

Background: Methylation of rRNA is a common resistance mechanism in antibiotic-producing bacteria.
Results: Thiostrepton-resistance methyltransferase (Tsr) amino-terminal domain induces RNA substrate conformational changes necessary for catalysis by its carboxyl-terminal domain.
Conclusion: RNA structural reorganization distal from the methylated nucleotide is implicated in specific substrate recognition by Tsr.
Significance: RNA substrate structure can directly regulate modification enzyme activity.
Keywords: Antibiotic Resistance, Enzyme Mechanism, Ribosomal Ribonucleic Acid (rRNA) (Ribosomal RNA), RNA Methyltransferase, RNA-Protein Interaction, RNase Structure Probing, UV Melting, Fluorescence Polarization, Hydroxyl Radical, Thiostrepton
Abstract
Ribosomal RNA (rRNA) post-transcriptional modifications are essential for ribosome maturation, translational fidelity, and are one mechanism used by both antibiotic-producing and pathogenic bacteria to resist the effects of antibiotics that target the ribosome. The thiostrepton producer Streptomyces azureus prevents self-intoxication by expressing the thiostrepton-resistance methyltransferase (Tsr), which methylates the 2′-hydroxyl of 23 S rRNA nucleotide adenosine 1067 within the thiostrepton binding site. Tsr is a homodimer with each protomer containing an L30e-like amino-terminal domain (NTD) and a SPOUT methyltransferase family catalytic carboxyl-terminal domain (CTD). We show that both enzyme domains are required for high affinity RNA substrate binding. The Tsr-CTD has intrinsic, weak RNA affinity that is necessary to direct the specific high-affinity Tsr-RNA interaction via NTDs, which have no detectable RNA affinity in isolation. RNA structure probing experiments identify the Tsr footprint on the RNA and structural changes in the substrate, induced specifically upon NTD binding, which are necessary for catalysis by the CTD. Additionally, we identify a key amino acid in each domain responsible for CTD-RNA binding and the observed NTD-dependent RNA structural changes. These studies allow us to develop a model for Tsr-RNA interaction in which the coordinated substrate recognition of each Tsr structural domain is an obligatory pre-catalytic recognition event. Our findings underscore the complexity of substrate recognition by RNA modification enzymes and the potential for direct involvement of the RNA substrate in controlling the process of its modification.
Introduction
Post-transcriptional modification of RNA is a conserved and essential process in all kingdoms of life. Although many details remain to be uncovered, important roles played by such modifications have steadily emerged for tRNAs and rRNAs, which are the best studied and among the most highly modified RNAs. Many tRNA modifications are essential in bacteria, for example, methylation of the tRNA anticodon stem loop at G37 by TrmD prevents frameshifting during translation (1). rRNA modifications are not essential for growth under laboratory conditions, but frequently exhibit cold sensitive growth phenotypes and a variety of translational defects (2). Deletion of RsmA (formally KsgA; 16 S rRNA m62A1518/m62A1519) disrupts 30 S biogenesis, whereas deletion of RsmB and RsmD (16S rRNA m5C967 and m2G966, respectively) disrupts translation initiation (2, 3). Additionally, in antibiotic-producing and resistant pathogenic bacteria, methylation of rRNA at antibiotic binding sites disrupts drug binding and thus their toxic effects (4, 5). The Escherichia coli 16 S and 23 S rRNAs contain 24 constitutively methylated nucleotides and the enzyme responsible for catalyzing each has been identified (6). Structural and biochemical studies of these enzymes and their homologs have provided significant insight into their interactions with the essential co-substrate S-adenosyl-l-methionine (AdoMet)3 and their catalytic mechanisms (7, 8). However, due to the relative paucity of structural studies methyltransferase-RNA substrate complexes, much less is understood about rRNA substrate recognition.
Thiostrepton is the prototypical member of the thiazole-containing class of antibiotics and is produced by a number of Streptomyces strains including Streptomyces azureus (9). Thiostrepton has been used in veterinary medicine to treat mastitis and as a topical agent for dogs but, due to its poor solubility and toxicity, has found only limited applications to date. However, there is renewed interest in the clinical use of thiostrepton as an antibiotic, and also as a therapy for cancer and malaria (10–12). Thiostrepton binds to a compact 58-nucleotide (nt) rRNA domain within the 23 S rRNA, which is also the binding site for ribosomal protein L11 (13, 14). This rRNA·protein complex forms part of the ribosomal functional center known as the GTPase center, which interacts with translational factors EF-G, EF-Tu, and RF3. Resistance to thiostrepton can arise from L11 loss or mutation, rRNA mutation, or the specific methylation of the rRNA that makes up the drug-RNA binding interface (15–17).
In S. azureus, resistance is specifically conferred by methylation of 23 S rRNA on the ribose 2′-hydroxyl of adenosine 1067 (A1067, E. coli numbering) by the thiostrepton resistance methyltransferase (Tsr) (15, 18). Tsr uses the co-substrate AdoMet to methylate the 23 S rRNA, presumably prior to the assembly of the 50 S subunit as the L11 and proposed Tsr binding surfaces are overlapping. The crystal structure of the Tsr·AdoMet complex definitively classified the enzyme as a member of the SPOUT family of methyltransferases (19, 20). As such, it is an obligate homodimer through interactions mediated by its carboxyl-terminal domain (CTD; Fig. 1A). The CTD also contains a characteristic trefoil knot structure that makes up the AdoMet binding site. Furthermore, the Tsr structure illustrated the organization of each CTD and its associated amino-terminal domain (NTD). The NTD is structurally similar to the yeast ribosomal protein L30e and is presumed to be involved in RNA substrate recognition. However, its specific contribution(s) to recognition and which structural elements of the 58-nt RNA domain (Fig. 1B) are essential for interaction with Tsr are unknown.
FIGURE 1.
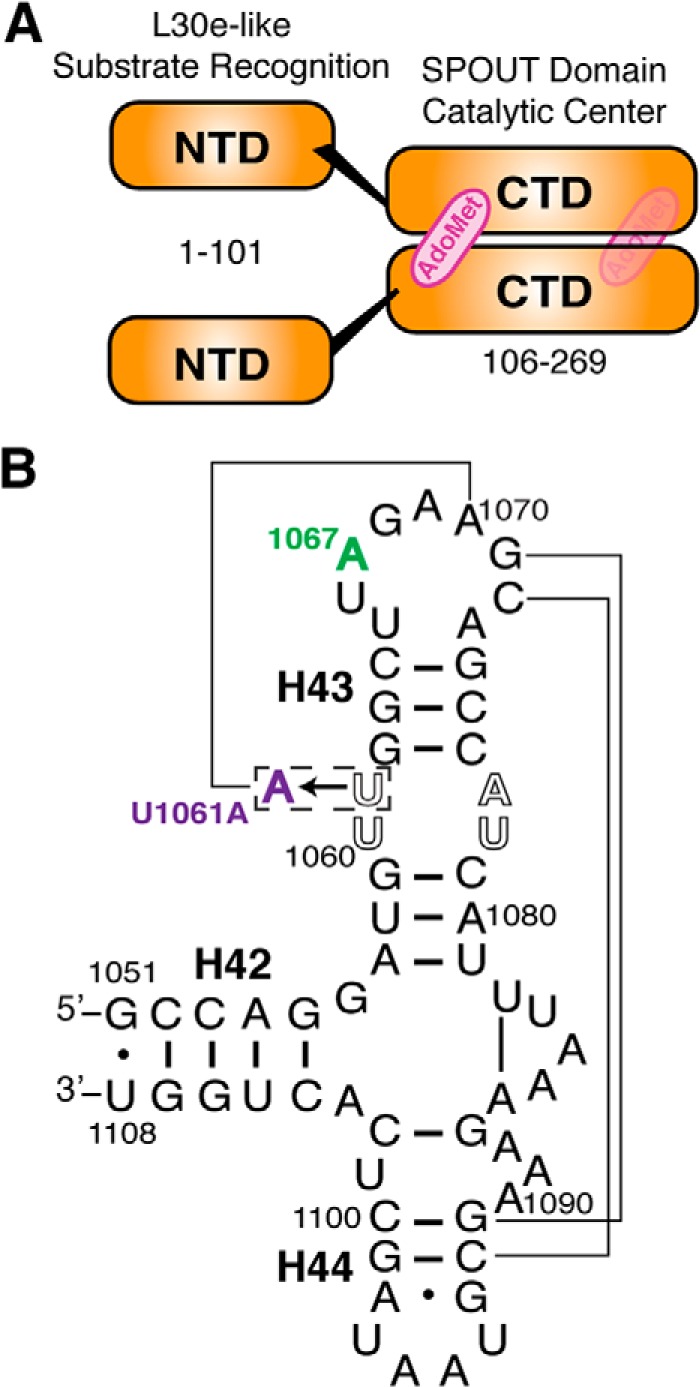
Tsr domain organization and 58-nt RNA substrate secondary structure. A, Tsr is a homodimer consisting of protomers each containing an NTD proposed to direct substrate recognition and a CTD containing the catalytic centers and AdoMet binding sites. B, secondary structure of the 58-nt RNA substrate comprising E. coli 23 S rRNA helices 42 to 44 (H42, H43, and H44). The Tsr target nucleotide (A1067, green) and tertiary structure stabilizing point mutation (U1061A, purple) are highlighted. Lines indicate key long-range interactions within the 58-nt domain tertiary fold.
