

Background: The mechanism of excised oligonucleotide processing during nucleotide excision repair is unknown.
Results: UV photoproduct-containing oligonucleotides associate with chromatin following the dual incisions. Inhibition of gap-filling activities results in an accumulation of RPA-bound small, excised, damaged DNA (sedDNA) fragments.
Conclusion: Gap filling-mediated dissociation of sedDNAs from RPA influences nucleotide excision repair rate.
Significance: sedDNA processing is important in the DNA damage response.
Keywords: DNA Damage, DNA Damage Response, DNA Repair, DNA-Protein Interaction, Nucleotide Excision Repair, Subcellular Fractionation
Abstract
Ultraviolet (UV) photoproducts are removed from genomic DNA by dual incisions in humans in the form of 24- to 32-nucleotide-long oligomers (canonical 30-mers) by the nucleotide excision repair system. How the small, excised, damage-containing DNA oligonucleotides (sedDNAs) are processed in cells following the dual incision event is not known. Here, we demonstrate that sedDNAs are localized to the nucleus in two biochemically distinct forms, which include chromatin-associated, transcription factor II H-bound complexes and more readily solubilized, RPA-bound complexes. Because the nuclear mobility and repair functions of transcription factor II H and RPA are influenced by post-incision gap-filling events, we examined how DNA repair synthesis and DNA ligation affect sedDNA processing. We found that although these gap filling activities are not essential for the dual incision/sedDNA generation event per se, the inhibition of DNA repair synthesis and ligation is associated with a decrease in UV photoproduct removal rate and an accumulation of RPA-sedDNA complexes in the cell. These findings indicate that sedDNA processing and association with repair proteins following the dual incisions may be tightly coordinated with gap filling during nucleotide excision repair in vivo.
Introduction
Nucleotide excision repair is a multi-protein repair system that is capable of removing a wide variety of DNA lesions from genomic DNA, including ultraviolet (UV) photoproducts and bulky base adducts that are induced by a number of chemical carcinogens and chemotherapeutic agents (1–3). Repair of these diverse forms of DNA damage involves a common mechanism that is defined by incision events at 20 ± 5 phosphodiester bonds 5′ and 6 ± 3 phosphodiester bonds 3′ to the lesion (4). These dual incisions are accomplished by the action of six core repair factors (RPA,3 XPA, TFIIH, XPC, XPF-ERCC1, and XPG) (5–7) and result in the production of a 24- to 32-nucleotide-long oligonucleotide that contains the damaged bases (4, 8, 9).
We recently developed methods to detect and quantify the primary excision product (nominal 30-mer) as well as its processed forms ranging in size from 30 to <10 nucleotides in length in mammalian cells (10, 11). Here, we will refer to the primary excision products as canonical 30-mers and the primary products along with its processed forms as small, excised, damage-containing DNA (sedDNA) oligonucleotides.
Our recent work has shown that sedDNAs are abundant in cells (1–2 × 105 per cell) following UV irradiation and stably associate with the core excision repair proteins TFIIH (transcription factor II-H) and RPA following the dual incision event (10–13). This association may impact TFIIH and RPA activity in new rounds of repair and in other DNA metabolic processes.
The biological fate and physiological functions of the sedDNA repair products have not been considered previously in experimental models of excision repair (14). However, there is increasing evidence that by-products of DNA repair have the potential to impact cell signaling events (15–18). Moreover, defects in the clearance of DNA oligonucleotides are associated with various autoimmune disorders (19–21), and abnormal localization of DNA in the cytosol may be sensed by the innate immune system (22, 23). Thus, the fate of the damage-containing sedDNA products of excision repair is an important issue that needs to be addressed.
In this report, we examined the subcellular localization and post-excision processing of sedDNAs during nucleotide excision repair. We find that sedDNAs remain largely in the nucleus following UV irradiation and exist in two distinct biochemical forms characterized by chromatin extractability and association with the excision repair proteins TFIIH and RPA. Furthermore, we have also uncovered a role for DNA repair synthesis and ligation, which are required to fill in the single-stranded DNA (ssDNA) gaps that remain in the duplex following the dual incisions, in the post-incision processing of sedDNAs. We find that though gap filling DNA synthesis and ligation are not required for the dual incision event, the inhibition of gap filling slows post-incision sedDNA processing and nucleotide excision repair rate and leads to an accumulation of sedDNAs that remain in complex with RPA. These findings provide valuable new information on the biochemical properties and processing of sedDNAs during the post-incision steps of nucleotide excision repair.
EXPERIMENTAL PROCEDURES
Cell Lines
Human A375 melanoma and U2OS osteosarcoma cells were cultured at 37 °C in a 5% CO2 humidified incubator in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin. To move cells into a quiescent state, U2OS cells were grown to near confluence and then maintained in DMEM containing 0.5% FBS for 4–5 days. Metaphase-arrested cells were prepared by addition of 50 ng/ml of nocodazole to the culture medium for 12–16 h. In experiments with inhibitors of DNA synthesis and ligation, quiescent cells were preincubated with the indicated compounds for 30 min prior to UV irradiation. To irradiate the cells, the culture medium was removed from the cells and set aside. Cells were then placed under a GE germicidal lamp that emits primarily 254-nm UV light (UV-C) connected to a digital timer. Following irradiation, the culture medium was replaced, and the cells were incubated for the indicated periods of time before harvesting. trypan blue staining was used to determine cell viability 24 h after irradiation.
Chemicals and Antibodies
Hydroxyurea (HU), cytosine β-d-arabinofuranoside (AraC), aphidicolin, and nocodazole were purchased from Sigma. The DNA ligase inhibitor L189 was obtained from Tocris Bioscience. Antibodies used for immunoprecipitation included anti-p62 (Santa Cruz Biotechnology catalogue no. sc-292) and anti-RPA34 (Calbiochem catalogue no. NA18). Antibodies used for immunoblotting included anti-MEK2 (catalogue no. 610235) from BD Transduction Laboratories; anti-RPA70 (catalogue no. A300–241A) and anti-phospho-RPA32 (Ser-33) (catalogue no. A300–246A) from Bethyl Laboratories; anti-XPB (catalogue no. sc-293) from Santa Cruz Biotechnology; and anti-phospho-H2AX (Ser-139) (catalogue no. 2577), anti-phospho-p53 (Ser-15) (catalogue no. 9284), anti-cleaved PARP (catalogue no. 5625), anti-calnexin (catalogue no. 2679), and anti-cleaved caspase-3 (catalogue no. 9664) from Cell Signaling Technology. Anti-(6-4)PP antibody was from Cosmo Bio, and anti-cyclobutane pyrimidine dimer (CPD) antibody was from Kamiya Biomedical. Secondary antibodies included horseradish peroxidase-linked anti-mouse and anti-rabbit IgG (catalogue nos. NA931V and NA934V) from GE Healthcare.
Cell Extracts and Fractionation
Following UV irradiation, cells were washed twice with cold PBS and then harvested by scraping the cells from the plate into cold PBS. Cells were then pelleted by gentle centrifugation (800 × g, 5 min, 4 °C). Cells were then either frozen on dry ice and stored at −80 °C or directly used for preparation of cell extracts. Whole cell lysates for immunoprecipitation were prepared by lysing the cells in Manley dialysis buffer (25 mm HEPES-KOH, pH 7.9, 100 mm KCl, 12.5% glycerol, 12 mm MgCl2, and 0.5 mm EDTA) containing 0.05% Nonidet P-40 substitute Igepal CA-630 and centrifugation at maximum speed in a microcentrifuge for 15 min at 4 °C. For preparation of nuclei, cells were resuspended in buffer A (10 mm HEPES, pH 7.9, 10 mm KCl, 1.5 mm MgCl2, 10% glycerol, 0.34 m sucrose) containing 50 μg/ml digitonin (Sigma). Cells were extracted three times with buffer A containing digitonin to separate the cytosolic material from the nuclei. Soluble cytosolic extracts were centrifuged for 15 min at maximum speed in a microcentrifuge at 4 °C to remove any contaminating nuclei. Chromatin fractions were also analyzed by using a similar fractionation procedure that employed 0.1% Triton X-100 in place of digitonin.
