

Background: The EGO complex activates yeast TORC1 and is somehow required for recovery from rapamycin, a potent inhibitor of TORC1.
Results: Rapamycin-insensitive activity of TORC1 partly depends on the EGO complex and supports residual cell proliferation that dilutes the metabolically stable drug among progeny cells.
Conclusion: TORC1 activity is required to dilute rapamycin.
Significance: Rapamycin only partly inhibits yeast TORC1.
Keywords: Cell Signaling, Drug Metabolism, Enzyme Inhibitor, TOR Complex (TORC), Yeast, EGO Complex, Proliferation-dependent Dilution, Rapamycin, Rapamycin-insensitive Function, Yeast
Abstract
The target of rapamycin complex 1 (TORC1) is a key conserved regulator of eukaryotic cell growth. The xenobiotic rapamycin is a potent inhibitor of the yeast complex. Surprisingly, the EGO complex, a nonessential in vivo activator of TORC1, is somehow required for yeast cells to recover efficiently from a period of treatment with rapamycin. Why? Here, we found that rapamycin is only a partial inhibitor of TORC1. We confirmed that saturating amounts of rapamycin do not fully inhibit proliferation of wild-type cells, and we found that the residual proliferation in the presence of the drug is dependent on the EGO complex and on the activity of TORC1. We found that this residual TORC1-dependent proliferation is key to recovery from rapamycin treatment. First, the residual proliferation rate correlates with the ability of cells to recover from treatment. Second, the residual proliferation rate persists long after washout of the drug and until cells recover. Third, the total observable pool of cell-associated rapamycin is extremely stable and decreases only with increasing cell number after washout of the drug. Finally, consideration of the residual proliferation rate alone accurately and quantitatively accounts for the kinetics of recovery of wild-type cells and for the nature and severity of the ego− mutant defect. Overall, our results revealed that rapamycin is a partial inhibitor of yeast TORC1, that persistence of the drug limits recovery, and that rapamycin is not detoxified by yeast but is passively diluted among progeny cells because of residual proliferation.
Introduction
Rapamycin, a macrocyclic lactone, inhibits the target of rapamycin complex 1 (TORC1),2 a highly conserved and central regulator of cell growth in eukaryotes (1). Yeast TORC1 is essential for cell growth and contains a core Tor1 protein kinase (which can be substituted by the partly redundant Tor2), the yeast Raptor homolog Kog1, Lst8, and the nonessential Tco89 (2, 3).
Rapamycin is a noncompetitive inhibitor of TORC1 and acts by first binding to the cis-trans-prolylisomerase FKBP (FKBP12 in humans and Fpr1 in yeast) (4). This binary rapamycin-FKBP complex then binds directly to the FRB domain of the Tor kinase in the TORC1 complex and thereby inhibits its activity (5). The potency of rapamycin as an allosteric inhibitor of TORC1 seems to vary across species. The drug is thought to fully inactivate the budding yeast TORC1, with rapamycin treatment driving cells into a quiescent/G0 state (6). By contrast, rapamycin only partially inactivates mTORC1 in mammalian cells (7, 8) and TORC1 of Schizosaccharomyces pombe (9, 10); the complexes are essential in both species, but treatment with rapamycin results in only slowed cell proliferation or has no effect on proliferation, respectively. The basis for this variation in potency of rapamycin is not understood.
Does rapamycin fully inhibit yeast TORC1? The answer is not known. It is clear that rapamycin does not fully inhibit proliferation of wild-type yeast cells, even when present at saturating concentrations (up to 50 ng/ml), i.e. concentrations in excess of the minimum inhibitory concentration (of ∼3–5 ng/ml) and that fully induce phenotypes associated with inactivated TORC1 (11). Rapamycin may fully inhibit TORC1, but where the residual proliferation rate seen in the presence of rapamycin is supported by TORC1-independent mechanisms. Alternatively, residual proliferation may rely on a rapamycin-insensitive function of TORC1, reminiscent of the situation in mammalian cells and in fission yeast.
TORC1 is activated by the presence of nutrients (in yeast and metazoan cells) and also by growth factor signaling (in metazoan cells) (1). How nutrients control yeast TORC1 is poorly understood but somehow involves an upstream activator located at the vacuolar membrane, the EGO complex (12–15). The heterotetrameric EGO complex in yeast includes the GTPases Gtr1 and Gtr2, homologs of the mammalian Rag GTPases (16), and the associated proteins Ego1 and Ego3 (14, 15), potential functional homologs of the mammalian Ragulator complex (16). Surprisingly, the EGO complex is not essential for growth or for TORC1 activity in the presence of nutrients, although subtle defects in nutrient activation of the complex and in sensitivity to rapamycin can be seen in mutants lacking the complex (12, 14, 15). Additional EGO-independent mechanisms must exist for maintaining activity of TORC1 in the presence of nutrients.
Mutants lacking the EGO complex do share a very dramatic phenotype: the inability to recover from a period of exposure to inhibitory concentrations of rapamycin (hence Exit from rapamycin-induced GrOwth arrest (14)). The ego− mutants fail to return to active proliferation upon washout of the drug. The basis for this dramatic recovery defect is not understood. However, some key parameters have been established. First, the ego− mutant cells do not lose viability when treated with rapamycin (11).3 Second, TORC1 activity itself appears to be limiting for recovery from rapamycin treatment. Genetic activation of TORC1 signaling can partly suppress the recovery defect of mutants lacking the EGO complex (12); second site mutations that partially suppress the ego− recovery defect share the ability to increase intracellular glutamine, a metabolite thought to be key to activating TORC1 (14, 17). Mutants lacking Tor1 or Tco89, both nonessential components of TORC1, also show a recovery defect (12, 18). Third, the recovery defect may be selective for, if not specific to, inactivation of TORC1 by rapamycin, because cells lacking the EGO complex recover normally when cultures are grown to stationary phase in synthetic medium, a condition that triggers physiological inactivation of TORC1 (19).
The ego− mutant cells appear to respond normally to rapamycin, entering a state indistinguishable from that of wild-type cells treated with the drug. For example, glycogen storage, autophagy, and expression of key genes are all induced normally in the mutants when treated with the drug (14). Furthermore, phosphorylation of eIF2α (14) and de-phosphorylation of Sch9 also occur equivalently in mutant and wild-type cells when treated (12). The EGO complex thus appears to be specifically required for the transition back to active proliferation.
Here we find that ego− mutants do not respond normally to rapamycin treatment. We uncover the basis for this altered response to the drug, and we show that this difference accounts for the recovery defect of the mutants. We find that saturating amounts of rapamycin inhibit proliferation of ego− mutant cells more profoundly than it does wild-type cells. We find that this difference is due to residual proliferation in saturating amounts of the drug being dependent on a previously unnoticed rapamycin-insensitive activity of TORC1. We find that residual proliferation is key to recovery from the drug. We find that rapamycin is very metabolically stable in yeast; its intracellular concentration decreases slowly with time and only via dilution among progeny cells.
EXPERIMENTAL PROCEDURES
Strains and Media
All strains were derived from the yeast deletion collection in the BY4743 background (20). Haploid cells containing kog1ts as the sole allele were created by transforming kog1Δ/KOG1 diploids with pRS313(kog1–105) (21). After sporulation, haploid kog1Δ pRS313(kog1–105) transformants were isolated by tetrad dissection using a standard protocol.
Strains were grown at room temperature and in standard rich YPD media with 2% glucose unless otherwise stated. For carbon and nitrogen starvation, cells were collected from mid-logarithmic cultures in YPD and resuspended in either YP or SD-N media (Difco) as described previously (22). All chemicals were from Sigma.
Spot Assay for Recovery
Cultures were grown to mid-logarithmic phase, and the densities were normalized to an A600 nm of 0.1 before drug addition. Cells were collected by centrifugation and washed three times in 2× volume of fresh medium before being resuspended in 1× volume of fresh medium. 10-Fold serial dilutions of resuspended cells were spotted (5 μl) onto YPD plates and incubated at 30 °C for 2 days.
