

Background: Cerebellar dysfunction leads to ataxia characterized by loss of balance and coordination.
Results: Lgr4 deficiency mice show an ataxia-like phenotype and impaired long term depression in cerebellum.
Conclusion: Lgr4-mediated cAMP-Creb signaling is required for motor coordination and cerebellar synaptic plasticity.
Significance: Lgr4 has potential implications for diagnosis or drug discovery of inherited cerebellar ataxia.
Keywords: 7-Helix Receptor, Ataxia, cAMP-response Element-binding Protein (Creb), Cerebellum, Signal Transduction
Abstract
Cerebellar dysfunction causes ataxia characterized by loss of balance and coordination. Until now, the molecular and neuronal mechanisms of several types of inherited cerebellar ataxia have not been completely clarified. Here, we report that leucine-rich G protein-coupled receptor 4 (Lgr4/Gpr48) is highly expressed in Purkinje cells (PCs) in the cerebellum. Deficiency of Lgr4 leads to an ataxia-like phenotype in mice. Histologically, no obvious morphological changes were observed in the cerebellum of Lgr4 mutant mice. However, the number of PCs was slightly but significantly reduced in Lgr4−/− mice. In addition, in vitro electrophysiological analysis showed an impaired long term depression (LTD) at parallel fiber-PC (PF-PC) synapses in Lgr4−/− mice. Consistently, immunostaining experiments showed that the level of phosphorylated cAMP-responsive element-binding protein (Creb) was significantly decreased in Lgr4−/− PCs. Furthermore, treatment with forskolin, an adenylyl cyclase agonist, rescued phospho-Creb in PCs and reversed the impairment in PF-PC LTD in Lgr4−/− cerebellar slices, indicating that Lgr4 is an upstream regulator of Creb signaling, which is underlying PF-PC LTD. Together, our findings demonstrate for first time an important role for Lgr4 in motor coordination and cerebellar synaptic plasticity and provide a potential therapeutic target for certain types of inherited cerebellar ataxia.
Introduction
Cerebellar ataxia is a motor disorder that manifests as problems with gait abnormalities, balance, and motor coordination (1). The origin of many forms of cerebellar ataxia, including a number of inherited disorders, can be traced to specific genetic mutations that cause the impairment of motor coordination (2). PCs,6 the sole output neurons of the cerebellar cortex, play a central role in the cerebellar circuitry. PCs integrate two types of excitatory synaptic inputs, the parallel fibers (PFs, the axons of granule cells) and the climbing fibers (CFs, originating from the inferior olive) (3, 4). As the major form of cerebellar plasticity, long term depression (LTD) at parallel fiber-PC (PF-PC) synapses has classically been assumed to be a neural correlate of cerebellar function (5, 6). Although many signaling molecules are involved in the regulation of cerebellar synaptic plasticity (7), cAMP-responsive element-binding protein (Creb), a transcription factor, has received particular attention due to its role in the late phase of cerebellar LTD (8). Creb plays a critical role in regulating intrinsic neuronal excitability, synaptic plasticity and long term memory formation in rodents (9, 10). The activation of Creb by phosphorylation is triggered by various signaling processes, including Gαs-induced adenylyl cyclase activation following G protein-coupled receptor stimulation, resulting in increased cAMP levels and PKA activation. Moreover, studies have observed the critical importance of cAMP signaling in ataxia, for example, modulation of cAMP-PKA signaling in the cerebellum played a critical role in sustained antagonism of ethanol-induced ataxia (11).
Lgr4, also called Gpr48, is one of the leucine-rich G protein-coupled receptors (Lgr4/Gpr48). Previously, we and others have demonstrated that Lgr4 activates Creb through cAMP-PKA signaling in different cell types and tissues (12, 13). Constitutively active mutant hLGR4 dramatically increased cAMP levels in vitro (14). Our results also showed that through cAMP-Creb signaling, Lgr4 down-regulated Pitx2 expression during ocular anterior segment development (15) and Atf4 expression in erythropoiesis during midgestation (16), whereas in macrophages, Lgr4 negatively regulates CD14 expression through the cAMP-Pka-Creb pathway (17). Recently, studies demonstrated that Lgr4 is one of the receptors for R-spondins, which function as amplifiers for Wnt signaling (18, 19). Lgr4 deficiency in rodents resulted in developmental abnormalities in both embryonic and postnatal stages as well as physiological dysfunction in multiple organs (20, 21). Using a gene trap strategy, we generated Lgr4 hypomorphic mice with low viability (60% mortality rate) but a normal life span. Our previous studies have demonstrated that Lgr4 plays important roles in various organs, including eye (15), bone (22), blood (16, 17), intestine (23), testis (24), mammary gland (25), and prostate (26). Although a previous report showed strong expression in neurons of many brain regions, especially in cerebellar PCs (27), the role of Lgr4 in the central nervous system has not been studied.
Here, we investigated the functional role of Lgr4 in cerebellum-related behavior and cerebellar LTD at PF-PC synapses using Lgr4−/− mice. Behavioral tests revealed that Lgr4−/− mice exhibited pronounced motor coordination defects and an ataxia-like phenotype. Using slice electrophysiological and immunochemical approaches, we demonstrated that hypomorphic mutant Lgr4 mice had impaired PF-PC LTD and a decreased level of phospho-Creb (pCreb) in PCs. However, pharmacological treatment of slices with forskolin successfully recovered the reduced p-Creb levels and restored LTD to levels seen in wild-type mice. To our knowledge, these results demonstrated for the first time that Lgr4 plays an essential role in cerebellum-related motor coordination and PF-PC LTD.
EXPERIMENTAL PROCEDURES
Mice
Lgr4−/− mice were initially generated on a mixed 129 ×C57BL/6 background as described previously (15) and were backcrossed with C57BL/6 mice for at least six generations at the beginning of this study. Heterozygous mice were intercrossed to generate homozygous Lgr4−/− mice and wild-type littermate controls. All animals were treated according to a protocol approved by the Institutional Animal Care and Use Committee.
Behavioral Analysis
All mice used for behavioral tests were 8–12 weeks old. Footprint analysis was carried out as described (28, 29). Briefly, after coating their hind feet with a nontoxic paint, mice were allowed to walk through a dark 50 cm long, 9 cm wide, 6 cm high tunnel. Then the footprint patterns made on the paper lining the floor were scored for three step parameters. Gait width is the average lateral distance between opposite left and right steps. The gait width was determined by measuring the perpendicular distance of a given step to a line connecting its opposite preceding and succeeding steps. Alternation coefficient, a value describing the uniformity of step alternation, was determined by calculating the mean of the absolute values of 0.5 minus the ratio of right-left step distance to right-right step distance for every right-left step pair taken in the tunnel. Linear movement, average change in angle between consecutive right-right steps, was calculated by drawing a line perpendicular to the direction of travel, starting at the first right footprint. The angle between this perpendicular line and each subsequent right footprint was determined, and differences in angle were calculated between each consecutive step pair. The absolute values of all angle differences were summed and divided by the number of steps scored.
For the accelerating rotarod test, the rotating rod apparatus (Med Associates Inc.) was used to measure the motor coordination and ability of mice to improve motor skill performance with training. Mice were placed on a rod (3 cm in diameter), and the rod was accelerated from 4 to 40 rpm in 5 min. The time periods that individual mice spent on the rod without falling were recorded. Three trials with a 30-min inter-trial interval were performed every day for 4 days.
In the balance beam test, mice were trained to walk from a start platform along a 28-cm wide, 1-m long square beam suspended 50 cm above bedding (30, 31). Mice were allowed to stay on the beam for a maximum of 60 s. The latency to traverse each beam and the number of times the hind feet slipped off each beam were recorded for each trial. Analysis of each measure was based on the mean scores of the two trials for each beam.
For the open field test, mice were placed in the center of a 30 × 30-cm open field and were allowed to move for 1 h. The total distance moved was recorded (Tru Scan System, Coulbourn Instruments).
In the hanging wire test, a mouse was placed on a wire cage lid and then the lid was turned upside down at a height of ∼20 cm above the cage litter to prevent the animal from climbing down. The time elapsed before falling off the wire lid was scored, with the cutoff time being set at 60 s.
In the forced swimming test, mice were placed in a glass cylinder (25 cm high × 15 cm wide) filled to a 15-cm depth with water at room temperature. The time of immobility during a 6-min test was calculated. The mice were then removed from the water, dried by the experimenter, and returned to their home cages.