Isolation and Detection of sedDNAs
Cell extracts and immunoprecipitates were incubated with RNase A for 5 min at room temperature and were then deproteinized by incubation with proteinase K for 30 min at 55 °C. Following phenol-chloroform extraction and ethanol precipitation, the sedDNAs were resuspended in 10 μl of 10 mm Tris, pH 8.5. To 3′-end label the sedDNAs, 5 μl of sedDNAs were incubated in a 10-μl reaction containing six units of terminal deoxynucleotidyl transferase (New England Biolabs), 0.25 mm CoCl2, and [α-32P]3′-deoxyadenosine 5′-triphosphate (cordycepin 5′-triphosphate; PerkinElmer Life Sciences) in 1× terminal deoxynucleotidyl transferase buffer (New England Biolabs) for 1 h at 37 °C. Following phenol-chloroform extraction and ethanol precipitation, sedDNAs were separated on urea-containing polyacrylamide gels and then detected with a phosphorimager. Radiolabeled oligonucleotides of known length were resolved on all gels as size markers. Excision repair activity was quantified using ImageQuant software (version 5.2, GE Healthcare) as described previously (10).
Immunoprecipitation
Cell extracts were incubated on a rotary device at 4 °C with 1–2 μg of the indicated antibody for 2–6 h and were then incubated with recombinant protein A/G PLUS agarose (Santa Cruz Biotechnology) for 2 h. Immunoprecipitations were washed three times with buffer Manley dialysis buffer containing 0.5% Igepal CA-630. To elute DNA from the immunoprecipitations, the immunocomplexes were incubated for 30 min at 55 °C in immunoprecipitation elution buffer (45 mm Tris, pH 8, 9 mm EDTA, 250 mm NaCl, 0.5% SDS) containing 50 μg of proteinase K. Eluted DNAs were phenol-chloroform extracted and ethanol-precipitated prior to radiolabeling as described above. For analysis of proteins in the immunoprecipitations, the immunocomplexes were boiled for 5 min in 1× SDS-PAGE sample buffer (50 mm Tris, pH 6.8, 100 mm DTT, 1% SDS, 5% glycerol, and 0.005% bromphenol blue).
Immunoblotting
Proteins from cell fractionations and immunoprecipitations were subjected to SDS-PAGE, transferred to Hybond ECL membranes (GE Healthcare), probed with the indicated antibodies, and detected with Clarity Western ECL Substrate (Bio-Rad) using a Molecular Imager Chemi-Doc XRS+ system (Bio-Rad).
Immunoslot Blot Analysis of UV Photoproduct Repair
Repair of UV photoproducts from genomic DNA was measured as described previously (13). (6-4) photoproduct ([6–4]PP) and CPD signals were detected as described for the immunoblotting approach above, and ImageQuant (version 5.2) was used to analyze the experimental results.
RESULTS
UV-generated sedDNAs Remain in the Nucleus Following the Dual Incisions
Although in vitro excision repair assays have demonstrated that the primary products of nucleotide excision repair are released from the gapped, duplex DNA following the dual incision step of repair (6, 12), the subcellular localization of the sedDNAs during repair in vivo is not known but is an important issue given that cytoplasmic or improperly localized DNAs have the potential to induce inflammatory and immune signaling responses (22, 23).
Using our recently developed methods for following sedDNA fate in vivo (10, 11), we therefore examined the subcellular distribution of sedDNAs in cultured human cells by biochemical fractionation. A375 melanoma cells were irradiated with UV light and then lysed in a hypotonic buffer containing the glycoside digitonin, which through the preferential removal of cholesterol from the relatively cholesterol-rich plasma membrane effectively separates nuclei from cytosolic material. As shown in Fig. 1, this fractionation procedure resulted in a clear separation of the cytoplasmic protein MEK2 from the majority of the primarily nuclear excision repair protein XPB (a component of TFIIH) and the nucleosome component H2AX. RPA, which has been shown to readily dissociate/leak from nuclei in hypotonic buffers (24, 25), was found in both the soluble cytosolic fraction and the nuclear pellet fraction. We also processed these subcellular fractions for isolation and detection of sedDNAs by 3′-radiolabeling, urea-PAGE, and phosphorimaging. Previous studies showed that these excised oligonucleotides, which range from ∼15 to 32 nt in length, contain UV photoproducts and are only generated in cells with a functional excision repair system (10, 11). Examination of the UV-generated sedDNA distribution revealed that nearly 90% of the sedDNAs were found in the nuclear fraction. Because the 10–15% of sedDNAs that are apparently cytosolic are associated with RPA (data not shown; see below), which is a protein that is known to readily leak from nuclei upon cell lysis (24, 25), we conclude that the dual incision sedDNA products of nucleotide excision repair remain primarily in the nucleus following the dual incision reaction.
FIGURE 1.
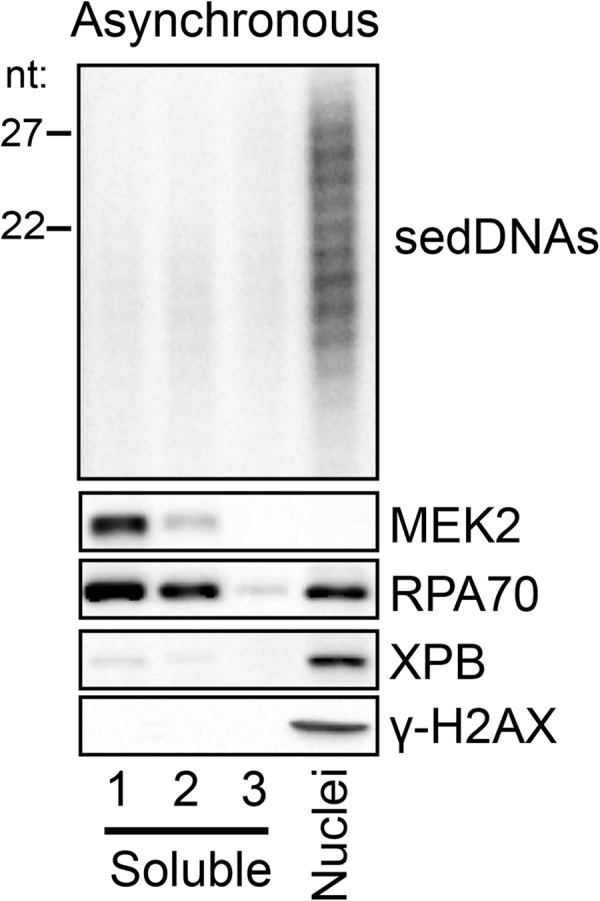
sedDNAs remain in the nucleus following the dual incision event. Asynchronously growing A375 cells were harvested 30 min following exposure to 10 J/m2 of UV and extracted three times with a hypotonic buffer containing digitonin. Equal volumes of these three soluble fractions (1, 2, 3) along with the nuclear pellet fraction were analyzed for the indicated proteins by immunoblotting and for sedDNA content by radiolabeling, urea-PAGE, and phosphorimaging. Densitometry was used to determine the distribution of sedDNAs among these fractions.
