

Background: Molecular mechanisms translating effects of folate on metastasis are not clear.
Results: Folate restriction inhibits methylation and membrane translocation of Rho GTPases in A549 cancer cells and provides survival advantage in mice with lung cancer.
Conclusion: Folate enhances migratory ability and invasiveness of cancer cells through Rho GTPase pathways and promotes metastasis.
Significance: This study highlights the interaction between nutrients and metastasis-related signaling.
Keywords: Cell Motility, Cofilin, Folate, Rho GTPases, Tumor Metastases
Abstract
Folate, an important nutrient in the human diet, has been implicated in cancer, but its role in metastasis is not established. We have shown previously that the withdrawal of medium folate leads to the inhibition of migration and invasion of A549 lung carcinoma cells. Here we have demonstrated that medium folate regulates the function of Rho GTPases by enabling their carboxyl methylation and translocation to plasma membrane. Conversely, the lack of folate leads to the retention of these proteins in endoplasmic reticulum. Folate also promoted the switch from inactive (GDP-bound) to active (GTP-bound) GTPases, resulting in the activation of downstream kinases p21-activated kinase and LIM kinase and phosphorylation of the actin-depolymerizing factor cofilin. We have further demonstrated that in A549 cells two GTPases, RhoA and Rac1, but not Cdc42, are immediate sensors of folate status: the siRNA silencing of RhoA or Rac1 blocked effects of folate on cofilin phosphorylation and cellular migration and invasion. The finding that folate modulates metastatic potential of cancer cells was confirmed in an animal model of lung cancer using tail vein injection of A549 cells in SCID mice. A folate-rich diet enhanced lung colonization and distant metastasis to lymph nodes and decreased overall survival (35 versus 63 days for mice on a folate-restricted diet). High folate also promoted epithelial-mesenchymal transition in cancer cells and experimental mouse tumors. Our study provides experimental evidence for a mechanism of metastasis promotion by dietary folate and highlights the interaction between nutrients and metastasis-related signaling.
Introduction
Metastasis is a complex process that includes several events collectively termed the invasion-metastasis cascade (1, 2). The entire process is under the control of specific molecular pathways operating in metastasizing cancers and is also regulated by interactions between neoplastic and stromal cells (2). Invasion of individual tumor cells can proceed via the integrin-dependent (mesenchymal invasion) or integrin-independent and Rho/Rho-associated protein kinase-dependent (amoeboid invasion) mechanism (3).
Small Rho family GTPases control multiple cellular functions such as adhesion, spreading, migration, and division and are involved in all stages during cancer progression (4). One of the mechanisms of Rho GTPase-dependent regulation of cellular motility and migration is associated with the control of dynamic reorganization of the actin cytoskeleton and the mediation of the formation of specific actin structures (5). GTPases undergo a complex post-translational processing that includes prenylation of a cysteine at the C terminus, proteolytic cleavage of three C-terminal amino acids, and carboxyl methylation of the prenylated cysteine (6). The prenyl group allows anchoring GTPases to ER3 or plasma membrane: prenylation itself targets proteins to ER, whereas methylation enables their extraction from ER to cytoplasm with subsequent translocation to the plasma membrane (6). In the plasma membrane, GTPases cycle between inactive GDP-bound and active GTP-bound conformations. In their active conformation, GTPases interact with effector proteins, thus inducing downstream signaling events. The transition between inactive and active forms as well as extraction of protein from ER or plasma membrane is tightly regulated by three groups of proteins: guanine nucleotide exchange factors, GTPase-activating proteins, and guanine nucleotide dissociation inhibitors (GDIs) (7). The molecular mechanisms that control precise targeting of Rho GTPases are not completely understood, but methylation-dependent localization to the cellular membrane is required for their activation and proper initiation of downstream signaling events, and the lack of the corresponding methyltransferase, isoprenylcysteine carboxylmethyltransferase, results in mislocalization of the proteins to the cytosol (8, 9).
One of the actin-related downstream targets of Rho GTPases is the actin-depolymerizing factor cofilin, a protein that regulates actin dynamics through depolymerization and severing of actin filaments (10). Cofilin activity is controlled by phosphorylation at a single serine residue by two LIM kinases (LIMK1 and LIMK2) or testis-specific kinases (11, 12). The activity of LIMKs, the primary kinases responsible for cofilin phosphorylation, is regulated by a phosphorylation cascade under the control of Rho GTPases (13). This cascade has been demonstrated in experiments with stimulation of cells by growth factors that led to activation of receptor tyrosine kinases and the subsequent recruitment of Rho GTPase and Rho-associated protein kinase to phosphorylate LIMK and then cofilin (14). Of note, cofilin is involved in the regulation of migration, locomotion, and metastasis of cancer cells (11), although the precise relationship between cofilin function and metastasis progression remains obscure.
A growing body of evidence also indicates a role for the human diet in cancer diseases and metastasis (15, 16). Direct effects of food components on signaling pathways as well as a general concept of nutrient-genome interactions have found experimental support in modern days (17–19). The role of one of the essential nutrients, folate, in cancer has been recognized for decades. Indeed, folate abundance is beneficial for proliferation of cancer cells, which require efficient folate pathways to support de novo nucleotide biosynthesis and methylation processes. This is the basis for treatment of malignancies with antifolate drugs targeting folate pathways (20). For years, it has also been believed that folate supplementation exerts antitumorigenic effects, although later epidemiological studies have failed to provide a definite conclusion regarding the role of folate intake in mediating cancer risk (21–24). There is also a lack of knowledge about the role of folate in metastatic disease. We have previously shown that medium folate regulates cellular motility through effects on cofilin-dependent actin dynamics (25). In the present study, we identified Rho GTPase signaling as an immediate downstream sensor of the folate status in the regulation of cofilin-dependent motility. Our study also demonstrated correlation between dietary folate and metastasis in a mouse model and suggested the Rho/LIMK/cofilin pathway as a mechanism for such effect.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
Media and dialyzed FBS were from Invitrogen. FBS was purchased from Atlanta Biologicals. PDGF-β and EGF were obtained from R&D Systems. Other reagents were from Sigma unless otherwise indicated.
Plasmids and Transfection
pEGFP-C3 plasmids carrying cDNA for Rac1, RhoA, or Cdc42 were provided by Dr. Philips (New York University Cancer Institute); intramolecular biosensor plasmid Raichu-1011x (carrying Rac1) was a gift from Dr. Matsuda (Kyoto University). p3XFLAG-CMV-7.1 vector was purchased from Sigma. Rac1 cDNA was cloned from pEGFP-C3-Rac1 into p3XFLAG-CMV-7.1 plasmid and confirmed by sequencing. pCMV6-AC-CALR-mRFP was purchased from Origene. Cells were transfected using Neon Nucleofector (Invitrogen) according to the manufacturer's manual.
Western Blotting and Immunoprecipitation
Cellular lysates in radioimmune precipitation assay buffer containing a protease inhibitor mixture were normalized by the level of total protein and analyzed by SDS-PAGE and immunoblotting with corresponding antibodies. Rac1/RhoA and Cdc42 antibodies (1:200) were from Chemicon and Cytoskeleton, Inc., respectively; all other antibodies were from Sigma. Secondary antibodies conjugated with horseradish peroxidase and the ECL substrate were from Amersham Biosciences and Thermo Scientific, respectively. For immunoprecipitation, cellular fractions (cytosolic and membrane) were incubated with 2 μg of RhoGDI antibody (Sigma) for 1 h at 4 °C followed by overnight incubation with Protein G-Sepharose 4 Fast Flow (50 μl of a 50% slurry) (Amersham Biosciences). Resin was washed three to five times, and pulled down proteins were analyzed by SDS-PAGE and Western blotting with Rac1 antibody.