Western Blot to Determine Induction of Autophagy
Transformants containing the PGK1-GFP construct (23) were grown to mid-logarithmic phase and 0.25 A600 nm units of cells collected by centrifugation. Proteins were extracted as described in Ref. 23 and separated on a 12% NuPAGE BisTris gel (Invitrogen). Protein separation and transfer to an Immobilon PVDF membrane (Millipore) were carried out according to the recommended protocol (Invitrogen). The Immobilon membrane was blocked in 5% nonfat milk in TBS for 30 min at room temperature with agitation before being exposed to a mouse anti-GFP primary antibody (Santa Cruz Biotechnology) at a 1:100 concentration in 5% nonfat milk/TBS overnight at 4 °C with agitation. The membrane was washed five times in TBS-T for 10 min each then blocked again for 30 min in 5% nonfat milk/TBS at room temperature with agitation. A sheep anti-mouse IgG-HRP antibody (GE Healthcare) was used for the secondary antibody at a concentration of 1:10,000 in 5% nonfat milk/TBS and incubated for 1 h at room temperature with agitation. The membrane was washed five times with TBS-T for 10 min each before being developed using an ECL development kit (GE Healthcare) according to manufacturer's instructions.
Quantifying Cell Number
The number of cells per ml of sonicated culture was quantified using a Z2 Beckman Coulter Counter (High Wycombe, UK) according to the manufacturer's instructions and set for particle sizes between 2.5 and 8.58 μm. Samples were sonicated for 5 s prior to Coulter counting. All cultures were maintained in exponential growth by the addition of the appropriate fresh medium when necessary. Raw Coulter counts were adjusted by any dilution factor as appropriate.
Determining Recovery Time
Cultures were grown to mid-logarithmic phase in YPD, and the densities normalized to an A600 nm of 0.1. Cultures were then treated or not with rapamycin (20–200 ng/ml as appropriate) for 2 h with agitation. Cell pellets were collected by centrifugation, washed three times in 2× volume of fresh medium lacking rapamycin before being resuspended into fresh medium lacking any drug and incubated with agitation. Aliquots were removed with time, and the concentration of cells was determined by Coulter counting as above. The recovery time was taken as the time at which the growth rate of the recovering culture switched from that characteristic of cultures in the continuous presence of rapamycin to that characteristic of untreated cultures. This recovery time was determined by plotting the log2 of cell number per ml as a function of time after washout of the drug and determining by extrapolation when the slope of the straight line semi-log plots switched from that characteristic of treatment with rapamycin to that characteristic of untreated cultures.
Mass Spectrometry Determination of Rapamycin
Cultures were grown to mid-logarithmic phase in YPD with agitation and treated or not with rapamycin (400 ng/ml) for 4 h. Cell pellets equivalent to 10 A600 nm units were collected by centrifugation. The cell pellet was washed three times in ice-cold water (1 ml) before resuspension in ice-cold water (200 μl) and lysed using an equal volume of glass beads by six cycles of vortexing for 30 s followed by 30 s on ice. Rapamycin was extracted into ethyl acetate (250 μl) (Fisher) by vortexing for 30 s prior to centrifugation for 2 min at 16,000 × g and collecting the organic layer; this was repeated five times, and all organic layers were pooled.
Extracted sample (10 μl) was injected into an Ultimate 3000 RLSC system (Thermo Fisher Scientific, Hemel Hempstead, UK) equipped with an Acclaim 5-μm 2.1 × 150-mm C18 column. The separation gradient ran from 5% acetonitrile, 95% water to 50% acetonitrile, 50% water in 20 min, followed by a wash at 95% acetonitrile, 5% water for 6 min, and a 6-min re-equilibration at 95% water, 5% acetonitrile. Mass spectrometry detection was performed on a Q-Exactive (Thermo Fisher Scientific) in negative ionization mode at 70,000 resolution. Identity of rapamycin was confirmed by retention time, mass, and fragment pattern matching to an authentic standard. Quantification was performed using Quan Browser version 2.2 (Thermo Fisher Scientific).
RESULTS
EGO Complex Is Selectively Required for Recovery from Rapamycin
The EGO complex is required for recovery from rapamycin treatment, but not for recovery from growth to saturation in synthetic medium, a condition that inactivates TORC1 (19). To extend this analysis, we assayed the ability of wild-type and congenic ego− cells to recover from a variety of other environmental triggers or chemical treatments that lower TORC1 activity. Liquid cultures of haploid wild-type, ego1Δ, and gtr2Δ cells in the BY4743 strain background (20) were grown to mid-logarithmic phase in rich medium and then exposed to a period of acute short term starvation for a carbon or nitrogen source, grown to stationary phase in rich medium for 7 days, or treated with inhibitory concentrations of caffeine or rapamycin sufficient to inactivate TORC1 (4, 24–27). Cells were collected, washed, and spotted onto rich medium to allow recovery. Wild-type cells recovered from all treatments (Fig. 1). The ego− mutant cells likewise recovered efficiently from all treatments tested except from treatment with rapamycin (Fig. 1). The EGO complex appears to be selectively required for recovery from rapamycin and not from other treatments that inactivate TORC1.
FIGURE 1.
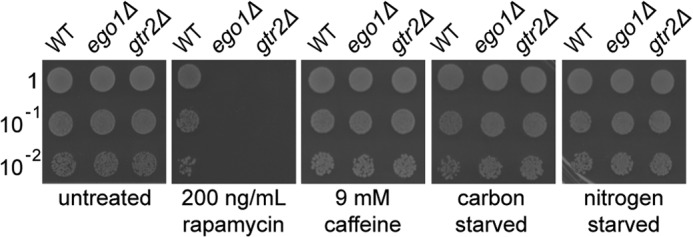
ego− mutant defect is selective for recovery from rapamycin. Cultures of WT, ego1Δ, or gtr2Δ cells in mid-logarithmic phase were diluted to an A600 nm of ∼0.1 and treated or not for 2 h with rapamycin (200 ng/ml) or caffeine (9 mm) or deprived of carbon or nitrogen for 2 h at room temperature. Samples were washed in fresh YPD medium, and 10-fold serial dilutions were spotted (5 μl) onto YPD plates and incubated at 30 °C for 2 days prior to scanning.
Rapamycin Does Not Fully Inhibit Proliferation of Wild-type Cells
Rapamycin is a potent inhibitor of TORC1 (4) and, in the BY4743 strain background, inhibits proliferation above a minimum threshold of ∼3–5 ng/ml (11). Indeed, we find that all concentrations tested of rapamycin in the range of 20–200 ng/ml are equally effective at inducing autophagy (Fig. 2A), a key downstream consequence of lowered TORC1 activity (14, 28, 29), and at inhibiting proliferation (Fig. 2B) in wild-type cells. However, we confirmed and extended the work of Neklesa and Davis (11) and found that even highly supersaturating concentrations of rapamycin do not fully inhibit the growth of wild-type cultures (Fig. 2, B and C). The residual rapamycin-insensitive growth rate is stable over time starting at 2 h after addition of the drug (Fig. 2C) and is stable over the full range of saturating concentrations of rapamycin tested (20–200 ng/ml) (Fig. 2B) (up to 1 μg/ml; data not shown). These data indicate that rapamycin does not fully inhibit the proliferation of wild-type cells, even when present in supersaturating concentrations in the medium.
FIGURE 2.