For the tail suspension test, the protocol was carried out as described (32). Mice were suspended by their tail to a metal bar fixed about 15 cm above the surface of a table covered with a soft cloth in a soundproof room. The time of immobility during a 6-min test was calculated.
In the novel object recognition test, the protocol was similar to that published previously (33). Mice were individually handled for 3 days and then habituated to an open field box (40 × 40 × 20 cm) for 1 day. The object recognition task consisted of training and retention sessions. During the training session, two objects were placed equidistant from the center of the box, and each mouse was given 5 min to explore the box. The amount of time spent exploring each object was recorded. Then the mouse was returned to its home cage for 1 day. During retention tests, the trained mice were again placed individually in the box, in which one of the familiar objects was replaced by a novel object, and given 5 min to explore. The ratio of the amount of time spent exploring the novel object over the total time spent exploring both objects (discrimination index) was calculated for each mouse.
In the cued fear-conditioning test, the protocol was the same as those described previously (34). The mice were handled for 3 days and then habituated to the training chamber for 5 min 1 day before the training began. The conditioned stimulus used was an 85-db sound at 2800 Hz, and the unconditioned stimulus was one time-continuous scrambled foot shock at 0.75 mA for 2 s. After the conditioned stimulus/unconditioned stimulus paring, the mouse was allowed to stay in the chamber for another 30 s for the measurement of immediate freezing. The mice were then returned to their home cages for 1 day. During the recall test, each mouse was put into a novel chamber and monitored for 2 min (pretone freezing) and then subjected to 3 min of conditioned stimulus tone exposure. The freezing responses were recorded.
β-Galactosidase (LacZ) Staining
Mouse brains were fixed in ice-cold LacZ fixture buffer (2% formaldehyde, 0.2% glutaraldehyde, 0.02% Nonidet P-40 in PBS) for 2 h at 4 °C on a shaking platform. After washing twice in LacZ washing buffer (2 mm MgCl2, 0.01% deoxycholate, 0.02% Nonidet P-40 in PBS, pH 8.0), tissues successively moved to 15 and then 30% sucrose solutions (in PBS, pH 7.4) for cryoprotection. Brains were then embedded in Tissue Tek OCT medium (Sakura) and cryosectioned at 10–12 μm. Sections were incubated in LacZ staining buffer (0.5 mg/ml X-gal dissolved in LacZ wash buffer) overnight at room temperature. Sections were then fixed in 4% paraformaldehyde and counterstained with nuclear fast red.
Histological Analysis
Mice were anesthetized and perfused transcardially with buffered 4% paraformaldehyde in PBS, and their brains removed and postfixed in the same fixative overnight at 4 °C. For immunohistochemistry, paraffin sections (5 μm) were dewaxed, rehydrated, and boiled for 30 min in 10 mm citrate buffer, pH 6.0. Sections were incubated in primary antibody, monoclonal anti-calbindin D-28K (1:400; Sigma). Sections were then treated with 3,3-N-diaminobenzidine tetrahydrochloride (Miao Tong Biological Technology) and counterstained with hematoxylin (Sigma). For quantification, the average number of PCs, the measurement was the same as those described previously (35). Calbindin-positive PCs on five successive mediosagittal sections prepared from the indicated age of the mice were counted. Each average number of PCs per section for each genotype at the indicated time point was calculated from a total of 25 sections prepared from five mice. For proliferation assays, the sections were stained with anti-PCNA (1:1000, PC10, Cell Signaling Technology). The TUNEL assay was used for apoptosis assay according to the manufacturer's instructions using the ApopTag peroxidase in situ apoptosis detection kit (catalog no. 7100, Chemicon). The average number of PCNA-positive or TUNEL-positive PCs was counted similarly as described above. For immunofluorescence, brains or acute slices from adult mice were transcardially perfused with the same fixative as above. After a 2-h immersion in the fixative at 4 °C, both brains and slices were successively moved to 15 and then 30% sucrose solutions (in PBS, pH 7.4) for cryoprotection. Brains were embedded in Tissue Tek OCT medium (Sakura) and cryosectioned at 10–12 μm. The sections were then processed for immunolabeling using standard procedures. Mouse anti-calbindin D-28K (1:200; Sigma) and rabbit anti-phosphorylated Creb (1:100; CST) were used as primary antibodies. Nuclei were stained with DAPI (Invitrogen). Images were captured using a confocal system (Leica). All imaging and analysis were performed in blinded manner. The detailed morphology, including the entire dendritic areas and the density of PC dendritic spines, was then analyzed using ImageJ software (National Institutes of Health) by an investigator who was blind to genotype. The entire dendritic areas were calculated by closely outlining the entire dendritic tree and cell body of individual PCs, and the spines from 3 to 5 dendritic segments per cell were calculated for the density of dendritic spine measurements. The average number of pCreb-positive PCs was counted similarly as described above.
Field Potential Recordings
Thin sagittal slices (370 μm) were cut from the cerebellar vermis of 8–12-week-old mice and then kept for at least 1 h at room temperature in artificial cerebral spinal fluid (containing 124 mm NaCl, 5 mm KCl, 1.25 mm Na2HPO4, 2 mm MgSO4, 2 mm CaCl2, 26 mm NaHCO3, 10 mm d-glucose, and 100 μm picrotoxin aerated with 95% O2 and 5% CO2) before the experiments. PFs were stimulated by a bipolar electrode in the molecular layer at about two-thirds of the distance between the PC layer and the pial surface, and extracellular field potentials were recorded in the PC layer using a glass microelectrode (3–5 megohms, filled with 2 m NaCl). CFs were stimulated though a bipolar electrode placed in the granule cell layer and moved around. Test responses were evoked at 0.03 Hz. PF-PC LTD was induced by pairing PF and CF stimulation at 1Hz for 5 min. Forskolin was purchased from Sigma.
Whole-cell Patch clamp Recordings
Cerebellar slices (320 μm) were placed in a recording chamber on the fixed stage of a BX51W1 microscope (Olympus) equipped with infrared differential interference contrast optics for visualization. Whole-cell patch clamp recordings were taken from visualized Purkinje cell using a Multiclamp 700B amplifier (Axon Instruments, Foster City, CA) at room temperature (22–24 °C). Electrodes of 3–5 megohms were pulled from borosilicate glass capillaries (Sutter Instruments, Novato, CA) using a Flaming-Brown-type horizontal puller (PC-97; Sutter Instruments, Novato, CA) and filled with a solution containing 145 mm potassium gluconate, 5 mm NaCl, 1 mm MgCl2, 0.2 mm EGTA, 10 mm HEPES, 2 mm Mg-ATP, and 0.1 mm Na3-GTP. pH was adjusted to 7.2 with KOH. The holding potential in the whole-cell voltage clamp mode was set to −70 mV. Data acquisition was digitized at 20 kHz using Digidata 1440A (Axon Instruments). Analysis was performed using pClamp-10 (Axon Instruments) and MiniAnalysis (Synaptosoft Inc., Fort Lee, NJ) software; prior analysis data were filtered at 1 kHz. For miniature excitatory postsynaptic current (mEPSC) recording, 1 μm tetrodotoxin was added to block voltage-dependent Na+ currents in the perfusion solution. Picrotoxin (100 μm) was always present to block GABAA receptor-mediated inhibitory synaptic currents in all experiments.
Data Analyses and Statistics
Results are shown as mean ± S.E. For behavioral analysis, Student's t test was used for comparison of two groups. For comparisons with multiple data sets, one-way analysis of variance was used followed by a post hoc Fisher's PLSD analysis. Differences were considered statistically significant at p < 0.05. sEPSCs and mEPSCs were detected semi-automatically using MiniAnalysis (Synaptosoft Inc., Fort Lee, NJ) with specific parameters as described previously (36). The Kolmogorov-Smirnov test was used to determine whether the two distributions were different, using a criterion of p < 0.05.