Gelatin Zymography Assay
MMP-2 and MMP-9 activities were measured using a gelatin zymography assay as described (26). Conditioned media were concentrated, diluted (1:1) with 2× non-reducing SDS-PAGE loading buffer, and resolved in a 10% (w/v) polyacrylamide gel impregnated with 2 mg/ml gelatin (Bio-Rad). After SDS removal, gels were incubated in developing buffer followed by staining with Coomassie Brilliant Blue R-250 and stain removal until the bands became clear.
RT-PCR
The isolation of mRNA and cDNA synthesis were performed as described (27). Primer pairs used for amplification of target transcripts in RT-PCR are shown in Table 1.
TABLE 1.
Primer pairs for amplification of target transcripts in RT-PCR
G3PDH, glyceraldehyde-3-phosphate dehydrogenase; Col, collagen.
| Gene | Primers from 5′- to 3′-end (forward/reverse) |
|---|---|
| Col I | ACGTCCTGGTGAAGTTGGTC |
| ACCAGGGAAGCCTCTCTCTC | |
| Col III | AGCCTCATTAGTCCTGATGCTTCTCG |
| CTTCTCAGCACTAGAATCTGTCCACC | |
| Rac1 | CCCTGATACAGGCCATC |
| GAACGATGGGCTGAGAC | |
| RhoA | ATGGCTGCCATCCGGAAG |
| TCACAAGACAAGGCACC | |
| Cdc42 | GCAATGCAGACAAAG |
| GGCCCTCATAGCACCTTG | |
| G3PDH | TGAAGGTCGGAGTCAACGGATTTGGT |
| CATGTGGGCCATGAGGTCCACCAC |
Active GTPase Pulldown Assay
The activated Rho GTPase proteins were measured using the Active RhoA, Rac1, and Cdc42 Pulldown and Detection kit (Pierce) according to the manufacturer's directions. Briefly, A549 cells grown on folate-deficient or -proficient media were stimulated with PDGF-β (Rac1), EGF (Rho), or calpeptin (Cdc42) for 1 h. Active GTPases were pulled down from cell lysates with GST-Rhotekin Rho-binding domain (Rho) or GST-PAK1 binding domain (Rac1 and Cdc42). Individual GTPases were detected in the pulled down fractions by SDS-PAGE/Western blotting with corresponding antibody.
Transwell Migration and Invasion Assays
Cells were seeded into the upper chamber of a Transwell insert (BD Bioscience) in serum-free medium at a density of 50,000 cells/well. For migration assays, inserts were precoated with 5 μg/ml fibronectin (Fisher Scientific). Medium containing 10% FBS was placed in the lower chamber as a chemoattractant, and cells were incubated for 24 h in CO2 incubator. Non-migrating cells were removed from the upper chamber by scraping; the remaining cells were stained with Diff-Quick (Dade Behring, Inc.). Stained cells were counted in 10 random optical fields in three different inserts. Alternatively, adherent cells were fixed with 3% glutaraldehyde and stained with 0.1% crystal violet. After that, cells were lysed with 10% acetic acid (100–200 μl/well) and quantified by colorimetry at 560 nm. Invasion assays were carried out in a similar manner using inserts precoated with extracellular matrix. For these experiments, a Cell Invasion Assay kit (Chemicon) was used according to the manufacturer's protocol.
Confocal Microscopy
Cells transfected with pEGFP-C3 plasmids carrying Rac1, RhoA, or Cdc42 cDNA were stimulated with corresponding Rho GTPase activator (Rac1, 50 ng/ml PDGF-β for 10 min; RhoA, 100 ng/ml calpeptin for 20 min; Cdc42, 100 ng/ml EGF for 2 min) after overnight serum starvation. In colocalization experiments, A549 cells grown under folate-proficient or -deficient conditions were co-transfected with pEGFP-C3-Rac1 and pCMV6-AC-CALR-mRFP (endoplasmic reticulum marker). Live cell images were captured and processed using an Olympus FluoView FV10i LIV laser-scanning confocal microscope at the Medical University of South Carolina Cell and Molecular Imaging Core Facility.
Analysis of Base-labile Methylation
Analysis was done essentially as described (28). A549 cells (70% confluence) were maintained for 3 days on folate-deficient or folate-proficient media and placed in RPMI 1640 medium lacking methionine (Invitrogen) for 1 h. Cells were transfected with pCMV-Rac1-FLAG, seeded in Met-free RPMI 1640 medium supplemented with l-[3H-methyl]methionine (50 μCi/ml) (PerkinElmer Life Sciences), harvested after 24 h, and lysed in radioimmune precipitation assay buffer. Lysates cleared by centrifugation were incubated with anti-FLAG resin for 6 h at 4 °C. FLAG-Rac1 was eluted with FLAG peptide. The eluate was resolved in a 15% SDS-polyacrylamide gel, and gel lanes were sliced into 2-mm strips and treated with 1 n NaOH for in-gel hydrolysis of esterified labile methyl groups. Radioactivity released into the liquid phase (alkali-labile methyl groups) was quantified using a liquid scintillation counter (Beckman). Remaining gel slices were neutralized with 1 n acetic acid, and the total protein-incorporated radioactivity (non-labile) was quantified.
Fluorescence Resonance Energy Transfer (FRET) Analysis
Active GTPases were monitored in vivo using FRET analysis as described (29). To quantify FRET of intramolecular biosensors, a simple ratiometry technique was used. A549 cells grown on folate-proficient or -deficient media were transiently transfected with biosensor Raichu-1011x plasmid carrying Rac1. Following overnight serum starvation, time-lapsed imaging was performed. After 6 min of steady state imaging, TGF-containing (10 ng/μl) conditioned medium was added to the cells, and data acquisition continued for 32 min. The biosensors containing CFP and YFP as the donor and acceptor, respectively, were excited at a wavelength of 440 nm. The images at 480 (CFP emission) and 535 nm (YFP emission due to FRET) were taken using a Zeiss LSM 510 NLO laser-scanning confocal microscope with multiphoton excitation. Images were analyzed using MetaMorph 7.0 software.
siRNA
Stealth siRNA targeting RhoA, Rac1, and Cdc42 as well as negative control Stealth duplexes (Table 2) were purchased from Invitrogen. For each protein, two siRNA duplexes targeting different sequences were tested. They were transfected in A549 cells by electroporation with Amaxa Nucleofector (Lonza) according to the manufacturer's instructions. Cells were collected at 24–120 h post-transfection, lysed, and analyzed by conventional RT-PCR and Western blot assays for the presence of Rho proteins.
TABLE 2.
Rho GTPase DNA sequences targeted by siRNA
Two sequences were targeted for each protein.
| Gene | Targeted sequence |
|---|---|
| Rac1 | CAACAGCAGGCATTTTCTCTTCCT (1) |
| CACCTCAGGATACCACTTTGCACG (2) | |
| Cdc42 | TTTGAGTCCCAACAAGCAAGAAAG (1) |
| CAGCGGTCGTAATCTGTCATAATC (2) | |
| RhoA | AAGCAGATGAGAATGACGTCGGTG (1) |
| TGCCCAGCTGTGTCCCACAAAGCC (2) |
Tail Vein Injection and Bioluminescence Imaging
Five-week-old male SCID/beige mice (CB17.Cg-Prkdcscid Lystbg/Crl, Charles River) were segregated in two groups, 10 animals each. Mice were placed on folate-deficient (0.01 ppm folic acid) or folate-proficient (6 ppm folic acid) diet (Harlan Tekland) 1 week prior to the tail vein injection with A549-luc-C8 cells (Caliper; 5 × 106 cells in a volume of 0.1 ml). Total tumor burden in mice was assessed by in vivo bioluminescence measured weekly using an IVIS Lumina Imaging System (Xenogen) 2 min after intraperitoneal luciferin injection. Relative intensities of emitted light were presented as a pseudocolor image ranging from blue (least intense) to red (most intense). Igor Pro software was used to overlay color images of the tumor and the corresponding mouse. Imaging was performed at the Medical University of South Carolina Cell and Molecular Imaging Core Facility. All animals were housed in the Medical University of South Carolina facility, which is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care and the United States Department of Agriculture. All mice were maintained according to the guidelines established by the United States Department of Agriculture and the American Association for Accreditation of Laboratory Animal Care. This project was approved by the Institutional Animal Care and Use Committee of the Medical University of South Carolina.