Rapamycin-insensitive proliferation of wild-type yeast cells is partly dependent on the EGO complex. A, mid-logarithmic cultures of WT cells carrying a plasmid expressing PGK1-GFP were treated or not with rapamycin (20, 50, 100, and 200 ng/ml) for 6 h. A Western blot was carried out using whole cell protein extraction and a primary antibody against GFP. The Pgk1-GFP fusion protein is degraded by autophagy (23); the GFP component is resistant to vacuolar proteases, and hence, accumulation of a GFP fragment reflects the level of autophagy that occurred because of the addition of rapamycin. B, steady-state growth rates of logarithmically growing cultures of WT and ego1Δ cells at a starting A600 nm of ∼0.025 in the absence or continuous presence of different concentrations of rapamycin were determined by Coulter counting between 2 and 12 h after addition of the drug. Cultures were grown at room temperature. Growth rates were calculated relative to the average of the untreated wild-type controls. Error bars denote S.E.; *, p < 0.05 for treated ego1Δ relative to the equivalent treated WT. C, mid-logarithmic cultures of WT cells were treated or not with rapamycin (200 ng/ml) at t = 0 h and at a starting A600 nm of ∼0.025. Cell number was measured by Coulter counting over the subsequent 12-h incubation at room temperature, and the log2 values of the increase in cell number with time relative to t = 0 h are shown. Error bars denote S.E. D, logarithmically growing cultures of wild-type cells were treated with rapamycin (200 ng/ml) for 24 h after which cells were washed with fresh media and plated onto plain YPD to recover. Recovered wild-type cells were treated again with rapamycin (200 ng/ml) for a further 24 h, washed, and recovered on plain YPD. Naive wild-type cells (never exposed to rapamycin) and those that had recovered from 1 or 2 rounds of rapamycin treatment were grown to mid-logarithmic phase, diluted to an A600 nm of ∼0.04, and treated or not with rapamycin (200 ng/ml). The steady state growth rate was determined by monitoring the increase in A600 nm between 3 and 6 h after the introduction of the drug. Cultures were grown at 28 °C. Growth rates are shown relative to those of the untreated controls. Error bars denote S.E. E, mid-logarithmic cultures of ego1Δ cells were treated or not with rapamycin (200 ng/ml) at t = 0 h and at a starting A600 nm of ∼0.025. Cell number was measured by Coulter counting over the subsequent 12 h, and the log2 values of the increase in cell number with time relative to t = 0 h are shown. Error bars denote S.E. F, logarithmically growing cultures of WT, ego3Δ, gtr1Δ, and gtr2Δ cells were treated or not with rapamycin (200 ng/ml), and the relative steady-state growth rate was determined as for B. Error bars denote S.E.; *, p < 0.05 relative to continuously treated WT. Inset, recovery of WT, ego3Δ, and gtr1Δ cells from rapamycin treatment was determined as for Fig. 1.
The residual rapamycin-insensitive proliferation rate seen for wild-type cells was not due to selection for rapamycin-resistant mutants. First, the residual growth rate is stable with time in the presence of the drug (Fig. 2C), consistent with the majority of cells in the culture proliferating at the same slow rate throughout. Second, the same slow residual growth rate is seen for cultures of all samples and isolates of isogenic wild-type cells tested (data not shown). Third, all wild-type haploid microcolonies generated by tetrad dissection onto medium containing saturating concentrations of rapamycin proliferate at the same slow rate (see Fig. 3D). Finally, the rapamycin-insensitive growth rate of wild-type cultures derived from cells that had previously been recovered from rapamycin treatment is identical to that of naive cells that had not previously been exposed to the drug (Fig. 2D).
FIGURE 3.

Rapamycin-insensitive proliferation is dependent on the presence of TORC1 components. A, relative steady-state growth rate of cultures of WT, tco89Δ, and tor1Δ cells in the presence and absence of rapamycin (200 ng/ml) was determined as for Fig. 2B. Error bars denote S.E.; *, p < 0.05 relative to treated WT. Inset recovery of WT, tco89Δ, and tor1Δ cells from rapamycin treatment was determined as for Fig. 1. B, cultures of WT and kog1Δ-pkog1ts cells were grown for 2 days at either permissive (22 °C) or nonpermissive (37 °C) temperatures during which they were diluted to maintain logarithmic growth and subsequently treated or not with rapamycin (rap) (200 ng/ml). The relative steady-state growth rates were determined as for Fig. 2B. Error bars denote S.E.; *, p < 0.05. C, relative steady-state growth rates of WT, ego1Δ, gtr2Δ, tco89Δ, and kog1Δ-pkog1ts cultures treated or not with rapamycin (200 ng/ml), at a starting A600 nm of 0.05 for untreated or 0.1 for rapamycin-treated cultures, were determined as for Fig. 2B. Cultures were grown at 28 °C. Error bars denote S.E.; *, p < 0.05 relative to equivalent WT treatment. D, heterozygous KOG1/kog1Δ::KanMX diploids were sporulated, and the tetrads (10 and 9, respectively) were dissected onto solid medium comprising YPD or YPD supplemented with rapamycin (200 ng/ml). The resulting plates were incubated at 30 °C. The dissected spores were monitored by microscopy for 3 days, and their ability to proliferate was scored. The percentage and fraction (in parentheses) of total spores that failed to double, doubled three times or fewer, or formed colonies (too many cells to accurately estimate the number of doublings) on each medium are shown as a function of time after dissection. All tetrads resulted in only two visible colonies after 3 days, and these were all found to be wild type (i.e. sensitive to G418). We infer that the 50% of dissected spores that failed to form visible colonies were all kog1Δ::KanMX.
Rapamycin-insensitive Proliferation Requires the EGO Complex
The EGO complex is required for recovery from a period of treatment with a high, saturating concentration of rapamycin, typically 200 ng/ml (12, 14). To date, phenotypic analyses indicate that wild-type and ego− mutant cells enter identical states when treated with saturating concentrations of the drug (12, 14). Is this also true for the residual proliferation rate? We compared the growth rates of cultures of wild-type and ego1Δ cells in the absence and presence of saturating concentrations of rapamycin. As shown in Fig. 2, B and E, the vegetative growth rates in the absence of rapamycin were identical for mutant and wild-type cultures. By contrast, the residual growth rate of ego1Δ cultures is significantly lower than that for wild-type cultures when grown in the continuous presence of saturating concentrations of rapamycin (Fig. 2, B and E). This slow residual growth rate of ego1Δ cultures is stable over the range of saturating drug concentrations tested (Fig. 2B) and over time (Fig. 2E), and it is shared with cultures of the other three ego− mutants: gtr1Δ, gtr2Δ, and ego3Δ (Figs. 2F and 3C). These observations indicate that the EGO complex is selectively required for residual rapamycin-insensitive proliferation.
Rapamycin-insensitive Proliferation Requires TORC1 Activity
The EGO complex activates TORC1 (12, 14). Why then do the ego− mutants proliferate more slowly than wild-type cells in the presence of rapamycin? It is possible that rapamycin fully inhibits TORC1 with residual proliferation requiring some novel role for the EGO complex. Alternatively, it is possible that rapamycin only partly inhibits TORC1, with residual proliferation requiring a rapamycin-insensitive activity of the complex. Is residual proliferation dependent on TORC1?
Tco89 is the only nonessential, specific component of TORC1 (3), and it is required for its activation by the EGO complex (12). Mutants lacking Tco89, like ego− mutants, show a rapamycin recovery defect (3) (e.g. see inset panel, Fig. 3A). We found that tco89Δ haploids, like ego− haploids, proliferate more slowly than do congenic wild-type cells but only in the presence of saturating concentrations of rapamycin (Fig. 3A). Tor1 is the central protein kinase component of TORC1 (16). Mutants lacking Tor1 are viable because the related Tor2 protein can partly substitute for it in its absence (30). Mutants lacking Tor1 show lowered TORC1 activity (18, 30) and a rapamycin-recovery defect(e.g. see inset panel, Fig. 3A) (18). We find that tor1Δ cells also proliferate more slowly than do congenic wild-type cells, again only in the presence of rapamycin (Fig. 3A). The slow residual growth rate of tco89Δ is stable over the range of saturating concentrations tested (Fig. 3C). Rapamycin-insensitive proliferation is thus partly dependent on both Tco89 and Tor1.