RESULTS
Lgr4−/− Mice Exhibit Impaired Motor Coordination
To explore the function of Lgr4 in the cerebellum, we first analyzed the cerebellar expression pattern of Lgr4 by lacZ staining in which the lacZ reporter gene is tightly controlled by the native Lgr4 promoter (15). In Lgr4+/− mice, a high level of β-galactosidase activity was detected in PCs and within some cell bodies of interneurons in the molecular layer (Fig. 1, A and B), which is consistent with a previous report (27). The PCs were identified by their large soma and characteristic morphology, and they were further confirmed by immunostaining with a specific PC marker, calbindin D28K (Fig. 1C). These experiments suggest a potential physiological function of Lgr4 in the cerebellum.
FIGURE 1.
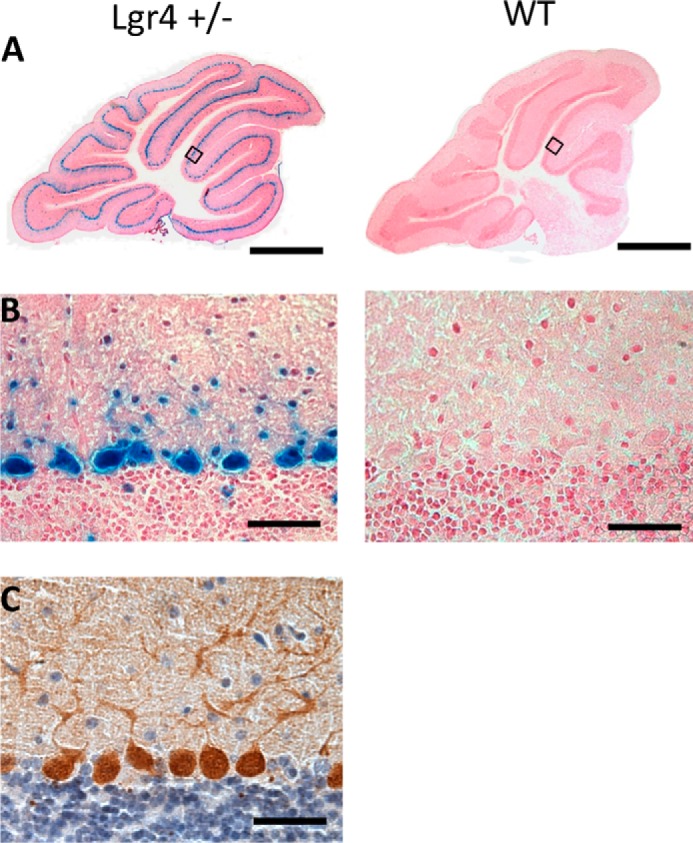
High expression of Lgr4 in cerebellar PCs. A, Lgr4 overview staining of a sagittal cerebellar section using β-galactosidase staining in Lgr4 heterozygous (left) and wild-type mice (right); sections were counterstained with nuclear fast red. B, higher magnification showed that Lgr4 was highly expressed in PCs. C, calbindin immunostaining of cerebellar PCs in Lgr4 heterozygous mice. Scale bar, 800 μm in A; 50 μm in B and C.
The cerebellum is involved in balance and the coordination of motor movement (37). For this purpose, we performed the accelerating rotarod test, which requires good motor coordination (38) on Lgr4−/− mice as well as wild-type littermate controls. Although both wild-type and mutant mice improved their performance over a 4-day training session, Lgr4−/− mice showed a significantly shorter latency to fall compared with wild-type littermates (Fig. 2A), suggesting that motor coordination was impaired in Lgr4−/− mice. We further assessed cerebellum-related balance and gait coordination in Lgr4−/− mice and their wild-type littermates. In a balance beam task, Lgr4−/− mice exhibited a significant increase in the time to traverse a beam and the number of hind foot missteps on the beam compared with their littermate controls (Fig. 2B). This indicated that cerebellum-related balance was impaired in Lgr4−/− mice. In addition, Lgr4−/− mice exhibited gait abnormalities. Instead of walking along a straight line with a smooth alternating gait as did wild-type littermates, Lgr4−/− mice weaved from side to side while moving through a tunnel, using a broader gait width in a gait that lacked a normal, uniform alternating left-right step pattern (Fig. 2, C–F). These data indicated that Lgr4−/− mice exhibited motor coordination deficits and an ataxia-like phenotype. To further test the genetic impact of Lgr4 mutation in cerebellum-related motor coordination, locomotor activity and grip/muscle strength were measured in Lgr4−/− mice with the open field and hanging wire test. As shown in Fig. 2, no significant differences were detected in general locomotor activity (Fig. 2G) and grip/muscle strength (Fig. 2H) between the two groups. Furthermore, emotion and forebrain-related memory was also evaluated in Lgr4−/− mice. Lgr4−/− mice displayed normal performance compared with wild-type mice in forced swimming, tail suspension, novel object recognition, and cued fear-conditioning tasks (Fig. 3). Together, these data suggested that impaired balance and motor coordination were not correlated with forebrain functions, and these forebrain functions were not affected by genetic defects in Lgr4. In conclusion, these data demonstrated that as a consequence of Lgr4 mutation, the Lgr4−/− mice exhibited an ataxia-like phenotype that showed deficits in balance and motor coordination, suggesting that Lgr4 plays an important role in cerebellum functions.
FIGURE 2.

Ataxia-like phenotype in Lgr4−/− mice. A, time before falling off the accelerating rotarod during the learning session. Lgr4−/− mice (n = 9) stayed on the rod for a significantly shorter period than wild-type littermates (n = 12). B, balance beam test. Lgr4−/− mice (n = 9) were significantly impaired in their ability to traverse the beam (B, left), and they had more hind foot missteps per trial on the beam (B, right) compared with wild-type littermates (n = 11). C, representative footprint patterns of wild-type and Lgr4−/− mice at 2 months. Footprint patterns were quantitatively assessed for gait width (D), step alternation (E), and linearity of movement (F). Lgr4−/− mice (n = 9) displayed a significantly broader gait width, higher alternation coefficient (indicating irregular step alternation), and increased linear movement measure (indicating nonlinear movement) as compared with wild-type mice (n = 10). G and H, rotarod and balance beam deficits are not caused by a lack of desire to move or grip/muscle strength, as Lgr4−/− mice (n = 10) moved a similar distance as wild-type littermates (n = 11) in the open-field test (G), and they revealed no significant muscle deficits in the hanging wire test (n = 10) compared with wild-type mice (n = 11) (H). Data are mean ± S.E. *, p < 0.05; **, p < 0.01.
FIGURE 3.
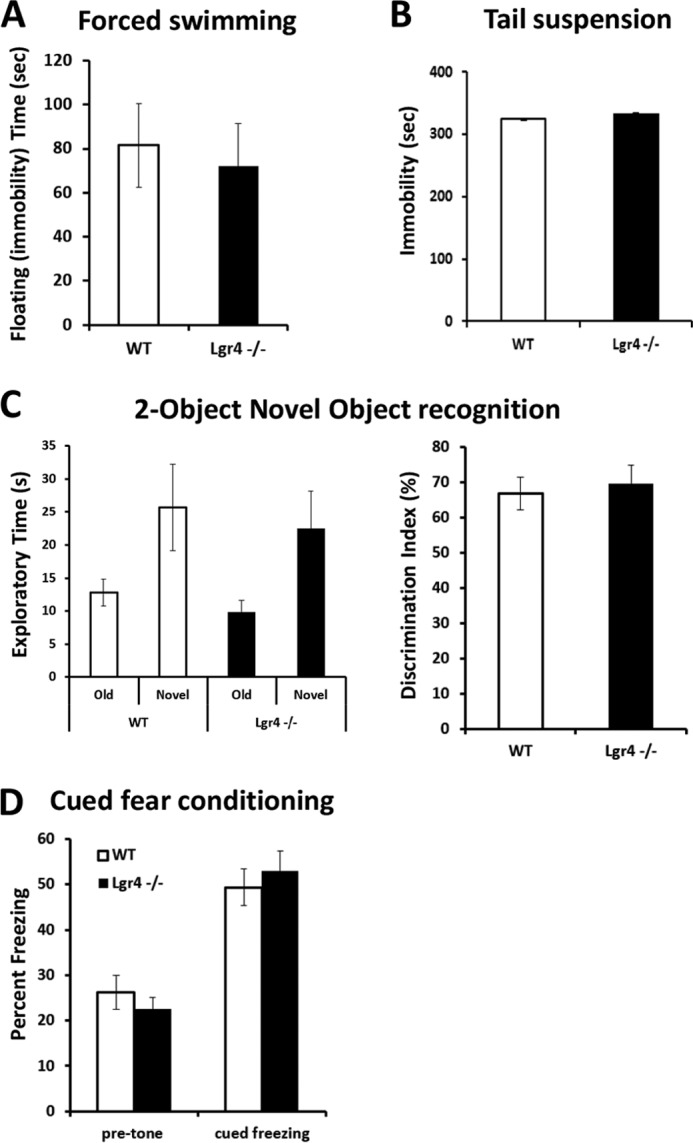
Lgr4−/− mice behave normally in despair and memory tests. A and B, forced swimming test (A) and tail suspension test (B) show a similar immobility time between Lgr4−/− mice (n = 10) and wild-type mice (n = 11). C, two-object novel object recognition test. Lgr4−/− and wild-type mice had a similar exploratory preference during training and retention sessions of the novel object recognition test (left); the discrimination index of Lgr4−/− mice (n = 10) also showed no difference compared with wild-type mice (n = 11, right). D, contextual fear-conditioning test showed no significant deficits between Lgr4−/− mice (n = 10) and wild-type mice (n = 11). Data are mean ± S.E.