Two lines of evidence from previous studies have suggested that unfolding the 58-nt RNA domain tertiary structure may be required in substrate recognition by Tsr. First, stabilization of the RNA tertiary structure decreased in vitro methylation of the 58-nt RNA by Tsr (21). Second, a model 29-nt RNA hairpin containing the target loop and its associated stem, but lacking the full tertiary structure, was more readily methylated than the full 58-nt domain. These observations suggest that there may be an energetic penalty paid by the enzyme to unfold the RNA tertiary structure prior to catalysis (20). We therefore sought to determine whether Tsr must alter the RNA structure as part of its substrate recognition mechanism. Here, we demonstrate that specific RNA recognition by Tsr involves docking of its CTD on the A1067 target loop, followed by engagement of one or both NTDs in a process that drives specific RNA conformational changes at a site distant from the target loop. Furthermore, this RNA structural change is an essential step for catalysis and may form part of a pre-catalytic recognition signal from the N-terminal RNA recognition domain. Collectively, these studies reveal new mechanistic details of the intricate process of specific substrate recognition by Tsr and suggest a direct role for the RNA substrate in control of catalysis.
EXPERIMENTAL PROCEDURES
Tsr Purification and Mutagenesis
Tsr was expressed from plasmid pET28a-Tsr in E. coli BL21(DE3)-pLysS as described previously (20), and purified using Ni2+ affinity, heparin affinity, and gel filtration chromatographies. Elution volume from the Superdex 200 10/300 gel filtration column (GE Healthcare) was calibrated using Gel Filtration Standards (Bio-Rad). Purified enzyme was flash frozen and stored at −80 °C.
Tsr mutagenesis was performed using the MEGAWHOP protocol (22) to modify pET28a-Tsr. The Tsr amino-terminal domain (Tsr-NTD) construct was created by inserting two stop codons after that coding for residue Arg-101. The Tsr-CTD construct was made by deletion of codons corresponding to amino acids 1–105. Individual domain proteins were expressed and purified as described for full-length (FL) Tsr. Each protein was expressed with a His6 tag and thrombin cleavage site, giving calculated molecular masses of 31 (62-kDa dimer), 19.5 (39-kDa dimer), and 13 kDa for the FL-Tsr, Tsr-CTD, and Tsr-NTD proteins, respectively.
RNA in Vitro Transcription
Wild-type and U1061A 58-nt RNAs (Fig. 1B) were in vitro transcribed from linearized plasmid DNA as previously described (23). Prior to use, RNA was annealed by incubation at 65 °C for 10 min and slowly cooled to 25 °C.
Hydroxyl Radical and Ribonuclease RNA Structure Probing
RNA was dephosphorylated by alkaline phosphatase treatment and then 32P-5′-end labeled using [γ-32P]ATP and T4 polynucleotide kinase. The products of the kinase reaction were resolved on a 12% (w/v) polyacrylamide, 50% urea denaturing sequencing gel, and full-length 32P-labeled RNA excised and recovered from gel slices by soaking in 0.3 m sodium acetate and subsequent ethanol precipitation.
Hydroxyl radical probing experiments contained FL-Tsr dialyzed overnight against assay buffer, 50 mm Tris, pH 7.5, 75 mm KCl, 5 mm MgCl2, at final concentrations of 20, 10, 5, 1, or 0.2 μm with annealed 32P-labeled RNA (50,000 cpm) in a 20-μl reaction. One microliter each of 50 mm Fe(SO4)2, 100 mm EDTA, 250 mm ascorbic acid, and 3% H2O2 were added to the side of the tube and pulsed in a microcentrifuge to mix rapidly. After 5 min on ice, the reaction was quenched by ethanol precipitation and the recovered RNA resuspended in 8 μl of denaturing loading dye. Radioactivity was quantified by liquid scintillation counting and equal counts were loaded for each reaction and resolved on a 12% (w/v) polyacrylamide, 50% urea denaturing sequencing gel. Gels were run at a constant 55 W for 1.5 h, dried, exposed to a phosphorscreen overnight and scanned using a Typhoon FLA 7000 laser scanner (GE Healthcare). Band intensities were quantified using ImageQuant TL software (GE Healthcare) applying the rubber band background subtraction method. Band intensities were normalized to the most intense band in 0 μm lane and normalized intensities were compared between 0 and 20 μm Tsr. A nucleotide was considered protected or enhanced if the difference in relative intensity was ±15%. An alkaline hydrolysis ladder (AH) and denaturing RNase T1 pattern (guanosine sequence) were used to identify the specific nucleotides cleaved.
For RNase enzymatic probing experiments, annealed 32P-labeled RNA (50,000 cpm) was mixed with assay buffer alone (supplemented with 10% glycerol) or a final concentration of 20 μm Tsr. The optimal concentration of each RNase (V1, T1, or A) was empirically identified by performing reactions with 10-fold serial dilutions of the RNase. Additional control reactions with RNA (± protein), but without RNase were also performed to ensure no contaminating RNases were present. Reactions were incubated for 10 min at 25 °C. The RNA was recovered by ethanol precipitation and resuspended in denaturing gel loading dye. Samples were analyzed by PAGE and recorded as described for the hydroxyl radical probing experiments.
RNA UV Melting
FL-Tsr and RNA were dialyzed overnight against 10 mm Hepes buffer, pH 7.5, 100 mm NH4Cl, 5 mm Mg2SO4, and 10% glycerol. After annealing, RNA (25 μg/sample) was melted alone or with an equal or half-molar ratio of Tsr. The UV absorbance at 260 and 280 nm was measured over a linear temperature gradient (18–65 °C) with a heating rate of 1 °C/min. The first derivative of each melting curve, referred to as its “melting profile,” was calculated as described previously (24, 25).
Fluorescence Polarization
RNA was 5′-end labeled with a fluorescein analog as described previously (26). Binding experiments were performed in the same assay buffer as used for structure probing experiments with a final concentration of 10 nm annealed RNA and protein concentrations ranging from 1 nm to 50 μm. Polarization was measured in black, non-binding surface, 96-well plates (Corning) using a Synergy4 plate reader running Gen5 software (BioTek). Data were background subtracted and non-linear curve fitting was performed in GraphPad Prism. Fits for all binding isotherms were compared between one-site and two-site specific binding, and the latter accepted only when the p value was <0.05.
Methylation Assays
Methylation assays using illustra MicroSpin G-25 columns (GE Healthcare) to separate 3H-labeled RNA and the remaining [3H]AdoMet were performed as described previously (20) with the following modifications. Assays were performed in 200-μl reactions in assay buffer supplemented with 10% glycerol. After 30 min at 37 °C, 3 × 50 μl samples (technical triplicates) were applied to the spin column and 40 μl of filtrate was counted in 2 ml of Ecoscint Ultra scintillation fluid (National Diagnostics). Assays were performed with three independent preparations of Tsr. Tritium incorporation values in counts per minute (cpm) were averaged and plotted with the associated mean ± S.E.
Electrophoretic Mobility Shift Assay
58-Nucleotide RNA (100 nm) was incubated with a range of concentrations of FL-Tsr or Tsr-CTD (2-fold dilutions from 10 μm to 78 nm) at room temperature for 15 min and then separated on a 10% acrylamide, Tris borate-EDTA (TBE) native gel. The gel was stained with SYBR Gold and imaged using a Typhoon Trio imager (GE Healthcare). For binding specificity experiments, 100 nm in vitro transcribed 58-nt RNA, HDV ribozyme, or tRNAAsn were incubated with FL-Tsr (1 or 10 μm) or Tsr-CTD (10 μm) and analyzed as above.
Partial Proteolysis
Purified wild-type and mutant Tsr proteins were treated with chymotrypsin at 1:100 (w/w) chymotrypsin:Tsr ratio using the same assay buffer as for probing experiments but supplemented with 10% glycerol. Samples were incubated for 30 min at room temperature. Reactions were stopped by adding SDS-loading dye and heating to 90 °C, and the products were resolved on a 10% SDS-PAGE gel and visualized by staining with Coomassie Brilliant Blue dye. Otherwise identical samples were run without chymotrypsin as control for each protein.