Kog1, the yeast homolog of mammalian Raptor (2), is the only essential and specific component of TORC1 (2, 3, 21). Is Kog1 required for rapamycin-insensitive proliferation? We exploited a mild temperature-sensitive allele of KOG1 (21) and monitored the proliferation rate of wild-type and kog1ts cells in the presence and absence of rapamycin and at both the permissive and the restrictive temperatures. Importantly, the mutant cultures grew more slowly than wild-type cultures at both permissive and restrictive temperatures when treated with saturating amounts of drug (Fig. 3B). This slow residual growth rate is also stable over the range of saturating concentrations tested (Fig. 3C). We conclude that residual rapamycin-insensitive proliferation also depends, at least partly, on Kog1.
Is rapamycin-insensitive proliferation fully dependent on TORC1? Viable, heterozygous kog1Δ::KanMX/KOG1 diploids were sporulated, tetrads dissected into rich medium, and incubated at 30 °C. The progress of microcolonies derived from 20 dissected tetrads was followed by regular microscopic examination (Fig. 3D). For each tetrad, only two spores produced visible G418-sensitive colonies, consistent with the remaining spores failing to form colonies and being of kog1Δ genotype (2). Microscopic examination indicated that the majority (80%) of kog1Δ spores did not proliferate even once in 3 days after dissection. The remaining 20% completed 1–3 doublings within the 1st day, with no subsequent proliferation noted (Fig. 3D). This early and limited burst of proliferation seen in the minority of mutant cells is likely to result from some active TORC1 being inherited through meiosis because none of 18 kog1Δ spores progressed past the one-cell stage when dissected onto medium containing an inhibitory concentration of rapamycin (Fig. 3D). By contrast, although dissection onto rapamycin significantly slowed proliferation of all wild-type haploids, it did not prevent them forming micro-colonies. These data indicate that Kog1, and thus TORC1, is absolutely required for proliferation of yeast cells. We conclude that TORC1 is absolutely required for proliferation of yeast cells and that rapamycin can only partly inhibit the complex.
Finally, caffeine selectively inhibits TORC1 and by a mechanism distinct from that used by rapamycin (26, 27). Is residual rapamycin-insensitive proliferation inhibited by caffeine? Note that for the following experiments, we chose a concentration of caffeine (3 mm) just below that sufficient to inhibit proliferation of wild-type cells. This “sub-inhibitory” concentration should mimic ego− mutations that cause no growth defect in the absence of rapamycin. This concentration should also lessen the likelihood of any possible off-target effects of caffeine complicating our analyses.
We treated wild-type cultures with saturating levels of rapamycin in the presence or absence of a sub-inhibitory concentration of caffeine. We found that caffeine alone did not slow proliferation, as expected for a sub-inhibitory concentration of the drug. However, the presence of caffeine significantly slowed the growth rate of cultures also treated with a saturating amount of rapamycin, i.e. the rapamycin-insensitive growth rate is selectively inhibited by caffeine (Fig. 4A). Taken together, our data indicate that the residual proliferation seen in the presence of saturating amounts of rapamycin is dependent on the activity and not merely on the presence of TORC1.
FIGURE 4.
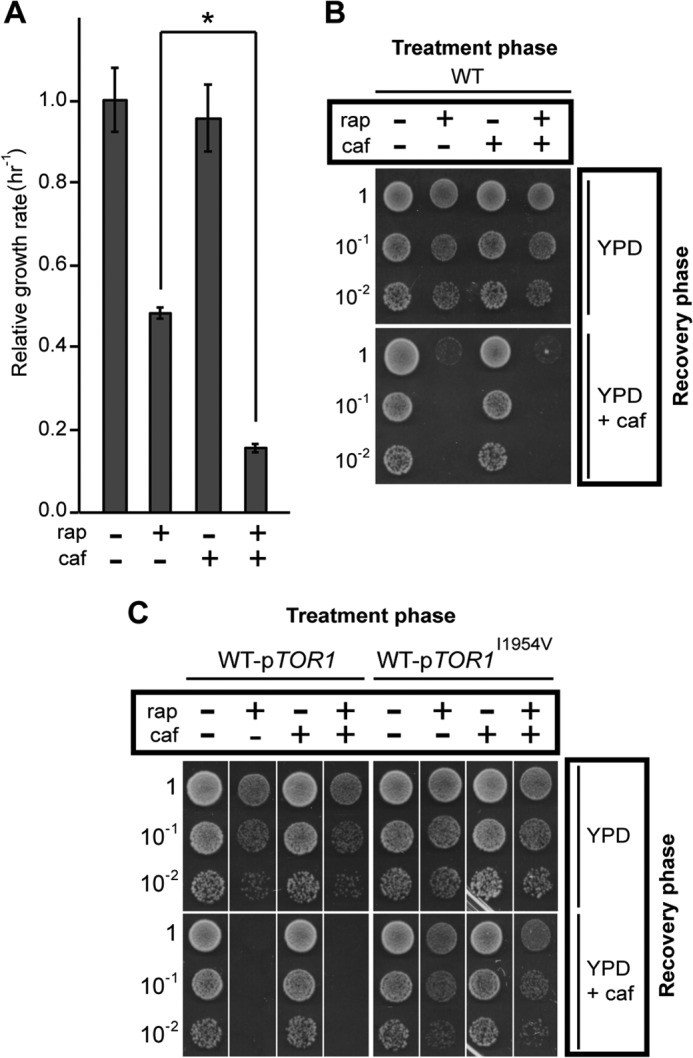
Caffeine treatment mimics the effects of ego− mutations. A, relative steady-state growth rate of WT cells treated or not with rapamycin (rap) (200 ng/ml), caffeine (caf) (3 mm), or both was determined as for Fig. 2B. Error bars denote S.E.; *, p < 0.05. B, logarithmically growing cultures of WT cells were diluted to an A600 nm of ∼0.1, treated or not for 2 h with rapamycin (rap) (200 ng/ml), caffeine (caf) (3 mm), or both. Cells were collected and washed with YPD or YPD containing caffeine (3 mm) as appropriate. 10-Fold dilutions were spotted onto plates containing YPD or YPD with caffeine (3 mm). Plates were incubated at 30 °C for 2 days. C, cultures of WT transformants containing a plasmid expressing either a wild-type TOR1 allele or a caffeine-resistant TOR1I1954V allele (26) were grown to mid-logarithmic phase in selective medium (SD-URA), transferred to YPD at an A600 nm of ∼0.1, and processed as for B.
Rapamycin-insensitive TORC1 Activity Is Required for Recovery from Rapamycin
Mutations that compromise rapamycin-insensitive TORC1 activity (and thus rapamycin-insensitive proliferation) also compromise recovery after washout of the drug, e.g. the correlation holds for the ego1Δ, ego3Δ, gtr1Δ, gtr2Δ, tco89Δ, or tor1Δ mutations (see above). To further explore this correlation, we exploited the fact that caffeine is a selective inhibitor of rapamycin-insensitive proliferation (Fig. 4A). We treated wild-type yeast cells with a saturating concentration of rapamycin (200 ng/ml), the sub-inhibitory concentration of caffeine (3 mm), or both for a “treatment” phase lasting 2 h. The cells were then washed and spotted onto solid “recovery” medium containing rich medium alone or rich medium supplemented with caffeine at the sub-inhibitory concentration of 3 mm (Fig. 4B). We found that cells recovered efficiently from treatment with caffeine alone. As expected, cells recovered efficiently from treatment with a saturating concentration of rapamycin alone (Fig. 4B). By contrast, co-treatment with rapamycin and caffeine, followed by recovery in the presence of caffeine induced a strong recovery defect (Fig. 4B). This recovery defect is dependent on caffeine acting on Tor1 and not on any off-target effects, because the presence of a caffeine-resistant allele of TOR1 (26) but not of the wild-type allele on a plasmid restores the ability of the cells to recover from treatment with both drugs (Fig. 4C). We conclude that rapamycin-insensitive TORC1 activity is sensitive to caffeine and is required for residual proliferation rate in the presence of rapamycin and for recovery from the drug.