Lgr4−/− Mice Exhibit a Decreased PC Population
Ataxia commonly arises from cerebellar dysfunction and is often observed in mutants with immature or degenerated PCs, so we examined whether the morphology or cell number of PCs was altered in Lgr4-deficient mice. Grossly, no obvious morphological change was observed in the cerebellum between Lgr4 mutant and wild-type controls by H&E staining (Fig. 4, A and B). Results showed that Lgr4−/− mice exhibited a normal foliation pattern in the cerebellum (Fig. 4, A–D). The PC population was examined by counting calbindin D28K-positive cells in both wild-type and Lgr4−/− mice. A significant reduction in PC number was observed in Lgr4−/− mice at 4, 8, and 28 weeks (loss of ≈22, 27, and 14%, respectively; Fig. 4, E–G). Furthermore, proliferation and apoptosis were examined using PCNA and TUNEL staining in Lgr4−/− and wild-type mice (Fig. 5). Results showed that the number of TUNEL-positive PCs was significantly increased at 8 weeks, but no difference was observed in PCNA-positive cells between the two genotypes at P5. These results suggested that the loss of PC in Lgr4−/− mice might be caused by apoptosis. Moreover, no significant changes were detected in PC dendritic arborization and spine density in Lgr4−/− mice (Fig. 4, H–K), suggesting that the loss of Lgr4 did not significantly affect the cyto-architecture of PCs.
FIGURE 4.

Number of PCs decreased in Lgr4−/− mice. A–D, hematoxylin staining of cerebellar sections of wild-type (A and C) and Lgr4−/− (B and D) mice reveals a normal overall morphology of the cerebellum of Lgr4−/− mice. C and D, enlarged views of the boxed areas indicated in (A and B). E and F, calbindin staining of wild-type (E) and Lgr4−/− (F) cerebellum reveals that the number of PCs decreased in Lgr4−/− mice. G, cell counts quantitating the numbers of PCs in wild-type and Lgr4−/− mice at different adult ages. The number of PC per section was obtained from five adjacent midsagittal sections/mouse and five mice/group. Values were normalized to the average PC count in the corresponding control group. H–K, analysis of calbindin-labeled PCs reveals no differences in the total dendritic areas (J) or spine density (K) between wild-type (n = 10) and Lgr4−/− (n = 9) mice (H and I). Data are mean ± S.E.; Student's t test was used for statistical analysis, *, p < 0.05; **, p < 0.01. Scale bar, 800 μm in A and B; 50 μm in C and D; 100 μm in E and F; 50 μm in H and I.
FIGURE 5.
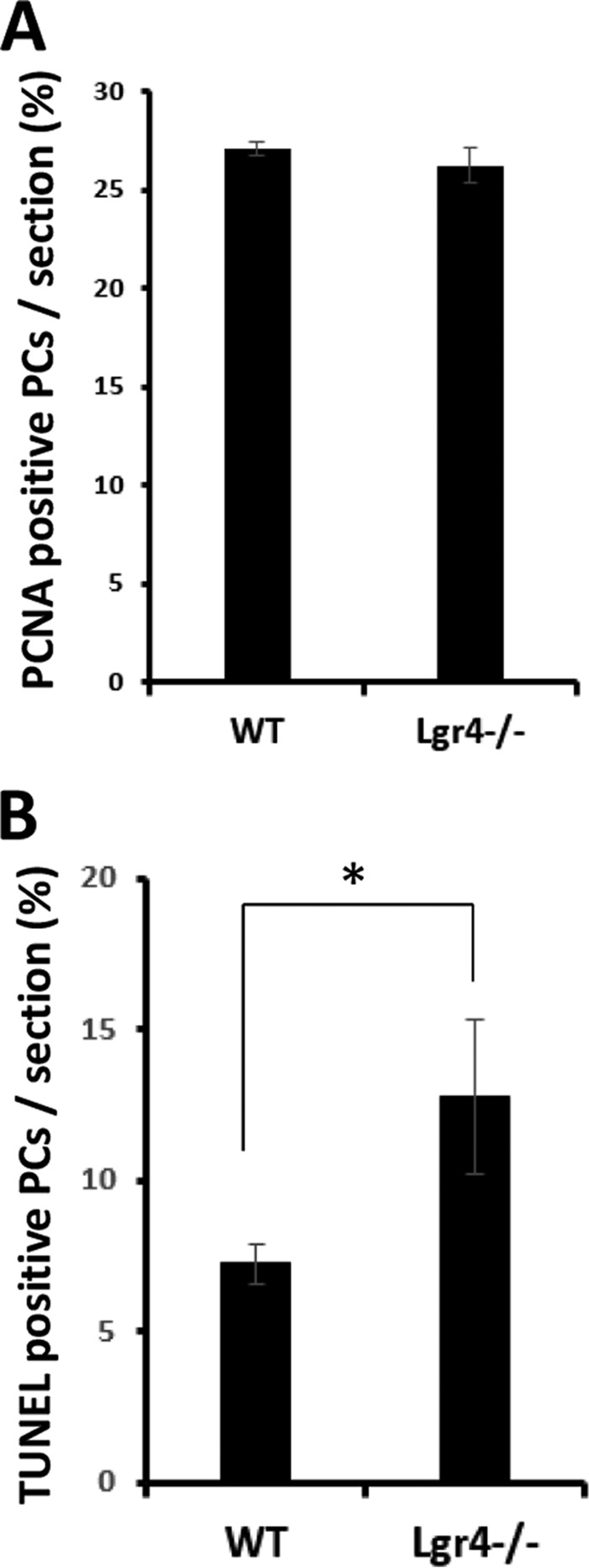
A, proliferation assays showed that the numbers of proliferating (PCNA-positive) PCs are similar in wild-type and Lgr4−/− mice at P5. B, quantitative analysis shows that a significant increase of apoptotic PCs was detected in Lgr4−/− mice at 2 months. The statistical data were obtained from five adjacent midsagittal sections/mouse and five mice/genotype. Data are mean ± S.E.; Student's t test was used for statistical analysis, *, p < 0.05.
Unaffected Basal Synaptic Transmission in PF-PC Synapses of Lgr4−/− Mice
To investigate the effects of Lgr4 deficiency on PF-PC synaptic plasticity, which has been assumed to be a neural correlate of cerebellar function (7), in vitro electrophysiological recording was performed on cerebellar slices from Lgr4−/− mice. First, we investigated the basal synaptic transmission and short term plasticity at PF-PC synapses in the Lgr4−/− mice. The input-output relationship at PF-PC synapses showed no statistically significant difference between Lgr4−/− mice and their wild-type littermates (Fig. 6A; Lgr4−/−, n = 8 slices/6 mice; WT, n = 8 slices/7 mice). Furthermore, as shown in Fig. 6B, paired-pulse facilitation of PF-PC synapses in wild-type littermates and Lgr4−/− cerebellar slices was equivalent at pulse intervals ranging from 20 to 1000 ms (Lgr4−/−, n = 10 slices/7 mice; WT, n = 10 slices/9 mice); likewise, the paired-pulse depression ratio of climbing fiber-PC (CF-PC) synapses showed no difference between wild-type littermates and Lgr4−/− mice at 20–1000-ms inter-stimulus intervals (Fig. 6C; Lgr4−/−, n = 5 slices/4 mice; WT, n = 5 slices/5 mice). These data suggest that the basal synaptic transmission and presynaptic function at PF-PC and CF-PC synapses were unaffected in Lgr4−/− mice. To further confirm the results, we tested sEPSCs and mEPSCs in PCs using whole-cell patch clamp recordings. PCs were identified by their morphological properties and electrical characterization with typical current traces as shown in Fig. 7, A and B. No significant difference was detected in frequency and amplitude of sEPSCs (Fig. 7, C and E; Lgr4−/−, n = 7 slices/5 mice; WT, n = 7 slices/6 mice) and mEPSCs (Fig. 7, D and F; Lgr4−/−, n = 7 slices/5 mice; WT, n = 7 slices/7 mice) between two groups, suggesting that the capability of presynaptic excitatory neurotransmitter release was not changed in Lgr4−/− mice. Taken together, these results indicated that the basal synaptic transmission and presynaptic function at PF-PC synapses were not altered in Lgr4−/− mice.