Assays of Reduced Folate Pools
Fifty milligrams of tissues were homogenized on ice in 1 ml of 50 mm Tris-HCl buffer, pH 7.4 containing 50 mm sodium ascorbate using a Dounce homogenizer and lysed by heating for 3 min in a boiling water bath. In the case of whole blood, 50 μl of sample were mixed with 450 μl of the same buffer and treated as above. Lysates were chilled on ice and centrifuged for 5 min at 17,000 × g at 4 °C. Folate pools were measured in tissue lysates by the ternary complex assay method as we described previously (30, 31). Folate levels were calculated per milligram of protein measured by Bradford assay or per milliliter of whole blood.
Histopathological and Immunohistochemical Examination of Tumors from Folate-proficient and Folate-deficient Mice
Mouse tissues harvested at the end point of the experiment were fixed in 10% neutral buffered formalin for 24 h and processed for histological analysis. Five-micrometer sections were mounted on glass slides and stained with hematoxylin and eosin or with specific antibody. Mouse antibodies (1:200) for staining of human nucleus (Millipore), phosphocofilin, cofilin, or vimentin (1:100; all from Sigma) were used. Images were captured at 400× using a Nikon Eclipse 80i microscope and NIS-Elements Basic Research software. Images were analyzed with Adobe Photoshop CS5 software, and the number of pixels containing neoplastic cells was measured. Prism 5.0 software was used for statistical analysis.
Statistical Analysis
Statistical analysis was done by the paired Student's t test, χ2 test, or two-way analysis of variance test. Overall survival was estimated using a Kaplan-Meier approach, and differences were tested using a log rank test. A p value (calculated using Excel, GraphPad Prism 4, or SAS V9.3) less than 0.05 was considered significant.
RESULTS
Membrane Translocation and Activation of Small Rho GTPases Are Controlled by Medium Folate
To examine whether folate regulates cellular migration through the control of Rho GTPases, we transfected A549 cells with GFP-tagged RhoA, Rac1, or Cdc42 and treated them with calpeptin, PDGF-β, or EGF. Confocal microscopy showed that GTPases were localized to cellular membrane in stimulated cells grown on folate-proficient media but remained diffusely distributed in cells grown in folate-depleted media (Fig. 1A). Cellular fractionation experiments confirmed that folate promotes the translocation of endogenous Rho GTPases to the plasma membrane: levels of Rac1, Cdc42, and RhoA associated with the membrane fraction were strongly decreased in cells on folate-depleted media compared with cells on regular media (Fig. 1B). In agreement with the impairment of the membrane translocation, a dramatic decrease in the activation of all three GTPases was evident in folate-deficient cells (Fig. 1C).
FIGURE 1.

Effects of medium folate on Rho GTPases. A, fluorescent images of A549 cells transiently transfected with GFP-GTPase fusions (translocation to plasma membrane was induced by recombinant PDGF-β for Rac1, EGF for Cdc42, and calpeptin for Rho). B, distribution of endogenous GTPases (Western blot) between the cytosol (C) and plasma membrane (M). C, folate depletion decreases active (GTP-bound) GTPases. D, carboxyl methylation of Rac1 (base-labile methyl groups) is dramatically impaired in cells kept on folate-deficient media. E, distribution of endogenous Rac1 between cytoplasm and ER (top and middle panels). The bottom panel shows cytoplasm-localized GDI-bound Rac1 (visualized by Western blotting (WB) with Rac1-specific antibody after immunoprecipitation (IP) with GDI-specific antibody). F, folate depletion leads to ER retention of RhoA, Rac1, and Cdc42. A549 cells kept on normal and folate-depleted media for 3 days were co-transfected with pEGFP-C3-GTPase (green) and pCMV6-AC-CALR-mRFP (red). Images (confocal microscopy) were taken 48 h post-transfection. Error bars represent S.D. −FA, folate-free medium; +FA, 2.2 μm folic acid.
Folate Depletion Impairs Carboxyl Methylation of Rac1 and Leads to Its Retention in ER
To test whether folate depletion affects Rho GTPase carboxyl methylation, we analyzed the FLAG-tagged Rac1 protein transiently expressed in folate-depleted and control A549 cells. FLAG-Rac1 was pulled down on FLAG-specific antibody after exposure of cells to l-[3H-methyl]methionine in methionine-free medium overnight, and its carboxyl methylation was evaluated as the inclusion of base-labile methyl groups (28). We observed a drop in radioactivity eluted from the FLAG-Rac1 SDS-PAGE band for the protein pulled down from cells kept on folate-free media (Fig. 1D), indicating a strong decline in the carboxyl methylation. The loss of carboxyl methylation should prevent the interaction of Rho GTPases with RhoGDI and the extraction from ER into cytoplasm resulting in redistribution of the proteins between the two compartments (32). In agreement with this notion, we observed a strong shift in the subcellular distribution of Rac1 in cells kept on folate-free media compared with cells kept on standard folate-proficient media with a much higher presence of the protein in ER of the former cells (Fig. 1E, upper panel). Furthermore, the pulldown assay of Rac1 on GDI showed that these proteins co-immunoprecipitate in folate-proficient but not folate-deficient cells (Fig. 1E, lower panel). The retention of Rho GTPases in ER of folate-deficient cells was further confirmed by confocal microscopy (Fig. 1F).
Direct Monitoring of Rac1 Activation in Vivo upon Different Folate Supplementation
To directly address the spatiotemporal activation of Rho GTPases as the function of folate status, we carried out FRET assays using an intramolecular biosensor consisting of YFP-linked Rac1 and CFP-linked binding domain of the Rac1 downstream target PAK (Fig. 2A) (29). In this biosensor, the activation-induced conformational changes of the Rac1 portion of the molecule enable its intramolecular interaction with the PAK domain, bringing CFP in close proximity to YFP. Resulting FRET directly reflects the level of Rac1 activation in TGF-β-treated cells expressing the biosensor (Fig. 2A). Analysis of time lapse images in our experiments revealed a strong energy transfer in folate-proficient A549 cells within 2 min of TGF-β induction with the maximum FRET:CFP signal ratio of 2.1 achieved within 8 min, indicating rapid and robust activation of Rac1 (Fig. 2, B and C). In contrast, in A549 cells cultured in folate-free media, the response was much slower and weaker, reaching the maximum FRET:CFP signal ratio of 1.65. Of note, Rac1-PAK binding showed diffuse distribution of Rac1 protein in broad areas of the cell (Fig. 2C). Thus, our data indicated the impaired activation of Rac1 upon folate withdrawal.
FIGURE 2.

FRET upon interaction of Rac1 with binding domain of PAK. A, individual FRET and CFP images were obtained using 60× magnification. The FRET:CFP ratio images are shown in intensity modulated display mode. Scale bar, 10 μm. The inset shows the schematic organization of expressed protein construct. B, time lapse images of FRET:CFP ratio in folate-deficient (−FA; upper panel) and folate-proficient (+FA; lower panel) A549 cells. The upper and lower limits of the FRET:CFP ratio range are shown on the right in the colored key. C, time course of FRET:CFP ratio averaged from three experiments for folate-proficient (+FA; red curve) and folate-deficient (−FA; blue curve) conditions. Rac1 activation was induced by TGF-β (10 ng/μl).