TORC1 Activity during the Recovery Phase Is Key to Recovery
When is TORC1 activity required to allow recovery from rapamycin? First, we assayed recovery of wild-type cells from treatment with rapamycin but with the sub-inhibitory level of caffeine (3 mm) present only during the treatment phase, i.e. both drugs present during the treatment phase and neither drug present during the recovery phase. The cells recovered normally (Fig. 4, B and C). We conclude that rapamycin-insensitive TORC1 activity during the treatment phase per se is not necessary for subsequent recovery from rapamycin. Second, we assayed recovery of wild-type cells from treatment with rapamycin but with caffeine present only during the recovery phase, i.e. rapamycin present alone during the treatment phase and caffeine present alone during the recovery phase. The cells displayed a strong recovery defect (Fig. 4, B and C). Caffeine treatment during the recovery phase and at sub-inhibitory levels (with respect to vegetative proliferation) is thus sufficient to prevent recovery from prior treatment with rapamycin. These data indicate that normal TORC1 activity is required during the recovery phase for recovery from prior treatment with rapamycin. The cell somehow remembers its prior exposure to rapamycin.
Proliferation Rate in Rapamycin Persists after Washout
It is possible that TORC1 is reactivated only slowly after washout of rapamycin. Does the rapamycin-insensitive proliferation rate characteristic of cells in saturating concentrations of rapamycin persist after washout of the drug? We grew cultures of congenic wild-type and ego− mutant cells to mid-logarithmic phase, treated them for 2 h with a saturating concentration of rapamycin (40 ng/ml), washed out the drug, and monitored the growth rate of the cultures with time after washout for up to 12 h. We found that each culture grew at the same steady rate throughout this initial 12 h of recovery phase (Fig. 5, A and B) and at a rate characteristic to that seen in the continuous presence of rapamycin (Figs. 5, A and B, versus 2, C and E). The proliferation rate characteristic of treated cells thus persists long after washout of rapamycin, at least for the concentration of drug used here. Even wild-type cultures appear to recover surprisingly slowly from a period of treatment with rapamycin.
FIGURE 5.
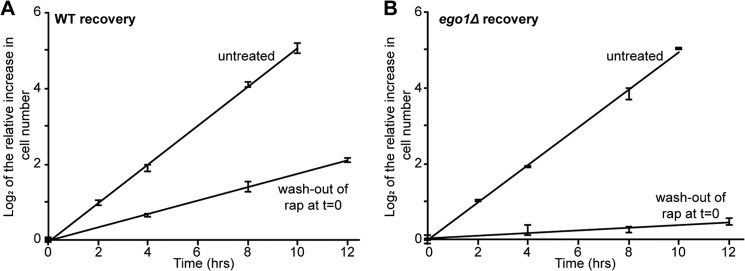
Residual growth rate seen in the presence of rapamycin persists after washout of the drug. A, logarithmically growing cultures of WT cells were diluted to an A600 nm of ∼0.1 and treated or not with rapamycin (40 ng/ml) for 2 h at room temperature after which cells were washed in fresh medium and transferred to fresh medium to recover at room temperature. Cell number was monitored by Coulter counting over the course of 12 h. The log2 of the increase in cell number relative to t = 0 (i.e. time of drug washout) was determined, and trend lines were plotted. Error bars denote S.E. B, logarithmically growing cultures of ego1Δ cells were treated and analyzed as for A.
Cell-associated Rapamycin Is Diluted Slowly by Proliferation
Does rapamycin persist in cells after washout of the drug from the medium? We determined the fate of rapamycin in wild-type cells as a function of time after washout of the drug, i.e. during recovery.
We adapted a quantitative assay for rapamycin using mass spectrometry to detect the cell-associated pool of the drug in yeast cells (see under “Experimental Procedures”) (31). This assay detects the parent ion of rapamycin and thus only chemically unmodified drug. Furthermore, to maximize the uptake of the drug, we treated cultures with a high concentration of the drug (400 ng/ml) and for a long period (4 h). Cells respond appropriately under these conditions as follows: wild-type cells recover efficiently from treatment with rapamycin (200–400 ng/ml) for 4 h (Fig. 6A), whereas ego1Δ mutants do not (Fig. 6A).
FIGURE 6.

Rapamycin is stable in wild-type cells and decreases only with residual proliferation during recovery. Cultures of WT and ego1Δ cells in mid-logarithmic phase were diluted to an A600 nm of ∼0.1 and treated or not for 4 h with rapamycin (rap) (either 200 or 400 ng/ml) at room temperature. Cells were washed in YPD and serial dilutions were spotted (5 μl) onto YPD plates and incubated at 30 °C for 2 days. Scans of the plates are shown in A. Exponentially growing cultures of WT cells in YPD at an A600 nm of 0.6 were treated with rapamycin (400 ng/ml) for 4 h at room temperature after which cells were washed three times and transferred into fresh YPD to recover. Samples were taken at 0, 7, and 20 h following drug removal and diluted as appropriate to prevent saturation. B, relative cell-associated signal for rapamycin in WT cells as a function of recovery time and expressed relative to the mass spectrometry signal at t = 0. C, relative increase in culture density (by A600 nm) for WT cells as a function of recovery time. D, relative cell-associated rapamycin signal for WT cells as a function of recovery time (from B) and corrected for growth of the culture (from C). Error bars denote S.E.; *, p < 0.05 relative to t = 0.
We found that the amount of cell-associated rapamycin is remarkably stable, reducing to only approximately a third of its initial value for a given amount of cells by 20 h after drug removal (Fig. 6B). Under the conditions of our experiment, the cells had proliferated very slowly over the 20-h recovery period (Fig. 6C). Surprisingly, this residual proliferation accounted for the apparent reduction in the observed concentration of cell-associated rapamycin; the total amount of cell-associated rapamycin in the culture was stable over the period examined (Fig. 6D). Rapamycin does not persist in wild-type cells after washout of the drug from the medium, but the concentration of the drug falls with the rate of residual cell proliferation. To a first approximation, it appears that yeast may not be able to detoxify rapamycin: rather, the size of the drug pool may fall slowly with time by dilution among progeny cells as the culture grows.
The recovery defect of ego− mutant cells could then be due to slower rapamycin-insensitive proliferation diluting the drug less efficiently among progeny cells. Consistent with this model, we find that the initial size of the intracellular drug pool is the same in wild-type and in ego1Δ mutant cells when treated with rapamycin (Fig. 7A). However, the concentration of cell-associated rapamycin decreased even more slowly in ego1Δ cells after washout of the drug (Fig. 7B) but at the slower rate of residual proliferation (Fig. 7, C and D). The observable pool of rapamycin is thus reduced in both WT and ego− cells by the process of residual proliferation; to a first approximation, the difference in rapamycin-insensitive proliferation rate accounts for the different rates of drug dilution in the strains and may thus underpin their different capacities to recovery from the drug.
FIGURE 7.

Rapamycin does not hyper-accumulate in ego1Δ cells and decreases only with residual proliferation during recovery. Exponentially growing cultures of wild-type and ego1Δ cultures in YPD at an A600 nm of 0.6 were treated with rapamycin (rap) (400 ng/ml) for 4 h at room temperature after which cells were washed three times, collected by centrifugation and rapamycin extracted from the cell pellet. The mass spectrometry signal for rapamycin extracted from equivalent amounts of WT and ego1Δ cells (normalized for A600 nm) was determined side-by-side. The signal from the ego1Δ sample relative to the WT sample for each run is shown in A. Exponentially growing cultures of ego1Δ cells in YPD at an A600 nm of 0.6 were treated with rapamycin (400 ng/ml) for 4 h at room temperature after which cells were washed three times and transferred into fresh YPD to recover. Samples were taken at 0, 7, and 20 h following drug removal and processed for mass spectrometry as for Fig. 6. B, relative cell-associated signal for rapamycin in ego1Δ cells as a function of recovery time and expressed relative to the mass spectrometry signal at t = 0. C, relative increase in culture density (by A600 nm) for ego1Δ cells as a function of recovery time. D, relative cell-associated rapamycin signal for ego1Δ cells as a function of recovery time (from B) and corrected for growth of the culture (from C). Error bars denote S.E.; *, p < 0.05 relative to t = 0.