RESULTS
The Tsr NTDs Aid in rRNA Binding and Are Necessary for Catalysis
We predicted that the Tsr NTD and CTD domains might form stable and correctly folded proteins in isolation as homologous proteins are known, e.g. L30e and TrmL (a minimal SPOUT methyltransferase), and inspection of the Tsr NTD-CTD interface revealed it to be comprised primarily of polar and charged amino acids with almost no hydrophobic residues that would be exposed to solvent. Thus, to define the contribution of each Tsr domain to rRNA substrate recognition and catalysis, proteins corresponding to FL-Tsr (Fig. 1A), the NTD (Tsr-NTD; amino acids 1–101), and CTD (Tsr-CTD; amino acids 106–269) were expressed, purified, and their binding and catalytic activities quantified. Each isolated domain was indeed soluble and could be purified identically to FL-Tsr. In support of their correct, stable folding, FL-Tsr and Tsr-CTD eluted from the gel filtration column at a volume corresponding to the dimeric proteins (62 and 39 kDa, respectively), whereas Tsr-NTD was monomeric (13 kDa) (Fig. 2A). Furthermore, in each case the target protein eluted as a highly symmetrical peak indicative of well folded proteins suitable for subsequent biochemical analysis.
FIGURE 2.

The Tsr NTDs increase RNA binding affinity and are necessary for catalysis. A, chromatograms of the final gel filtration purification for full-length Tsr (FL-Tsr, black), Tsr-CTD (purple), and Tsr-NTD (teal). Gel filtration standards are shown (gray) and the approximate molecular mass (kDa) indicated above each peak. B, FP analysis of FL-Tsr (black), Tsr-CTD (purple), and Tsr-NTD (teal) binding to the wild-type (WT) 58-nt RNA. Arrowheads (open and solid) shown in panels B–D indicate Tsr concentrations (1 and 10 μm, respectively) used in the control EMSAs of panel E. C, EMSA of FL-Tsr (0.078 to 10 μm, left to right lanes) interaction with 58-nt RNA. Free 58 nt (*), Tsr·58-nt RNA complex (solid circle), and higher molecular weight complexes (open circle) are indicated. The dashed box indicates the region of the gel shown (bottom) at greater exposure. D, as in C but for Tsr-CTD. E, control EMSAs comparing FL-Tsr (1 or 10 μm) and Tsr-CTD (10 μm) affinity for the 58-nt substrate and two unrelated in vitro transcribed RNAs: the HDV ribozyme and tRNAAsn. F, relative methylation of 58-nt RNA by FL-Tsr and Tsr-CTD, relative to background in the absence of RNA substrate.
The binding dissociation constant (KD) of FL-Tsr for the 58-nt RNA was determined using fluorescence polarization (FP; Fig. 2B). Interaction of FL-Tsr and the 58-nt RNA was best fit using a two-site binding model yielding KD values of 160 nm and ∼10 μm (Table 1). Two binding events were previously observed for Tsr and other SPOUT family members using electrophoretic mobility shift assays (EMSAs) and the shifted bands were attributed to dimeric and tetrameric complexes of enzyme with RNA (7, 20, 27). We performed EMSAs using a similar range of Tsr concentrations as the FP experiments and confirmed the presence of these same complexes and their formation consistent with the KD values determined by FP (Fig. 2C).
TABLE 1.
Dissociation constants for Tsr and RNA mutants
| Protein | 58-nt RNA | KD | Fig. |
|---|---|---|---|
| μm | |||
| FL-Tsr | Wild-type | 1) 0.16 ± 0.06 | 2B |
| 2) 10 ± 6.7 | |||
| Tsr-CTD | Wild-type | 6.1 ± 1.6 | 2B |
| Tsr-NTD | Wild-type | NBa | 2B |
| FL-Tsr | U1061A | 1.6 ± 0.3 | 6A |
| Tsr-R26A | Wild-type | 4.6 ± 1.1 | 7G |
| Tsr-R162A | Wild-type | 18 ± 9.4 | 7G |
a NB, no binding detected.
We next performed FP binding assays for each Tsr domain protein. In isolation, the monomeric Tsr-NTD had no measurable affinity for the RNA (Fig. 2B) suggesting that the CTD dimer is necessary to properly position both NTDs to achieve high affinity substrate binding. In contrast, the isolated Tsr-CTD dimer retained the ability to bind the 58-nt RNA but with >30-fold weaker affinity than FL-Tsr (Table 1), and the data were best fit with a one-site binding model. However, an EMSA performed with the Tsr-CTD protein (Fig. 2D) clearly indicated that the isolated domain forms equivalent complexes to FL-Tsr, in a protein concentration-dependent manner, albeit with reduced affinities. Fit of the Tsr-CTD FP data using the same two-site binding model as for FL-Tsr yielded KD values of ∼3 and ∼13 μm. We note that these values are entirely consistent with the appearance of protein-RNA complexes in the Tsr-CTD EMSA (Fig. 2D), but the errors associated with the fit values are unacceptably large (presumably due to the transient nature of the faster migrating band with respect to protein concentration and the closeness of the KD values compared with FL-Tsr). Most importantly, however, the FP and EMSA experiments together demonstrate that the catalytic CTD of Tsr interacts with the 58-nt RNA substrate similarly whether part of FL-Tsr or as the isolated Tsr-CTD but with substantially reduced affinity in the latter case. This observation is also reinforced by the ribonuclease probing experiments described below, which clearly demonstrate that Tsr-CTD binds the RNA substrate in an identical manner as when it is part of full-length Tsr. Specifically, Tsr-CTD is able to confer protection of a subset of the same residues in the RNA as the full-length enzyme.
The proposed RNA-binding cleft of Tsr (20) contains positively charged amino acids on the CTD surface that could potentially promote nonspecific interactions with any nucleic acid. We therefore sought to explore the specificity of RNA binding by Tsr and Tsr-CTD using EMSAs with the 58-nt RNA and two other structured RNAs with stem-loops: hepatitis δ virus (HDV) ribozyme and tRNAAsn. With both concentrations of FL-Tsr and 10 μm Tsr-CTD, all of the 58-nt RNA was bound and its mobility retarded, resulting in the absence of a band with mobility corresponding to the free RNA (Fig. 2E). Conversely, neither FL-Tsr nor Tsr-CTD shifted all HDV RNA, suggesting that both proteins have significantly weaker affinity for HDV than the 58-nt RNA. A similar result was obtained with tRNAAsn, except that the highest FL-Tsr concentration, but not Tsr-CTD, resulted in a significant fraction of shifted RNA. These results indicate that although Tsr does have a propensity to bind other nucleic acids, binding of both Tsr and Tsr-CTD to the 58-nt RNA substrate is of higher affinity and specificity compared with other structured RNAs.
Collectively, these RNA binding data for FL-Tsr and the domain constructs indicate that the CTD plays a direct role in binding the RNA substrate and is critical for optimally positioning the NTDs to fulfill their essential role in forming a specific, high-affinity complex. As Tsr-CTD preserves its dimeric state, binds the 58-nt RNA and, most importantly, maintains both intact AdoMet binding pockets and active sites, we next asked whether Tsr-CTD additionally retains any ability to methylate the 58-nt RNA. We compared the ability of full-length Tsr and Tsr-CTD to methylate the 58-nt RNA substrate and found that deletion of the NTDs, reduces Tsr activity to background (Fig. 2D). Collectively, from these binding and activity assays we conclude that the CTD has weak RNA binding affinity, but proper recognition of the substrate by the NTDs is necessary for substrate specificity and stimulation of catalysis of methyl transfer by its CTD.
Identification of the RNA Binding Surface and Binding-induced Perturbations in the RNA Structure by Tsr
To identify the RNA surface contacted by Tsr we performed solution hydroxyl radical probing of 32P-5′-end labeled wild-type 58-nt RNA in the absence and presence of various concentrations of full-length Tsr. Comparison of probing experiments performed without Tsr or in the presence of 20 μm Tsr revealed the protection of nucleotides 1058, 1059, and 1061–1066 in Helix 43 (H43) from hydroxyl radical-mediated strand scission, defining at least part of the Tsr-RNA “footprint” (Fig. 3). This region is smaller than expected and most likely does not reflect the entirety of the interaction surface between Tsr and the 58-nt RNA. One complication in this analysis is that the compact tertiary structure of the 58-nt RNA protects many nucleotides from strand scission (28) and as a result such residues might concomitantly be relieved of protection by RNA-RNA contacts but protected by newly formed RNA-protein interactions. In support of this are observations of multiple regions of enhancement of hydroxyl radical cleavage and Tsr-mediated protection from RNase cleavage outside of this observed footprint region (described below and in the next section, respectively).