FIGURE 6.
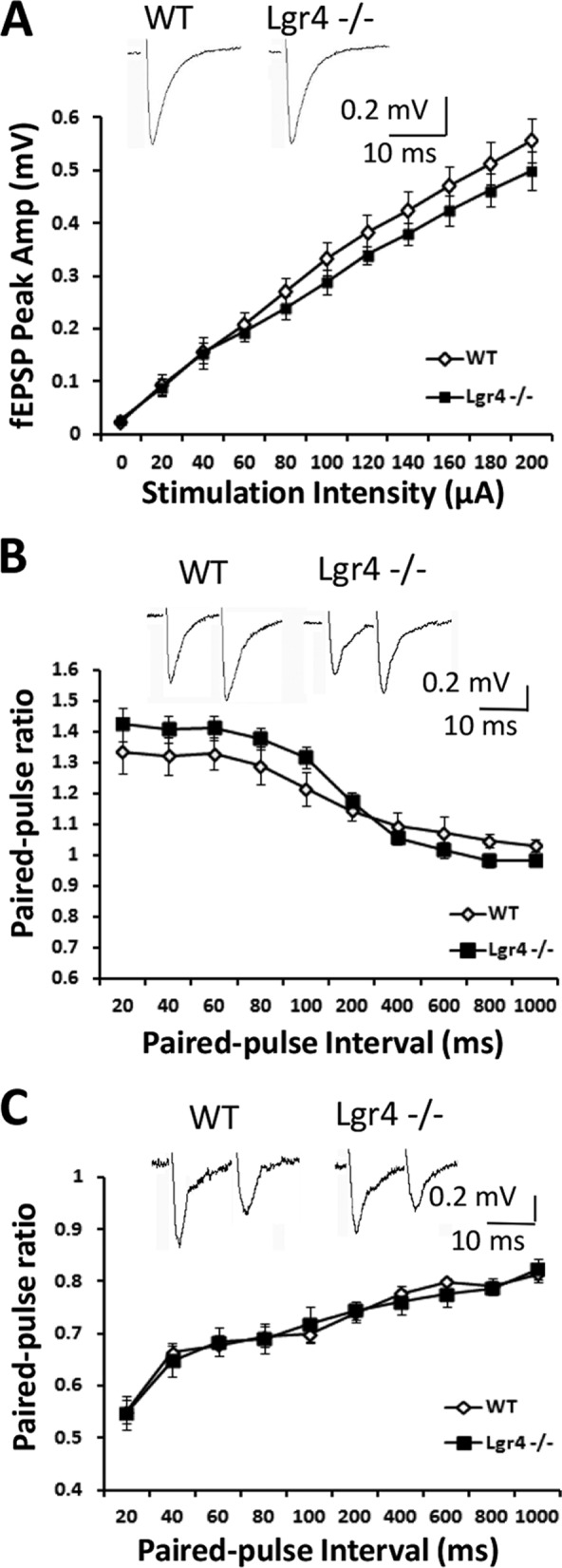
Parallel fiber-PC synaptic transmission is normal in Lgr4−/− mice. A, mean peak amplitudes of field EPSPs are not significantly different between wild-type and Lgr4−/− mice (Lgr4−/−, n = 8 slices/6 mice; WT, n = 8 slices/7 mice). B, paired-pulse ratios of PF-EPSPs are not significantly different between wild-type and Lgr4−/− mice (Lgr4−/−, n = 10 slices/7 mice; WT, n = 10 slices/9 mice). C, paired-pulse ratios of CF-EPSPs are not significantly different between wild-type and Lgr4−/− mice (Lgr4−/−, n = 5 slices/4 mice; WT, n = 5 slices/5 mice). Data are mean ± S.E., repeated-measures analysis of variance was used for statistical analysis.
FIGURE 7.

sEPSCs and mEPSCs of PCs are unaffected in Lgr4−/− mice. A, typical traces of sEPSCs recorded in PCs from wild-type and Lgr4−/− mice. B, typical traces of mEPSCs recorded in the presence of tetrodotoxin (100 μm) in PCs from wild-type and Lgr4−/− mice. C, cumulative probabilities of interval (left) and mean peak frequency (right) of sEPSCs recorded in PCs from wild-type mice (gray line; n = 7 slices/6 mice) and Lgr4−/− mice (black line; n = 7 slices/5 mice). D, cumulative probabilities of interval (left) and mean peak frequency (right) of mEPSCs recorded in PCs from wild-type mice (gray line; n = 7 slices/7 mice) and Lgr4−/− mice (black line; n = 7 slices/5 mice). E, cumulative probabilities of amplitude (left) and mean peak amplitude (right) of sEPSCs recorded in PCs from wild-type mice (gray line; n = 7 slices/6 mice) and Lgr4−/− mice (black line; n = 7 slices/5 mice). F, cumulative probabilities of amplitude (left) and mean peak amplitude (right) of mEPSCs recorded in PCs from wild-type mice (gray line; n = 7 slices/7 mice) and Lgr4−/− mice (black line; n = 7 slices/5 mice). The membrane potential was held at −70 mV.
LTD at PF-PC Synapses Is Impaired in Lgr4−/− Mice
To explore the potential causes of the observed cerebellum-related behavioral deficits in Lgr4−/− mice, we investigated the role of Lgr4 in PF-PC LTD. In control mice, 5 min of conjunctive PF and CF stimulation at 1 Hz (39, 40) reliably induced significant LTD at PF-PC synapses. However, the magnitude of LTD was significantly reduced in Lgr4−/− mice (Fig. 8, A and B; Lgr4−/−, 80 ± 3%, n = 5 slices/4 mice; WT, 63 ± 5%, n = 5 slices/5 mice, p < 0.05, Student's t test). These results suggested that Lgr4 was required for PF-PC LTD.
FIGURE 8.

Impaired parallel fiber-PC LTD in Lgr4−/− mice. A, induction of LTD at the parallel fiber to PC synapse was significantly impaired in slices of Lgr4−/− mice compared with those of controls (WT, n = 5 slices/5 mice; Lgr4−/−, n = 5 slices/4 mice; WT + forskolin, n = 5 slices/5 mice; Lgr4−/− + forskolin, n = 5 slices/4 mice). B, statistical analysis shows the effects of forskolin on LTD in Lgr4−/− slices. LTD was induced by paired PF and CF stimulation at 1 Hz for 5 min. Data are mean ± S.E., Student's t test was used for statistical analysis. *, p < 0.05.