If residual proliferation in rapamycin drives dilution of the drug, then ego− mutant cells should recover more slowly from treatment with the drug than do the wild-type cells, but the defect should not be absolute. We predict that ego− mutant cells should be able to recover, albeit slowly, from treatment with lower, but still inhibitory, concentrations of rapamycin. Using the spot assay for recovery, we indeed found that the recovery defect of ego− mutants is concentration-dependent, with efficient recovery occurring from treatment with 20 ng/ml of drug for 2 h but not from treatment with higher concentrations for the same period (50–200 ng/ml) (Fig. 8).
FIGURE 8.
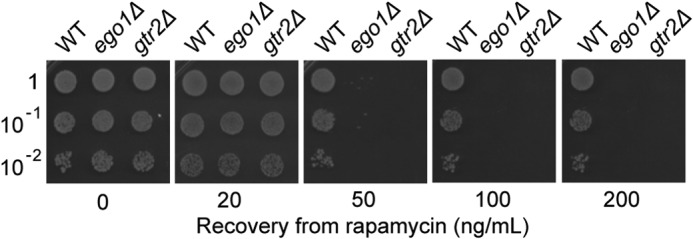
ego− mutants can recover from treatment with a lower saturating concentration of rapamycin. Cultures of WT, ego1Δ, or gtr2Δ cells in mid-logarithmic phase were treated or not for 2 h with different concentrations of rapamycin (20–200 ng/ml). Recovery was determined as for Fig. 1.
Rapamycin-insensitive Proliferation Determines Recovery from Rapamycin
Is the biologically active pool of the drug detoxified by residual cell proliferation? Does proliferation rate dictate the time it takes for wild-type cells to recover from rapamycin? Does the slower residual proliferation rate of ego− mutants quantitatively account for their recovery defect? To address these questions, we explored the detailed kinetics by which cells recover from a period of rapamycin treatment.
Liquid cultures of wild-type and congenic ego1Δ mutant cells were treated, or not, for 2 h with different inhibitory concentrations of rapamycin (20–200 ng/ml). The drug was washed out, and the progress of each culture was monitored by Coulter counting as a function of time after washout. We estimated the time it took for each culture to switch its growth rate from that characteristic of the continuous presence of rapamycin to that of untreated cultures, i.e. the recovery (e.g. see Fig. 9, A and B). We found that the recovery time for wild-type cultures varies dramatically as a function of rapamycin concentration (Fig. 9C), and it asymptotically approaches a maximum of ∼20 h for recovery from the highest concentrations of the drug tested (200 ng/ml). We conclude that the recovery time of wild-type cells increases with increasing size of the drug pool. Recovery can be extremely slow after treatment with high concentrations of rapamycin.
FIGURE 9.

Residual proliferation in the presence of rapamycin can explain the kinetics of recovery of wild-type and ego1Δ mutant cells. A, logarithmically growing culture of WT cells in YPD at an A600 nm of ∼0.1 and growing at room temperature was split and left untreated, continuously treated with rapamycin (50 ng/ml), or treated with rapamycin (100 ng/ml) for 2 h at room temperature after which the drug was washed out and cells were transferred to fresh medium to recover at room temperature (t = 0). Cell number was monitored by Coulter counting over the course of 48 h. The log2 of the increase in cell number relative to t = 0 (time of rapamycin washout) was determined for each sample. Trend lines were plotted for untreated and continuously treated samples. Recovery was determined as the time at which the growth rate of the cultures switched to being comparable with that of cells in the continuous presence of the drug to that of untreated cells. B, logarithmically growing culture of ego1Δ cells in YPD at an A600 nm of ∼0.1 and growing at room temperature was split and left untreated, continuously treated with rapamycin (20 ng/ml), or treated with rapamycin (30 ng/ml) for 2 h after which the drug was washed out and cells were transferred to fresh medium to recover at room temperature (t = 0). Cell number was monitored by Coulter counting over the course of 48 h as for A. C, logarithmically growing cultures were treated with rapamycin (20, 40, 50, 100, or 200 ng/ml for WT and 20, 30, or 40 ng/ml for ego1Δ) for 2 h, after which rapamycin was washed out, and cells were transferred to fresh medium to recover. The time at which the growth rate of the recovering cultures switched from that comparable with continuously treated to that of untreated cells was determined for each concentration of rapamycin (rap) tested as described in A. Error bars denote S.E. A predicted recovery time was calculated and plotted (gray lines) using the following equation: y = observed doubling time in constant rap × (log2[rap]) + b, where b is a variable chosen for best fit to the initial trajectory of each experimentally determined recovery time versus concentration of rapamycin plot.
By contrast, ego1Δ mutant cells can recover only from treatment with low concentrations of the drug (Figs. 8 and 9, B and C). Their recovery time increases very dramatically with increasing drug concentration and became difficult to measure accurately beyond the ∼34 h needed for recovery from treatment with 40 ng/ml rapamycin for 2 h (Fig. 9C). It thus appears that ego1Δ mutant cells recover more slowly than do wild-type cells from treatment with any given inhibitory concentration of rapamycin. The recovery defect of ego− mutant cells is one of degree.
If the active pool of rapamycin is diluted by residual proliferation, then doubling the size of the intracellular drug pool should extend the recovery time of a culture by one doubling time of that culture (i.e. at the rapamycin-insensitive growth rate). Assuming that the maximum intracellular concentration of rapamycin reached during the treatment phase varies linearly with the extracellular concentration of the drug and using the measured doubling times in the continuous presence of rapamycin, we can predict a recovery time versus concentration of rapamycin plot for wild-type and ego− cultures. As shown in Fig. 9C, these predicted plots (gray lines) fit the experimentally observed recovery time versus concentration plots extremely well, particularly for lower concentrations of rapamycin during the treatment phase (up to 100 ng/ml for wild-type and 40 ng/ml for mutant cultures).
Indeed, if the cultures are recovering by proliferation-dependent dilution of the intracellular drug pool, then we can calculate the theoretical doubling times of the wild-type and ego− cell cultures that would account for the initial trajectories (up to 100 ng/ml of rapamycin) of the experimentally observed recovery time versus concentration plots. These theoretical doubling times were determined as the slopes of the linear recovery time versus log2[rapamycin] plots (data not shown). We find that these theoretical doubling times are, within error, identical to the experimentally observed doubling times of the wild-type and ego− cultures determined on the same day and in the constant presence of saturating amounts of rapamycin (Table 1). The proliferation rates observed in the constant presence of saturating concentrations of rapamycin are sufficient to explain, quantitatively, the recovery times for wild-type cultures and the magnitude of the ego− recovery defect. We conclude that yeast cells do not detoxify rapamycin; the biologically active pool of drug is reduced predominantly, if not solely, by proliferation-dependent dilution.
TABLE 1.
Proliferation rates in the continuous presence of rapamycin can quantitatively explain the times-to-recovery from rapamycin treatment
Comparison of the calculated initial slopes of the time-to-recovery data for WT and ego1Δ cultures derived from Fig. 1E but re-plotted as a function of log2 of rapamycin concentration and the experimentally determined doubling times of WT and ego1Δ cells determined in the continuous presence of rapamycin (20 ng/ml) and on the same day. Results are shown means ± S.D.
| Strain | Initial slope of recovery time vs. log2 of rapamycin concentration (hours), data from Fig. 1E | Observed doubling time in the constant presence of rapamycin (20 ng/ml) (hours) |
|---|---|---|
| WT | 6.85 ± 2.02 | 5.32 ± 0.17 |
| ego1Δ | 22.25 ± 1.22 | 23.79 ± 5.51 |
DISCUSSION
Extensive functional genomics screening in yeast has failed to identify a detoxification mechanism for this macrocyclic lactone (32–34). Our results provide an explanation. Rapamycin is not detoxified; rather, the drug concentration in the cell falls passively with time by a process that is selectively dependent on activity of the drug target itself, TORC1. Hence, the genetic fingerprint of an altered drug pool is inextricably linked to that of altered TORC1 signaling.