Folate Regulates Motility of A549 Cells through RhoA and Rac1 but Not Cdc42
We evaluated the contribution of RhoA, Rac1, and Cdc42 in the regulation of motility of A549 cells in response to folate status. In these experiments, we silenced the proteins using siRNA (two commercial duplexes were tested in each case; Table 2). Strong down-regulation of Rac1 and RhoA mRNA was observed when duplexes 2 and 1, respectively, were used, whereas both duplexes were effective in the case of Cdc42 (Fig. 3A). These duplexes (duplex 1 was selected for Cdc42) also allowed a strong down-regulation of GTPases at the protein level (Fig. 3B). Of note, GTPase protein levels as well as the efficiency of silencing were not affected by the folate status in the media (Fig. 3A). We found that silencing of RhoA or Rac1 resulted in a strong drop in the level of phosphorylated cofilin, whereas the silencing of Cdc42 did not produce a notable effect (Fig. 3B). Furthermore, A549 cells with silenced RhoA or Rac1 demonstrated decreased ability to migrate as well as decreased invasion potential (Fig. 3C). In contrast, in Cdc42-silenced cells, these parameters were not changed (Fig. 3C). In agreement with this finding, Cdc42-silenced cells responded to the medium folate withdrawal by a decrease in the level of phosphocofilin and inhibition of migration and invasion in a manner similar to Cdc42-proficient cells, whereas RhoA- or Rac1-silenced cells became insensitive to the lack of medium folate (Fig. 3, B and C).
FIGURE 3.

Role of Rac1, RhoA, and Cdc42 in the regulation of motility by folate. A, silencing of Rac1, RhoA, and Cdc42 by siRNA at the mRNA (top panel) and protein (bottom panel) levels. Initial experiments (top panel) were done using two Stealth siRNA duplexes, 1 and 2, for each protein (targeted sequences are shown in Table 2). The most efficient duplexes were selected for further experiments (Rac1, duplex 2; RhoA, duplex 1; Cdc42, duplex 1). B, levels of cofilin and phosphocofilin (p-Cofilin) (Western blot) in A549 cells with silenced Rac1, RhoA, or Cdc42. C, migration (top panel) and invasion (bottom panel) assays of A549 cells with silenced Rac1, RhoA, and Cdc42 in the presence and absence of folate in media (*, p < 0.001; p values calculated by two-tailed Student's t test). −FA, folate-free media; +FA, media with 2.2 μm folic acid. The number of cells in the optical field (evaluated as described under “Experimental Procedures”) is an indicator of migration and invasion. Error bars represent S.D. Scr, scrambled.
Regulation of the Cascade from Rho GTPases to Cofilin Phosphorylation
We have previously demonstrated that medium folate controls the migratory potential of cultured cancer cells through the regulation of phosphorylated cofilin, a major calcium-independent regulator of actin dynamics (25). In the present study, we have shown that cells on folate-deficient media lack the ability to phosphorylate (activate) LIMK, the key kinase directly phosphorylating cofilin; accordingly, they have extremely low levels of phosphorylated cofilin (Fig. 4, A and B). The inability to activate LIMK was associated with the lack of activation of the upstream kinase PAK, which remained mainly non-phosphorylated (inactive) in cells grown on folate-deficient media (Fig. 4, A and B). In contrast, PAK and LIMK1 were phosphorylated in cells on folate-proficient media upon TGF-β treatment (Fig. 4, A and B). Likewise, levels of phosphorylated proteins in folate-deficient cells were restored upon folate repletion (Fig. 4, A and B). In these experiments, we applied prolonged treatment with TGF-β (24–72 h) to evaluate the sustained effect of folate on the PAK/LIMK/cofilin pathway. Interestingly, in A549 cells, this treatment activated the phosphorylation of LIMK1 but not LIMK2 (Fig. 4A), whereas both isoforms can be phosphorylated in response to TGF-β (33–35).
FIGURE 4.

Folate is required for the activation of EMT in cultured cells. A, Western blot assay of A549 cells kept on regular (2.2 μm folic acid; +FA) or folate-free (−FA) media. Replete, cells supplemented with 2.2 μm folic acid after growing on folate-free media. Cells were stimulated with 10 ng/ml TGF-β. B, calculation of the intensity of bands from A normalized to actin. C, morphology of cells grown in the absence (−FA) or presence (+FA) of folic acid upon treatment with TGF-β. Levels of collagen I and III mRNA (D) and activity of MMP-2 and MMP-9 (E) for cells grown with (+FA) and without (−FA) folic acid and treated with TGF-β are shown. Error bars represent S.D. EDA, extra domain A; Fn, fibronectin; Col, collagen; p-, phospho-; AU, arbitrary units.
Folate Promotes Epithelial-Mesenchymal Transition (EMT)
EMT is viewed as one of the characteristics of the cellular capacity for invasion and metastasis (36). We induced EMT in A549 cells with different folate status by prolonged treatment with TGF-β (24–72 h), a common approach in these type of experiments (26, 37). The expression of the epithelial markers E-cadherin and cytokeratin 19 was steady in cells kept on folate-free media but was significantly suppressed in cells on folate-rich media (Fig. 4, A and B). Conversely, the mesenchymal markers fibronectin and vimentin were elevated in folate-proficient cells in a time-dependent manner upon TGF-β treatment, whereas folate-deficient cells did not demonstrate the same trait (Fig. 4, A and B). Accordingly, folate repletion restored levels of EMT markers (Fig. 4, A and B). Cells grown in folate-depleted media did not show notable morphological changes in response to TGF-β and maintained a classic cobblestone (pebble-like) epithelial morphology and cell-cell adhesion (Fig. 4C). In contrast, cells cultured in regular media acquired more elongated fibroblast-like morphology accompanied by decreased cell-cell contacts upon TGF-β treatment (Fig. 4C). One of the characteristics of mesenchymal cells is their ability to synthesize fibrillar collagen (38). An evident increase in mRNA levels of collagens I and III was observed in folate-proficient but not -deficient cells in response to TGF-β (Fig. 4D). Gelatin zymography assays further showed the increased expression of two matrix metalloproteinases, MMP-2 and MMP-9, indicative of EMT in folate-proficient cells (Fig. 4E). Overall, our experiments indicated that folate supports acquisition of a mesenchymal-like phenotype, whereas its deficiency inhibits EMT.
Dietary Folate Promotes Lung Colonization by Cancer Cells and Decreases Survival
The above findings prompted us to hypothesize that dietary folate might influence metastasis formation. Specifically, they indicated that the dietary folate restriction should inhibit metastasis. To study the effect of dietary folate on metastasis, we injected the lateral tail vein of SCID/beige mice with human lung adenocarcinoma A549 cells stably expressing luciferase. This approach allows the monitoring of tumor progression in live animals with the level of luminescence reflecting the tumor burden (39). Two groups of mice were used: one kept on a standard rodent chow (6 ppm folic acid) and another kept on a folate-free chow (0.01 ppm folate). A folate-deficient diet significantly decreases blood and intracellular folate levels within the 2-week period (40). Four reduced folate forms represent the majority of the folate pool in mammalian cells (tetrahydrofolate (THF), 5-methyltetrahydrofolate (5-CH3-THF), 5,10-methylenetetrahydrofolate (5,10-CH2-THF), and 10-formyltetrahydrofolate (10-CHO-THF)), whereas in plasma, 5-CH3-THF comprises more than 90% of the reduced folate (41). We measured reduced folate pools in blood, lung, and liver using a technique allowing the measurement of major folate forms independent of their polyglutamylation status (30, 31). We observed that the total folate in blood, represented mostly by 5-CH3-THF, was decreased 2.2-fold in mice kept on folate-free chow compared with mice on a typical folate diet (Fig. 5A). An even higher drop in reduced folates (2.7-fold) was observed in lung of folate-deficient mice (Fig. 5A). The drop in two pools, 5-CH3-THF (involved in methylation pathways) and 10-CHO-THF (involved in the purine biosynthesis pathway), contributed to this effect (Fig. 5A). In contrast, the total reduced folate in liver of mice on the folate-deficient diet was essentially preserved (Fig. 5A; 18% decrease).
