

Background: Mathematical simulations of apoptosome formation and mechanisms of caspase-9 activation were conducted and validated against experimental data.
Results: Only simulations that assume allosteric caspase-9 activation can accurately reproduce all experimental data.
Conclusion: A mechanism of allosteric caspase-9 activation is favored over homodimerization-based activation.
Significance: Our findings challenge the dogma that all initiator caspases are activated by homodimerization.
Keywords: Apoptosis, Caspase, Cell Death, Computational Biology, Systems Biology, Apaf-1, Apoptosome, Caspase-3, Caspase-9
Abstract
The protease caspase-9 is activated on the apoptosome, a multiprotein signal transduction platform that assembles in response to mitochondria-dependent apoptosis initiation. Despite extensive molecular research, the assembly of the holo-apoptosome and the process of caspase-9 activation remain incompletely understood. Here, we therefore integrated quantitative data on the molecular interactions and proteolytic processes during apoptosome formation and apoptosis execution and conducted mathematical simulations to investigate the resulting biochemical signaling, quantitatively and kinetically. Interestingly, when implementing the homodimerization of procaspase-9 as a prerequisite for activation, the calculated kinetics of apoptosis execution and the efficacy of caspase-3 activation failed to replicate experimental data. In contrast, assuming a scenario in which procaspase-9 is activated allosterically upon binding to the apoptosome backbone, the mathematical simulations quantitatively and kinetically reproduced all experimental data. These data included a XIAP threshold concentration at which apoptosis execution is suppressed in HeLa cervical cancer cells, half-times of procaspase-9 processing, as well as the molecular timer function of the apoptosome. Our study therefore provides novel mechanistic insight into apoptosome-dependent apoptosis execution and suggests that caspase-9 is activated allosterically by binding to the apoptosome backbone. Our findings challenge the currently prevailing dogma that all initiator procaspases require homodimerization for activation.
Introduction
Members of the cysteine aspartate-specific proteases (caspases) protein family are important mediators of apoptosis signaling and fulfill key roles during the initiation and execution phases of apoptosis. The zymogens (procaspases) of the initiator caspases-2, -8, -9, and -10 are expressed as inactive monomers and bind to specific activation platforms upon apoptosis induction. It is generally believed that these activation platforms serve to locally accumulate initiator procaspase monomers, resulting in their proximity-induced homodimerization, activation, and auto-proteolytic processing (1). The PIDDosome and the death-inducing signaling complex have been identified as activation platforms for initiator caspases-2 or -8/-10, respectively (2, 3), whereas the apoptosome provides the activation platform for initiator caspase-9 (4–6). The apoptosome consists of apoptotic protease-activating factor 1 (Apaf-1)2 and the co-factors dATP/ATP and cytochrome c (4, 7, 8). Apaf-1 is a member of the STAND (signal transduction ATPases with numerous domains) family (9) and comprises three functional domains: a NH2-terminal caspase recruitment domain (CARD), a nucleotide-binding and oligomerization domain, and a COOH-terminal WD40 repeat domain (4, 10). Upon activation of the mitochondrial apoptosis pathway, cytochrome c is released into the cytosol and activates Apaf-1, and active Apaf-1 then homo-oligomerizes into the heptameric apoptosome backbone (11, 12). In the fully assembled complex, the CARDs of Apaf-1 form a ring structure that sits above the central hub of the apoptosome (13), and interactions with the CARD in the procaspase-9 prodomain recruit procaspase-9 into the apoptosome (14). Binding of procaspase-9 to the apoptosome results in its activation (7, 15). As for other initiator caspases and in contrast to effector caspases, intra-chain cleavage of procaspase-9 is not required for activation but rather a consequence of autocatalysis following activation (16). Processed caspase-9 can be released from the apoptosome and thereby is inactivated. The apoptosome therefore acts as a proteolytic molecular timer that provides transient caspase-9 activities (17). Active caspase-9 cleaves and activates the main proteases of the apoptosis execution phase, effector caspases-3 and -7 (18).
The molecular mechanism of caspase-9 activation on the apoptosome is still incompletely understood. Two hypotheses for possible activation mechanisms have been proposed. The dimerization hypothesis suggests that binding to the apoptosome facilitates the activation of procaspase-9 by homodimerization (19, 20). Experiments in which the CARD of procaspase-9 was replaced with a leucine zipper domain demonstrated that procaspase-9 indeed can be activated by forced dimerization (21). A similar finding was also made when dimerizing procaspase-9 in presence of high concentrations of kosmotropic salts (20). However, in contrast to all other caspases, the caspase-9 dimer is asymmetric and contains only one active catalytic site (19). The allosteric activation hypothesis instead suggests that binding to the heptameric Apaf-1 complex may allosterically induce a conformational change in procaspase-9, which results in the activation of the latter (22). In support of this hypothesis, it was found that procaspase-9 and processed caspase-9 are active when bound to the apoptosome, whereas cytosolic processed caspase-9 is inactive and monomeric (22, 23). Furthermore, caspase-9 bound to the apoptosome processes its physiological substrate procaspase-3 significantly more efficiently than force-dimerized free caspase-9 does (24). Despite a substantial body of published data on and quantitative investigations of apoptosome formation and caspase-9 processing, it therefore is still unclear whether caspase-9 is activated primarily by homodimerization or allosterically on the apoptosome backbone (25). We addressed this fundamental question in the current study by investigating both scenarios by a systems biology-based approach. First, we integrated the available quantitative biochemical knowledge on the signaling kinetics and molecular interactions during Apaf-1 activation and oligomerization and from this generated a cell physiologically relevant mathematical model for the formation of the apoptosome backbone. This core model was subsequently extended to compare the signaling processes during apoptosis execution for scenarios of allosteric and homodimerization-based caspase-9 activation. The results of these mathematical simulations were then validated against the body of published in vitro and in cellulo experimental data.
EXPERIMENTAL PROCEDURES
Model Implementation and Parameterization
All models were implemented as ordinary differential equations. An overview of all model components, reactions, and their parameterization can be found in the supplemental tables. MATLAB 2007b (The Mathworks, Cambridge, UK) was used as the programming and modeling environment. The reaction systems were solved using the ODE15s solver that implements a backward differentiation formula for numerical integration.
Determination of Kd for Procaspase-9 Binding to Apaf-1
The Kd value for binding of procaspase-9 to Apaf-1 was determined from an IC50 value reported by Palacios-Rodriguez et al. (26). The approach is described in brief in the following. Palacios-Rodriguez et al. initiated the assembly of the heptameric Apaf-1 complex in vitro by addition of dATP and cytochrome c, and in the presence of procaspase-9, which the latter binding to the apoptosome via CARD-CARD interactions. A catalytically inactive procaspase-9 mutant was added a competitive inhibitor in these assays. LEHD-afc was used as a fluorigenic caspase-9 substrate at a concentration of 100 μm, and the Km value of caspase-9 toward the substrate was reported as 686 μm (24). The IC50 from these measurements therefore can be used to determine the dissociation constant of procaspase-9, using the Cheng-Prusoff equation (27). An IC50 of 0.8 μm (26) therefore leads to a Kd of 0.7 μm for procaspase-9 binding to the apoptosome.