The fate of rapamycin in living cells has received scant attention to date. Here, we find that the observable pool of rapamycin associated with yeast cells is very stable in vivo (Figs. 6 and 7) and declines very slowly with time and only at the rate of population growth as cells proliferate (Figs. 6 and 7). Residual cell proliferation also accounts for the behavior of the biologically active pool of the drug, the pool that slows cell proliferation (Fig. 9C). Residual proliferation drives reduction in rapamycin by physical dilution. Proliferation-dependent dilution is not unusual per se; it must contribute to the reduction in the intracellular pool of any drug that does not fully inhibit cell proliferation and is sufficiently stable to persist at some level within the cell for at least one doubling. What is unusual is that such an inherently slow mechanism is the predominant or sole mode of clearing a drug. Rapamycin clearly presents an unusual toxicological and metabolic challenge to yeast cells.
Rapamycin behaves as a partial inhibitor of the budding yeast TORC1. First, the consequence for a cell losing TORC1 activity is much more severe than that of treatment with saturating amounts of rapamycin; TORC1 must therefore have functions that are not inhibited by rapamycin. Yeast TORC1, like mTORC1, appears to be essential for cell growth and proliferation (2). Our data extend and support that view and indicate that proliferation is not possible in the absence of Kog1 (and by inference TORC1) (Fig. 3D). Yet, significant residual proliferation occurs in the continuous presence of high concentrations of rapamycin in wild-type yeast cells (Fig. 2, B and C). Second, mutations or chemical treatments that directly or indirectly compromise TORC1 activity, e.g. loss of the EGO complex, loss of Tco89, loss of Tor1, a temperature-sensitive allele of KOG1 or treatment with caffeine, also compromise the residual proliferation rate seen in saturating amounts of rapamycin (Figs. 2, B, E, and F, 3, A and B, and 4A). The simplest explanation for these results is that some TORC1 activity is insensitive to rapamycin and that this rapamycin-insensitive activity is dependent on normal in vivo controls, e.g. on the upstream EGO complex. Third, rapamycin is known to only partly inhibit TORC1 in both mammalian cells (7, 8) and in fission yeast (9, 10). Our findings thus bring the budding yeast complex more in line with its homologs in these species and not in opposition to them.
Incomplete inhibition by rapamycin-Fpr1 is likely to be an inherent and universal characteristic of TORC1s: a consequence of the mode of action of the drug. The rapamycin-FKBP complex is a noncompetitive, “allosteric” inhibitor of TORC1s (35). Noncompetitive inhibitors do not directly compete for substrate binding and thus do not necessarily fully inactivate their target enzyme even when present at saturating levels. However, rapamycin-insensitive TORC1 activity may have a more trivial origin; a fraction of TORC1 may not bind to rapamycin-Fpr1, and this fraction could provide residual activity that supports residual proliferation. This possibility is very unlikely. First, it is possible that Fpr1 is limiting in amount. However, Fpr1 is one of the most abundant 10% of proteins in the yeast cell, whereas the dedicated components of TORC1, Tor1, Kog1, and Tco89,are among the least abundant 25% of proteins, as estimated by quantitative proteomics (36). The estimated ratio of Frp1 to TORC1 in yeast is ∼300:1. Fpr1 availability is not limiting. Second, it is possible that the active pool of rapamycin itself never reaches a sufficient threshold within the cell to bind to all of the TORC1, i.e. rapamycin is limiting. Our data do not support such a model. We find that the residual proliferation rate and, by deduction, the residual TORC1 activity is stable over a wide range of saturating concentrations of rapamycin (Figs. 2B and 3C). Yet the time to recovery after washout of the drug varies hugely over this same concentration range. For example, it takes wild-type cells up to 20 h, or ∼4 cell doublings, to recover from treatment with rapamycin (200 ng/ml for 2 h) via proliferation-dependent dilution of the drug pool (Fig. 9C). The initial concentration of rapamycin in the treated cell must therefore reach up to ∼24 or ∼16 times higher than the minimal inhibitory threshold sufficient to inactivate TORC1. The intracellular pool of rapamycin is not limiting. Finally, elevated levels of reactive oxygen species (ROS) can cause oxidative damage to yeast TORC1 itself, rendering a fraction of the complex less able to bind to rapamycin-Fpr1 under stressed conditions (11). Could a basal fraction of oxidized TORC1 explain the rapamycin-insensitive proliferation we see in budding yeast? Probably not. ROS damage to TORC1 has not been reported under normal basal conditions. Furthermore, we find that overexpression of superoxide dismutase alone or in combination with overexpression of catalase does not change the rapamycin-insensitive proliferation rates we measure.3 Overall, rapamycin-Fpr1 is likely to be a partial inhibitor of the TORC1 molecules to which it binds.
Mutants lacking components of the EGO complex proliferate normally in the absence of rapamycin. Yet these mutants show a strong proliferation defect (relative to wild-type cells) in the presence of rapamycin. Why? First, all mutations and chemical treatments tested that directly or indirectly compromise TORC1 activity, e.g. ego1Δ, ego3Δ, gtr1Δ, gtr2Δ, tco89Δ, tor1Δ, kog1ts, and caffeine, also selectively compromise rapamycin-insensitive proliferation (Figs. 2, B, and F, 3, A and B, and 4A). The selectivity thus appears to be a universal consequence of lowered TORC1 activity. Second, mutants lacking the EGO complex display substantially reduced Sch9 phosphorylation under normal vegetative conditions (and thus lowered TORC1 activity), yet show no appreciable proliferation defect (Fig. 2, B and F) (12). The simplest explanation for these observations is that TORC1 activity is not limiting for proliferation under normal growth conditions. However, treatment with rapamycin lowers TORC1 activity sufficiently to now make its residual activity limiting for cell proliferation.
Our results reveal that yeast cannot detoxify rapamycin, explain the origin of the ego− mutant phenotype, and uncover novel aspects of rapamycin action in yeast. The study of rapamycin-insensitive proliferation should provide a powerful new window onto novel aspects of the in vivo roles and regulators of TORC1.
Acknowledgments
We thank C. De Virgilio, Y. Kamada, and M. Thumm for kindly supplying GTR1, kog1ts, and PGK1 plasmids, respectfully; S. Weidt for technical assistance; C. McInerny for the use of the Coulter counter; and members of the Gray laboratory for discussions.
This work was supported by a departmental training award Ph.D. studentship from the Biotechnology and Biological Sciences Research Council (to S. K. E.) and a research award from the School of Life Sciences (to J. V. G.).