Primary and Partially Degraded sedDNAs Are Differentially Extractable from the Chromatin-enriched Fraction of Cells
To further examine the biochemical properties of the sedDNAs, we used the non-ionic detergent Triton X-100, which permeabilizes both the cell and nuclear membranes and can disrupt protein-protein interactions. Immunoblot analysis of the Triton-soluble and -resistant fractions of UV-irradiated cells demonstrated that >90% of RPA and ∼50% of XPB were extractable with Triton X-100 (Fig. 2A). Interestingly, the fractionation of UV-irradiated cells under these conditions led to a remarkable separation of two sedDNA species, with >90% of the 24- to 32-nt-long primary excision products remaining with the Triton-resistant chromatin fraction and >95% of the shorter species ∼15 to 23 nt in length present in the Triton-soluble fractions (Fig. 2A). Similar results were obtained when the non-ionic detergent Igepal CA-630 was used in place of Triton X-100 and at detergent concentrations ranging from 0.1 to 1% (data not shown). Because sedDNAs are known to undergo nucleolytic digestion following dual incisions both in vitro in cell-free extracts (8, 12) and in vivo (10, 11), we consider the longer species to be the primary, full-length sedDNA repair products (canonical 30-mers) and the shorter species to be secondary, partially degraded sedDNAs. These results indicate that these two sedDNA species have different biochemical properties with regards to their association with the chromatin-enriched fraction of cells.
FIGURE 2.

TFIIH- and RPA-sedDNA complexes are differentially extractable from chromatin. A, asynchronously growing A375 cells were harvested 30 min following exposure to 10 J/m2 of UV and extracted three times with a hypotonic buffer containing 0.1% Triton X-100. These three soluble fractions along with the chromatin pellet were analyzed for the indicated proteins by immunoblotting and for sedDNA content. B, 30 min following exposure to 20 J/m2 of UV, A375 cells were treated with formaldehyde on ice to cross-link proteins to DNA or were left untreated. Following preparation of a whole cell lysate, cross-links were reversed, and sedDNA content was analyzed. C, Triton-soluble and Triton-resistant (chromatin) fractions were prepared as in A and then immunoprecipitated with anti-RPA or anti-TFIIH antibodies. sedDNAs and the indicated proteins were analyzed.
We next wished to use an alternative approach to confirm the unique biochemical characteristics of the two sedDNA species with regards to their localization within nuclei. We therefore used formaldehyde to covalently cross-link proteins to DNA prior to extraction of the cells with a buffer capable of solubilizing both the primary and secondary sedDNAs. After cross-link reversal and deproteinization, the sedDNAs were purified and radiolabeled. As shown in Fig. 2B, formaldehyde treatment prevented the recovery of the primary, full-length sedDNA species from the cells. In contrast, the majority (70%) of the secondary, degraded sedDNAs were recoverable in the soluble cell lysate following deproteinization. Analysis of the insoluble chromatin-enriched fraction was not possible in these experiments because of the large amount of genomic DNA fragments that are present in sheared chromatin. We conclude from these results that the primary sedDNA products of nucleotide excision repair may remain in close contact with chromatin but that the secondary products are only loosely associated with chromatin following the dual incision event.
TFIIH- and RPA-sedDNA Complexes Are Differentially Associated with Chromatin
Immediately following the actions of the XPF and XPG nucleases, which generate the dual incisions required for removal of UV photoproduct-containing oligonucleotides from the genome, the primary excision products are found in a tight complex with the transcription/repair factor TFIIH (12). The TFIIH-bound oligonucleotides are released from TFIIH in an ATP-dependent but ATP hydrolysis-independent manner and then become bound by the single-stranded DNA binding protein RPA and undergo limited or full nucleolytic degradation (12). Our observation that the primary full-length and secondary partially degraded sedDNAs are differentially extractable from chromatin suggested that these sedDNAs may be bound to TFIIH and RPA, respectively.
To test this hypothesis, we immunoprecipitated TFIIH and RPA from both the Triton-soluble and Triton-insoluble factions of UV-irradiated A375 cells and then examined the relative sedDNA enrichment. As shown in Fig. 2C, RPA-bound sedDNAs were found solely in the Triton-soluble fraction along with the majority of the cellular RPA. In contrast, even though approximately half of the total cellular TFIIH is Triton-soluble (Fig. 2, A and C), nearly all of the TFIIH-bound sedDNAs were found in the Triton-resistant chromatin fraction. Repeated rounds of immunoprecipitation to isolate all of the TFIIH and RPA from the cell lysates demonstrated that nearly 100% of the sedDNAs in the cell lysates are bound to these two factors alone (data not shown). Moreover, these results indicate that the two biochemically distinct sedDNA species are largely correlated with their association with the excision repair proteins TFIIH and RPA. Finally, these data suggest that following the dual incisions, TFIIH-sedDNA complexes remain in close contact with chromatin before the sedDNAs dissociate from TFIIH and bind to the more readily solubilized RPA.
Inhibition of Excision Gap-filling Slows UV Photoproduct Removal
Coincident with the dual incision events that generate the sedDNAs, ssDNA gaps ∼30-nt in length (26–28) are generated in the duplex DNA that must be filled by the action of DNA polymerases and then the remaining nicks sealed by a DNA ligase. Using the ribonucleotide reductase inhibitor HU in combination with the chain-terminating nucleotide analog AraC to limit the extent of DNA repair synthesis in the gaps, early studies on excision repair in human cells reported a maximum of 1–2 × 105 breaks in DNA could be generated at any given time following UV irradiation (29–31). Interestingly, we recently reported nearly identical values for the maximum number of sedDNAs that can be generated in the cell following UV irradiation (11). The similarity in the number of sedDNAs and excision gaps indicates that post-incision processing of the sedDNAs may be coordinated with gap-filling synthesis during nucleotide excision repair.
To begin to investigate this issue, we first examined how the inhibition of gap-filling synthesis with HU/AraC treatment impacts the removal of (6-4)PPs and CPDs from the genomic DNA of UV-irradiated cells. To ensure the maximum effectiveness of HU/AraC on the inhibition of gap filling, we used cells that were grown to confluence and maintained in medium containing a low serum concentration to keep cells in a quiescent state prior to treatment with HU/AraC and UV irradiation. Treatment with HU/AraC under these conditions has been reported to reduce the amount of UV-induced DNA repair synthesis by 60–90% (32–35), which indicates that only a low level or rate of repair synthesis takes place in the presence of these inhibitors.
We therefore isolated genomic DNA at various time points following UV irradiation from quiescent cells that were pretreated or not with HU/AraC and then analyzed the DNA for (6-4)PP and CPD content by immunoslot blotting using anti-(6-4)PP and anti-CPD antibodies. As shown in Fig. 3, A and B, the rates of both (6-4)PPs and CPDs was slowed by about a factor of two in the presence of HU/AraC. These results therefore demonstrate that the inhibition of gap-filling synthesis has an inhibitory effect on the rate of UV photoproduct removal from genomic DNA, which is consistent with previous reports using alternative approaches for measuring UV photoproduct removal (29, 32, 36–38).
FIGURE 3.

Inhibition of gap-filling DNA synthesis slows UV photoproduct removal. A, quiescent U2OS cells were left untreated or were pretreated for 30 min with 10 mm HU and 100 μm AraC before exposure to 10 J/m2 of UV. At the indicated time points, genomic DNA was isolated from the cells and analyzed by immunoslot blotting with an anti-(6-4)PP antibody. Following immunoblotting, the membranes were stained with Sybr Gold to detect total DNA content. For quantification, (6-4)PP signals were normalized to Sybr Gold and then plotted as a function of time. Results show the average and S.D. from two independent experiments. B, genomic DNA from UV-irradiated cells was analyzed as in A except that an anti-CPD antibody was used for immunoblotting.