FIGURE 3.

Hydroxyl radical probing identifies the Tsr footprint and cleavage enhancements upon Tsr binding. A, representative hydroxyl radical probing gel of wild-type 58-nt RNA complexed with an increasing concentration of Tsr (left to right: 0.2, 1, 5, 10, and 20 μm). Untreated RNA (U), partial alkaline hydrolysis nucleotide ladder (AH), and RNase T1 digestion under denaturing conditions (T1) allow nucleotide identification (marked on left); FL is the full-length 58-nt RNA band, also shown with lighter exposure in the top panel. B, quantification of each band in the lanes without Tsr and with Tsr at 20 μm (noted * at top). Cleavage protections and enhancements are shown as blue and orange shading, respectively. Changes in sensitivity to hydroxyl radicals are mapped as protections (blue) and enhancements (orange) onto C, the 58-nt RNA secondary structure, and D, the x-ray crystal structure (PDB ID 1HC8).
Enhancement of hydroxyl radical cleavage upon protein binding could arise through Tsr-mediated distortion(s) of the RNA backbone that make it more susceptible to radical cleavage at specific nucleotides. In the presence of Tsr, enhancements are observed at three disparate locations: U1083/A1084 of the helical junction; A1087, located opposite the Tsr footprint surface at the base of H44; and U1094/A1095 at the apex of H44 (Fig. 3). The enhancements in H44 are likely due to structural rearrangement of the target loop or H43, which are necessary to orient the target nucleotide A1067 into the catalytic site of Tsr. However, the induced changes to hydroxyl radical sensitivity more distant from the target nucleotide offer a first experimental indication of a more global RNA structural alteration, potentially unfolding of the RNA tertiary structure, induced by Tsr binding.
Given that the 58-nt RNA has a unique, compact tertiary structure with H44 juxtaposed to H43, and the identified role of the NTDs in activation of catalysis, we sought further evidence that Tsr could be unfolding the RNA structure. The 58-nt RNA has been extensively characterized by UV melting analysis and its melting profile includes a low temperature unfolding transition (Tm ∼45 °C), observed at 260 nm but invisible at 280 nm (Fig. 4A), that has been definitively demonstrated to correspond to the RNA tertiary structure (29, 30). The remaining RNA secondary structures unfold in a single apparent transition (with apparent Tm ∼63 °C), corresponding to multiple two-state unfolding transitions. We tested whether Tsr is capable of altering the RNA structure upon binding, with the anticipation that stabilization or destabilization of the RNA tertiary structure would manifest as a higher or lower unfolding Tm, corresponding to a rightward or leftward shift in the unfolding transition, respectively (Fig. 4B). At 0.5 or 1 molar ratio of Tsr to 58-nt RNA, the tertiary structure unfolding transition in the melting profile was partially or fully eliminated, respectively, over the temperature range 20–40 °C. The relatively low unfolding Tm (∼40 °C) and subsequent precipitation of Tsr does not allow for this experiment to definitively distinguish whether the RNA tertiary structure is stabilized or destabilized. However, it can nonetheless be concluded that the stability of the 58-nt RNA tertiary structure is indeed altered in a Tsr concentration-dependent manner. Together, the hydroxyl radical cleavage enhancements and UV melting experiments in the presence of Tsr provide strong evidence that Tsr directly perturbs the RNA tertiary structure upon binding.
FIGURE 4.
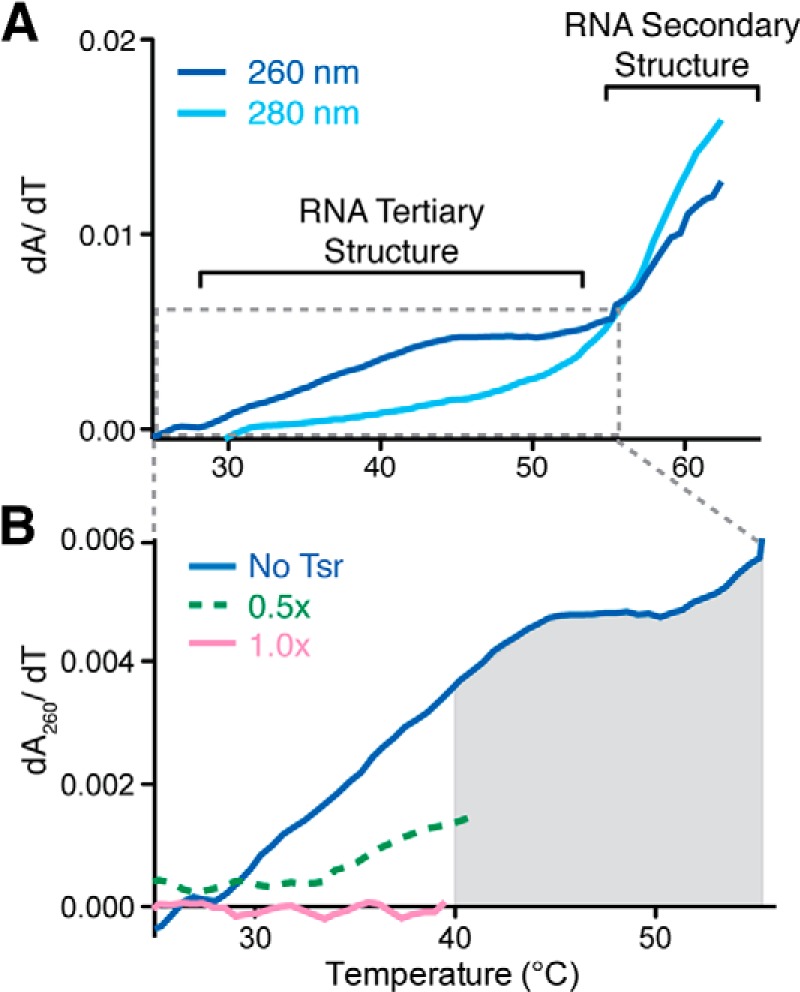
UV melting of the wild-type 58-nt RNA with and without full-length Tsr. A, melting profile of the 58-nt RNA at 260 (solid line) and 280 nm (dashed line). B, RNA melting profiles at 260 nm in the presence of 0 (solid blue line), 0.5 (dashed green line), and 1:1 (solid pink line) molar equivalents of Tsr. Tsr precipitation is denoted by the gray shaded region of the plot.
RNA Conformational Changes Induced by Tsr Are Dependent on Its NTD
To examine the Tsr-induced RNA conformational changes in more detail, we assessed the relative sensitivities to RNases V1, T1, and A of the wild-type 58-nt RNA in isolation and when complexed with either full-length Tsr or Tsr-CTD (Fig. 5). These enzymes preferentially cleave the RNA sugar-phosphate backbone at double-stranded/stacked nucleotides, single-stranded G, or single-stranded C/U nucleotides, respectively.
FIGURE 5.

RNase probing identifies RNA structural changes and cleavage protections induced upon binding full-length Tsr or Tsr-CTD. A, representative RNase structure probing gel of 58-nt RNA only, and complexes of 58-nt RNA with full-length Tsr or Tsr-CTD. Untreated RNA (U) and partial alkaline hydrolysis nucleotide ladder (AH) are also shown for nucleotide identification (numbering noted on left). Sites of cleavage are noted for RNase V1 (squares), RNase A (circles), RNase T1 (diamonds). Changes in sensitivity to RNases (boxed on the gel) are mapped as protections (blue) and enhancements (orange) onto B, the 58-nt RNA secondary structure, and C, two views of x-ray crystal structure (PDB ID 1HC8) related by a 90° rotation around the vertical axis.
With the exception of G1062, the wild-type 58-nt RNA is almost entirely resistant to RNase V1 cleavage (Fig. 5A, lane 3). Although much of the RNA is base paired, this result is not entirely unexpected given the highly complex nature of the RNA tertiary structure and distortion of helices from regular A-form RNA (31). In complex with Tsr, new strong cleavages are observed at nucleotides 1078–1080, whereas G1062 is protected (Fig. 5A, compare lanes 3 and 4). Protections from RNase cleavage could be due to physical occlusion of the RNase from its target site (analogous to protections from hydroxyl radicals) or, alternatively, Tsr-induced alteration of the RNA structure so that it no longer meets the RNase substrate specification. In the case of G1062, which was also protected from hydroxyl radical cleavage (Fig. 3), the observed protection further confirms the surrounding region as part of the direct Tsr binding surface. The strongly enhanced RNase V1 cleavage of nucleotides 1078–1080 is unexpected and particularly noteworthy as it indicates that these nucleotides are substantially remodeled, presumably becoming more base-stacked, upon Tsr binding. Mapping the changes induced by full-length Tsr in RNase V1 sensitivities onto the 58-nt RNA structure (PDB code 1HC8) reveals that they cluster around an unusual RNA backbone conformation (Fig. 5C). Given their colocalization around this structurally unique region, protection of G1062 and enhancement of RNase V1 cleavage at 1078–1080 likely arise in concert through their interaction with a specific region of Tsr.