FIGURE 5.

Dietary folate enhances metastatic colonization of lung in SCID mice/tail vein injection model. A, levels of the total reduced folate pool in tissues of mice kept on a standard (folate-proficient) or folate-deficient chow. Levels of two specific folate pools are shown for lung (5m-THF, 5-methyltetrahydrofolate; 10f-THF, 10-formyltetrahydrofolate) in addition to the total folate. B, bioluminescence imaging of mice after tail vein injection of A549-luc-C8 cells (5 × 106 cells in 0.1 ml) in folate-free (FD) and standard folate-proficient (FP) diet groups (the total number of animals was 10 for each group). Images were taken at the indicated time points (weeks). The scale for the bioluminescence intensity is shown on the right. C, bioluminescence calculated from B (average values of bioluminescence ±S.D. were calculated using all mice in a group). D, survival of mice in folate-proficient and folate-deficient groups (Kaplan-Meier analysis). Error bars represent S.D. ph, photons; sr, steradian.
One week after injection low but detectable bioluminescence intensity signals (2.6 × 105–5.6 × 105 photons/s/cm2/steradian) were recorded in the lungs of animals in both groups with a gradual elevation of the number of luminescent cells in the lung area observed between weeks 1 and 4 (Fig. 5B). These experiments revealed much heavier tumor burden in mice kept on the folate-rich diet (a bioluminescence intensity signal of 3.7 × 108–5.9 × 108 photons/s/cm2/steradian) than in mice kept on the folate-free diet (a bioluminescence intensity signal below 4.5 × 107 photons/s/cm2/steradian) at week 5 (Fig. 5, B and C). Overall, based on the luminescence intensity, folate-proficient mice developed a 10–20-fold larger tumor burden in lungs than folate-deficient mice in the period of time of 3–4 weeks (Fig. 5C). The Kaplan-Meier analysis showed a highly statistically significant (p = 0.0005) difference in the overall survival between the two groups with a median life span of 63 days for the mice kept on folate-deficient diet, a 28-day survival advantage over mice that received the folate-fortified diet (Fig. 5D).
Tumors in Folate-proficient Mice Display Invasive Phenotype and Metastasize to Lymph Nodes
Histopathological examination confirmed that folate-proficient mice developed much bigger and more actively dividing lung tumors (Fig. 6, A and B). In addition, three of four examined animals on the folate-proficient diet displayed metastases to the popliteal lymph nodes. These metastases were characterized by widespread infiltration of tumor cells in the subcapsular, cortical, and medullary areas and occupied 75–100% of the analyzed lymph nodes (Fig. 6C). In contrast, only one folate-restricted mouse had a much smaller tumor in the lymph node (Fig. 6C). In agreement with cell culture experiments, cancer cells in mice fed the folate-deficient diet lost phosphocofilin, whereas the total levels of the protein were not changed (Fig. 6, D and E). Furthermore, the mesenchymal markers vimentin and fibronectin were significantly higher in tumors from the lungs of folate-proficient compared with folate-deficient mice. In contrast, expression of cytokeratin 19, a marker of an epithelial phenotype, was almost completely suppressed in the lungs of folate-proficient mice, whereas its expression was prominent in tumors from folate-deficient mice (Fig. 6, D and E).
FIGURE 6.

Histological analysis of tumors from folate-deficient and -proficient SCID mice. A, microscopy tissue images of lung showing H&E staining (×20 and ×200) and human-specific anti-nuclear antibody staining (contoured area represents tumors formed by A549 human cells; ×10). B, comparison of weight, tumor area, and nucleus to cytoplasm ratio for lungs from folate-proficient and -deficient mice. The tumor area was quantified by analyzing (Nikon NIS-Elements Basic Research 3.0 software) two low magnification (×40) random fields within two different histologic sections separated by 600 μm per mouse. The nucleus to cytoplasm ratio was derived from computation of corresponding areas. Statistical analysis was conducted using the Student's t test with Welch correction for heterogeneous variances (n = 7 for each group). C, tumor area in lymph nodes of folate-deficient and -proficient mice; the graph shows statistical analysis (three and four animals per respective group were analyzed). D and E, immunohistochemical (×200) and Western blot analysis of lung tissues from mice on folate-deficient and folate-proficient diets. Two mice from each group are shown for the Western blot (the graph shows quantification of the Western blot assay). Error bars represent S.D. FD, folate-deficient; FP, folate-proficient; Cof, cofilin; p-Cofilin/p-Cof, phosphocofilin; Ker, keratin; Fn, fibronectin; Vim, vimentin; AU, arbitrary units.
DISCUSSION
The relationship between dietary folate and cancer is a complex process with the ultimate outcome being a superposition of numerous factors including the form of the consumed vitamin and individual genetic variations (42, 43). In line with this complexity, epidemiological studies indicated direct as well as reverse correlation between folate consumption and tumorigenesis (44). In recent years, however, a concern has been raised that folate supplementation can promote cancer disease (45). Despite the strong attention in the literature to the role of dietary folate in cancer disease, not much is known about the effects of folate on metastasis. Numerous cellular pathways involved in migration depend on the nucleotide supply and methylation processes, which require folate. For example, in rodents, the depletion of folate leads to the hypermethylation and inactivation of the p16 gene (46), a tumor suppressor implicated in EMT and metastasis (47, 48). In line with the above assumption, a positive correlation between the migratory ability of cancer cells and folate supplementation as well as the inhibitory effect on such ability of antifolates has been demonstrated (49, 50). Contrary to those reports, a recent study highlighted the inhibitory effect of folate on the invasiveness of HCT116 cells by the activation of sonic hedgehog pathways (51). One puzzling result in this study was the promoting effect on migration by the antifolate methotrexate. Although this controversy could be explained by a differential effect depending on cancer type or even perhaps genotypic patterns, it emphasizes the urgent necessity to study the role of dietary folate in metastatic disease in more detail.
Our study has demonstrated that dietary folate affects metastasis through the regulation of the carboxyl methylation of Rho family GTPases RhoA and Rac1 (schematically depicted in Fig. 7). In this mechanism, folate supplementation promotes metastasis, whereas the dietary folate restriction inhibits this process. Specifically, folate availability allows proper methylation of Rho GTPases, a process enabling their plasma membrane translocation and activation. This is an initial step in a cascade regulating actin-dependent cellular motility. In this cascade, the activation of Rho GTPases leads to phosphorylation and activation of LIMK1, a kinase responsible for the phosphorylation of the actin-depolymerizing factor cofilin. We have shown previously that the loss of phosphocofilin is a key step in the inhibition of migration/invasion by folate stress (25). The balance between phosphorylated and non-phosphorylated cofilin and the constant cycling between these two forms regulated by LIMKs and several phosphatases affect metastatic potential (11, 52). In our study, the lack of medium folate disabled the cascade, resulting in the total loss of active (phosphorylated) LIMK1. This in turn prevented rephosphorylation of cofilin, a necessary step to support actin dynamics (53). Phosphorylation of another member of the LIM kinase family, LIMK2, was only marginally changed upon folate withdrawal, and it was not responsive to TGF-β treatment. This, however, could be a result of cell type-specific function of the two kinases.
FIGURE 7.