Model Adaptation for the Study of the Molecular Timer Function of the Apoptosome
Conditions as detailed in supplemental Table 11 were used to investigate whether APOPTO-ALL or APOPTO-DIM variants can replicate the molecular timer behavior of the apoptosome. The mathematical models were parameterized to resemble the in vitro preincubation of Apaf-1 with dATP, cytochrome c, procaspase-9, and a caspase-3 substrate. Protein degradation and production were switched off. Procaspase-3 was added with a delay (5, 10, 20, or 30 min). The velocity of procaspase-3 activation was determined increases in cleaved caspase-3 substrate 1 min after procaspase-3 addition. These conditions replicate the in vitro experimental conditions used by Malladi et al. (17).
RESULTS
Successful Development of a Core Model of Apaf-1 Activation and Oligomerization
To investigate possible caspase-9 activation mechanisms, we first developed a core model that reflects Apaf-1 activation and the formation of the heptameric apoptosome backbone. This core model was subsequently used to generate extended models for the investigation of allosteric and homodimerization based caspase-9 activation.
The apoptosome consists of seven molecules of Apaf-1, which in healthy cells reside as inactive monomers in the cytosol. Subsequent to mitochondrial outer membrane permeabilization (MOMP), cytosolic cytochrome c binds to Apaf-1 and in presence of ATP or dATP induces a conformational change in Apaf-1. This conformational change allows multiple activated Apaf-1 molecules to assemble into a stable heptameric apoptosome backbone on which caspase-9 can be activated. Cyt-c was modeled to rapidly enter the cytosol (t½ = 1.5 min), as shown experimentally before (28) and, together with ATP or dATP, to bind to inactive Apaf-1. For simplicity, dATP and ATP were implemented as one species in the model. Because the binding order of cytochrome c and dATP to Apaf-1 is interchangeable (29), both reaction sequences were implemented in the core model. Affinities and rate constants for the interaction of cytochrome c and Apaf-1 were published previously (30, 31). kon and koff values for the interaction of dATP and Apaf-1 were calculated from the published Kd (32) and the rate of dATP turnover by Apaf-1 (29). Furthermore, we allowed activated Apaf-1 monomers to fall back into their auto-inhibited state (29). A list of all reactions and their parameterization is provided as supplemental Tables 1–4. From these data, we calculated the profiles for cytosolic cytochrome c accumulation and Apaf-1 activation using reactant quantities found in HeLa cervical cancer cells. Our simulations demonstrate that Apaf-1 activation is a rapid process that reaches completion within ∼2.5 min of onset of cytochrome c release (t½ = 1.2 min) (Fig. 1A).
FIGURE 1.

Development of a core model of Apaf-1 activation and oligomerisation. A, signaling kinetics for Apaf-1 activation as calculated from the interactions of inactive Apaf-1 with cytochrome c (cyt-c) and dATP/ATP. B, local sensitivity analysis of apoptosome assembly kinetics and quantities. Using a mathematical model comprising the interactions of cytochrome c, dATP/ATP and Apaf-1, kon/koff pairs for Apaf-1 oligomerization were screened. Calculated kinetics for the apoptosome formation process were plotted. C, for the optimal Apaf-1 kon/koff pair, quantities and formation kinetics of active Apaf-1 in all oligomeric species are shown. In correspondence with previous experimental data, 65% of the activated Apaf-1 assembled into hepatmeric apoptosomes within 4–5 min. D, The efficiency of Apaf-1 heptamerisation is shown for the concentration range of Apaf-1 as reported for various human cell lines.
We next implemented the oligomerisation process by which activated Apaf-1 forms stable heptamers. Upon activating Apaf-1 by adding cytochrome c to native cytosolic extracts or in in vitro reconstitution experiments, fully functional apoptosomes assemble within 5–10 min (6, 32–37). Based on the available literature, we determined that in HeLa cells and other widely used human cell line models at least 60–70% of Apaf-1 oligomerizes into the heptameric apoptosome backbone (6, 17, 33, 34, 38). For the mathematical simulations, we allowed all possible combinations of intermediate Apaf-1 oligomers to be reversibly generated prior to heptamer formation and assumed the fully formed heptamer to be stable. Because the Kd for Apaf-1 oligomerization has not been described yet, we first computationally screened for Kd values, which allowed for >60% apoptosome formation (data not shown). For these Kd values, we next screened for Apaf-1 kon/koff pairs, which provide 65% apoptosome formation within 5 min within the context of the overall signaling system (from MOMP-induced cytochrome c release to Apaf-1 heptamerization) (Fig. 1B). Simulations for HeLa cell conditions, using the optimal Apaf-1 kon/koff pair, demonstrate the trend toward higher order Apaf-1 oligomers, with heptamers dominating (Fig. 1C). Changes in Apaf-1 amounts within the expression range that is typically found in human cell line systems (300–600 nm) (39–41) do not notably alter the efficiency of heptamer formation (Fig. 1D). An overview of all reactions of the oligomerization process and its parameterization is provided in supplemental Tables 5 and 6.
Taken together, the core model for the formation of the apoptosome backbone developed here accurately remodels experimental data on apoptsome formation kinetics and Apaf-1 oligomerization efficiency. This core model subsequently served to implement extensions that represent scenarios of caspase-9 activation based on homodimerization on the apoptosome (APOPTO-DIM) or based on allosteric activation by the apoptosome backbone (APOPTO-ALL).
Model Extensions to Investigate Allosteric or Homo-dimerization-based Activation of Caspase-9 on the Apoptosome
Based on the above core model of apoptosome formation, we next developed two model variants that reflect scenarios of allosteric activation of procaspase-9 on the apoptosome (APOPTO-ALL) or activation of procaspase-9 by homodimerization on the apoptosome (APOPTO-DIM) (Fig. 2). For both APOPTO-DIM and APOPTO-ALL implementations, the apoptosome was modeled to provide seven binding sites for procaspase-9. In the APOPTO-DIM variant, procaspase-9 dimerization and activation was assumed to occur as soon as two procaspase-9 monomers bind to the apoptosome. As it is not required for these monomers to bind side-by-side in APOPTO-DIM, the simulation results of APOPTO-DIM reflect the maximum efficiency for a dimerization-based caspase-9 activation scenario. As demonstrated in the crystal structure of the processed caspase-9 hetero-tetramer (19), caspase-9 dimers were modeled to possess only one active site. In the APOPTO-ALL model variant, the binding of procaspase-9 monomers to the apoptosome is sufficient for allosteric activation. Cleavage of procaspase-3 and caspase-9 auto-processing were implemented identically in both model variants, taking into account differences in the affinity of active procaspase-9 and active processed caspase-9 to procaspase-3 (17). Both model variants were also linked to reactions which describe the activity of effector caspase-3, the role of the caspase inhibitor XIAP, the XIAP antagonist Smac, and the half-times of all proteins. The latter reactions were implemented as described for a previously validated model of downstream apoptosis execution signaling (41). The output of activated effector caspase-3 and the associated substrate cleavage by caspase-3 provided the possibility to compare the simulation results obtained with APOPTO-ALL and APOPTO-DIM to legacy experimental kinetics and quantities of substrate cleavage in living cells. A detailed overview of all reactions and rate constants is provided in supplemental Tables 7 and 8.