In contrast to the observed changes upon full-length Tsr binding, Tsr-CTD fails to induce the same pattern of changes in sensitivity to RNase V1 despite maintaining interaction with the RNA. Tsr-CTD neither protects G1062 nor induces the strong new cleavage sites at 1078–1080 (Fig. 5A, lanes 3 and 5). This result indicates that the Tsr NTD is in proximity of G1062 and directly implicates this domain as the primary driver of the conformational changes induced in the RNA.
RNase A treatment of the 58-nt RNA revealed six sites of sensitivity at nucleotides U1066, U1072, U1079, U1083, U1094, and U1097 (Fig. 5A, lane 6). Binding of full-length Tsr protects U1066 within the target loop, enhances cleavage of U1083 at the central helical junction, but has no effect on the sensitivities of U1072, U1079, U1094, and U1097 to RNase A cleavage (Fig. 5A, lanes 6 and 7). In contrast, binding of Tsr-CTD conferred only partial protection of U1066 and failed to induce enhancement of cleavage at U1083 (Fig. 5A, lanes 6 and 8). These results indicate, as might be expected given the location of the Tsr catalytic centers, that the CTD binds at the A1067 target loop directly occluding U1066 from cleavage in both the full-length and Tsr-CTD complexes. Additionally, the specificity of enhanced RNase A cleavage at U1083 within the helical junction to full-length Tsr provides further evidence, correlating with the hydroxyl radical probing results, that the structure of this region of the RNA is modified upon Tsr binding in a process dependent on the Tsr NTDs.
Subjecting the same samples to RNase T1 treatment revealed a similar pattern. The 58-nt RNA in isolation was cleaved three times, at residues G1068, G1071, and G1087 (Fig. 5A, lane 9). Sensitivity at all three sites was reduced by binding of full-length Tsr, whereas only G1087 was protected by the Tsr-CTD (Fig. 5A, lanes 9–11). In the tertiary structure of the RNA, G1087 is adjacent to 1078–1080 where cleavage was enhanced when probed with RNase V1 (Fig. 5C). This protection could be due to the CTD blocking RNase A from accessing this nucleotide. Interestingly, whereas G1068 is immediately adjacent to the target nucleotide A1067, it is only strongly protected by full-length Tsr, suggesting that its environment and/or conformation is only significantly changed in a catalytically competent complex and this change in the target loop is dependent on the distal interactions made by the Tsr-NTDs.
In summary, RNase probing of the isolated 58-nt RNA and its complexes with full-length Tsr or Tsr-CTD have identified unique RNA structural changes that occur upon Tsr binding and clearly identify the Tsr NTDs as the primary drivers of the RNA structural rearrangements necessary for specific recognition and methylation of A1067 by Tsr.
Stabilizing the 58-nt RNA Tertiary Structure Does Not Interfere with RNA Conformational Changes Induced by RNase V1
UV melting analysis and both hydroxyl radical and RNase structure probing indicate that binding of full-length Tsr induces RNA conformational changes, most likely partial unfolding of the tertiary structure. We next asked, whether a point mutation (U1061A) known to specifically stabilize the 58-nt RNA tertiary structure (>10 °C to Tm ∼58 °C) (32) and to reduce Tsr activity (20, 21) effects methylation by blocking this unfolding event. We confirmed that Tsr activity was substantially reduced against the U1061A RNA correlating with a reduced binding affinity of 1.56 ± 0.33 μm, ∼10-fold weaker than for the wild-type 58-nt RNA (Table 1, Fig. 6, A and B).
FIGURE 6.

Stabilizing the RNA tertiary structure decreases Tsr binding affinity and catalytic activity independent of RNA structural changes. A, FP binding analysis of U1061A RNA and full-length (FL) Tsr interaction. B, relative methylation activity of full-length Tsr on the wild-type (WT) and U1061A mutant 58-nt RNAs. C, representative RNase structure probing gel of U1061A RNA in the absence and presence of full-length Tsr. Untreated RNA (U) and partial alkaline hydrolysis nucleotide ladder (AH) are also shown for nucleotide identification (numbering noted on left). Sites of cleavage are noted for RNase V1 (squares), RNase A (circles), and RNase T1 (diamonds); cleavage sites unique to the U1061A mutant are shown as outlined symbols. Changes in sensitivity to RNase are mapped as protections (blue) and enhancements (orange) onto the 58-nt RNA. D, secondary structure, and E, x-ray crystal structure (PDB ID 1HC8).
To investigate whether Tsr is still able to induce structural changes in the U1061A RNA, we probed the isolated RNA and its complex with full-length Tsr with RNases as before. U1061A RNA is generally a more sensitive RNase V1 with weak cleavages throughout both strands of H43, presumably because this helix is more ordered in this RNA tertiary structure stabilized mutant. Two pronounced cuts were observed with U1061A alone, G1062 and U1082, the latter unique to U1061A (Fig. 6C, lane 3). When bound by Tsr, both G1062 and U1082 were protected from cleavage, whereas strong enhancements were also observed at U1078-A1080 (Fig. 6C, lane 4), as seen with the wild-type 58-nt RNA. These strong cleavage enhancements at nucleotides 1078–1080 demonstrate that Tsr is still able to change the tertiary structure of this stabilized RNA. The pattern of U1061A RNA sensitivity to RNase T1 was also very similar to the wild-type 58-nt RNA with three strong cleavages at G1067, G1071, and G1087. As for wild-type 58-nt RNA, these cleavages were protected in the presence of full-length Tsr (Fig. 6C, lanes 7 and 8).
When U1061A RNA was probed with RNase A, eight sites of strong strand scission were observed at U1060, U1066, C1072, C1075, C1079, U1083, U1094, and U1097. The cleavages at U1060, C1072, C1075, and C1079 are much stronger in U1061A RNA (Fig. 6C, lane 5) compared with the wild-type 58-nt RNA, probably because these nucleotides are stabilized within this deformed A-helix and better recognized by RNase A. In the presence of full-length Tsr, U1066 and U1094 are protected from cleavage (Fig. 6C, lanes 5 and 6), the latter unique to U1061A RNA. These protections of U1066 and U1094 are most likely due to direct occlusion of the RNase by Tsr as they are near the target nucleotide (Fig. 6, D and E). In contrast, the cleavages at C1072, C1075, U1083, and C1079 were enhanced, most likely due to the unfolding of the RNA structure in the presence of Tsr.
These data identify nucleotides with differing environments between the wild-type and U1061A mutant 58-nt RNAs that result in partially different RNase cleavage sensitivities. In addition, these data also clearly demonstrate that full-length Tsr can induce similar RNA tertiary conformational changes. However, the energetic penalty associated with unfolding this stabilized structure, reflected in the ∼10-fold weaker binding affinity, most likely results in the dramatically reduced methylation efficiency compared with the wild-type 58-nt RNA.
Tsr Mutants R162A and R26A Discriminate between RNA Binding and Induced Conformational Changes Necessary for Catalysis
We sought to identify key Tsr amino acids responsible for driving the RNA tertiary structure unfolding through mutagenesis and subsequent analysis of the ability of the mutant protein to induce the RNase V1-dependent cleavage of nucleotides 1078–1080. Potential targets for mutation in Tsr were selected based on their conservation among SPOUT family members with an L30e-like NTD and by inspection of their location in the modeled Tsr·58-nt RNA complex (20). Seven single alanine point mutations were created, each resulting in a mutant enzyme containing two alanine mutations, one on each protomer of the Tsr homodimer. Each mutant protein was expressed, found to be soluble and purified as for Tsr. Two arginine to alanine Tsr mutants, R26A and R162A (Fig. 7, A and B), were found to be deficient in their ability to promote strongly enhanced RNase V1 cleavage of nucleotides 1078–1080 in the wild-type 58-nt RNA despite retaining the native Tsr fold as assessed by partial proteolysis (Fig. 7, C and D). These two Tsr mutants were therefore selected for further analysis.
FIGURE 7.