Inhibition of Gap Filling Enhances DNA Damage Checkpoint Signaling and Induces Cell Death
Inhibition of gap-filling synthesis has been reported to lead to enlargement of excision gaps in human cells (39) and to enhanced DNA damage checkpoint signaling by the ATR kinase (40, 41), which likely occurs through the recruitment of the ATR kinase to RPA-coated ssDNA excision gaps (42–45). To confirm these findings and validate our experimental conditions, we therefore monitored the phosphorylation status of a number of protein substrates of the ATR kinase in UV-irradiated quiescent cells in the absence or presence of HU/AraC to limit gap filling. As shown in Fig. 4A, phosphorylation of p53, RPA2, and H2AX was elevated following UV exposure in cells that were treated with HU/AraC. Though the physiological role of H2AX and RPA phosphorylation on cell fate following UV irradiation is unclear (46–48), particularly in non-replicating quiescent cells, the enhanced phosphorylation of p53 on Ser-15 is linked to the promotion of apoptotic forms of cell death (49, 50).
FIGURE 4.

Inhibition of gap-filling DNA synthesis results in enhanced checkpoint signaling and cell death. A, quiescent U2OS cells were left untreated or were pre-treated for 30 min with 10 mm HU and 100 μm AraC before exposure to 10 J/m2 of UV. At the indicated time points, whole cell lysates were prepared from the cells and were analyzed by immunoblotting with antibodies against the indicated proteins. B, quiescent U2OS cells were treated at in A. Twenty-four h after irradiation, cells were harvested, stained with trypan blue, and counted under a microscope using a hemocytometer. The percentage of cells that stained positive with trypan blue, which indicates non-viable cells, is indicated. The results show the average and S.D. from three independent experiments. C, quiescent U2OS cells were treated at in A. Twenty-four h after irradiation, whole cell lysates were prepared from the cells and were analyzed by immunoblotting with antibodies against the cleaved forms of PARP and caspase-3.
Consistent with this increase in proapoptotic p53 phosphorylation, when we examined the viability of cells 24 h after UV irradiation by trypan blue staining, we observed a nearly 3-fold increase in non-viable cells when gap filling was inhibited with HU/AraC (Fig. 4B). To determine whether this increase in cell death was through an apoptotic mechanism, we examined the status of the apoptosis markers PARP and caspase-3. As shown in Fig. 4C, an enrichment of the cleaved forms of both PARP and caspase-3 was found in UV-irradiated cells exposed to HU/AraC in comparison with cells that were allowed to undergo normal gap filling. The lack of significant cell death or apoptotic signaling in non-irradiated cells treated with HU/AraC demonstrates that the effect of HU/AraC on cell death was UV-dependent and therefore not due to potential other effects of HU and AraC on general cell physiology.
We conclude from these findings that the inhibition of gap-filling DNA synthesis with HU/AraC is correlated with enhanced DNA damage checkpoint signaling and cell death and that this phenotype is associated with inhibition in the rate of removal of DNA damage. Thus, efficient gap filling is required for quiescent, non-replicating cells to effectively deal with UV-induced DNA damage.
Inhibition of Gap Filling Results in an Accumulation of RPA-bound sedDNAs
The slower rate of UV photoproduct removal in the presence of HU/AraC in Fig. 3 suggested that the level of sedDNAs that are generated in the presence of HU/AraC following UV irradiation may also be reduced. Thus, to determine how gap filling affects the generation of sedDNAs during excision repair, we treated quiescent cells with HU/AraC to slow gap filling following UV irradiation and then isolated the (6-4)PP-containing sedDNAs by immunoprecipitation with anti-(6-4)PP antibodies. As shown in Fig. 5A, pretreatment with HU/AraC did not prevent or inhibit the generation of (6-4)PP-containing sedDNAs and instead led to a modest increase in the level of (6-4)PP-sedDNAs. Additional analyses of (6-4)PP-sedDNA levels at various time points following irradiation showed that HU/AraC treatment was associated with an average increase in (6-4)PP-sedDNA levels of ∼40% in comparison with cells that were allowed to undergo normal gap filling (data not shown). These results therefore demonstrate that the inhibition of gap-filling synthesis does not prevent the dual incision reaction but may instead impact some other aspect of post-excision sedDNA processing that ultimately affects UV photoproduct removal rate.
FIGURE 5.

RPA-sedDNA complexes accumulate when gap-filling DNA synthesis is inhibited with HU/AraC. A, quiescent U2OS cells were left untreated or were pretreated for 30 min with 10 mm HU and 100 μm AraC before exposure to 10 J/m2 of UV. At the indicated time points, cells were harvested for isolation of sedDNAs, which were further immunoprecipitated with an anti-(6-4)PP antibody prior to radiolabeling, urea-PAGE, and phosphorimager analysis. B, whole cell lysates were prepared from quiescent U2OS cells with the indicated treatments and then subjected to immunoprecipitation with an anti-p62 (TFIIH) antibody. Protein content was analyzed by immunoblotting, and total sedDNA content was analyzed by radiolabeling, urea-PAGE, and phosphorimager analysis. Results from four independent experiments (average and S.D.) are graphed. C, whole cell lysates were prepared as in B but were subjected to immunoprecipitation with an anti-RPA32 (RPA) antibody.
Because sedDNAs are associated with TFIIH and RPA following the dual incision reaction, we next used protein immunoprecipitation and sedDNA analysis to determine whether the inhibition of gap filling affected the levels of sedDNAs that were associated with TFIIH and RPA following UV irradiation. As shown in Fig. 5B, HU/AraC treatment led to a small, 20–25% increase in the level of total UV photoproduct-containing sedDNAs that were bound to TFIIH. However, there was a greater than 2-fold increase in sedDNA association with RPA under these conditions (Fig. 5C). These results indicate that the normal post-excision processing of sedDNAs is altered following UV irradiation when gap filling is inhibited, which leads to an accumulation of sedDNAs that are in complex with RPA. This accumulation may prevent RPA from functioning in new rounds of excision repair at other sites of UV damage, which would explain the slower rate of UV photoproduct removal that we and others have observed in HU/AraC-treated cells (Fig. 3). Consistent with this hypothesis, studies using fluorescence microscopy to examine the ability of RPA to migrate to new sites of DNA damage following UV irradiation in cells was reported to be inhibited in cells treated with HU/AraC (36).
DNA Polymerase and Ligase Inhibition Slow UV Photoproduct Removal and Lead to an Accumulation of sedDNAs in Complex with RPA
Aphidicolin is a reversible inhibitor of DNA polymerases δ and ϵ, which have been implicated in filling in the excision gaps in during nucleotide excision repair in vivo (51–53). Early work on excision repair in human cells indicated that aphidicolin inhibited UV photoproduct removal in human cells (54, 55). Thus, to confirm our findings with HU/AraC, we pretreated quiescent cells with aphidicolin prior to exposure to UV and then monitored (6-4)PP and CPD removal from genomic DNA. The rate of removal of both types of UV photoproducts was slowed in the presence of aphidicolin (Fig. 6, A and B). To determine whether this slow rate of UV photoproduct removal was correlated with an increase of sedDNAs in complex with RPA as was observed with HU/AraC, we immunoprecipitated RPA from cells that were exposed to different combinations of UV and aphidicolin. As shown in Fig. 6C, we observed a nearly 2-fold increase in sedDNA association with RPA. We attribute the less dramatic effect of aphidicolin relative to HU/AraC on UV photoproduct removal rate and RPA-sedDNA accumulation to the fact that the aphidicolin-resistant DNA polymerase κ has been implicated in filling in a portion of DNA excision repair gaps (52, 56).
FIGURE 6.