FIGURE 2.
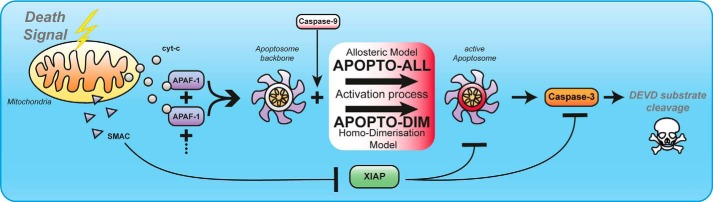
Model extension strategies to investigate allosteric or dimerization-based activation mechanisms on the apoptosome. The core model of Apaf-1 activation and oligomerization was extended in two variants. These reflect allosteric (APOPTO-ALL) or homodimerization based (APOPTO-DIM) caspase-9 activation. Both variants also entail additional reactions which reflect the overall network of apoptosis execution, including the key players Smac, XIAP, effector caspase-3, and the resulting cleavage of effector caspase-3 substrates. The latter model output can serve for direct comparison with cleavage kinetics and quantities measured experimental in individual cells. cyt-c, cytochrome c.
Mathematical Simulations of Allosteric Caspase-9 Activation Replicate Experimental Data on Apoptosis Execution Signaling, Quantitatively and Kinetically
Procaspase-9 binds via CARD-CARD interactions to the apoptosome backbone (7). For the parameterization of the CARD-CARD interaction, we took into account that procaspase-9 binds to the Apaf-1 CARD with a Kd of ∼0.7 μm, as calculated from Palacios-Rodriguez et al. (26) (see “Experimental Procedures”) and that procaspase-9 binds ∼10-fold more efficient to the apoptosome than processed caspase-9 (17). To obtain the related kon and koff rate constants, we conducted sensitivity analyses with both APOPTO-ALL and APOPTO-DIM, aiming to reproduce experimental data on caspase-3-dependent substrate cleavage in HeLa cervical cancer cells.
In HeLa cells, caspase-3 was reported to fully cleave recombinantly expressed FRET substrates within 15 min of MOMP (41–43). From the sensitivity analysis using the APOPTO-ALL implementation, a koff value of 2 min−1 with a corresponding kon of 2.85 (μm × min)−1 was determined as optimal to replicate these substrate cleavage profiles, quantitatively and kinetically (Fig. 3, A and B). APOPTO-ALL also replicated a previously described XIAP threshold concentration (41) at which caspase-3 substrate cleavage and apoptosis execution are efficiently suppressed (Fig. 3C).
FIGURE 3.

Mathematical modeling of allosteric caspase-9 activation replicates experimental data on apoptosis execution signaling. A, using the APOPTO-ALL variant of the mathematical model, comprising the entire signal transduction network from initiation of apoptosis execution to the resulting substrate cleavage, a local sensitivity analysis was conducted to identify a kon/koff pair for the interaction of caspase-9 with the apoptosome. Calculated kinetics for caspase-3-dependent substrate cleavage were plotted against time. B, for the optimal kon/koff pair, the substrate cleavage kinetic by effector caspase-3 was calculated for conditions found in HeLa cervical cancer cells. For comparison, previously reported experimental data (41) are shown in gray and demonstrate that the simulation results closely correlate with in cellulo signaling kinetics. C, XIAP concentrations were screened in APOPTO-ALL to identify whether a previously reported threshold concentration at which substrate cleavage at HeLa cell conditions becomes submaximal and sublethal (41) can be replicated. cyt-c, cytochrome c.
We next repeated the sensitivity analysis with the APOPTO-DIM model implementation. Surprisingly, the analysis failed to determine a suitable pair of rate constants because no kon/koff pair could provide the experimentally described rapid cleavage kinetics of effector caspase-3 substrates (Fig. 4A). Within 15 min of MOMP, no more than 25% of the substrate was calculated to be cleaved (Fig. 4B), indicating insufficient amounts of active caspase-9 early upon apoptosome formation. Rather than excluding caspase-9 homodimerization as a molecular mechanism required for activation on the apoptosome, we investigated whether the APOPTO-DIM parameterization could be modified justifiably to replicate experimental data. To accelerate caspase-9 activation, we allowed binding of procaspase-9 also to active Apaf-1 monomers and Apaf-1 oligomers smaller than the heptameric apoptosome backbone (additional reactions are listed in supplemental Tables 9 and 10). Procaspase-9 monomers therefore could also homodimerize at earlier times on Apaf-1 oligomers ranging from dimers to hexamers. However, the acceleration of caspase-9 activation was insignificant (data not shown). Next, we therefore considered a tighter binding of procaspase-9 to the apoptosome by modifying the Kd value itself. Strikingly, the Kd had to be reduced 10-fold from the experimentally determined value to obtain the condition where rapid and full substrate cleavage was achieved (Fig. 4C). With a koff value of 0.2 min−1 and a corresponding kon of 2.86 (μm × min)−1, it was possible to reproduce caspase-3 substrate cleavage kinetics for HeLa cell conditions (Fig. 4D) as well as the reported XIAP threshold behavior (Fig. 4E).
FIGURE 4.

Mathematical modeling of dimerization-based caspase-9 activation requires substantial parameter fitting to replicate experimental data on apoptosis execution signaling. A and B, using the APOPTO-DIM variant of the mathematical model, comprising the entire signal transduction network from initiation of apoptosis execution to the resulting substrate cleavage, a local sensitivity analysis was conducted to identify a kon/koff pair for the interaction of caspase-9 with the apoptosome. Calculated kinetics for caspase-3-dependent substrate cleavage for protein expression profiles found in HeLa cells were plotted against time. Simulation results demonstrate that rapid and complete substrate cleavage could not be replicated. C, as described in A, a local sensitivity analysis was conducted for a Kd 10-fold lower than the experimentally reported value. The adjustment the Kd value promoted a more rapid and complete substrate cleavage. D, from the sensitivity analysis conducted with an adjusted Kd value (as shown in C), an optimal kon/koff pair could be identified that allowed to replicate experimentally measured substrate cleavage kinetics and amounts. E, XIAP concentrations in the fitted APOPTO-DIM variant were screened to identify whether a previously reported threshold concentration at which substrate cleavage at HeLa cell conditions becomes submaximal and sublethal can be replicated. cyt-c, cytochrome c.
In summary, APOPTO-ALL, the systems model of apoptosis execution, which assumes a mechanism of allosteric caspase-9 activation, replicated experimental data on substrate cleavage by effector caspase-3 and on the XIAP cell death threshold, quantitatively and kinetically. In contrast, APOPTO-DIM required a considerable fitting of the procaspase-9/Apaf-1 affinity (APOPTO-DIMfit) to replicate experimental data on apoptosis execution signaling.