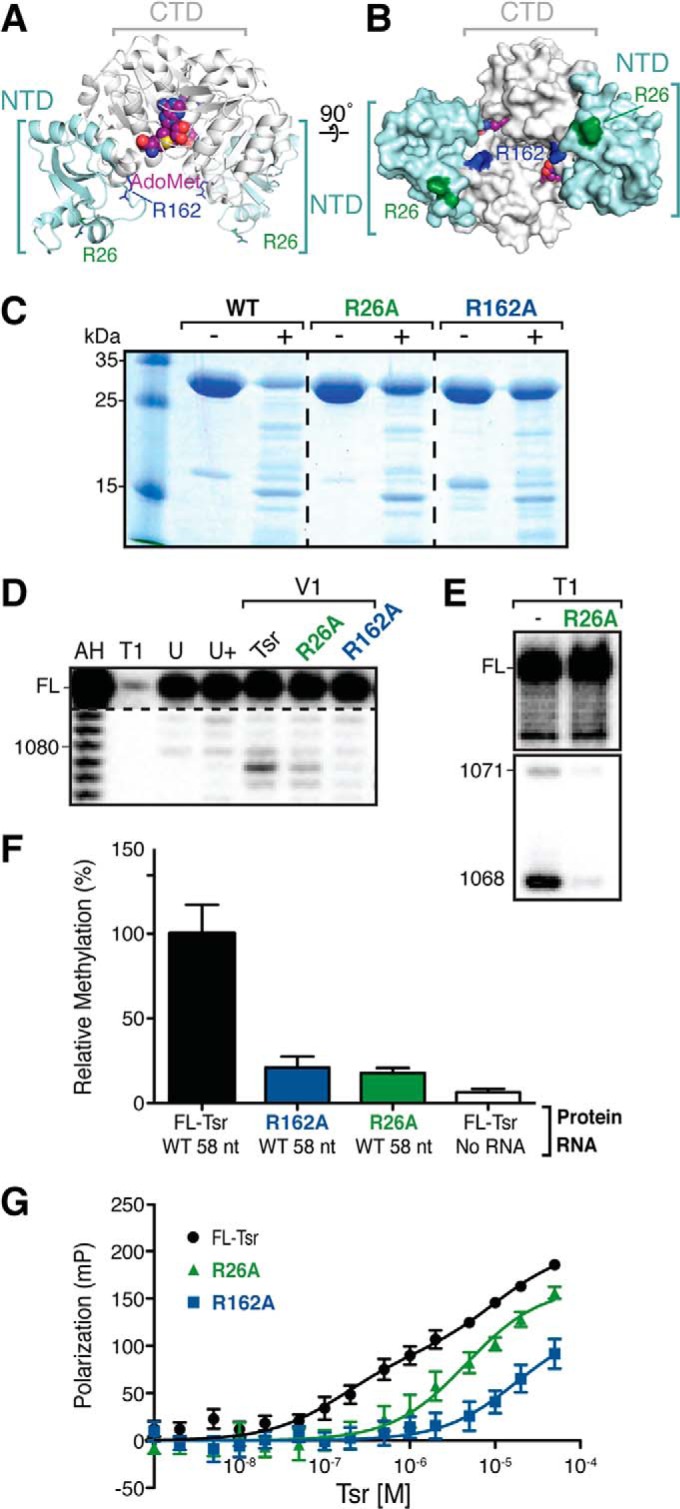
Tsr-NTD modification of the RNA structure is necessary for catalysis. A, schematic representation of Tsr-AdoMet complex (PDB ID 3GYQ) with sites of mutation indicated as sticks: Arg-26 (green) and Arg-162 (blue). The AdoMet co-substrate (magenta) is also shown. B, surface representation of the same complex rotated 90° around the horizontal axis. The color scheme is the same as in panel A. C, SDS-PAGE analysis of protein samples following incubation without (−) and with (+) chymotrypsin. Partial proteolytic cleavage of wild-type and mutant full-length Tsr proteins demonstrates that each adopts the same, native fold. D, RNase V1 probing of the wild-type 58-nt RNA in complex with wild-type and each mutant Tsr. Additional lanes are: partial alkaline hydrolysis nucleotide ladder (AH), denaturing RNase T1 digest (T1), untreated RNA (U), and Tsr-RNA complex with no added RNase V1 (U+). E, RNase T1 sensitivity of two cleavage sites within the 58-nt RNA in the absence (−) or presence of Tsr-R26A mutant. F, relative enzymatic activity, and G, FP binding analysis of the indicated Tsr mutants with wild-type 58-nt RNA.
We tested whether these mutations disrupted binding to and/or methylation of the wild-type 58-nt RNA (Fig. 7, F and G). Using the FP binding assay as before, the Tsr-R162A CTD mutant was found to bind with ∼100-fold weaker affinity than wild-type Tsr, with a KD of 18 ± 9.4 μm (close to the limit of measurement in this assay; Table 1). Thus, the R162A mutation dramatically reduces RNA binding, resulting in an ∼3-fold weaker binding affinity than for Tsr-CTD, which lacks the entire NTD. Methylation by Tsr-R162A was effectively ablated and comparable with background levels, correlating with the weakened affinity for the substrate. Because the RNA binding ability of Tsr-R162A is disrupted, this mutant cannot unfold the RNA structure. These data further implicate the CTD in an initial RNA docking event prior to the engagement of the NTDs, and demonstrate the critical role of Arg-162 in this process.
Binding of the Tsr-R26A NTD mutant to wild-type 58-nt RNA was also reduced but more modestly with a ∼30-fold weaker KD (4.6 ± 1.1 μm), comparable with the affinity for Tsr-CTD (Table 1, Fig. 7G). Although Tsr-R26A is unable to promote the RNase V1-sensitive RNA structural changes (Fig. 7D), this mutant maintains its interaction with the RNA under the conditions used as demonstrated by the protection of nucleotides G1068 and G1071 from cleavage by RNase T1 (Fig. 7E). Correlating with the lack of structural rearrangement, methylation of the wild-type 58-nt RNA by Tsr-R26A was reduced to near background levels. These data demonstrate the critical importance of residue Arg-26 for interaction of the Tsr NTDs with the 58-nt RNA and specifically in promoting the RNA structural conformational changes necessary for activation of catalysis. Furthermore, these two mutants establish a decoupling between the CTD docking on the RNA target loop and distal RNA structural changes induced by the NTD necessary for catalysis.
DISCUSSION
Modifications of rRNA are important for translational fitness and resistance against ribosome-targeting antibiotics. However, there is limited knowledge about how the enzymes responsible for their incorporation recognize their modification targets. We therefore sought to biochemically elucidate how the resistance conferring enzyme Tsr recognizes its substrate rRNA domain. Our data show that the Tsr NTD in isolation does not bind the RNA, but must be delivered to the RNA via the Tsr-CTD-RNA interaction to achieve a specific, high affinity complex. An essential part of the substrate recognition mechanism for Tsr involves the precise reorganization of the RNA tertiary and secondary structures coordinated by the accessory NTD in a process that is indispensable for catalysis in the distant CTD active site. Collectively, our results suggest a two-step model for substrate recognition (Fig. 8) and a direct role for the rRNA in control of Tsr activity on its substrate.
FIGURE 8.
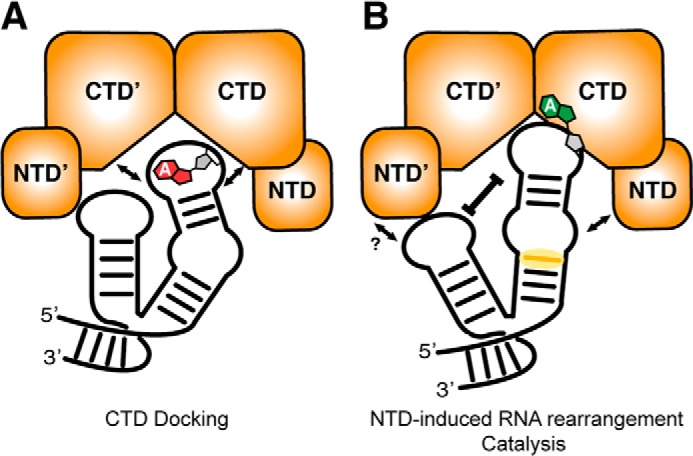
Model of the Tsr substrate recognition mechanism. A, the Tsr CTD mediates initial docking on the RNA substrate, with binding driven largely by the critical residue Arg-162. B, once bound, the Tsr NTDs are engaged and induce a rearrangement of the RNA structure (yellow shaded region is the most pronounced RNase V1 enhancement) that is signaled to the CTD to stimulate catalysis.