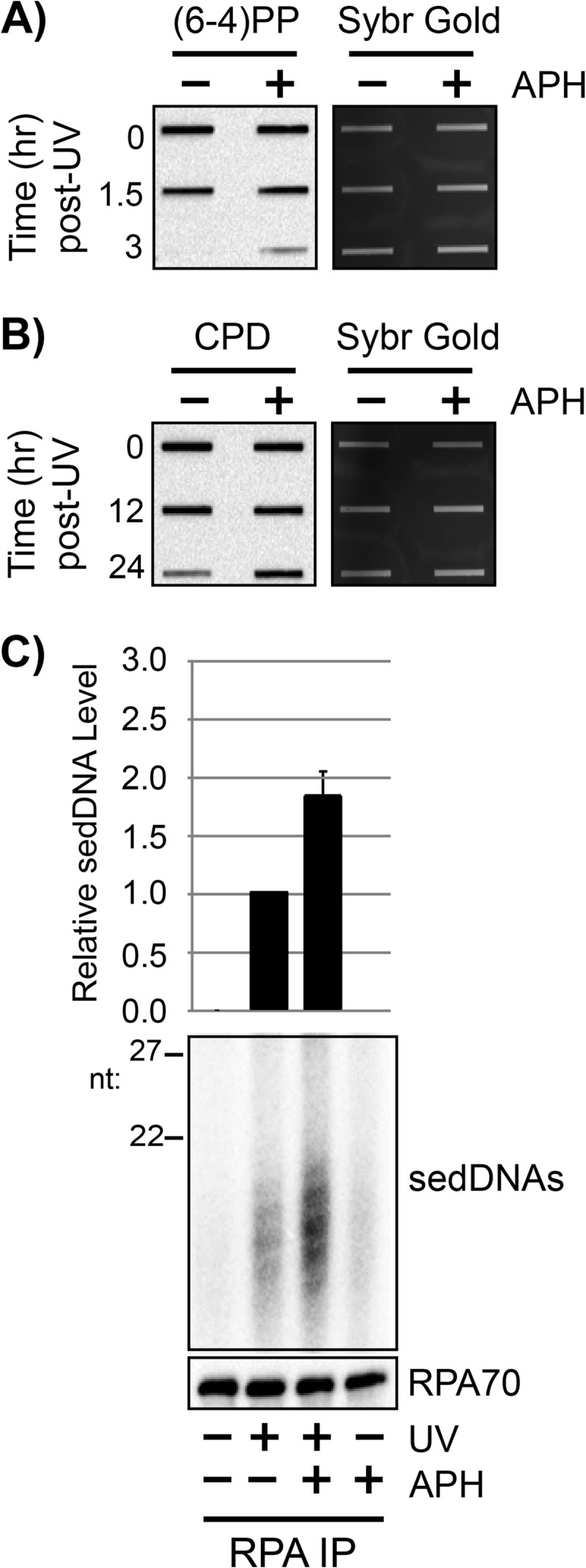
RPA-sedDNA complexes accumulate when gap-filling DNA polymerases are inhibited with aphidicolin. A, quiescent U2OS cells were left untreated or were pretreated for 30 min with 1 μg/ml aphidicolin (APH) prior to exposure to 10 J/m2 of UV. At the indicated time points, genomic DNA was isolated from the cells and analyzed by immunoslot blotting with an anti-(6-4)PP antibody. B, genomic DNA from UV-irradiated cells was analyzed as in A except that an anti-CPD antibody was used for immunoblotting. C, whole cell lysates were prepared from quiescent U2OS cells with the indicated treatments and then subjected to immunoprecipitation with an anti-RPA34 (RPA) antibody. Protein content was analyzed by immunoblotting, and total sedDNA content was analyzed by radiolabeling, urea-PAGE, and phosphorimager analysis. Results from three independent experiments (average and S.D.) are graphed.
DNA ligase action is also required to complete nucleotide excision repair by sealing the nicks that remain in the DNA following DNA repair synthesis. Furthermore, chemical inhibition or genetic knockdown of DNA ligase function has been reported to slow UV photoproduct removal and RPA nuclear mobility following UV irradiation (36, 37). We therefore used a small molecule inhibitor of DNA ligases I, III, and IV (57) to examine how DNA ligase inhibition affects RPA-sedDNA accumulation. As shown in Fig. 7A and B, although we were unable to detect a change in (6-4)PP removal rate with the DNA ligase inhibitor L189, we were able to observe a decrease in the rate of CPD removal with this compound (Fig. 7B). Furthermore, and consistent with the experiments with HU/AraC and aphidicolin, the level of sedDNAs in complex with RPA were increased following UV irradiation (Fig. 7C). These results indicate that DNA ligase inhibition has a similar effect as slowing DNA repair synthesis on sedDNA processing during nucleotide excision repair.
FIGURE 7.
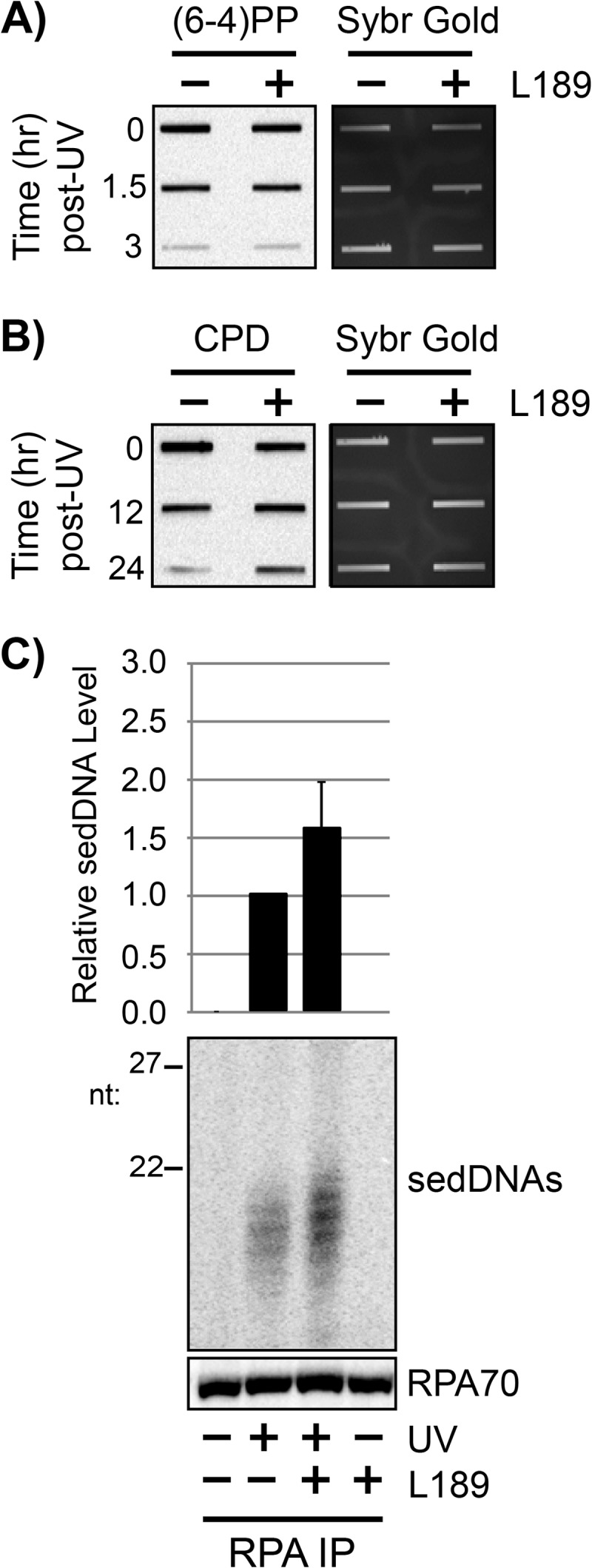
RPA-sedDNA complexes accumulate when DNA ligation is inhibited. A, quiescent U2OS cells were left untreated or were pretreated for 30 min with 100 μm L189 prior to exposure to 10 J/m2 of UV. At the indicated time points, genomic DNA was isolated from the cells and analyzed by immunoslot blotting with an anti-(6-4)PP antibody. B, genomic DNA from UV-irradiated cells was analyzed as in A except that an anti-CPD antibody was used for immunoblotting. C, whole cell lysates were prepared from quiescent U2OS cells with the indicated treatments and then subjected to immunoprecipitation with an anti-RPA34 (RPA) antibody. Protein content was analyzed by immunoblotting, and total sedDNA content was analyzed by radiolabeling, urea-PAGE, and phosphorimager analysis. Results from three independent experiments (average and S.D.) are graphed.