Mathematical Simulations of Allosteric Caspase-9 Activation Replicate Experimental Data on Procaspase-9 Processing
To further investigate which of the two model implementations provides biologically more meaningful results, we determined the profiles of procaspase-9 processing that result from scenarios of allosteric (APOPTO-ALL) and dimerization-based (APOPTO-DIMfit) caspase-9 activation and compared these with experimental data. The reported half times for procaspase-9 cleavage in cytochrome c activated native cell extracts range from 4 to 10 min (15, 36, 37, 44). The half-time of procaspase-9 processing in the APOPTO-ALL model variant was determined as 8 min, therefore falling into the experimentally determined range (Fig. 5A). When investigating procaspase-9 processing in APOPTO-DIMfit, we found that procaspase-9 processing proceeded significantly slower (t½ = 18 min) than reported experimentally (Fig. 5B). Therefore, APOPTO-DIMfit failed to reproduce experimental kinetics of procaspase-9 processing, whereas the molecular mechanism of allosteric caspase-9 activation, implemented in APOPTO-ALL, provides kinetics of procaspase-9 processing which match experimental findings. We furthermore conducted a sensitivity analysis to determine the influence of the procaspase-9 concentration on its own processing. Although 10-fold changes in the procaspase-9 concentration did not notably influence its half-time in APOPTO-ALL, such changes significantly affected procaspase-9 half-times during homodimerization based activation (APOPTO-DIMfit) (Fig. 5, C and D). Given the consistent findings of procaspase-9 half-times in the minutes range in various experimental systems (15, 36, 37, 44), an allosteric procaspase-9 activation mechanism may therefore be favored over a homodimerization-based mechanism.
FIGURE 5.

Mathematical modeling of allosteric caspase-9 activation replicates experimental data on procaspase-9 processing. A, procaspase-9 processing kinetics calculated for the allosteric activation scenario (APOPTO-ALL). A rectangle indicates experimentally determined half-times (15, 36, 37, 44). B, procaspase-9 processing kinetics calculated for the fitted homodimerization-based activation scenario (APOPTO-DIMfit). A rectangle indicates experimentally determined half-times. C, the procaspase-9 half-times for an allosteric activation mechanism are robust to changes in the procaspase-9 concentration. D, the procaspase-9 half-times calculated with APOPTO-DIMfit are highly sensitive to changes in the procaspase-9 concentration. cyt-c, cytochrome c.
Mathematical Simulations of Allosteric Caspase-9 Activation Replicate the Molecular Timer Function of the Apoptosome
The apoptosome functions as a molecular timer, which manifests in a transient capacity to process and activate procaspase-3 in in vitro apoptosome reconstitution experiments (17). This has been attributed to caspase-9 turnover on the apoptosome, where processed caspase-9 is released and then replaced continuously by procaspase-9. Once the pool of procaspase-9 becomes depleted, procaspase-3 activation becomes inefficient (17).
We implemented the in vitro experimental conditions used by Malladi et al. (17) in our in silico models (see “Experimental Procedures” and supplemental Table 11) and then tested whether APOPTO-ALL or APOPTO-DIMfit can replicate the molecular timer function of the apoptosome. In these experiments, cytochrome c, dATP, Apaf-1, and procaspase-9 were preincubated to allow for apoptosome formation and caspase-9 activation. Procaspase-3 was added after defined times (5, 10, 20, and 30 min) (Fig. 6A), and the resulting caspase-3 activity was determined. Using the APOPTO-ALL model variant, we found that delays in procaspase-3 addition resulted in reduced amounts of caspase-3 activity (Fig. 6B). These results closely correlated with the reported experimental data (see also reproduced data displays in Fig. 6A) (17). Repeating these simulations in presence of an uncleavable procaspase-9 mutant highlighted that the molecular timer function was abolished (Fig. 6C), an effect that can be attributed to the retention of procaspase-9 at the apoptosome. Again, the elimination of the timer function was indeed reported previously for conditions in which an uncleavable procaspase-9 mutant was used (see also reproduced data displays in Fig. 6A) (17). In contrast, dimerization-based caspase-9 activation, implemented in APOPTO-DIMfit, failed to replicate the reported experimental results (Fig. 6, D and E). Therefore, only the allosteric caspase-9 activation mechanism, implemented in APOPOTO-ALL, allowed us to quantitatively and kinetically replicate the catalytic timer function of the apoptosome.
FIGURE 6.

Mathematical modeling of allosteric caspase-9 activation replicates the molecular timer function of the apoptosome. A, visualization of an experimental setting by which the molecular timer function of the apoptosome can be detected. Delays in addition of procaspase-3 to reconstituted apoptosomes results in a significantly reduced capacity to activate caspase-3. The timer function is not observed when uncleavable procaspase-9 is used in the assay. Data insets are reproduced with kind permission from (17). B, the time-dependent capacity to activate caspase-3, as calculated by APOPTO-ALL, is shown. The timer function is clearly visible. C, calculations as described in B were repeated for conditions in which uncleavable procaspase-9 was used. The timer function is lost. D and E, the time-dependent capacity to activate caspase-3, as calculated by APOPTO-DIMfit, is shown for conditions in which procaspase-9 (D) or uncleavable procaspase-9 (E) was used. The molecular timer function cannot be detected. cyt c, cytochrome c.
Fitting and Modeling Dimerization-based Caspase-9 Activation with Catalytic Site Adjustment Fails to Replicate Half-times of Procaspase-9 and the Apoptosome Molecular Timer
Crystallization showed that the processed caspase-9 dimer possesses only one catalytic site (19). Because the catalytic mechanism of caspases is highly conserved within this protease family and because all crystal structures for other caspases show two catalytic sites in a conformation compatible with substrate binding and cleavage, it could be argued that the inactive catalytic site reported for caspase-9 may represent an intermediate in which one catalytic site was trapped in an inactive conformation. We therefore repeated all simulations with a variant of APOPTO-DIM in which two catalytic sites were assumed for dimerized caspase-9.
Similar to results reported for APOPTO-DIM in Fig. 3, also the variant with two catalytic sites per caspase-9 dimer initially failed to replicate experimental data on rapid and efficient apoptosis execution (Fig. 7, A and B). The Kd of procaspase-9 binding to the apoptosome had to be adjusted to ∼30% of its experimentally determined value before a kon/koff pair could be identified that allowed to replicate apoptosis execution kinetics (Fig. 7, C and D). As for APOPTO-DIMfit (Fig. 3), also for this variant of APOPTO-DIM (termed APOPTO-DIMfit2X), we implemented that irrespective of the binding site on the apoptosome, caspase-9 molecules instantaneously dimerize with 100% efficiency. Our simulations therefore reflect overestimation limits for the efficacy of a biological system that builds on dimerization-based caspase-9 activation.
FIGURE 7.

