. Bruenger E., Kowalak J. A., Kuchino Y., McCloskey J. A., Mizushima H., Stetter K. O., Crain P. F. (1993) 5S rRNA modification in the hyperthermophilic archaea Sulfolobus solfataricus and Pyrodictium occultum. FASEB J. 7, 196–200 [DOI] [PubMed] [Google Scholar]
- 41. Gupta R. (1984) Halobacterium volcanii tRNAs. Identification of 41 tRNAs covering all amino acids and the sequences of 33 class I tRNAs. J. Biol. Chem. 259, 9461–9471 [PubMed] [Google Scholar]
- 42. Noon K. R., Bruenger E., McCloskey J. A. (1998) Posttranscriptional modifications in 16 S and 23 S rRNAs of the archaeal hyperthermophile Sulfolobus solfataricus. J. Bacteriol. 180, 2883–2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Waters K., Yang A. Z., Reinke V. (2010) Genome-wide analysis of germ cell proliferation in C. elegans identifies VRK-1 as a key regulator of CEP-1/p53. Dev. Biol. 344, 1011–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Peter A., Schöttler P., Werner M., Beinert N., Dowe G., Burkert P., Mourkioti F., Dentzer L., He Y., Deak P., Benos P. V., Gatt M. K., Murphy L., Harris D., Barrell B., Ferraz C., Vidal S., Brun C., Demaille J., Cadieu E., Dreano S., Gloux S., Lelaure V., Mottier S., Galibert F., Borkova D., Miñana B., Kafatos F. C., Bolshakov S., Sidén-Kiamos I., Papagiannakis G., Spanos L., Louis C., Madueño E., de Pablos B., Modolell J., Bucheton A., Callister D., Campbell L., Henderson N. S., McMillan P. J., Salles C., Tait E., Valenti P., Saunders R. D., Billaud A., Pachter L., Klapper R., Janning W., Glover D. M., Ashburner M. (2002) Mapping and identification of essential gene functions on the X chromosome of Drosophila. EMBO Rep. 3, 34–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lv J., Liu H., Wang Q., Tang Z., Hou L., Zhang B. (2003) Molecular cloning of a novel human gene encoding histone acetyltransferase-like protein involved in transcriptional activation of hTERT. Biochem. Biophys. Res. Commun. 311, 506–513 [DOI] [PubMed] [Google Scholar]
- 46. Chimnaronk S., Suzuki T., Manita T., Ikeuchi Y., Yao M., Suzuki T., Tanaka I. (2009) RNA helicase module in an acetyltransferase that modifies a specific tRNA anticodon. EMBO J. 28, 1362–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shen Q., Zheng X., McNutt M. A., Guang L., Sun Y., Wang J., Gong Y., Hou L., Zhang B. (2009) NAT10, a nucleolar protein, localizes to the midbody and regulates cytokinesis and acetylation of microtubules. Exp. Cell Res. 315, 1653–1667 [DOI] [PubMed] [Google Scholar]
- 48. Larrieu D., Britton S., Demir M., Rodriguez R., Jackson S. P. (2014) Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science 344, 527–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tan T. Z., Miow Q. H., Huang R. Y., Wong M. K., Ye J., Lau J. A., Wu M. C., Bin Abdul Hadi L. H., Soong R., Choolani M., Davidson B., Nesland J. M., Wang L. Z., Matsumura N., Mandai M., Konishi I., Goh B. C., Chang J. T., Thiery J. P., Mori S. (2013) Functional genomics identifies five distinct molecular subtypes with clinical relevance and pathways for growth control in epithelial ovarian cancer. EMBO Mol. Med. 5, 983–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pérez-Fernández J., Román A., De Las Rivas J., Bustelo X. R., Dosil M. (2007) The 90 S preribosome is a multimodular structure that is assembled through a hierarchical mechanism. Mol. Cell. Biol. 27, 5414–5429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Holcik M., Sonenberg N. (2005) Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 6, 318–327 [DOI] [PubMed] [Google Scholar]
- 52. McCarthy J. E. (1998) Posttranscriptional control of gene expression in yeast. Microbiol Mol. Biol. Rev. 62, 1492–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cai L., Sutter B. M., Li B., Tu B. P. (2011) Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol. Cell 42, 426–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McLuckey S. A., Van Berkel G. J., Glish G. L. (1992) Tandem mass-spectrometry of small, multiply charged oligonucleotides. J. Am. Soc. Mass Spectrom. 3, 60–70 [DOI] [PubMed] [Google Scholar]