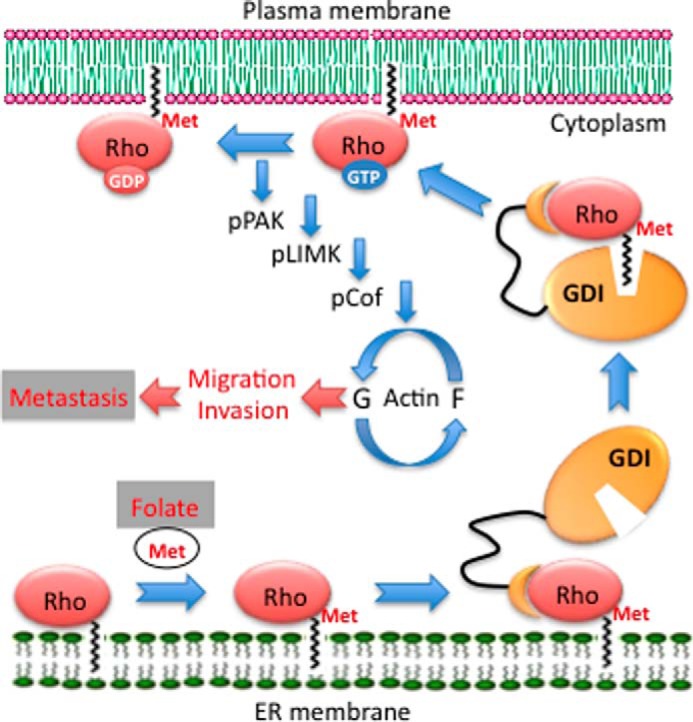
Pathway regulating metastasis by folate. Rho GTPases anchored in ER membrane through a prenyl group undergo methylation at the C-terminal cysteine by isoprenylcysteine carboxylmethyltransferase enzyme. This process depends on folate, which provides methyl groups (Met) for the reaction through methionine and S-adenosylmethionine biosynthesis. Methylation enables the extraction of GTPases by GDIs from ER membrane and its translocation to plasma membrane upon GTP loading. The GTP/GDP exchange in response to stimuli activates a cascade, which includes subsequent phosphorylation (p) of PAK, LIMK, and cofilin (Cof). The balance between phosphorylated and non-phosphorylated cofilin regulates the reorganization of actin cytoskeleton (G/F-actin cycling) responsible for cellular migration and invasion, key processes in metastasis.
Of note, another common Rho GTPase, Cdc42, was dispensable in the translation of the folate status to the effects on cellular migration, although its subcellular distribution and activation were as well affected by folate supplementation. RhoA, Rac1, and Cdc42 are prototypical members of the Rho family representing three canonical subgroups (4, 54). Different Rho GTPases can produce independent as well as redundant effects (4, 54). Specifically, Cdc42 and Rac1 revealed redundant effects toward cell polarization and lamellipodium formation, characteristic processes of mesenchymal motility (55). At the same time, the function of Rho subfamily members, which control amoeboid motility, antagonizes the effects of Rac and Cdc42 (4, 54). Of note, stress fiber assembly and contraction are predominantly controlled by the Rho subfamily through the downstream effector Rho-associated serine/threonine kinase (56), whereas both Rac1 and Cdc42, but not RhoA, activate PAK (4, 54). In line with the actin fiber formation demonstrated in our previous study, phosphorylation of PAK was strongly affected by folate status in the present study. It is possible, however, that the involvement of particular GTPases in the mediation of folate status is cell type-specific. In support of this conclusion, the regulation of the LIMK/cofilin pathway through different GTPases has been reported (33, 57–60).
Because folate metabolism controls both the de novo purine biosynthesis and methylation reactions (61), it could affect the Rho GTPase switch between an active GTP-bound and inactive GDP-bound form (5, 55) as well as the reaction catalyzed by isoprenylcysteine carboxylmethyltransferase, the enzyme responsible for the carboxyl methylation of these proteins. The potential role of folate in Rho pathways was encompassed by a study that demonstrated that the antifolate methotrexate leads to a dramatic decrease of Ras methylation and its mislocalization to cytosol, resulting in a decrease in activation of MAPK/Akt in cancer cells (62). In line with this study, folic acid prevented the ethanol-induced RhoGDI and gelsolin depletion, thus positively modulating Rho-dependent actin motility (63). Of note, a recent report demonstrated a different mode of interaction of folic acid with Rho pathways through the effect of the folate receptor on c-Src/RhoGDI (64). This mechanism, which is downstream of GTPase post-translational modification, inhibited RhoA and associated cellular migration.
Interestingly, the inhibitory effect of the dietary folate restriction on lung colonization was observed at conditions when only a partial reduction in the tissue folate took place. Notably, a rather marginal decrease in the total folate pool was observed in liver, the main organ of folate metabolism. The preservation of the folate pool in the liver indicates that animals on a folate-free diet still have active folate-dependent metabolic pathways. It is well known, however, that the withdrawal of folate from the laboratory diet is not sufficient to deplete this vitamin in rodents because they also obtain folate from their gut microflora, and the induction of severe folate deficiency in mice requires the use of an antibiotic to inhibit intestinal bacteria (65). Because we did not use an antibiotic in this study, our findings indicate that the metastasis promotion might not be associated with folate per se but rather with the dietary folate oversupplementation.
In addition to stronger lung colonization, the enhanced proliferation of tumor cells in folate-proficient mice could partially account for the increased tumor burden. Indeed, the increased nucleus:cytoplasmic ratio, a well known phenomenon in folate deficiency also observed in our experiments, results from delayed maturation of nuclei associated with the impaired DNA synthesis. However, neoplasia in lymph nodes observed in this study is most likely the result of metastasis of lung tumors, which further argues in support of the effect of folate on metastasis. Of note, our experiments showed that high dietary folate favors EMT, the process commonly viewed as the crucial mechanism for progression of carcinomas to a metastatic stage (2). EMT is required for local invasion of primary tumors, one of the initial key steps in metastasis formation. The dependence of EMT on folate was further confirmed in experiments with cultured cells that demonstrated that epithelial markers were down-regulated and mesenchymal markers were up-regulated in cells grown on folate-rich media compared with cells grown on folate-free media. Of note, in mammalian cells, folate depletion has been shown to produce hypermethylation of the promoter region of the H-cadherin gene, resulting in its transcriptional silencing (66). Importantly, in our experiments, secondary metastases to lymph nodes were observed in mice on a folate-proficient but not a folate-deficient diet.
Overall, our study provides experimental evidence for a mechanism of metastasis promotion by dietary folate and underscores the potential danger of folate oversupplementation for cancer patients. Although it is unlikely that dietary folate serves as a metastasis-initiating factor per se, its availability could be an essential factor to support metastasis-related processes. The extent to which this mechanism affects metastasis perhaps can vary in different tumor types and can be modified by other folate-dependent or folate-independent mechanisms. On a more general note, these findings highlight the interaction between nutrients and metastasis-related signaling.
Acknowledgments
We thank Drs. Philips and Matsuda for providing corresponding plasmids. The Cell and Molecular Imaging Core Facility was supported in part by National Institutes of Health Cancer Center Grant P30 CA138313 to the Hollings Cancer Center, Medical University of South Carolina.
This work was supported, in whole or in part, by National Institutes of Health Grants DK54388 and CA095030 (to S. A. K.).
- ER
- endoplasmic reticulum
- THF
- tetrahydrofolate
- EMT
- epithelial-mesenchymal transition
- LIMK
- LIM kinase
- PAK
- p21-activated kinase
- GDI
- guanine nucleotide dissociation inhibitor
- MMP
- matrix metalloproteinase
- CFP
- cyan fluorescent protein.