Tsr is a member of the SPOUT family of methyltransferases. These enzymes contain a common SPOUT domain catalytic core that may be decorated by several different RNA binding structural elements, ranging from a few helices as in TrmH (tRNA Gm18) (33, 34) or larger structural domains like the L30e domain of Tsr or PUA domain of RsmE (16 S rRNA m3U1498) (35, 36). Our results showed that without its N-terminal RNA binding domain, Tsr is catalytically inactive despite retaining an ability to bind the RNA substrate with ∼30-fold weaker affinity than the full-length enzyme. Such intrinsic RNA binding ability is also exhibited by the SPOUT family tRNA methyltransferase TrmL (formerly YibK; tRNA Um34/Cm34), which does not contain an accessory RNA-binding domain but instead interacts with its tRNA substrate via a flexible, positively charged patch of amino acids located on the non-catalytic protomer near the active site (27). The initial step of Tsr-CTD docking on its RNA substrate proposed in our model for Tsr action is thus reminiscent of the ability of this minimal SPOUT methyltransferase to bind its tRNA substrate, despite Tsr containing a defined RNA-binding domain.
Although the Tsr CTD binds RNA, it is remarkable that a single point mutation in the CTD, R162A, weakens the RNA binding affinity 500-fold compared with full-length Tsr. This residue is absolutely conserved in the closely related nosiheptide resistance methyltransferase (Nhr; 23 S rRNA Am1067) and the avilamycin-resistance conferring methyltransferase AviRb (23 S rRNA Um2479), and functionally conserved in other SPOUT family members (8, 37). The dramatic decrease in binding affinity is readily rationalized by examining the position of each Arg-162 residue in the dimeric Tsr structure. Each Arg-162 is solvent exposed and positioned ∼20 Å apart on opposite sides of a cleft made by the anti-parallel orientation of each Tsr CTD where the RNA must bind to access the AdoMet co-substrate. Therefore, the alanine mutation disrupts two essential interactions with the RNA that may be essential for recognizing the target loop backbone geometry.
Our studies have revealed that the NTD of Tsr is an essential component of the RNA substrate recognition mechanism by both promoting high affinity RNA binding and activation of catalysis by the CTD. In the absence of the NTD, Tsr is catalytically inactive despite possessing the intact AdoMet binding sites and catalytic center. The Tsr-CTD dimer binds the RNA ∼30-fold more weakly than the wild-type enzyme and, most critically, is unable to promote the NTD-dependent RNA conformational change. The Tsr NTD is structurally similar to the yeast ribosomal protein L30e, which binds both yeast 26 S rRNA (nucleotides 1711–1733) and its own pre-mRNA for transcriptional autoregulation. Both RNA domains contain a unique RNA structural motif known as a kink-turn (38). Although not geometrically identical, the 58-nt RNA also contains an unusual backbone secondary structure that reverses the RNA backbone in a similar way. Despite these structural similarities in both protein and RNA target, the Tsr-NTD in isolation showed no detectable affinity for the 58-nt RNA domain. We conclude, therefore that the weak intrinsic affinity of the CTD is essential for it to deliver and correctly position the Tsr NTDs for them to productively contribute to high affinity, specific recognition of the RNA substrate. This coordinated binding of the CTD and NTD positioning may allow for potential cross-talk between the two domains to relay a signal to perform catalysis.
Tsr recognition and subsequent methylation of its substrate requires significant perturbations in the 58-nt RNA structure. Hydroxyl radical and RNase structure probing of the Tsr-RNA complex revealed unique cleavage enhancements throughout H43 distant from the target loop, suggesting that the RNA is being unfolded, presumably to allow for proper orientation of A1067 into the catalytic center. The most significant specific structural change occurs at nucleotides 1078–1080 and is driven by the Tsr NTD. We have identified arginine 26 as a critical residue for NTD-mediated RNA recognition and the RNase V1-sensitve RNA structural change. This amino acid is conserved in the L30e-SPOUT methyltransferases AviRb, Nhr, RrmA (23 S rRNA m1G745) and RlmB (23 S rRNA Gm2251). In a previously published model of the Tsr·58-nt RNA complex, Arg-26 is positioned near U1058 (20). Given its sequence conservation, potential role recognizing the rRNA, and demonstrated importance in promoting the RNase V1-sensitive structural change, we propose that Arg-26 directly recognizes RNA nucleotides near 1058–1060. In doing so, this residue enhances base stacking of nucleotides on the opposite strand (1078–1080), increasing their sensitivity to RNase V1. As the catalytic activity of the Tsr-R26A mutant is drastically decreased compared with wild-type, we hypothesize that this RNA structural reorganization at the base of H43 may be relayed to induce changes in the target loop necessary for proper orientation of A1067 into the active site of Tsr. In further support of this concept is the observation that the RNase T1 protection of the adjacent nucleotide, G1068, is also dependent on the presence of the Tsr NTD.
There is precedent for the direct involvement of RNA structure and structural reorganization in the process of substrate recognition by rRNA modification enzymes. The crystal structure of RlmD (formerly RumA; 23 S rRNA m5U1939) in complex with a 29-nt model RNA (nucleotides 1932–1961) revealed that flipping of U1939 into the RlmD catalytic site was facilitated and stabilized by a major reorganization of the RNA (39). Additionally, two nucleotides fill the space the flipped target nucleotide 1939 would otherwise occupy, stabilizing the target nucleotide in the catalytic pocket. In contrast, the crystal structure of the aminoglycoside-resistance conferring enzyme NpmA (16 S rRNA m1A1408)·30 S ribosomal subunit complex showed minimal RNA structural changes induced upon recognition, with the striking exception of the flipping of the target nucleotide out of its RNA helix (40). Perhaps, as RlmD and Tsr recognize rRNA prior to subunit maturation, their RNA substrates are structurally more plastic and thus reorganization of the RNA is an effective mechanism to accomplish the necessary level of discrimination during substrate recognition.
Acknowledgments
We are grateful to Dr. Christine Dunham for critical reading of this manuscript and members of the Conn and Dunham laboratories for many useful discussions during the course of this work.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-AI088025 and T32-GM008367.
- AdoMet
- S-adenosyl-l-methionine
- CTD
- carboxyl-terminal domain
- FL-Tsr
- full-length Tsr protein
- FP
- fluorescence polarization
- HDV
- hepatitis δ virus (ribozyme RNA)
- NTD
- amino-terminal domain
- SPOUT
- SpoU/TrmD (methyltransferase enzyme family)
- Tsr-NTD
- isolated NTD protein
- Tsr-CTD
- isolated CTD protein
- nt
- nucleotide
- AH
- alkaline hydrolysis
- PDB
- Protein Data Bank.