Fitting and modeling dimerization-based caspase-9 activation with catalytic site adjustment. A and B, using an APOPTO-DIM (APOPTO-DIMfit2X) model variant, modified to reflect two catalytic sites per caspase-9 dimer, a local sensitivity analysis was conducted to identify a kon/koff pair for the interaction of caspase-9 with the apoptosome. Calculated kinetics for caspase-3-dependent substrate cleavage were plotted against time and demonstrate that experimental results on rapid and complete substrate cleavage could not be replicated. C and D, as described in A and B, a local sensitivity analysis was conducted with a Kd of ∼30% of the experimentally reported value. The fitting of Kd, kon and koff allowed to replicate rapid and complete substrate cleavage. E, XIAP concentrations in the fitted model variant were screened to identify whether a previously reported threshold concentration at which substrate cleavage at HeLa cell conditions becomes submaximal and sublethal can be replicated. F, APOPTO-DIMfit2X failed to replicate experimentally reported half-times of procaspase-9. G and H, As in Fig. 6, the time-dependent capacity to activate caspase-3, as calculated by APOPTO-DIMfit2X, is shown for conditions in which procaspase-9 (D) or uncleavable procaspase-9 (E) was used. cyt-c, cytochrome c.
As for APOPTO-DIMfit, also APOPTO-DIMfit2X could replicate submaximal substrate cleavage for elevated XIAP concentrations (Fig. 7E) but failed to reproduce experimentally described half-times of procaspase-9 (Fig. 7F). Analyzing the molecular timer function demonstrated that APOPTO-DIMfit2X calculated considerable substrate cleavage rates at 30 min after apoptosome formation (∼55 or 60% of the starting value) (Fig. 7G), whereas the experimentally reported half-time was <10 min (17). Uncleavable procaspase-9 remained stably bound to the apoptosome (Fig. 7H). Taken together, these results therefore demonstrate that while APOPTO-DIMfit2X performs better than APOTPTOfit, not all key features of apoptosis execution could be replicated. Simulations building on the assumption of an allosteric activation mechanism (APOPTO-ALL), without additional model fitting, instead replicated all tested characteristics of apoptosis execution signaling.
DISCUSSION
Here, we report the first systems biological model of apoptosis execution that entails all key signaling events of apoptosome formation and caspase-9 activation. Our simulations demonstrate that linking the individual reactions involved in Apaf-1 activation and oligomerization, parameterized based on available experimental data, is sufficient to accurately replicate apoptosome formation quantitatively and kinetically. Our study also provides strong support for an allosteric activation mechanism for procaspase-9 on the apoptosome backbone, therefore challenging the prevailing concept that all initiator caspases are exclusively activated by homodimerization.
Even though apoptosis signal transduction has been investigated comprehensively by mathematical modeling (45–47), details on the process of apoptosome formation have typically been omitted from such models. Instead, direct inputs of active caspase-9 or 1:1 interactions between Apaf-1 and procaspase-9 served as triggers to investigate the interplay of caspase-9, XIAP, Smac, and executioner caspases and to understand how this interplay controls apoptotic cell death decisions (41, 43, 48, 49). Prior modeling studies omitted details on apoptosome formation for justifiable reasons and at the cost of not being able to investigate caspase-9 activation mechanisms. First, biochemical data of sufficient quality and quantity to meaningfully simulate and analyze apoptosome formation by systems modeling were not available at the time. Second, although in most investigated cell lines, typically only 15–20 min are required for apoptosis signaling to proceed from MOMP to the completion of substrate cleavage by effector caspases (40–43, 50), the majority of Apaf-1 oligomerization and procaspase-9 processing occurs within the first 1–2 min following MOMP (35, 37). Because the kinetics of mitochondrial cytochrome c release, which can readily be measured in living cells (51), resemble the kinetic of apoptosome formation, this allowed to circumvent the need for a detailed implementation of apoptosome formation and caspase-9 activation. Third, the high number of intermediate oligomeric fractions between cytochrome c, dATP/ATP and Apaf-1 prior to formation of the heptameric apoptosome backbone would have introduced a significant combinatorial complexity into prior mathematical models. Indeed, in our study a total of 61 core reactions describe 222 reactant entities to capture the formation of the Apaf-1 heptamer and the subsequent activation and regulation of caspases. In contrast, only 51 reactions and 23 reactants were necessary to describe apoptosis execution when omitting apoptosome formation (41). Our implementation of the core reactions of apoptosome formation, their parameterization with published rate constants, and the determination of additional rate constants from published biochemical data were sufficient to calculate apoptosome formation quantities and kinetics that replicated experimental measurements without further parameter optimization or model training. This indicates that our simulations provide biologically meaningful insight into the complexity of this signaling process and also provides the possibility to study scenarios in which apoptosome formation or procaspase-9 recruitment may be physiologically or pathophysiologically suppressed (52–54).
The question of whether caspase-9 is activated by homodimerization or instead by an allosteric activation mechanism on the apoptosome backbone has been asked repeatedly (20–22, 25, 55). We therefore investigated the plausibility of both mechanisms in the context of the overall signaling system and found that only an allosteric activation mechanism can replicate all tested characteristics of apoptosis execution signaling without further parameter fitting. These included apoptosis execution kinetics, apoptosis suppression by XIAP, procaspase-9 half-times, and the molecular timer function of the apoptosome. Our findings therefore contradict the currently prevailing opinion that all initiator caspases are activated by homodimerization on specific activation platforms. The notion that caspase-9 homodimerization is a prerequisite for activation substantially builds on experimental conditions in which procaspase-9 dimerization was promoted artificially and in absence of Apaf-1. For example, the overexpression of procaspase-9 results in its auto-aggregation and activation, both in pro- and eukaryotic cells. Likewise, the stabilization of procaspase-9 dimers in presence of kosmotropic salts results in its activation (20), as does the forced dimerization of procaspase-9 in which the CARD domain was replaced by a leucine zipper domain (21). However, caspase-9 activities obtained in absence of Apaf-1 are ∼20-fold lower than those measured in presence of the apoptosome (56), indicating that forced dimerization may result in non-physiological caspase-9 activities. In further support of dimerization-based activation, it was brought forward that procaspase-9 mutants cannot be activated upon apoptosis induction in mammalian cells when amino acid substitutions are introduced into the dimerization interface (20). However, these substitutions may also abrogate the capacity of CARD-recruited caspase-9 monomers to further interact with the apoptosome backbone, and thereby may prevent the conformational changes that are required for allosteric caspase-9 activation. Finally, it was suggested that allosteric caspase-9 activation could be ruled out as replacing the procaspase-8 death effector domains with the caspase-9 CARD was sufficient to activate caspase-8 on the apoptosome (21). However, this finding provides only information on the propensity of caspase-8 to dimerize rather than excluding that caspase-9 requires the apoptosome as an allosteric activator.