Creb Signaling in PCs Is Impaired in Lgr4−/− Mice
Because Lgr4 has been identified as the receptor for R-spondins to amplify Wnt signaling (18, 19), we tried to determine whether downstream Wnt effectors were impaired in the Lgr4 mutant cerebellum. To our surprise, the number of PCs with nuclearly localized β-catenin was not altered in Lgr4−/− mice compared with wild-type controls (data not shown), suggesting that the defects in Lgr4−/− mice might not arise from the impairment of Wnt signaling in cerebellar PCs. Previously, we and others demonstrated that Lgr4 could also activate cAMP-Creb signaling, although the endogenous ligands responsible remain unknown (15, 16). As the cAMP-Creb cascade plays a critical role in the regulation of LTD at PF-PC synapses (8), we investigated this pathway in Lgr4 mutants. To clarify the functional role of Lgr4 in Creb signaling, which is involved in PF-PC LTD, we performed immunostaining for pCreb and calbindin. The protein level of Creb in PCs was not changed between wild-type and Lgr4 mutants (data not shown); however, phosphorylated Creb (the active form) was significantly down-regulated (Fig. 9, A–F) in PCs, which was normal in some surrounding cells. These results suggested that Lgr4 deficiency selectively attenuated the activity of Creb in PCs. To further verify whether the impaired PF-PC LTD in Lgr4 mutant mice was due to the attenuation of Creb activity in Lgr4-deficient PCs, we applied forskolin (50 μm for 10 min) to the live cerebellar slice to restore the endogenous cAMP level in Lgr4 mutant tissues. As shown in Fig. 9, J–L, the protein level of nuclear pCreb in Lgr4−/− PCs was dramatically increased in forskolin-treated slices. Furthermore, after treatment with forskolin, the LTD deficiency was reversed in Lgr4−/− slices; no significant difference in LTD magnitude was observed in Lgr4−/− cerebellar slices stimulated by forskolin compared with WT slices (Fig. 8, A and B; Lgr4−/− with forskolin, 63 ± 4%, n = 5 slices/4 mice; WT with forskolin, 58 ± 3%, n = 5 slices/5 mice; t test, p > 0.05). In conclusion, these data demonstrated that Lgr4 deficiency impaired LTD at PF-PC synapses through the aberrant modulation of the cAMP-Creb signaling pathway.
FIGURE 9.

Decreased activation of Creb in PCs of Lgr4−/− mice. Double immunofluorescence staining for anti-phosphorylated Creb antibody (green) and anti-calbindin (red) antibody in the cerebellar slices of wild-type (A–C) and Lgr4−/− (D–F) mice. Wild-type (G–I) and Lgr4−/− (J–L) slices were perfused for 10 min with 50 μm forskolin. Similar staining patterns were observed throughout the cerebellar cortices of wild-type and Lgr4−/− mice. M, statistical analysis showed that pCreb in PCs was selectively decreased in Lgr4−/− mice. The statistical data were obtained from five adjacent midsagittal sections/mouse and five mice/genotype. Data are mean ± S.E., Student's t test was used for statistical analysis, ***, p < 0.001. Scale bar, 50 μm.
DISCUSSION
Using Lgr4 hypomorphic gene-trap mice (15), we demonstrated that Lgr4 mutant mice displayed an ataxia-like phenotype. We also found a detectable decrease in the PC population, and the impaired LTD at PF-PC synapses correlate with attenuation of pCreb levels in PCs. Upon activation of cAMP-Creb signaling by application of an adenylyl cyclase activator, the decreased level of pCreb was recovered, and the impaired LTD was rescued in Lgr4−/− PCs. To our knowledge, this study reports for the first time that Lgr4 plays a crucial role in motor coordination and cerebellar synaptic plasticity in mice.
Human cerebellar ataxia is a motor disorder that manifests as gait abnormalities and problems with balance and motor coordination (1). The molecular pathogenesis of many forms of inherited cerebellar ataxia is caused by specific gene mutations (2). In accord with previous studies (27), we showed that Lgr4 was highly enriched in cerebellar PCs. In a battery of well established behavior tests, Lgr4−/− mice showed balance and motor coordination deficits, and gait abnormalities, suggesting that loss of Lgr4 results in severe ataxia-like behaviors in mice, which might be ascribed primarily to cerebellar dysfunction. Thus, mutations of LGR4 could potentially contribute to the pathogenesis of inherited cerebellar ataxia in humans, which needs to be confirmed by additional research. A further understanding of the normal physiological function of Lgr4 and how this function is altered in inherited cerebellar ataxia will be interesting for the development of therapeutic strategies.
One of the possible mechanisms of the observed defects in motor coordination behaviors could be a direct link to the loss of PC population (41, 42). Ataxia animal models that result from PC death per se (SV4 mice and T147 transgenic mice) exhibit considerable neuropathology only after there is a 50–75% loss of the PCs (43–45). However, our data showed that only 20–30% of the PC population was lost in Lgr4−/− mice, and this loss might be due to apoptosis at the adult stage, suggesting that the slightly decreased PC population might not be the main reason for the defects in Lgr4−/− mice. Therefore, changes in the synaptic function of mutant PCs lacking Lgr4 were tested.
Interestingly, the electrophysiological results revealed that Lgr4 deletion showed no difference in basal synaptic transmission and presynaptic function, but significantly impaired LTD at PF-PC synapses in adult mice. As LTD at PF-PC synapses is conventionally accepted as a neuronal correlate of cerebellar function, we assumed that this impaired LTD at PF-PC synapses at the adult stage might be the other major possible mechanism underlying the cerebellum-related behavioral defects in Lgr4−/− mice. In addition, the PF-PC LTD induced by our protocols is known to be expressed postsynaptically (39, 46), indicating there may be a change in postsynaptic signaling involved in the impaired cerebellar LTD at PF-PC synapses.
Our data and previous studies have observed that the soma of PCs is strongly labeled by lacZ staining, and some other soma of interneurons in the molecular layer are also labeled with lacZ (27). As PCs are the sole output neurons in the cerebellar cortex, the high expression of Lgr4 in PCs indicates a potentially important role of Lgr4 in the cerebellum. Considering that some interneurons in the molecular layer also express Lgr4, the upstream signaling of PC is also worth investigating. One candidate of Lgr4 downstream signaling is the Wnt/β-catenin signaling, which is triggered by R-spondin proteins in different organs (19, 23, 24). The Wnt/β-catenin signaling has also been shown to play an important role in the development of the central nervous system (47, 48), but its function in cerebellar development is not well defined. Wnt/β-catenin signaling is active in a highly dynamic pattern during cerebellum development (49). Using different Cre promoters, studies have found that conditional knock-out of β-catenin at early stages of embryogenesis led to agenesis of the cerebellum (50) or premature neural precursor cell fate commitment (51, 52). A recent report has observed that knock-out of β-catenin after E12.5 caused abnormal cerebellar foliation and lamination but did not have a cell autonomous role in developmental properties of major cerebellar cell types (52). These findings raise the possibility that the main function of Wnt/β-catenin signaling in the cerebellum is in regulation of developmental processes. Surprisingly, our data showed that the foliation and lamination of the cerebellum in Lgr4−/− mice is normal, and no significant difference in the β-catenin activation level was observed in Lgr4−/− PCs compared with their control littermates (data not shown), suggesting that the defects in Lgr4−/− mice might not be caused by an impairment of Wnt/β-catenin signaling in cerebellar PCs.
In addition to potentiating Wnt signaling following R-spondin binding, Lgr4 can trigger Creb signaling by the cAMP pathway following binding of an as-yet undiscovered ligand (15, 16). This cAMP-Creb signaling pathway is heavily implicated in different forms of synaptic plasticity in various brain regions. In hippocampal neurons, both Creb phosphorylation and the induction of a CRE-driven lacZ reporter construct were found in CA1 pyramidal neurons in late phase long term potentiation (9, 53, 54). Furthermore, it has also been reported that the late phase of LTD is blocked in cultured PCs with an expression vector encoding a dominant inhibitory form of Creb (8). Indeed, in Lgr4−/− mice, immunoreactivity of p-Creb was significantly reduced in PCs and could be rescued by forskolin, an adenylyl cyclase activator. These results provided strong evidence that Lgr4 loss impaired Creb signaling. In addition, our experiments also showed that forskolin successfully rescued the impaired LTD at PF-PC synapses from Lgr4−/− cerebellar slices, indicating that the impairment of LTD in Lgr4−/− mice is mainly due to the attenuation of cAMP-Creb signaling downstream of Lgr4.
In addition, the cerebellum has long been viewed as a machine-like structure designed for fine-tuning of sensorimotor gains and fast adaptation of motor output in response to changing behavioral needs using compensatory eye movement and classical eye-blink conditioning tests (55). Whether Lgr4 is involved in these cerebellar functions should be further addressed. Additionally, the expression of Lgr4 was observed in the hippocampus (27). It is notable that the hippocampus plays a critical role in the formation of memory (56, 57), and the function of Lgr4 in the forebrain will need to pay more attention in the near future.