REFERENCES
- 1. O'Dwyer K., Watts J. M., Biswas S., Ambrad J., Barber M., Brulé H., Petit C., Holmes D. J., Zalacain M., Holmes W. M. (2004) Characterization of Streptococcus pneumoniae TrmD, a tRNA methyltransferase essential for growth. J. Bacteriol. 186, 2346–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Burakovsky D. E., Prokhorova I. V., Sergiev P. V., Milón P., Sergeeva O. V., Bogdanov A. A., Rodnina M. V., Dontsova O. A. (2012) Impact of methylations of m2G966/m5C967 in 16 S rRNA on bacterial fitness and translation initiation. Nucleic Acids Res. 40, 7885–7895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Connolly K., Rife J. P., Culver G. (2008) Mechanistic insight into the ribosome biogenesis functions of the ancient protein KsgA. Mol. Microbiol. 70, 1062–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wilson D. N. (2014) Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 12, 35–48 [DOI] [PubMed] [Google Scholar]
- 5. Cundliffe E. (1989) How antibiotic-producing organisms avoid suicide. Annu. Rev. Microbiol. 43, 207–233 [DOI] [PubMed] [Google Scholar]
- 6. Machnicka M. A., Milanowska K., Osman Oglou O., Purta E., Kurkowska M., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K. M., Helm M., Bujnicki J. M., Grosjean H. (2013) MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res. 41, D262–D267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Watanabe K., Nureki O., Fukai S., Ishii R., Okamoto H., Yokoyama S., Endo Y., Hori H. (2005) Roles of conserved amino acid sequence motifs in the SpoU (TrmH) RNA methyltransferase family. J. Biol. Chem. 280, 10368–10377 [DOI] [PubMed] [Google Scholar]
- 8. Yang H., Wang Z., Shen Y., Wang P., Jia X., Zhao L., Zhou P., Gong R., Li Z., Yang Y., Chen D., Murchie A. I., Xu Y. (2010) Crystal structure of the nosiheptide-resistance methyltransferase of Streptomyces actuosus. Biochemistry 49, 6440–6450 [DOI] [PubMed] [Google Scholar]
- 9. Cundliffe E. (1978) Mechanism of resistance to thiostrepton in the producing-organism Streptomyces azureus. Nature 272, 792–795 [DOI] [PubMed] [Google Scholar]
- 10. Bhat U. G., Halasi M., Gartel A. L. (2009) Thiazole antibiotics target FoxM1 and induce apoptosis in human cancer cells. PLoS One 4, e5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hegde N. S., Sanders D. A., Rodriguez R., Balasubramanian S. (2011) The transcription factor FOXM1 is a cellular target of the natural product thiostrepton. Nature chemistry 3, 725–731 [DOI] [PubMed] [Google Scholar]
- 12. Aminake M. N., Schoof S., Sologub L., Leubner M., Kirschner M., Arndt H. D., Pradel G. (2011) Thiostrepton and derivatives exhibit antimalarial and gametocytocidal activity by dually targeting parasite proteasome and apicoplast. Antimicrob. Agents Chemother. 55, 1338–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thompson J., Cundliffe E., Stark M. (1979) Binding of thiostrepton to a complex of 23 S rRNA with ribosomal protein L11. Eur. J. Biochem. 98, 261–265 [DOI] [PubMed] [Google Scholar]
- 14. Ryan P. C., Lu M., Draper D. E. (1991) Recognition of the highly conserved GTPase center of 23 S ribosomal RNA by ribosomal protein L11 and the antibiotic thiostrepton. J. Mol. Biol. 221, 1257–1268 [DOI] [PubMed] [Google Scholar]
- 15. Cundliffe E., Thompson J. (1979) Ribose methylation and resistance to thiostrepton. Nature 278, 859–861 [DOI] [PubMed] [Google Scholar]
- 16. Cameron D. M., Thompson J., Gregory S. T., March P. E., Dahlberg A. E. (2004) Thiostrepton-resistant mutants of Thermus thermophilus. Nucleic Acids Res. 32, 3220–3227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wienen B., Ehrlich R., Stöffler-Meilicke M., Stöffler G., Smith I., Weiss D., Vince R., Pestka S. (1979) Ribosomal protein alterations in thiostrepton- and micrococcin-resistant mutants of Bacillus subtilis. J. Biol. Chem. 254, 8031–8041 [PubMed] [Google Scholar]
- 18. Thompson J., Schmidt F., Cundliffe E. (1982) Site of action of a ribosomal RNA methylase conferring resistance to thiostrepton. J. Biol. Chem. 257, 7915–7917 [PubMed] [Google Scholar]
- 19. Anantharaman V., Koonin E. V., Aravind L. (2002) SPOUT: a class of methyltransferases that includes spoU and trmD RNA methylase superfamilies, and novel superfamilies of predicted prokaryotic RNA methylases. J. Mol. Microbiol. Biotechnol. 4, 71–75 [PubMed] [Google Scholar]
- 20. Dunstan M. S., Hang P. C., Zelinskaya N. V., Honek J. F., Conn G. L. (2009) Structure of the thiostrepton resistance methyltransferase·S-adenosyl-l-methionine complex and its interaction with ribosomal RNA. J. Biol. Chem. 284, 17013–17020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bechthold A., Floss H. G. (1994) Overexpression of the thiostrepton-resistance gene from Streptomyces azureus in Escherichia coli and characterization of recognition sites of the 23S rRNA A1067 2′-methyltransferase in the guanosine triphosphatase center of 23 S ribosomal RNA. Eur. J. Biochem. 224, 431–437 [DOI] [PubMed] [Google Scholar]
- 22. Chen G. J., Qiu N., Karrer C., Caspers P., Page M. G. (2000) Restriction site-free insertion of PCR products directionally into vectors. BioTechniques 28, 498–500, 504–5 [DOI] [PubMed] [Google Scholar]
- 23. Linpinsel J. L., Conn G. L. (2012) General protocols for preparation of plasmid DNA template, RNA in vitro transcription, and RNA purification by denaturing PAGE. Methods Mol. Biol. 941, 43–58 [DOI] [PubMed] [Google Scholar]
- 24. Dunstan M. S., Guhathakurta D., Draper D. E., Conn G. L. (2005) Coevolution of protein and RNA structures within a highly conserved ribosomal domain. Chem. Biol. 12, 201–206 [DOI] [PubMed] [Google Scholar]
- 25. Draper D. E., Bukhman Y. V., Gluick T. C. (2001) Thermal methods for the analysis of RNA folding pathways. Curr. Protoc. Nucleic Acid Chem. Chapter 11, Unit 11.3 [DOI] [PubMed] [Google Scholar]
- 26. Zearfoss N. R., Ryder S. P. (2012) End-labeling oligonucleotides with chemical tags after synthesis. Methods Mol. Biol. 941, 181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu R. J., Zhou M., Fang Z. P., Wang M., Zhou X. L., Wang E. D. (2013) The tRNA recognition mechanism of the minimalist SPOUT methyltransferase, TrmL. Nucleic Acids Res. 41, 7828–7842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Conn G. L., Gittis A. G., Lattman E. E., Misra V. K., Draper D. E. (2002) A compact RNA tertiary structure contains a buried backbone-K+ complex. J. Mol. Biol. 318, 963–973 [DOI] [PubMed] [Google Scholar]
- 29. Laing L. G., Gluick T. C., Draper D. E. (1994) Stabilization of RNA structure by Mg ions: specific and non-specific effects. J. Mol. Biol. 237, 577–587 [DOI] [PubMed] [Google Scholar]
- 30. Xing Y., Draper D. E. (1995) Stabilization of a ribosomal RNA tertiary structure by ribosomal protein L11. J. Mol. Biol. 249, 319–331 [DOI] [PubMed] [Google Scholar]
- 31. Conn G. L., Draper D. E., Lattman E. E., Gittis A. G. (1999) Crystal structure of a conserved ribosomal protein-RNA complex. Science 284, 1171–1174 [DOI] [PubMed] [Google Scholar]
- 32. Lu M., Draper D. E. (1994) Bases defining an ammonium and magnesium ion-dependent tertiary structure within the large subunit ribosomal RNA. J. Mol. Biol. 244, 572–585 [DOI] [PubMed] [Google Scholar]
- 33. Tkaczuk K. L., Dunin-Horkawicz S., Purta E., Bujnicki J. M. (2007) Structural and evolutionary bioinformatics of the SPOUT superfamily of methyltransferases. BMC Bioinformatics 8, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nureki O., Watanabe K., Fukai S., Ishii R., Endo Y., Hori H., Yokoyama S. (2004) Deep knot structure for construction of active site and cofactor binding site of tRNA modification enzyme. Structure 12, 593–602 [DOI] [PubMed] [Google Scholar]
- 35. Zhang H., Wan H., Gao Z. Q., Wei Y., Wang W. J., Liu G. F., Shtykova E. V., Xu J. H., Dong Y. H. (2012) Insights into the catalytic mechanism of 16 S rRNA methyltransferase RsmE (m3U1498) from crystal and solution structures. J. Mol. Biol. 423, 576–589 [DOI] [PubMed] [Google Scholar]
- 36. Forouhar F., Shen J., Xiao R., Acton T. B., Montelione G. T., Tong L. (2003) Functional assignment based on structural analysis: crystal structure of the yggJ protein (HI0303) of Haemophilus influenzae reveals an RNA methyltransferase with a deep trefoil knot. Proteins 53, 329–332 [DOI] [PubMed] [Google Scholar]
- 37. Mosbacher T. G., Bechthold A., Schulz G. E. (2005) Structure and function of the antibiotic resistance-mediating methyltransferase AviRb from Streptomyces viridochromogenes. J. Mol. Biol. 345, 535–545 [DOI] [PubMed] [Google Scholar]
- 38. Klein D. J., Schmeing T. M., Moore P. B., Steitz T. A. (2001) The kink-turn: a new RNA secondary structure motif. EMBO J. 20, 4214–4221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee T. T., Agarwalla S., Stroud R. M. (2005) A unique RNA Fold in the RumA-RNA-cofactor ternary complex contributes to substrate selectivity and enzymatic function. Cell 120, 599–611 [DOI] [PubMed] [Google Scholar]
- 40. Dunkle J. A., Vinal K., Desai P. M., Zelinskaya N., Savic M., West D. M., Conn G. L., Dunham C. M. (2014) Molecular recognition and modification of the 30 S ribosome by the aminoglycoside-resistance methyltransferase NpmA. Proc. Natl. Acad. Sci. U.S.A. 111, 6275–6280 [DOI] [PMC free article] [PubMed] [Google Scholar]