Evidence that may instead support an allosteric activation mechanism for procaspase-9 can be found in the published literature. The initial suggestion that procaspase-9 may be activated allosterically was based on the observation that caspase-9 activity increases significantly in presence of the apoptosome (22, 57), but the possibility that caspase-9 homodimerizes in this setting was not ruled out. However, if dimers form on the apoptosome, these dimers must be very unstable because caspase-9, once released from the apoptosome, is monomeric and inactive (22, 58). In contrast, commercially available caspase-9, recombinantly overexpressed in and purified from Escherichia coli, provides stable activity, suggesting that it may differ from caspase-9 activated in living mammalian cells. Although the apoptosome provides seven binding sites for pro-/caspase-9 monomers through CARD-CARD interactions with Apaf-1 (6), saturating the apoptosome with excess caspase-9 results in activities that reflect only one to two catalytically active sites, as found in two independent studies (17, 21). Although initially this was suggested to be attributable to inefficient apoptosome formation (21), this could indicate that the apoptosome activates only a fraction of the bound pool of caspase-9 (one to two molecules) rather than triggering efficient caspase-9 dimerization. Stoichiometric data on Apaf-1 and procaspase-9 expression in human cancer cell lines further supports a mechanism of allosteric caspase-9 activation. For example, HeLa cervical cancer cells express ∼12-fold higher amounts of Apaf-1 than procaspase-9 (41), indicating that the likelihood of two procaspase-9 molecules to bind side-by-side on the apoptosome is very low. Nevertheless, caspase-9 activation and apoptosis execution is highly efficient in these cells. Similar unfavorable ratios for a dimerization-based activation process were reported in various other cell lines, including MCF-7 breast cancer cells, human colon carcinoma cell lines (HCT-116, Lovo, DLD-1, HT29), and panels of 11 glioblastoma and 11 melanoma cell lines (39, 40, 59). Structural analysis based on electron cryo-microscopy data indicated that the seven Apaf-1 CARD domains may form a flexible procaspase-9 recruitment platform in the center of the apoptosome complex (60). Of note, three-dimensional reconstructions without symmetry restraints indicated that only a single caspase-9 may interact with the apoptosome through the Apaf-1 nucleotide-binding domain, whereas the other caspase-9 molecules are flexibly tethered to the apoptosome backbone. This asymmetry may therefore likewise be indicative of an allosteric activation of procaspase-9 monomers.
Despite our results and the supportive findings described above, we cannot formally exclude the possibility that caspase-9 may also homodimerize on the apoptosome. However, we suggest that such a homodimerization may not be required for the initial activation event and/or that it may occur subsequent to activation. To unequivocally reject a dimerization-based activation mechanism and thereby to confirm a purely allosteric activation mechanism will pose a tremendous experimental challenge, because ultimately it would be required to engineer apoptosomes that possess only a single procaspase-9 binding site. Our study may therefore direct future research to decipher one of the most fundamental processes in apoptosis signal transduction. A deeper understanding of the mechanisms of this process may also assist in directing drug design and intervention strategies for human pathologies that are associated with excessive or unwanted caspase-9-dependent apoptosis, including acute settings such as stroke, but also neurodegenerative diseases, or ototoxicity during platinum drug-based chemotherapy (61–64). Due to the strongly overlapping substrate specificities in the caspase family (65), identifying highly specific inhibitors of caspase-9 may have no prospect of success. Likewise, compounds that interfere with Apaf-1 directly to prevent apoptosome formation (66) may also negatively affect non-apoptotic roles of Apaf-1 (67, 68). Inhibitors that instead prevent the recruitment of procaspase-9 to the apoptosome or, if required for allosteric activation, the interaction with the Apaf-1 nucleotide binding domain following recruitment (60), could provide a mechanism-based and more specific route toward identifying potent suppressors of apoptotic cell death.
Supplementary Material
Acknowledgment
We thank Prof. Kelvin Cain (University of Leicester) for helpful discussions.
This work was supported by the European Union Framework Programme 7 (FP7 APO-DECIDE and FP7 SYS-MEL), the National Biophotonics and Imaging Platform (HEA PRTLI Cycle 4), and Science Foundation Ireland (SFI 09/RFP/BIC2375).

This article contains supplemental Tables 1–11 and additional references.
- Apaf-1
- apoptotic protease-activating factor 1
- CARD
- caspase recruitment domain
- MOMP
- mitochondrial outer membrane permeabilization
- XIAP
- X-linked inhibitor of apoptosis protein.
REFERENCES
- 1. Pop C., Salvesen G. S. (2009) Human caspases: activation, specificity, and regulation. J. Biol. Chem. 284, 21777–21781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Janssens S., Tinel A. (2012) The PIDDosome, DNA-damage-induced apoptosis and beyond. Cell Death Differ. 19, 13–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dickens L. S., Powley I. R., Hughes M. A., MacFarlane M. (2012) The 'complexities' of life and death: death receptor signalling platforms. Exp. Cell Res. 318, 1269–1277 [DOI] [PubMed] [Google Scholar]
- 4. Zou H., Henzel W. J., Liu X., Lutschg A., Wang X. (1997) Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90, 405–413 [DOI] [PubMed] [Google Scholar]
- 5. Yuan S., Akey C. W. (2013) Apoptosome structure, assembly, and procaspase activation. Structure 21, 501–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cain K., Brown D. G., Langlais C., Cohen G. M. (1999) Caspase activation involves the formation of the aposome, a large (approximately 700 kDa) caspase-activating complex. J. Biol. Chem. 274, 22686–22692 [DOI] [PubMed] [Google Scholar]
- 7. Li P., Nijhawan D., Budihardjo I., Srinivasula S. M., Ahmad M., Alnemri E. S., Wang X. (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–489 [DOI] [PubMed] [Google Scholar]
- 8. Liu X., Kim C. N., Yang J., Jemmerson R., Wang X. (1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157 [DOI] [PubMed] [Google Scholar]
- 9. Leipe D. D., Koonin E. V., Aravind L. (2004) STAND, a class of P-loop NTPases including animal and plant regulators of programmed cell death: multiple, complex domain architectures, unusual phyletic patterns, and evolution by horizontal gene transfer. J. Mol. Biol. 343, 1–28 [DOI] [PubMed] [Google Scholar]
- 10. Riedl S. J., Salvesen G. S. (2007) The apoptosome: signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 8, 405–413 [DOI] [PubMed] [Google Scholar]