DISCUSSION
In this study, we have addressed several issues regarding the post-dual incision fate of sedDNAs during nucleotide excision repair, including the subcellular localization of sedDNAs and the role of gap filling in excision and sedDNA processing.
sedDNA Association with Chromatin
We previously showed using in vitro excision assays with cell-free extract that the primary excision products are released from duplex DNA in complex with TFIIH and then dissociate from TFIIH to form complexes with RPA or are degraded by nucleases that are present in the extract (12). Our data here show that sedDNAs remain associated with the chromatin-enriched fraction following the dual incisions in vivo in two distinct biochemical complexes that are defined by the ease of extraction from chromatin and the association with the excision repair factors TFIIH and RPA (Figs. 1 and 2).
Our results further indicate that under normal conditions following UV irradiation, only a small fraction of the sedDNAs that are detected with our labeling methodology are present in the cytosolic fraction of cells. Because cytoplasmic DNAs may stimulate inflammatory and innate immune signaling pathways (22, 23), the normal post-excision processing of sedDNAs likely minimizes this aberrant immune signaling by keeping the sedDNAs on chromatin within the nucleus.
Role of DNA Synthesis in the Dual Incisions
Based in part on the slower rate of removal of pyrimidine dimers from acid-insoluble DNA relative to the rate of incorporation of new bases into genomic DNA, early “patch-and-cut” models of mammalian nucleotide excision repair (58, 59) posited that DNA repair synthesis begins before, or is required for, the second incision event that occurs 3′ to the lesion by the action of the XPG nuclease. Consistent with this model, a recent study reported observing DNA repair synthesis and post-incision repair protein recruitment to damage sites in an XP-G patient cell line that was artificially engineered to express a nuclease mutant version of XPG (60). However, the use of reconstituted excision repair systems in vitro has shown that DNA synthesis is not necessary for the dual incision reaction to take place (5–7). Thus, our data here demonstrate that the same principle holds true in vivo: DNA repair synthesis is not essential for the dual incision event that removes the damage-containing oligonucleotide from the duplex.
Nonetheless, the immunoslot blot data we presented here (Figs. 3, 6, and 7) showed that the inhibition of gap filling was associated with a slower rate of UV photoproduct removal from genomic DNA and with an accumulation of RPA-bound sedDNAs (Figs. 5–7). These findings complement and extend previous analyses using alternative methods for measuring excision repair and post-incision processing of excision repair proteins (29, 32, 36, 37). For example, early work showed that the use of the DNA polymerase inhibitor aphidicolin or the ribonucleotide reductase inhibitor HU in combination with the chain-terminating nucleotide analogue AraC allowed for the visualization of UV-dependent incision events that are normally rapidly filled and ligated and hence not observable by alkaline sucrose sedimentation of genomic DNA from UV-irradiated cells (29, 32, 54, 55). Furthermore, using an assay that directly measured removal of preincorporated, radiolabeled thymine residues from acid-precipitable DNA, it was similarly reported that removal of pyrimidine dimers was nearly completely prevented by aphidicolin or HU/AraC treatment (29, 55). More recently, through the use of immunofluorescence microscopy to monitor (6-4)PP content in genomic DNA, it was reported that HU/AraC treatment inhibited (37) or slowed (36, 38) the rate of (6-4)PP removal. The variations in the absolute excision repair rates in these studies are likely due to differences in the cell lines, culture conditions, and proliferation states, all of which can affect dNTP levels and UV-induced DNA synthesis in quiescent cells (35, 55, 61, 62). Our results show that the inhibition of gap filling only slows the rate of UV photoproduct removal.
Moreover, in light of our observation that UV photoproduct-containing sedDNAs remain associated with the chromatin fraction following the dual incision event, it should be noted that some of the previous approaches that have been used to monitor excision may have a number of limitations. For example, chromatographic measurements of pyrimidine dimer content in genomic DNA involved trichloroacetic acid extraction of whole cells to precipitate genomic DNA from acid-soluble thymine dimers (29). However, oligonucleotides greater than ∼10 to 17 nt in length are insoluble in acid (63). Thus, the sedDNAs that are initially generated during the dual incisions and which are readily detectable with our 3′-labeling methodology would be expected to co-precipitate with genomic DNA in TCA and hence would not be detected as repair events with this classical DNA repair assay. Furthermore, the immunological detection of (6-4)PP loss from genomic DNA by fluorescence microscopy (36, 37) used fixation techniques that crosslink the primary sedDNA products to chromatin (Fig. 2A), which similarly prevents their detection as repair events. Thus, the ability of certain methodologies to accurately monitor repair (as defined by UV photoproduct removal by dual incisions) must be carefully considered when interpreting experimental results.
Fate of sedDNAs
The ultimate fate of the sedDNAs following the association with TFIIH and RPA is not known. Our previous observations and quantitative measurements of sedDNA abundance following UV irradiation indicate that sedDNAs do not accumulate above a maximum level of ∼1–2 × 105 per cell (11), which indicates that sedDNA processing and degradation may be regulated processes that are coordinated with the completion of repair. Further work is needed to clarify this issue and to identify the nucleases that degrade the sedDNAs following UV irradiation. It will also be important to determine whether there are conditions under which improper post-excision processing of sedDNAs leads to abnormal cellular localization that may negatively impact cellular physiology.
Similarly, the observation that the inhibition of gap filling leads to both an accumulation of RPA-sedDNA complexes (Figs. 5–7) and to enhanced ATR signaling (Fig. 4) indicates a possible regulatory role for sedDNAs or TFIIH/RPA-sedDNA complexes in the ATR-mediated DNA damage checkpoint response (64, 65). Alternatively, our finding of a 3- to 4-fold increase in cell death by HU/AraC treatment of UV-irradiated cells relative to cells that are simply UV irradiated indicate that caution must be exercised in interpreting excision repair data in HU/AraC-treated cells because of the complication arising from counting dead cells.
Acknowledgment
We thank Laura Lindsey-Boltz for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants GM32833 (to A. S.) and K99ES022640 (to S. G.).
- RPA
- replication protein A
- TFIIH
- transcription factor II H
- ATR
- ataxia telangiectasia-mutated and Rad3-related
- sedDNA
- small, excised, damage-containing DNA oligonucleotide
- AraC
- cytosine β-d-arabinofuranoside
- PP
- photoproduct
- PARP
- poly(ADP-ribose) polymerase.