S. K. Evans and J. V. Gray, unpublished observations.
- TORC1
- target of rapamycin complex 1
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Wullschleger S., Loewith R., Hall M. N. (2006) TOR signaling in growth and metabolism. Cell 124, 471–484 [DOI] [PubMed] [Google Scholar]
- 2. Loewith R., Jacinto E., Wullschleger S., Lorberg A., Crespo J. L., Bonenfant D., Oppliger W., Jenoe P., Hall M. N. (2002) Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 10, 457–468 [DOI] [PubMed] [Google Scholar]
- 3. Reinke A., Anderson S., McCaffery J. M., Yates J., 3rd, Aronova S., Chu S., Fairclough S., Iverson C., Wedaman K. P., Powers T. (2004) TOR complex 1 includes a novel component, Tco89p (YPL180w), and cooperates with Ssd1p to maintain cellular integrity in Saccharomyces cerevisiae. J. Biol. Chem. 279, 14752–14762 [DOI] [PubMed] [Google Scholar]
- 4. Heitman J., Movva N. R., Hall M. N. (1991) Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253, 905–909 [DOI] [PubMed] [Google Scholar]
- 5. Schmelzle T., Hall M. N. (2000) TOR, a central controller of cell growth. Cell 103, 253–262 [DOI] [PubMed] [Google Scholar]
- 6. Barbet N. C., Schneider U., Helliwell S. B., Stansfield I., Tuite M. F., Hall M. N. (1996) TOR controls translation initiation and early G1 progression in yeast. Mol. Biol. Cell 7, 25–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thoreen C. C., Kang S. A, Chang J. W., Liu Q., Zhang J., Gao Y., Reichling L. J., Sim T., Sabatini D. M., Gray N. S. (2009) An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 284, 8023–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feldman M. E., Apsel B., Uotila A., Loewith R., Knight Z. A, Ruggero D., Shokat K. M. (2009) Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 7, e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakashima A., Sato T., Tamanoi F. (2010) Fission yeast TORC1 regulates phosphorylation of ribosomal S6 proteins in response to nutrients, and its activity is inhibited by rapamycin. J. Cell Sci. 123, 777–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takahara T., Maeda T. (2012) TORC1 of fission yeast is rapamycin-sensitive. Genes Cells 17, 698–708 [DOI] [PubMed] [Google Scholar]
- 11. Neklesa T. K., Davis R. W. (2008) Superoxide anions regulate TORC1 and its ability to bind Fpr1:rapamycin complex. Proc. Natl. Acad. Sci. U.S.A. 105, 15166–15171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Binda M., Péli-Gulli M.-P., Bonfils G., Panchaud N., Urban J., Sturgill T. W., Loewith R., De Virgilio C. (2009) The Vam6 GEF controls TORC1 by activating the EGO complex. Mol. Cell 35, 563–573 [DOI] [PubMed] [Google Scholar]
- 13. Bonfils G., Jaquenoud M., Bontron S., Ostrowicz C., Ungermann C., De Virgilio C. (2012) Leucyl-tRNA synthetase controls TORC1 via the EGO complex. Mol. Cell 46, 105–110 [DOI] [PubMed] [Google Scholar]
- 14. Dubouloz F., Deloche O., Wanke V., Cameroni E., De Virgilio C. (2005) The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol. Cell 19, 15–26 [DOI] [PubMed] [Google Scholar]
- 15. Gao M., Kaiser C. A. (2006) A conserved GTPase-containing complex is required for intracellular sorting of the general amino-acid permease in yeast. Nat. Cell Biol. 8, 657–667 [DOI] [PubMed] [Google Scholar]
- 16. Loewith R., Hall M. N. (2011) Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics 189, 1177–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Crespo J. L., Powers T., Fowler B., Hall M. N. (2002) RTG1, and RTG3 are regulated in response to intracellular levels of glutamine. Proc. Natl. Acad. Sci. U.S.A. 99, 6784–6789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zurita-Martinez S. A, Puria R., Pan X., Boeke J. D., Cardenas M. E. (2007) Efficient Tor signaling requires a functional class C Vps protein complex in Saccharomyces cerevisiae. Genetics 176, 2139–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Powers R. W., 3rd, Kaeberlein M., Caldwell S. D., Kennedy B. K., Fields S. (2006) Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 20, 174–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shoemaker D. D., Lashkari D. A., Morris D., Mittmann M., Davis R. W. (1996) Quantitative phenotypic analysis of yeast deletion mutants using a highly parallel molecular bar-coding strategy. Nat. Genet. 14, 450–456 [DOI] [PubMed] [Google Scholar]
- 21. Nakashima A., Maruki Y., Imamura Y., Kondo C., Kawamata T., Kawanishi I., Takata H., Matsuura A., Lee K. S., Kikkawa U., Ohsumi Y., Yonezawa K., Kamada Y. (2008) The yeast Tor signaling pathway is involved in G2/M transition via polo-kinase. PLoS One 3, e2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krause S. A., Gray J. V. (2002) The protein kinase C pathway is required for viability in quiescence in Saccharomyces cerevisiae. Curr. Biol. 12, 588–593 [DOI] [PubMed] [Google Scholar]
- 23. Welter E., Thumm M., Krick R. (2010) Quantification of nonselective bulk autophagy in S. cerevisiae using Pgk1-GFP. Autophagy 6, 794–797 [DOI] [PubMed] [Google Scholar]
- 24. Beck T., Schmidt A., Hall M. N. (1999) Starvation induces vacuolar targeting and degradation of the tryptophan permease in yeast. J. Cell Biol. 146, 1227–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schmidt A., Beck T., Koller A., Kunz J., Hall M. N. (1998) The TOR nutrient signalling pathway phosphorylates NPR1 and inhibits turnover of the tryptophan permease. EMBO J. 17, 6924–6931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reinke A., Chen J. C., Aronova S., Powers T. (2006) Caffeine targets TOR complex I and provides evidence for a regulatory link between the FRB and kinase domains of Tor1p. J. Biol. Chem. 281, 31616–31626 [DOI] [PubMed] [Google Scholar]
- 27. Wanke V., Cameroni E., Uotila A., Piccolis M., Urban J., Loewith R., De Virgilio C. (2008) Caffeine extends yeast lifespan by targeting TORC1. Mol. Microbiol. 69, 277–285 [DOI] [PubMed] [Google Scholar]
- 28. Shin C.-S., Huh W.-K. (2011) Bidirectional regulation between TORC1 and autophagy in Saccharomyces cerevisiae. Autophagy 7, 854–862 [DOI] [PubMed] [Google Scholar]
- 29. Umekawa M., Klionsky D. J. (2012) Ksp1 kinase regulates autophagy via the target of rapamycin complex 1 (TORC1) pathway. J. Biol. Chem. 287, 16300–16310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Urban J., Soulard A., Huber A., Lippman S., Mukhopadhyay D., Deloche O., Wanke V., Anrather D., Ammerer G., Riezman H., Broach J. R., De Virgilio C., Hall M. N., Loewith R. (2007) Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol. Cell 26, 663–674 [DOI] [PubMed] [Google Scholar]
- 31. Geda P., Patury S., Ma J., Bharucha N., Dobry C. J., Lawson S. K., Gestwicki J. E., Kumar A. (2008) A small molecule-directed approach to control protein localization and function. Yeast 25, 577–594 [DOI] [PubMed] [Google Scholar]
- 32. Butcher R. A., Bhullar B. S., Perlstein E. O., Marsischky G., LaBaer J., Schreiber S. L. (2006) Microarray-based method for monitoring yeast overexpression strains reveals small-molecule targets in TOR pathway. Nat. Chem. Biol. 2, 103–109 [DOI] [PubMed] [Google Scholar]
- 33. Hillenmeyer M. E., Fung E., Wildenhain J., Pierce S. E., Hoon S., Lee W., Proctor M., St Onge R. P., Tyers M., Koller D., Altman R. B., Davis R. W., Nislow C., Giaever G. (2008) The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320, 362–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Parsons A. B., Lopez A., Givoni I. E., Williams D. E., Gray C. A, Porter J., Chua G., Sopko R., Brost R. L., Ho C.-H., Wang J., Ketela T., Brenner C., Brill J. A., Fernandez G. E., Lorenz T. C., Payne G. S., Ishihara S., Ohya Y., Andrews B., Hughes T. R., Frey B. J., Graham T. R., Andersen R. J., Boone C. (2006) Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell 126, 611–625 [DOI] [PubMed] [Google Scholar]
- 35. Benjamin D., Colombi M., Moroni C., Hall M. N. (2011) Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 10, 868–880 [DOI] [PubMed] [Google Scholar]
- 36. Wang M., Weiss M., Simonovic M., Haertinger G., Schrimpf S. P., Hengartner M. O., von Mering C. (2012) PaxDb, a database of protein abundance averages across all three domains of life. Mol. Cell. Proteomics 11, 492–500 [DOI] [PMC free article] [PubMed] [Google Scholar]