REFERENCES
- 1. Fidler I. J. (2003) The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis revisited. Nat. Rev. Cancer 3, 453–458 [DOI] [PubMed] [Google Scholar]
- 2. Valastyan S., Weinberg R. A. (2011) Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Friedl P., Wolf K. (2003) Tumour-cell invasion and migration: diversity and escape mechanisms. Nat. Rev. Cancer 3, 362–374 [DOI] [PubMed] [Google Scholar]
- 4. Tybulewicz V. L., Henderson R. B. (2009) Rho family GTPases and their regulators in lymphocytes. Nat. Rev. Immunol. 9, 630–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Etienne-Manneville S., Hall A. (2002) Rho GTPases in cell biology. Nature 420, 629–635 [DOI] [PubMed] [Google Scholar]
- 6. Winter-Vann A. M., Casey P. J. (2005) Post-prenylation-processing enzymes as new targets in oncogenesis. Nat. Rev. Cancer 5, 405–412 [DOI] [PubMed] [Google Scholar]
- 7. Bos J. L., Rehmann H., Wittinghofer A. (2007) GEFs and GAPs: critical elements in the control of small G proteins. Cell 129, 865–877 [DOI] [PubMed] [Google Scholar]
- 8. Bergo M. O., Leung G. K., Ambroziak P., Otto J. C., Casey P. J., Young S. G. (2000) Targeted inactivation of the isoprenylcysteine carboxyl methyltransferase gene causes mislocalization of K-Ras in mammalian cells. J. Biol. Chem. 275, 17605–17610 [DOI] [PubMed] [Google Scholar]
- 9. Cushman I., Casey P. J. (2011) RHO methylation matters: a role for isoprenylcysteine carboxylmethyltransferase in cell migration and adhesion. Cell Adh. Migr. 5, 11–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gungabissoon R. A., Bamburg J. R. (2003) Regulation of growth cone actin dynamics by ADF/cofilin. J. Histochem. Cytochem. 51, 411–420 [DOI] [PubMed] [Google Scholar]
- 11. Wang W., Eddy R., Condeelis J. (2007) The cofilin pathway in breast cancer invasion and metastasis. Nat. Rev. Cancer 7, 429–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Scott R. W., Olson M. F. (2007) LIM kinases: function, regulation and association with human disease. J. Mol. Med. 85, 555–568 [DOI] [PubMed] [Google Scholar]
- 13. Bishop A. L., Hall A. (2000) Rho GTPases and their effector proteins. Biochem. J. 348, 241–255 [PMC free article] [PubMed] [Google Scholar]
- 14. Kobayashi M., Nishita M., Mishima T., Ohashi K., Mizuno K. (2006) MAPKAPK-2-mediated LIM-kinase activation is critical for VEGF-induced actin remodeling and cell migration. EMBO J. 25, 713–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ross S. A. (2010) Evidence for the relationship between diet and cancer. Exp. Oncol. 32, 137–142 [PubMed] [Google Scholar]
- 16. Meadows G. G. (2012) Diet, nutrients, phytochemicals, and cancer metastasis suppressor genes. Cancer Metastasis Rev. 31, 441–454 [DOI] [PubMed] [Google Scholar]
- 17. Stover P. J. (2009) One-carbon metabolism-genome interactions in folate-associated pathologies. J. Nutr. 139, 2402–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yuan H. X., Xiong Y., Guan K. L. (2013) Nutrient sensing, metabolism, and cell growth control. Mol. Cell 49, 379–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reece R. J., Beynon L., Holden S., Hughes A. D., Rebora K., Sellick C. A. (2006) Nutrient-regulated gene expression in eukaryotes. Biochem. Soc. Symp. 85–96 [DOI] [PubMed] [Google Scholar]
- 20. Goldman I. D., Chattopadhyay S., Zhao R., Moran R. (2010) The antifolates: evolution, new agents in the clinic, and how targeting delivery via specific membrane transporters is driving the development of a next generation of folate analogs. Curr. Opin. Investig. Drugs 11, 1409–1423 [PubMed] [Google Scholar]
- 21. Ulrich C. M., Potter J. D. (2007) Folate and cancer—timing is everything. JAMA 297, 2408–2409 [DOI] [PubMed] [Google Scholar]
- 22. Lucock M., Yates Z. (2009) Folic acid fortification: a double-edged sword. Curr. Opin. Clin. Nutr. Metab. Care 12, 555–564 [DOI] [PubMed] [Google Scholar]
- 23. Smith A. D., Kim Y. I., Refsum H. (2008) Is folic acid good for everyone? Am. J. Clin. Nutr. 87, 517–533 [DOI] [PubMed] [Google Scholar]
- 24. Vollset S. E., Clarke R., Lewington S., Ebbing M., Halsey J., Lonn E., Armitage J., Manson J. E., Hankey G. J., Spence J. D., Galan P., Bønaa K. H., Jamison R., Gaziano J. M., Guarino P., Baron J. A., Logan R. F., Giovannucci E. L., den Heijer M., Ueland P. M., Bennett D., Collins R., Peto R., B-Vitamin Treatment Trialists' Collaboration (2013) Effects of folic acid supplementation on overall and site-specific cancer incidence during the randomised trials: meta-analyses of data on 50,000 individuals. Lancet 381, 1029–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oleinik N. V., Krupenko N. I., Krupenko S. A. (2010) ALDH1L1 inhibits cell motility via dephosphorylation of cofilin by PP1 and PP2A. Oncogene 29, 6233–6244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kasai H., Allen J. T., Mason R. M., Kamimura T., Zhang Z. (2005) TGF-β1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 6, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krupenko N. I., Dubard M. E., Strickland K. C., Moxley K. M., Oleinik N. V., Krupenko S. A. (2010) ALDH1L2 is the mitochondrial homolog of 10-formyltetrahydrofolate dehydrogenase. J. Biol. Chem. 285, 23056–23063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bergo M. O., Leung G. K., Ambroziak P., Otto J. C., Casey P. J., Gomes A. Q., Seabra M. C., Young S. G. (2001) Isoprenylcysteine carboxyl methyltransferase deficiency in mice. J. Biol. Chem. 276, 5841–5845 [DOI] [PubMed] [Google Scholar]
- 29. Aoki K., Matsuda M. (2009) Visualization of small GTPase activity with fluorescence resonance energy transfer-based biosensors. Nat. Protoc. 4, 1623–1631 [DOI] [PubMed] [Google Scholar]
- 30. Oleinik N. V., Krupenko N. I., Reuland S. N., Krupenko S. A. (2006) Leucovorin-induced resistance against FDH growth suppressor effects occurs through DHFR up-regulation. Biochem. Pharmacol. 72, 256–266 [DOI] [PubMed] [Google Scholar]
- 31. Hoeferlin L. A., Oleinik N. V., Krupenko N. I., Krupenko S. A. (2011) Activation of p21-dependent G1/G2 arrest in the absence of DNA damage as an antiapoptotic response to metabolic stress. Genes Cancer 2, 889–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cushman I., Casey P. J. (2009) Role of isoprenylcysteine carboxylmethyltransferase-catalyzed methylation in Rho function and migration. J. Biol. Chem. 284, 27964–27973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee-Hoeflich S. T., Causing C. G., Podkowa M., Zhao X., Wrana J. L., Attisano L. (2004) Activation of LIMK1 by binding to the BMP receptor, BMPRII, regulates BMP-dependent dendritogenesis. EMBO J. 23, 4792–4801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vardouli L., Moustakas A., Stournaras C. (2005) LIM-kinase 2 and cofilin phosphorylation mediate actin cytoskeleton reorganization induced by transforming growth factor-β. J. Biol. Chem. 280, 11448–11457 [DOI] [PubMed] [Google Scholar]