REFERENCES
- 1. Sancar A. (1996) DNA excision repair. Annu. Rev. Biochem. 65, 43–81 [DOI] [PubMed] [Google Scholar]
- 2. Wood R. D. (1997) Nucleotide excision repair in mammalian cells. J. Biol. Chem. 272, 23465–23468 [DOI] [PubMed] [Google Scholar]
- 3. Reardon J. T., Sancar A. (2005) Nucleotide excision repair. Prog. Nucleic Acid Res. Mol. Biol. 79, 183–235 [DOI] [PubMed] [Google Scholar]
- 4. Huang J. C., Svoboda D. L., Reardon J. T., Sancar A. (1992) Human nucleotide excision nuclease removes thymine dimers from DNA by incising the 22nd phosphodiester bond 5′ and the 6th phosphodiester bond 3′ to the photodimer. Proc. Natl. Acad. Sci. U.S.A. 89, 3664–3668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mu D., Park C. H., Matsunaga T., Hsu D. S., Reardon J. T., Sancar A. (1995) Reconstitution of human DNA repair excision nuclease in a highly defined system. J. Biol. Chem. 270, 2415–2418 [DOI] [PubMed] [Google Scholar]
- 6. Mu D., Hsu D. S., Sancar A. (1996) Reaction mechanism of human DNA repair excision nuclease. J. Biol. Chem. 271, 8285–8294 [DOI] [PubMed] [Google Scholar]
- 7. Evans E., Moggs J. G., Hwang J. R., Egly J. M., Wood R. D. (1997) Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 16, 6559–6573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang J. C., Sancar A. (1994) Determination of minimum substrate size for human excinuclease. J. Biol. Chem. 269, 19034–19040 [PubMed] [Google Scholar]
- 9. Svoboda D. L., Taylor J. S., Hearst J. E., Sancar A. (1993) DNA repair by eukaryotic nucleotide excision nuclease. Removal of thymine dimer and psoralen monoadduct by HeLa cell-free extract and of thymine dimer by Xenopus laevis oocytes. J. Biol. Chem. 268, 1931–1936 [PubMed] [Google Scholar]
- 10. Hu J., Choi J. H., Gaddameedhi S., Kemp M. G., Reardon J. T., Sancar A. (2013) Nucleotide excision repair in human cells: fate of the excised oligonucleotide carrying DNA damage in vivo. J. Biol. Chem. 288, 20918–20926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Choi J. H., Gaddameedhi S., Kim S. Y., Hu J., Kemp M. G., Sancar A. (2014) Highly specific and sensitive method for measuring nucleotide excision repair kinetics of ultraviolet photoproducts in human cells. Nucleic Acids Res. 42, e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kemp M. G., Reardon J. T., Lindsey-Boltz L. A., Sancar A. (2012) Mechanism of release and fate of excised oligonucleotides during nucleotide excision repair. J. Biol. Chem. 287, 22889–22899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gaddameedhi S., Kemp M. G., Reardon J. T., Shields J. M., Smith-Roe S. L., Kaufmann W. K., Sancar A. (2010) Similar nucleotide excision repair capacity in melanocytes and melanoma cells. Cancer Res. 70, 4922–4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kemp M. G., Sancar A. (2012) DNA excision repair: where do all the dimers go? Cell Cycle 11, 2997–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jazayeri A., Balestrini A., Garner E., Haber J. E., Costanzo V. (2008) Mre11-Rad50-Nbs1-dependent processing of DNA breaks generates oligonucleotides that stimulate ATM activity. EMBO J. 27, 1953–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boldogh I., Hajas G., Aguilera-Aguirre L., Hegde M. L., Radak Z., Bacsi A., Sur S., Hazra T. K., Mitra S. (2012) Activation of Ras signaling by 8-oxoguanine DNA glycosylase bound to its excision product 8-oxoguanine. J. Biol. Chem. 287, 20769–20773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. German P., Szaniszlo P., Hajas G., Radak Z., Bacsi A., Hazra T. K., Hegde M. L., Ba X., Boldogh I. (2013) Activation of cellular signaling by 8-oxoguanine DNA glycosylase-1-initiated DNA base excision repair. DNA Repair 12, 856–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hajas G., Bacsi A., Aguilera-Aguirre L., Hegde M. L., Tapas K. H., Sur S., Radak Z., Ba X., Boldogh I. (2013) 8-Oxoguanine DNA glycosylase-1 links DNA repair to cellular signaling via the activation of the small GTPase Rac1. Free Radic. Biol. Med. 61, 384–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yasutomo K., Horiuchi T., Kagami S., Tsukamoto H., Hashimura C., Urushihara M., Kuroda Y. (2001) Mutation of DNASE1 in people with systemic lupus erythematosus. Nat. Genet. 28, 313–314 [DOI] [PubMed] [Google Scholar]
- 20. Stetson D. B., Ko J. S., Heidmann T., Medzhitov R. (2008) Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 134, 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fye J. M., Orebaugh C. D., Coffin S. R., Hollis T., Perrino F. W. (2011) Dominant mutation of the TREX1 exonuclease gene in lupus and Aicardi-Goutieres syndrome. J. Biol. Chem. 286, 32373–32382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Paludan S. R., Bowie A. G. (2013) Immune sensing of DNA. Immunity 38, 870–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barber G. N. (2011) Cytoplasmic DNA innate immune pathways. Immunol. Rev. 243, 99–108 [DOI] [PubMed] [Google Scholar]
- 24. Fairman M. P., Stillman B. (1988) Cellular factors required for multiple stages of SV40 DNA replication in vitro. EMBO J. 7, 1211–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Treuner K., Eckerich C., Knippers R. (1998) Chromatin association of replication protein A. J. Biol. Chem. 273, 31744–31750 [DOI] [PubMed] [Google Scholar]
- 26. Kaye J., Smith C. A., Hanawalt P. C. (1980) DNA repair in human cells containing photoadducts of 8-methoxypsoralen or angelicin. Cancer Res. 40, 696–702 [PubMed] [Google Scholar]
- 27. Cleaver J. E., Jen J., Charles W. C., Mitchell D. L. (1991) Cyclobutane dimers and (6-4) photoproducts in human cells are mended with the same patch sizes. Photochem. Photobiol. 54, 393–402 [DOI] [PubMed] [Google Scholar]
- 28. Reardon J. T., Thompson L. H., Sancar A. (1997) Rodent UV-sensitive mutant cell lines in complementation groups 6–10 have normal general excision repair activity. Nucleic Acids Res. 25, 1015–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Snyder R. D., Carrier W. L., Regan J. D. (1981) Application of arabinofuranosyl cytosine in the kinetic analysis and quantitation of DNA repair in human cells after ultraviolet irradiation. Biophys. J. 35, 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Squires S., Johnson R. T., Collins A. R. (1982) Initial rates of DNA incision in UV-irradiated human cells: differences between normal, xeroderma pigmentosum and tumour cells. Mutat. Res. 95, 389–404 [DOI] [PubMed] [Google Scholar]
- 31. Erixon K., Ahnström G. (1979) Single-strand breaks in DNA during repair of UV-induced damage in normal human and xeroderma pigmentosum cells as determined by alkaline DNA unwinding and hydroxylapatite chromatography: effects of hydroxyurea, 5-fluorodeoxyuridine and 1-β-d-arabinofuranosylcytosine on the kinetics of repair. Mutat. Res. 59, 257–271 [DOI] [PubMed] [Google Scholar]
- 32. Dunn W. C., Regan J. D. (1979) Inhibition of DNA excision repair in human cells by arabinofuranosyl cytosine: effect on normal and xeroderma pigmentosum cells. Mol. Pharmacol. 15, 367–374 [PubMed] [Google Scholar]
- 33. Mirzayans R., Enns L., Dietrich K., Barley R. D., Paterson M. C. (1996) Faulty DNA polymerase δ/ϵ-mediated excision repair in response to gamma radiation or ultraviolet light in p53-deficient fibroblast strains from affected members of a cancer-prone family with Li-Fraumeni syndrome. Carcinogenesis 17, 691–698 [DOI] [PubMed] [Google Scholar]