- 11. Reubold T. F., Wohlgemuth S., Eschenburg S. (2011) Crystal structure of full-length Apaf-1: how the death signal is relayed in the mitochondrial pathway of apoptosis. Structure 19, 1074–1083 [DOI] [PubMed] [Google Scholar]
- 12. Acehan D., Jiang X., Morgan D. G., Heuser J. E., Wang X., Akey C. W. (2002) Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol. Cell 9, 423–432 [DOI] [PubMed] [Google Scholar]
- 13. Yuan S., Yu X., Topf M., Ludtke S. J., Wang X., Akey C. W. (2010) Structure of an apoptosome-procaspase-9 CARD complex. Structure 18, 571–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Qin H., Srinivasula S. M., Wu G., Fernandes-Alnemri T., Alnemri E. S., Shi Y. (1999) Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature 399, 549–557 [DOI] [PubMed] [Google Scholar]
- 15. Srinivasula S. M., Ahmad M., Fernandes-Alnemri T., Alnemri E. S. (1998) Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol. Cell 1, 949–957 [DOI] [PubMed] [Google Scholar]
- 16. Stennicke H. R., Deveraux Q. L., Humke E. W., Reed J. C., Dixit V. M., Salvesen G. S. (1999) Caspase-9 can be activated without proteolytic processing. J. Biol. Chem. 274, 8359–8362 [DOI] [PubMed] [Google Scholar]
- 17. Malladi S., Challa-Malladi M., Fearnhead H. O., Bratton S. B. (2009) The Apaf-1*procaspase-9 apoptosome complex functions as a proteolytic-based molecular timer. EMBO J. 28, 1916–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Slee E. A., Harte M. T., Kluck R. M., Wolf B. B., Casiano C. A., Newmeyer D. D., Wang H. G., Reed J. C., Nicholson D. W., Alnemri E. S., Green D. R., Martin S. J. (1999) Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J. Cell Biol. 144, 281–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Renatus M., Stennicke H. R., Scott F. L., Liddington R. C., Salvesen G. S. (2001) Dimer formation drives the activation of the cell death protease caspase 9. Proc. Natl. Acad. Sci. U.S.A. 98, 14250–14255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boatright K. M., Renatus M., Scott F. L., Sperandio S., Shin H., Pedersen I. M., Ricci J. E., Edris W. A., Sutherlin D. P., Green D. R., Salvesen G. S. (2003) A unified model for apical caspase activation. Mol. Cell 11, 529–541 [DOI] [PubMed] [Google Scholar]
- 21. Pop C., Timmer J., Sperandio S., Salvesen G. S. (2006) The apoptosome activates caspase-9 by dimerization. Mol. Cell 22, 269–275 [DOI] [PubMed] [Google Scholar]
- 22. Rodriguez J., Lazebnik Y. (1999) Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev. 13, 3179–3184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bratton S. B., Walker G., Srinivasula S. M., Sun X. M., Butterworth M., Alnemri E. S., Cohen G. M. (2001) Recruitment, activation and retention of caspases-9 and -3 by Apaf-1 apoptosome and associated XIAP complexes. EMBO J. 20, 998–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yin Q., Park H. H., Chung J. Y., Lin S. C., Lo Y. C., da Graca L. S., Jiang X., Wu H. (2006) Caspase-9 holoenzyme is a specific and optimal procaspase-3 processing machine. Mol. Cell 22, 259–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Würstle M. L., Laussmann M. A., Rehm M. (2012) The central role of initiator caspase-9 in apoptosis signal transduction and the regulation of its activation and activity on the apoptosome. Exp. Cell Res. 318, 1213–1220 [DOI] [PubMed] [Google Scholar]
- 26. Palacios-Rodríguez Y., García-Laínez G., Sancho M., Gortat A., Orzáez M., Pérez-Payá E. (2011) Polypeptide modulators of caspase recruitment domain (CARD)-CARD-mediated protein-protein interactions. J. Biol. Chem. 286, 44457–44466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cheng Y., Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 [DOI] [PubMed] [Google Scholar]
- 28. Waterhouse N. J., Goldstein J. C., von Ahsen O., Schuler M., Newmeyer D. D., Green D. R. (2001) Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J. Cell Biol. 153, 319–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reubold T. F., Wohlgemuth S., Eschenburg S. (2009) A new model for the transition of APAF-1 from inactive monomer to caspase-activating apoptosome. J. Biol. Chem. 284, 32717–32724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Purring-Koch C., McLendon G. (2000) Cytochrome c binding to Apaf-1: the effects of dATP and ionic strength. Proc. Natl. Acad. Sci. U.S.A. 97, 11928–11931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Purring C., Zou H., Wang X., McLendon G. (1999) Stoichiometry, free energy, and kinetic aspects of cytochrome c: Apaf-1 binding in apoptosis. J. Am. Chem. Soc. 121, 7435–7436 [Google Scholar]
- 32. Jiang X., Wang X. (2000) Cytochrome c promotes caspase-9 activation by inducing nucleotide binding to Apaf-1. J. Biol. Chem. 275, 31199–31203 [DOI] [PubMed] [Google Scholar]
- 33. Cain K., Bratton S. B., Langlais C., Walker G., Brown D. G., Sun X. M., Cohen G. M. (2000) Apaf-1 oligomerizes into biologically active approximately 700-kDa and inactive approximately 1.4-MDa apoptosome complexes. J. Biol. Chem. 275, 6067–6070 [DOI] [PubMed] [Google Scholar]
- 34. Freathy C., Brown D. G., Roberts R. A., Cain K. (2000) Transforming growth factor-β1 induces apoptosis in rat FaO hepatoma cells via cytochrome c release and oligomerization of Apaf-1 to form a approximately 700-kd apoptosome caspase-processing complex. Hepatology 32, 750–760 [DOI] [PubMed] [Google Scholar]
- 35. Twiddy D., Brown D. G., Adrain C., Jukes R., Martin S. J., Cohen G. M., MacFarlane M., Cain K. (2004) Pro-apoptotic proteins released from the mitochondria regulate the protein composition and caspase-processing activity of the native Apaf-1/caspase-9 apoptosome complex. J. Biol. Chem. 279, 19665–19682 [DOI] [PubMed] [Google Scholar]
- 36. Saikumar P., Mikhailova M., Pandeswara S. L. (2007) Regulation of caspase-9 activity by differential binding to the apoptosome complex. Front Biosci. 12, 3343–3354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hill M. M., Adrain C., Duriez P. J., Creagh E. M., Martin S. J. (2004) Analysis of the composition, assembly kinetics and activity of native Apaf-1 apoptosomes. EMBO J. 23, 2134–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang X., Kim H. E., Shu H., Zhao Y., Zhang H., Kofron J., Donnelly J., Burns D., Ng S. C., Rosenberg S., Wang X. (2003) Distinctive roles of PHAP proteins and prothymosin-α in a death regulatory pathway. Science 299, 223–226 [DOI] [PubMed] [Google Scholar]