In summary, this study is the first to demonstrate that Lgr4 deficiency causes an ataxia-like phenotype and impairment of cerebellar PF-PC LTD. Using different strategies, we also demonstrated for the first time that Lgr4 modulated Creb activation by cAMP-Creb signaling in cerebellar PCs. Because the pathogenesis of several types of human inherited cerebellar ataxia is largely unknown, our observations provide deeper insights into inherited cerebellar ataxia and one more possible underlying neuronal and molecular mechanism. Therefore, LGR4 is important as a potential therapeutic target of human inherited cerebellar ataxia, as only limited therapeutic approaches are currently available.
This work was supported in part by State Key Development Programs of China Grants 2012CB910400 (to M. L.) and 2010CB945403 (to D. L.), National Natural Science Foundation of China Grants 31371455, 30930055, 31070993, and 31271200, and Science and Technology Commission of Shanghai Municipality Grants 12XD1406100 and 14140900300.
- PC
- Purkinje cell
- LTD
- long term depression
- PF
- parallel fiber
- Creb
- cAMP-responsive element-binding protein
- CF
- climbing fiber
- mEPSC
- miniature excitatory postsynaptic current
- sEPSC
- spontaneous EPSC
- PCNA
- proliferating cell nuclear antigen.
REFERENCES
- 1. Trouillas P., Takayanagi T., Hallett M., Currier R. D., Subramony S. H., Wessel K., Bryer A., Diener H. C., Massaquoi S., Gomez C. M., Coutinho P., Ben Hamida M., Campanella G., Filla A., Schut L., Timann D., Honnorat J., Nighoghossian N., Manyam B. (1997) International Cooperative Ataxia Rating Scale for pharmacological assessment of the cerebellar syndrome. The Ataxia Neuropharmacology Committee of the World Federation of Neurology. J. Neurol. Sci. 145, 205–211 [DOI] [PubMed] [Google Scholar]
- 2. Manto M., Marmolino D. (2009) Cerebellar ataxias. Curr. Opin. Neurol. 22, 419–429 [DOI] [PubMed] [Google Scholar]
- 3. Hansel C., Linden D. J., D'Angelo E. (2001) Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat. Neurosci. 4, 467–475 [DOI] [PubMed] [Google Scholar]
- 4. Ito M. (2002) The molecular organization of cerebellar long term depression. Nat. Rev. Neurosci. 3, 896–902 [DOI] [PubMed] [Google Scholar]
- 5. Jörntell H., Hansel C. (2006) Synaptic memories upside down: bidirectional plasticity at cerebellar parallel fiber-Purkinje cell synapses. Neuron 52, 227–238 [DOI] [PubMed] [Google Scholar]
- 6. Ito M. (2001) Cerebellar long term depression: characterization, signal transduction, and functional roles. Physiol. Rev. 81, 1143–1195 [DOI] [PubMed] [Google Scholar]
- 7. Evans G. J. (2007) Synaptic signalling in cerebellar plasticity. Biol. Cell 99, 363–378 [DOI] [PubMed] [Google Scholar]
- 8. Ahn S., Ginty D. D., Linden D. J. (1999) A late phase of cerebellar long term depression requires activation of CaMKIV and CREB. Neuron 23, 559–568 [DOI] [PubMed] [Google Scholar]
- 9. Lonze B. E., Ginty D. D. (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35, 605–623 [DOI] [PubMed] [Google Scholar]
- 10. Barco A., Marie H. (2011) Genetic approaches to investigate the role of CREB in neuronal plasticity and memory. Mol. Neurobiol. 44, 330–349 [DOI] [PubMed] [Google Scholar]
- 11. Dar M. S. (2011) Sustained antagonism of acute ethanol-induced ataxia following microinfusion of cyclic AMP and cpt-cAMP in the mouse cerebellum. Pharmacol. Biochem. Behav. 98, 341–348 [DOI] [PubMed] [Google Scholar]
- 12. Yi T., Weng J., Siwko S., Luo J., Li D., Liu M. (2014) Lgr4/Gpr48 inactivation leads to aniridia-genitourinary anomalies-mental retardation syndrome defects. J. Biol. Chem. 289, 8767–8780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang F., Zhang X., Wang J., Chen M., Fan N., Ma Q., Liu R., Wang R., Li X., Liu M., Ning G. (2014) LGR4 acts as a link between the peripheral circadian clock and lipid metabolism in liver. J. Mol. Endocrinol. 52, 133–143 [DOI] [PubMed] [Google Scholar]
- 14. Gao Y., Kitagawa K., Shimada M., Uchida C., Hattori T., Oda T., Kitagawa M. (2006) Generation of a constitutively active mutant of human GPR48/LGR4, a G-protein-coupled receptor. Hokkaido Igaku Zasshi 81, 101–105, 107, 109 [PubMed] [Google Scholar]
- 15. Weng J., Luo J., Cheng X., Jin C., Zhou X., Qu J., Tu L., Ai D., Li D., Wang J., Martin J. F., Amendt B. A., Liu M. (2008) Deletion of G protein-coupled receptor 48 leads to ocular anterior segment dysgenesis (ASD) through down-regulation of Pitx2. Proc. Natl. Acad. Sci. U.S.A. 105, 6081–6086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Song H., Luo J., Luo W., Weng J., Wang Z., Li B., Li D., Liu M. (2008) Inactivation of G-protein-coupled receptor 48 (Gpr48/Lgr4) impairs definitive erythropoiesis at midgestation through down-regulation of the ATF4 signaling pathway. J. Biol. Chem. 283, 36687–36697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Du B., Luo W., Li R., Tan B., Han H., Lu X., Li D., Qian M., Zhang D., Zhao Y., Liu M. (2013) Lgr4/Gpr48 negatively regulates TLR2/4-associated pattern recognition and innate immunity by targeting CD14 expression. J. Biol. Chem. 288, 15131–15141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Glinka A., Dolde C., Kirsch N., Huang Y. L., Kazanskaya O., Ingelfinger D., Boutros M., Cruciat C. M., Niehrs C. (2011) LGR4 and LGR5 are R-spondin receptors mediating Wnt/β-catenin and Wnt/PCP signalling. EMBO Rep. 12, 1055–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carmon K. S., Gong X., Lin Q., Thomas A., Liu Q. (2011) R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/β-catenin signaling. Proc. Natl. Acad. Sci. U.S.A. 108, 11452–11457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mazerbourg S., Bouley D. M., Sudo S., Klein C. A., Zhang J. V., Kawamura K., Goodrich L. V., Rayburn H., Tessier-Lavigne M., Hsueh A. J. (2004) Leucine-rich repeat-containing, G protein-coupled receptor 4 null mice exhibit intrauterine growth retardation associated with embryonic and perinatal lethality. Mol. Endocrinol. 18, 2241–2254 [DOI] [PubMed] [Google Scholar]
- 21. Mendive F., Laurent P., Van Schoore G., Skarnes W., Pochet R., Vassart G. (2006) Defective postnatal development of the male reproductive tract in LGR4 knockout mice. Dev. Biol. 290, 421–434 [DOI] [PubMed] [Google Scholar]
- 22. Luo J., Zhou W., Zhou X., Li D., Weng J., Yi Z., Cho S. G., Li C., Yi T., Wu X., Li X. Y., de Crombrugghe B., Höök M., Liu M. (2009) Regulation of bone formation and remodeling by G-protein-coupled receptor 48. Development 136, 2747–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu S., Qian Y., Li L., Wei G., Guan Y., Pan H., Guan X., Zhang L., Lu X., Zhao Y., Liu M., Li D. (2013) Lgr4 gene deficiency increases susceptibility and severity of dextran sodium sulfate-induced inflammatory bowel disease in mice. J. Biol. Chem. 288, 8794–8803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qian Y., Liu S., Guan Y., Pan H., Guan X., Qiu Z., Li L., Gao N., Zhao Y., Li X., Lu Y., Liu M., Li D. (2013) Lgr4-mediated Wnt/β-catenin signaling in peritubular myoid cells is essential for spermatogenesis. Development 140, 1751–1761 [DOI] [PubMed] [Google Scholar]
- 25. Wang Y., Dong J., Li D., Lai L., Siwko S., Li Y., Liu M. (2013) Lgr4 regulates mammary gland development and stem cell activity through the pluripotency transcription factor Sox2. Stem Cells 31, 1921–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luo W., Rodriguez M., Valdez J. M., Zhu X., Tan K., Li D., Siwko S., Xin L., Liu M. (2013) Lgr4 is a key regulator of prostate development and prostate stem cell differentiation. Stem Cells 31, 2492–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Van Schoore G., Mendive F., Pochet R., Vassart G. (2005) Expression pattern of the orphan receptor LGR4/GPR48 gene in the mouse. Histochem. Cell Biol. 124, 35–50 [DOI] [PubMed] [Google Scholar]