- 34. Mullenders L. H., van Kesteren-van Leeuwen A. C., van Zeeland A. A., Natarajan A. T. (1985) Analysis of the structure and spatial distribution of ultraviolet-induced DNA repair patches in human cells made in the presence of inhibitors of replicative synthesis. Biochim. Biophys. Acta 826, 38–48 [DOI] [PubMed] [Google Scholar]
- 35. Collins A. R., Squires S., Johnson R. T. (1982) Inhibitors of repair DNA synthesis. Nucleic Acids Res. 10, 1203–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Overmeer R. M., Moser J., Volker M., Kool H., Tomkinson A. E., van Zeeland A. A., Mullenders L. H., Fousteri M. (2011) Replication protein A safeguards genome integrity by controlling NER incision events. J. Cell Biol. 192, 401–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moser J., Kool H., Giakzidis I., Caldecott K., Mullenders L. H., Fousteri M. I. (2007) Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III α in a cell-cycle-specific manner. Mol. Cell. 27, 311–323 [DOI] [PubMed] [Google Scholar]
- 38. Overmeer R. M., Gourdin A. M., Giglia-Mari A., Kool H., Houtsmuller A. B., Siegal G., Fousteri M. I., Mullenders L. H., Vermeulen W. (2010) Replication factor C recruits DNA polymerase δ to sites of nucleotide excision repair but is not required for PCNA recruitment. Mol. Cell. Biol. 30, 4828–4839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Francis A. A., Blevins R. D., Carrier W. L., Smith D. P., Regan J. D. (1979) Inhibition of DNA repair in ultraviolet-irradiated human cells by hydroxyurea. Biochim. Biophys. Acta 563, 385–392 [DOI] [PubMed] [Google Scholar]
- 40. Matsumoto M., Yaginuma K., Igarashi A., Imura M., Hasegawa M., Iwabuchi K., Date T., Mori T., Ishizaki K., Yamashita K., Inobe M., Matsunaga T. (2007) Perturbed gap-filling synthesis in nucleotide excision repair causes histone H2AX phosphorylation in human quiescent cells. J. Cell Sci. 120, 1104–1112 [DOI] [PubMed] [Google Scholar]
- 41. Hanasoge S., Ljungman M. (2007) H2AX phosphorylation after UV irradiation is triggered by DNA repair intermediates and is mediated by the ATR kinase. Carcinogenesis 28, 2298–2304 [DOI] [PubMed] [Google Scholar]
- 42. Choi J. H., Lindsey-Boltz L. A., Kemp M., Mason A. C., Wold M. S., Sancar A. (2010) Reconstitution of RPA-covered single-stranded DNA-activated ATR-Chk1 signaling. Proc. Natl. Acad. Sci. U.S.A. 107, 13660–13665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lindsey-Boltz L. A., Reardon J. T., Wold M. S., Sancar A. (2012) In vitro analysis of the role of replication protein A (RPA) and RPA phosphorylation in ATR-mediated checkpoint signaling. J. Biol. Chem. 287, 36123–36131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lindsey-Boltz L. A., Kemp M. G., Reardon J. T., DeRocco V., Iyer R. R., Modrich P., Sancar A. (2014) Coupling of human DNA excision repair and the DNA damage checkpoint in a defined in vitro system. J. Biol. Chem. 289, 5074–5082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zou L., Elledge S. J. (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548 [DOI] [PubMed] [Google Scholar]
- 46. Cleaver J. E. (2011) γH2Ax: biomarker of damage or functional participant in DNA repair “all that glitters is not gold.” Photochem. Photobiol. 87, 1230–1239 [DOI] [PubMed] [Google Scholar]
- 47. Oakley G. G., Patrick S. M. (2010) Replication protein A: directing traffic at the intersection of replication and repair. Front. Biosci. (Landmark Ed) 15, 883–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Revet I., Feeney L., Bruguera S., Wilson W., Dong T. K., Oh D. H., Dankort D., Cleaver J. E. (2011) Functional relevance of the histone γH2Ax in the response to DNA damaging agents. Proc. Natl. Acad. Sci. U.S.A. 108, 8663–8667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sluss H. K., Armata H., Gallant J., Jones S. N. (2004) Phosphorylation of serine 18 regulates distinct p53 functions in mice. Mol. Cell. Biol. 24, 976–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sluss H. K., Gannon H., Coles A. H., Shen Q., Eischen C. M., Jones S. N. (2010) Phosphorylation of p53 serine 18 upregulates apoptosis to suppress Myc-induced tumorigenesis. Mol. Cancer Res. 8, 216–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nishida C., Reinhard P., Linn S. (1988) DNA repair synthesis in human fibroblasts requires DNA polymerase δ. J. Biol. Chem. 263, 501–510 [PubMed] [Google Scholar]
- 52. Ogi T., Limsirichaikul S., Overmeer R. M., Volker M., Takenaka K., Cloney R., Nakazawa Y., Niimi A., Miki Y., Jaspers N. G., Mullenders L. H., Yamashita S., Fousteri M. I., Lehmann A. R. (2010) Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell. 37, 714–727 [DOI] [PubMed] [Google Scholar]
- 53. Shivji M. K., Podust V. N., Hübscher U., Wood R. D. (1995) Nucleotide excision repair DNA synthesis by DNA polymerase ϵ in the presence of PCNA, RFC, and RPA. Biochemistry 34, 5011–5017 [DOI] [PubMed] [Google Scholar]
- 54. Snyder R. D., Regan J. D. (1981) Aphidicolin inhibits repair of DNA in UV-irradiated human fibroblasts. Biochem. Biophys. Res. Commun. 99, 1088–1094 [DOI] [PubMed] [Google Scholar]
- 55. Snyder R. D., Regan J. D. (1982) Differential responses of log and stationary phase human fibroblasts to inhibition of DNA repair by aphidicolin. Biochim. Biophys. Acta 697, 229–234 [DOI] [PubMed] [Google Scholar]
- 56. Ogi T., Lehmann A. R. (2006) The Y-family DNA polymerase κ (pol κ) functions in mammalian nucleotide-excision repair. Nat. Cell Biol. 8, 640–642 [DOI] [PubMed] [Google Scholar]
- 57. Chen X., Zhong S., Zhu X., Dziegielewska B., Ellenberger T., Wilson G. M., MacKerell A. D., Jr, Tomkinson A. E. (2008) Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res. 68, 3169–3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Williams J. I., Cleaver J. E. (1978) Excision repair of ultraviolet damage in mammalian cells. Evidence for two steps in the excision of pyrimidine dimers. Biophys. J. 22, 265–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ehmann U. K., Cook K. H., Friedberg E. C. (1978) The kinetics of thymine dimer excision in ultraviolet-irradiated human cells. Biophys. J. 22, 249–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Staresincic L., Fagbemi A. F., Enzlin J. H., Gourdin A. M., Wijgers N., Dunand-Sauthier I., Giglia-Mari G., Clarkson S. G., Vermeulen W., Schärer O. D. (2009) Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 28, 1111–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Downes C. S., Collins A. R. (1982) Effects of DNA replication inhibitors on UV excision repair in synchronised human cells. Nucleic Acids Res. 10, 5357–5368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Snyder R. D. (1988) Consequences of the depletion of cellular deoxynucleoside triphosphate pools on the excision-repair process in cultured human fibroblasts. Mutat. Res. 200, 193–199 [DOI] [PubMed] [Google Scholar]
- 63. Cleaver J. E., Boyer H. W. (1972) Solubility and dialysis limits of DNA oligonucleotides. Biochim. Biophys. Acta 262, 116–124 [DOI] [PubMed] [Google Scholar]
- 64. Sancar A., Lindsey-Boltz L. A., Unsal-Kaçmaz K., Linn S. (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85 [DOI] [PubMed] [Google Scholar]
- 65. Cimprich K. A., Cortez D. (2008) ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]