- 39. Passante E., Würstle M. L., Hellwig C. T., Leverkus M., Rehm M. (2013) Systems analysis of apoptosis protein expression allows the case-specific prediction of cell death responsiveness of melanoma cells. Cell Death Differ. 20, 1521–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schmid J., Dussmann H., Boukes G. J., Flanagan L., Lindner A. U., O'Connor C. L., Rehm M., Prehn J. H., Huber H. J. (2012) Systems analysis of cancer cell heterogeneity in caspase-dependent apoptosis subsequent to mitochondrial outer membrane permeabilization. J. Biol. Chem. 287, 41546–41559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rehm M., Huber H. J., Dussmann H., Prehn J. H. (2006) Systems analysis of effector caspase activation and its control by X-linked inhibitor of apoptosis protein. EMBO J. 25, 4338–4349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rehm M., Düssmann H., Prehn J. H. (2003) Real-time single cell analysis of Smac/DIABLO release during apoptosis. J. Cell Biol. 162, 1031–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Albeck J. G., Burke J. M., Aldridge B. B., Zhang M., Lauffenburger D. A., Sorger P. K. (2008) Quantitative analysis of pathways controlling extrinsic apoptosis in single cells. Mol. Cell 30, 11–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zou H., Yang R., Hao J., Wang J., Sun C., Fesik S. W., Wu J. C., Tomaselli K. J., Armstrong R. C. (2003) Regulation of the Apaf-1/caspase-9 apoptosome by caspase-3 and XIAP. J. Biol. Chem. 278, 8091–8098 [DOI] [PubMed] [Google Scholar]
- 45. Würstle M. L., Zink E., Prehn J. H., Rehm M. (2014) From computational modelling of the intrinsic apoptosis pathway to a systems-based analysis of chemotherapy resistance: achievements, perspectives and challenges in systems medicine. Cell Death Dis. 5, e1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lavrik I. N. (2014) Systems biology of death receptor networks: live and let die. Cell Death Dis. 5, e1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Spencer S. L., Sorger P. K. (2011) Measuring and modeling apoptosis in single cells. Cell 144, 926–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Legewie S., Blüthgen N., Herzel H. (2006) Mathematical modeling identifies inhibitors of apoptosis as mediators of positive feedback and bistability. PLoS Comput. Biol. 2, e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bentele M., Lavrik I., Ulrich M., Stösser S., Heermann D. W., Kalthoff H., Krammer P. H., Eils R. (2004) Mathematical modeling reveals threshold mechanism in CD95-induced apoptosis. J. Cell Biol. 166, 839–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. O'Connor C. L., Anguissola S., Huber H. J., Dussmann H., Prehn J. H., Rehm M. (2008) Intracellular signaling dynamics during apoptosis execution in the presence or absence of X-linked-inhibitor-of-apoptosis-protein. Biochim. Biophys. Acta 1783, 1903–1913 [DOI] [PubMed] [Google Scholar]
- 51. Goldstein J. C., Waterhouse N. J., Juin P., Evan G. I., Green D. R. (2000) The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat. Cell Biol. 2, 156–162 [DOI] [PubMed] [Google Scholar]
- 52. Kim J., Parrish A. B., Kurokawa M., Matsuura K., Freel C. D., Andersen J. L., Johnson C. E., Kornbluth S. (2012) Rsk-mediated phosphorylation and 14–3-3varepsilon binding of Apaf-1 suppresses cytochrome c-induced apoptosis. EMBO J. 31, 1279–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Niimi S., Arakawa-Takeuchi S., Uranbileg B., Park J. H., Jinno S., Okayama H. (2012) Cdc6 protein obstructs apoptosome assembly and consequent cell death by forming stable complexes with activated Apaf-1 molecules. J. Biol. Chem. 287, 18573–18583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim H. E., Jiang X., Du F., Wang X. (2008) PHAPI, CAS, and Hsp70 promote apoptosome formation by preventing Apaf-1 aggregation and enhancing nucleotide exchange on Apaf-1. Mol. Cell 30, 239–247 [DOI] [PubMed] [Google Scholar]
- 55. Shi Y. (2004) Caspase activation: revisiting the induced proximity model. Cell 117, 855–858 [DOI] [PubMed] [Google Scholar]
- 56. Chao Y., Shiozaki E. N., Srinivasula S. M., Rigotti D. J., Fairman R., Shi Y. (2005) Engineering a dimeric caspase-9: a re-evaluation of the induced proximity model for caspase activation. PLoS Biol. 3, e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shiozaki E. N., Chai J., Shi Y. (2002) Oligomerization and activation of caspase-9, induced by Apaf-1 CARD. Proc. Natl. Acad. Sci. U.S.A. 99, 4197–4202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shiozaki E. N., Chai J., Rigotti D. J., Riedl S. J., Li P., Srinivasula S. M., Alnemri E. S., Fairman R., Shi Y. (2003) Mechanism of XIAP-mediated inhibition of caspase-9. Mol. Cell 11, 519–527 [DOI] [PubMed] [Google Scholar]
- 59. Murphy Á. C., Weyhenmeyer B., Schmid J., Kilbride S. M., Rehm M., Huber H. J., Senft C., Weissenberger J., Seifert V., Dunst M., Mittelbronn M., Kögel D., Prehn J. H., Murphy B. M. (2013) Activation of executioner caspases is a predictor of progression-free survival in glioblastoma patients: a systems medicine approach. Cell Death Dis. 4, e629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yuan S., Yu X., Asara J. M., Heuser J. E., Ludtke S. J., Akey C. W. (2011) The holo-apoptosome: activation of procaspase-9 and interactions with caspase-3. Structure 19, 1084–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rigamonti D., Sipione S., Goffredo D., Zuccato C., Fossale E., Cattaneo E. (2001) Huntingtin's neuroprotective activity occurs via inhibition of procaspase-9 processing. J. Biol. Chem. 276, 14545–14548 [DOI] [PubMed] [Google Scholar]
- 62. Kanungo A. K., Hao Z., Elia A. J., Mak T. W., Henderson J. T. (2008) Inhibition of apoptosome activation protects injured motor neurons from cell death. J. Biol. Chem. 283, 22105–22112 [DOI] [PubMed] [Google Scholar]
- 63. Cozzolino M., Ferraro E., Ferri A., Rigamonti D., Quondamatteo F., Ding H., Xu Z. S., Ferrari F., Angelini D. F., Rotilio G., Cattaneo E., Carrì M. T., Cecconi F. (2004) Apoptosome inactivation rescues proneural and neural cells from neurodegeneration. Cell Death Differ. 11, 1179–1191 [DOI] [PubMed] [Google Scholar]
- 64. García-Berrocal J. R., Nevado J., Ramírez-Camacho R., Sanz R., González-García J. A., Sánchez-Rodríguez C., Cantos B., España P., Verdaguer J. M., Trinidad Cabezas A. (2007) The anticancer drug cisplatin induces an intrinsic apoptotic pathway inside the inner ear. Br. J. Pharmacol. 152, 1012–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. McStay G. P., Salvesen G. S., Green D. R. (2008) Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ. 15, 322–331 [DOI] [PubMed] [Google Scholar]
- 66. Vicent M. J., Pérez-Payá E. (2006) Poly-L-glutamic acid (PGA) aided inhibitors of apoptotic protease activating factor 1 (Apaf-1): an antiapoptotic polymeric nanomedicine. J. Med. Chem. 49, 3763–3765 [DOI] [PubMed] [Google Scholar]
- 67. Ferraro E., Pesaresi M. G., De Zio D., Cencioni M. T., Gortat A., Cozzolino M., Berghella L., Salvatore A. M., Oettinghaus B., Scorrano L., Pérez-Payà E., Cecconi F. (2011) Apaf1 plays a pro-survival role by regulating centrosome morphology and function. J. Cell Sci. 124, 3450–3463 [DOI] [PubMed] [Google Scholar]
- 68. Zermati Y., Mouhamad S., Stergiou L., Besse B., Galluzzi L., Boehrer S., Pauleau A. L., Rosselli F., D'Amelio M., Amendola R., Castedo M., Hengartner M., Soria J. C., Cecconi F., Kroemer G. (2007) Nonapoptotic role for Apaf-1 in the DNA damage checkpoint. Mol. Cell 28, 624–637 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.