- 35. Collazo J., Zhu B., Larkin S., Martin S. K., Pu H., Horbinski C., Koochekpour S., Kyprianou N. (2014) Cofilin drives cell-invasive and metastatic responses to TGF-β in prostate cancer. Cancer Res. 74, 2362–2373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kalluri R., Weinberg R. A. (2009) The basics of epithelial-mesenchymal transition. J. Clin. Investig. 119, 1420–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Salem S., Harris T., Mok J. S., Li M. Y., Keenan C. R., Schuliga M. J., Stewart A. G. (2012) Transforming growth factor-β impairs glucocorticoid activity in the A549 lung adenocarcinoma cell line. Br. J. Pharmacol. 166, 2036–2048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kalluri R., Zeisberg M. (2006) Fibroblasts in cancer. Nat. Rev. Cancer 6, 392–401 [DOI] [PubMed] [Google Scholar]
- 39. Kocher B., Piwnica-Worms D. (2013) Illuminating cancer systems with genetically engineered mouse models and coupled luciferase reporters in vivo. Cancer Discov. 3, 616–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schmitz J. C., Grindey G. B., Schultz R. M., Priest D. G. (1994) Impact of dietary folic acid on reduced folates in mouse plasma and tissues. Relationship to dideazatetrahydrofolate sensitivity. Biochem. Pharmacol 48, 319–325 [DOI] [PubMed] [Google Scholar]
- 41. Bunni M. A., Priest D. G. (1991) Human red blood cell-mediated metabolism of leucovorin [(R,S)5-formyltetrahydrofolate]. Arch. Biochem. Biophys. 286, 633–637 [DOI] [PubMed] [Google Scholar]
- 42. Strickland K. C., Krupenko N. I., Krupenko S. A. (2012) Molecular mechanisms underlying the potentially adverse effects of folate. Clin. Chem. Lab. Med. 51, 607–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Molloy A. M. (2012) Genetic aspects of folate metabolism. Subcell. Biochem. 56, 105–130 [DOI] [PubMed] [Google Scholar]
- 44. Baggott J. E., Oster R. A., Tamura T. (2012) Meta-analysis of cancer risk in folic acid supplementation trials. Cancer Epidemiol. 36, 78–81 [DOI] [PubMed] [Google Scholar]
- 45. Ebbing M., Bønaa K. H., Nygård O., Arnesen E., Ueland P. M., Nordrehaug J. E., Rasmussen K., Njølstad I., Refsum H., Nilsen D. W., Tverdal A., Meyer K., Vollset S. E. (2009) Cancer incidence and mortality after treatment with folic acid and vitamin B12. JAMA 302, 2119–2126 [DOI] [PubMed] [Google Scholar]
- 46. Pogribny I. P., James S. J. (2002) De novo methylation of the p16INK4A gene in early preneoplastic liver and tumors induced by folate/methyl deficiency in rats. Cancer Lett. 187, 69–75 [DOI] [PubMed] [Google Scholar]
- 47. Steinestel J., Cronauer M. V., Müller J., Al Ghazal A., Skowronek P., Arndt A., Kraft K., Schrader M., Schrader A. J., Steinestel K. (2013) Overexpression of p16(INK4a) in urothelial carcinoma in situ is a marker for MAPK-mediated epithelial-mesenchymal transition but is not related to human papillomavirus infection. PLoS One 8, e65189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen Y. W., Chu H. C., Ze-Shiang L., Shiah W. J., Chou C. P., Klimstra D. S., Lewis B. C. (2013) p16 Stimulates CDC42-dependent migration of hepatocellular carcinoma cells. PLoS One 8, e69389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Terzis A. J., Fiskerstrand T., Refsum H., Ueland P. M., Arnold H., Bjerkvig R. (1993) Proliferation, migration and invasion of human glioma cells exposed to antifolate drugs. Int. J. Cancer 54, 112–118 [DOI] [PubMed] [Google Scholar]
- 50. Siu M. K., Kong D. S., Chan H. Y., Wong E. S., Ip P. P., Jiang L., Ngan H. Y., Le X. F., Cheung A. N. (2012) Paradoxical impact of two folate receptors, FRα and RFC, in ovarian cancer: effect on cell proliferation, invasion and clinical outcome. PLoS One 7, e47201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang T. P., Hsu S. H., Feng H. C., Huang R. F. (2012) Folate deprivation enhances invasiveness of human colon cancer cells mediated by activation of sonic hedgehog signaling through promoter hypomethylation and cross action with transcription nuclear factor-κB pathway. Carcinogenesis 33, 1158–1168 [DOI] [PubMed] [Google Scholar]
- 52. Huang T. Y., DerMardirossian C., Bokoch G. M. (2006) Cofilin phosphatases and regulation of actin dynamics. Curr. Opin. Cell Biol. 18, 26–31 [DOI] [PubMed] [Google Scholar]
- 53. Oser M., Condeelis J. (2009) The cofilin activity cycle in lamellipodia and invadopodia. J. Cell. Biochem. 108, 1252–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Heasman S. J., Ridley A. J. (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 9, 690–701 [DOI] [PubMed] [Google Scholar]
- 55. Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T., Horwitz A. R. (2003) Cell migration: integrating signals from front to back. Science 302, 1704–1709 [DOI] [PubMed] [Google Scholar]
- 56. Fukata Y., Amano M., Kaibuchi K. (2001) Rho-Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol. Sci. 22, 32–39 [DOI] [PubMed] [Google Scholar]
- 57. Yang N., Higuchi O., Ohashi K., Nagata K., Wada A., Kangawa K., Nishida E., Mizuno K. (1998) Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393, 809–812 [DOI] [PubMed] [Google Scholar]
- 58. Thirone A. C., Speight P., Zulys M., Rotstein O. D., Szászi K., Pedersen S. F., Kapus A. (2009) Hyperosmotic stress induces Rho/Rho kinase/LIM kinase-mediated cofilin phosphorylation in tubular cells: key role in the osmotically triggered F-actin response. Am. J. Physiol. Cell Physiol. 296, C463–C475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jiang X. S., Wassif C. A., Backlund P. S., Song L., Holtzclaw L. A., Li Z., Yergey A. L., Porter F. D. (2010) Activation of Rho GTPases in Smith-Lemli-Opitz syndrome: pathophysiological and clinical implications. Hum. Mol. Genet. 19, 1347–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Peris B., Gonzalez-Granero S., Ballester-Lurbe B., García-Verdugo J. M., Pérez-Roger I., Guerri C., Terrado J., Guasch R. M. (2012) Neuronal polarization is impaired in mice lacking RhoE expression. J. Neurochem. 121, 903–914 [DOI] [PubMed] [Google Scholar]
- 61. Fox J. T., Stover P. J. (2008) Folate-mediated one-carbon metabolism. Vitam. Horm. 79, 1–44 [DOI] [PubMed] [Google Scholar]
- 62. Winter-Vann A. M., Kamen B. A., Bergo M. O., Young S. G., Melnyk S., James S. J., Casey P. J. (2003) Targeting Ras signaling through inhibition of carboxyl methylation: an unexpected property of methotrexate. Proc. Natl. Acad. Sci. U.S.A. 100, 6529–6534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. García-Rodríguez S., Argüelles S., Llopis R., Murillo M. L., Machado A., Carreras O., Ayala A. (2003) Effect of prenatal exposure to ethanol on hepatic elongation factor-2 and proteome in 21 d old rats: protective effect of folic acid. Free Radic. Biol. Med. 35, 428–437 [DOI] [PubMed] [Google Scholar]
- 64. Hou T. C., Lin J. J., Wen H. C., Chen L. C., Hsu S. P., Lee W. S. (2013) Folic acid inhibits endothelial cell migration through inhibiting the RhoA activity mediated by activating the folic acid receptor/cSrc/p190RhoGAP-signaling pathway. Biochem. Pharmacol. 85, 376–384 [DOI] [PubMed] [Google Scholar]
- 65. Balaghi M., Wagner C. (1995) Folate deficiency inhibits pancreatic amylase secretion in rats. Am. J. Clin. Nutr. 61, 90–96 [DOI] [PubMed] [Google Scholar]
- 66. Jhaveri M. S., Wagner C., Trepel J. B. (2001) Impact of extracellular folate levels on global gene expression. Mol. Pharmacol. 60, 1288–1295 [DOI] [PubMed] [Google Scholar]