- 28. Becker E. B., Oliver P. L., Glitsch M. D., Banks G. T., Achilli F., Hardy A., Nolan P. M., Fisher E. M., Davies K. E. (2009) A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc. Natl. Acad. Sci. U.S.A. 106, 6706–6711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Clark H. B., Burright E. N., Yunis W. S., Larson S., Wilcox C., Hartman B., Matilla A., Zoghbi H. Y., Orr H. T. (1997) Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J. Neurosci. 17, 7385–7395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maltecca F., Magnoni R., Cerri F., Cox G. A., Quattrini A., Casari G. (2009) Haploinsufficiency of AFG3L2, the gene responsible for spinocerebellar ataxia type 28, causes mitochondria-mediated Purkinje cell dark degeneration. J. Neurosci. 29, 9244–9254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carter R. J., Lione L. A., Humby T., Mangiarini L., Mahal A., Bates G. P., Dunnett S. B., Morton A. J. (1999) Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J. Neurosci. 19, 3248–3257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cryan J. F., O'Leary O. F., Jin S. H., Friedland J. C., Ouyang M., Hirsch B. R., Page M. E., Dalvi A., Thomas S. A., Lucki I. (2004) Norepinephrine-deficient mice lack responses to antidepressant drugs, including selective serotonin reuptake inhibitors. Proc. Natl. Acad. Sci. U.S.A. 101, 8186–8191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang D., Cui Z., Zeng Q., Kuang H., Wang L. P., Tsien J. Z., Cao X. (2009) Genetic enhancement of memory and long term potentiation but not CA1 long term depression in NR2B transgenic rats. PLoS One 4, e7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cao X., Wang H., Mei B., An S., Yin L., Wang L. P., Tsien J. Z. (2008) Inducible and selective erasure of memories in the mouse brain via chemical-genetic manipulation. Neuron 60, 353–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nishihara E., Yoshida-Komiya H., Chan C. S., Liao L., Davis R. L., O'Malley B. W., Xu J. (2003) SRC-1 null mice exhibit moderate motor dysfunction and delayed development of cerebellar Purkinje cells. J. Neurosci. 23, 213–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lonchamp E., Gambino F., Dupont J. L., Doussau F., Valera A., Poulain B., Bossu J. L. (2012) Pre- and post-synaptic NMDA effects targeting Purkinje cells in the mouse cerebellar cortex. PLoS One 7, e30180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Strick P. L. (1985) The cerebellum: the cerebellum and neural control. Science 229, 547. [DOI] [PubMed] [Google Scholar]
- 38. Jones B. J., Roberts D. J. (1968) The quantitative measurement of motor inco-ordination in naive mice using an acelerating rotarod. J. Pharm. Pharmacol. 20, 302–304 [DOI] [PubMed] [Google Scholar]
- 39. Hansel C., de Jeu M., Belmeguenai A., Houtman S. H., Buitendijk G. H., Andreev D., De Zeeuw C. I., Elgersma Y. (2006) αCaMKII Is essential for cerebellar LTD and motor learning. Neuron 51, 835–843 [DOI] [PubMed] [Google Scholar]
- 40. van Woerden G. M., Hoebeek F. E., Gao Z., Nagaraja R. Y., Hoogenraad C. C., Kushner S. A., Hansel C., De Zeeuw C. I., Elgersma Y. (2009) βCaMKII controls the direction of plasticity at parallel fiber-Purkinje cell synapses. Nat. Neurosci. 12, 823–825 [DOI] [PubMed] [Google Scholar]
- 41. Thomas J. D., Goodlett C. R., West J. R. (1998) Alcohol-induced Purkinje cell loss depends on developmental timing of alcohol exposure and correlates with motor performance. Brain Res. Dev. Brain Res. 105, 159–166 [DOI] [PubMed] [Google Scholar]
- 42. Gilman S., Sima A. A., Junck L., Kluin K. J., Koeppe R. A., Lohman M. E., Little R. (1996) Spinocerebellar ataxia type 1 with multiple system degeneration and glial cytoplasmic inclusions. Ann. Neurol. 39, 241–255 [DOI] [PubMed] [Google Scholar]
- 43. Mullen R. J., Eicher E. M., Sidman R. L. (1976) Purkinje cell degeneration, a new neurological mutation in the mouse. Proc. Natl. Acad. Sci. U.S.A. 73, 208–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Feddersen R. M., Ehlenfeldt R., Yunis W. S., Clark H. B., Orr H. T. (1992) Disrupted cerebellar cortical development and progressive degeneration of Purkinje cells in SV40 T antigen transgenic mice. Neuron 9, 955–966 [DOI] [PubMed] [Google Scholar]
- 45. Feddersen R. M., Clark H. B., Yunis W. S., Orr H. T. (1995) In vivo viability of postmitotic Purkinje neurons requires pRb family member function. Mol. Cell. Neurosci. 6, 153–167 [DOI] [PubMed] [Google Scholar]
- 46. Coesmans M., Weber J. T., De Zeeuw C. I., Hansel C. (2004) Bidirectional parallel fiber plasticity in the cerebellum under climbing fiber control. Neuron 44, 691–700 [DOI] [PubMed] [Google Scholar]
- 47. Kalani M. Y., Cheshier S. H., Cord B. J., Bababeygy S. R., Vogel H., Weissman I. L., Palmer T. D., Nusse R. (2008) Wnt-mediated self-renewal of neural stem/progenitor cells. Proc. Natl. Acad. Sci. U.S.A. 105, 16970–16975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wexler E. M., Paucer A., Kornblum H. I., Palmer T. D., Plamer T. D., Geschwind D. H. (2009) Endogenous Wnt signaling maintains neural progenitor cell potency. Stem Cells 27, 1130–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Selvadurai H. J., Mason J. O. (2011) Wnt/β-catenin signalling is active in a highly dynamic pattern during development of the mouse cerebellum. PLoS One 6, e23012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brault V., Moore R., Kutsch S., Ishibashi M., Rowitch D. H., McMahon A. P., Sommer L., Boussadia O., Kemler R. (2001) Inactivation of the β-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128, 1253–1264 [DOI] [PubMed] [Google Scholar]
- 51. Schüller U., Rowitch D. H. (2007) β-Catenin function is required for cerebellar morphogenesis. Brain Res. 1140, 161–169 [DOI] [PubMed] [Google Scholar]
- 52. Wen J., Yang H. B., Zhou B., Lou H. F., Duan S. (2013) β-Catenin is critical for cerebellar foliation and lamination. PLoS One 8, e64451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. De Zeeuw C. I., Yeo C. H. (2005) Time and tide in cerebellar memory formation. Curr. Opin. Neurobiol. 15, 667–674 [DOI] [PubMed] [Google Scholar]
- 54. Blom J. M., Tascedda F., Carra S., Ferraguti C., Barden N., Brunello N. (2002) Altered regulation of CREB by chronic antidepressant administration in the brain of transgenic mice with impaired glucocorticoid receptor function. Neuropsychopharmacology 26, 605–614 [DOI] [PubMed] [Google Scholar]
- 55. Schonewille M., Belmeguenai A., Koekkoek S. K., Houtman S. H., Boele H. J., van Beugen B. J., Gao Z., Badura A., Ohtsuki G., Amerika W. E., Hosy E., Hoebeek F. E., Elgersma Y., Hansel C., De Zeeuw C. I. (2010) Purkinje cell-specific knockout of the protein phosphatase PP2B impairs potentiation and cerebellar motor learning. Neuron 67, 618–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Squire L. R. (1992) Memory and the hippocampus: a synthesis from findings with rats, monkeys, and humans. Psychol. Rev. 99, 195–231 [DOI] [PubMed] [Google Scholar]
- 57. Tang Y. P., Shimizu E., Dube G. R., Rampon C., Kerchner G. A., Zhuo M., Liu G., Tsien J. Z. (1999) Genetic enhancement of learning and memory in mice. Nature 401, 63–69 [DOI] [PubMed] [Google Scholar]