

Background: The identification of novel molecular mechanisms for cardiac hypertrophy is of considerable interest.
Results: Cardiomyocyte-specific knockout of KLF4 enhanced β-adrenoreceptor agonist-induced cardiac hypertrophy by modulating myocardin expression and activity.
Conclusion: KLF4 is a novel regulator of cardiac hypertrophy.
Significance: Control of KLF4 is a potential therapeutic target for cardiac hypertrophy.
Keywords: Adrenergic Receptor, Angiotensin II, Cardiac Hypertrophy, Natriuretic Peptide, Transcription Factor, Serum Response Factor
Abstract
Kruppel-like factor 4 (KLF4) plays an important role in vascular diseases, including atherosclerosis and vascular injury. Although KLF4 is expressed in the heart in addition to vascular cells, the role of KLF4 in cardiac disease has not been fully determined. The goals of this study were to investigate the role of KLF4 in cardiac hypertrophy and to determine the underlying mechanisms. Cardiomyocyte-specific Klf4 knockout (CM Klf4 KO) mice were generated by the Cre/LoxP technique. Cardiac hypertrophy was induced by chronic infusion of the β-adrenoreceptor agonist isoproterenol (ISO). Results showed that ISO-induced cardiac hypertrophy was enhanced in CM Klf4 KO mice compared with control mice. Accelerated cardiac hypertrophy in CM Klf4 KO mice was accompanied by the augmented cellular enlargement of cardiomyocytes as well as the exaggerated expression of fetal cardiac genes, including atrial natriuretic factor (Nppa). Additionally, induction of myocardin, a transcriptional cofactor regulating fetal cardiac genes, was enhanced in CM Klf4 KO mice. Interestingly, KLF4 regulated Nppa expression by modulating the expression and activity of myocardin, providing a mechanical basis for accelerated cardiac hypertrophy in CM Klf4 KO mice. Moreover, we showed that KLF4 mediated the antihypertrophic effect of trichostatin A, a histone deacetylase inhibitor, because ISO-induced cardiac hypertrophy in CM Klf4 KO mice was attenuated by olmesartan, an angiotensin II type 1 antagonist, but not by trichostatin A. These results provide novel evidence that KLF4 is a regulator of cardiac hypertrophy by modulating the expression and the activity of myocardin.
Introduction
Cardiac hypertrophy is a common adaptive response of the heart to hemodynamic stress, acute myocardial injury, infection, or mutations in genes encoding sarcomeric proteins (1). It is characterized by increased cell size and reactivation of fetal cardiac genes, such as atrial natriuretic factor (NPPA), b-type natriuretic peptide (NPPB), and β-myosin heavy chain (MYH7) in ventricular myocytes. Although it initially occurs as a compensatory response, it can eventually lead to decompensation accompanied by heart failure, arrhythmias, and sudden death. Because it is a major source of mortality in Westernized societies, the identification of the novel molecular mechanisms underlying the development of cardiac hypertrophy is of considerable interest.
Kruppel-like factor 4 (KLF4) is a zinc finger transcription factor that plays a key role in multiple tissues, including the cardiovascular system (2). For example, KLF4 has been shown to repress smooth muscle cell (SMC)2 proliferation and also to suppress the expression of multiple SMC differentiation markers, such as SM α-actin (ACTA2) and SM22α (TAGLN), in cultured SMCs and in animal models in vivo (3–5). Indeed, although KLF4 is not normally expressed in differentiated SMCs, it is transiently induced in phenotypically modulated SMCs after vascular injury (3, 5). We showed that tamoxifen-inducible deletion of the Klf4 gene in mice resulted in the attenuation of down-regulation of SMC differentiation markers and the accelerated neointimal formation following carotid ligation injury (5). In endothelial cells, KLF4 is expressed constitutively and has been shown to play anti-inflammatory and antithrombotic roles (6–8). Indeed, results of recent studies showed that endothelial KLF4 protected against atherothrombosis in Apoe−/− background mice (7). In addition, Tek promoter-dependent deletion of Klf4 caused the enhancement of injury-induced neointimal formation, in part by the exaggerated expression of cell adhesion molecules such as VCAM1 and E-selectin, as well as the augmented infiltration of inflammatory cells into injured vessels (8). Moreover, KLF4 has been shown to play an important role in the maintenance of endothelial barrier function because KLF4 regulated VE-cadherin expression, and knockdown of Klf4 augmented lipopolysaccharide-induced lung injury and pulmonary edema in mice (9). As such, the results of the preceding studies provide evidence that KLF4 is a critical factor regulating the development of multiple vascular diseases.
Regarding the role of KLF4 in the heart, results of our previous studies showed that KLF4 was expressed in the heart from late embryonic development through adulthood, and some KLF4-positive cells were positive for GATA4, which is mainly expressed in cardiomyocytes (10). We also showed that smooth and cardiac muscle-selective Klf4 deletion under the control of the Tagln promoter resulted in postnatal death and growth retardation in mice (10). The phenotype of Tagln promoter-dependent Klf4 knockout mice was likely to be caused by a defect of Klf4 in the heart, rather than in SMCs, because no differences in the expression levels of SMC differentiation markers and in the morphology of the aorta and coronary vessels were found between conditional Klf4 knockout mice and control mice and because cardiac output was decreased significantly in Klf4-deficient mice, as determined by magnetic resonance imaging (10). However, TAGLN is not a specific marker for cardiomyocytes (11), and studies are needed to further define the role of KLF4 in the heart. In addition, the results of recent studies showed that mice deficient for Klf4 in cardiomyocytes were viable but highly sensitized to transverse aortic constriction, although the underlying mechanisms remain unidentified (12). To confirm and extend the preceding results, we here derived cardiomyocyte-specific Klf4 knockout (CM Klf4 KO) mice by breeding transgenic mice expressing Cre recombinase under the control of the α-myosin heavy chain (Myh6) promoter (Myh6-Cre mice) (13) and Klf4loxP mice (14) and examined the effects of Klf4 deletion on cardiac hypertrophy induced by chronic infusion of the β-adrenoreceptor agonist isoproterenol (ISO). We also sought to determine the mechanisms by which KLF4 regulated the reactivation of fetal cardiac genes during the development of cardiac hypertrophy. Finally, we examined whether KLF4 mediated the antihypertrophic effect of olmesartan and trichostatin A (TSA), which are an angiotensin II type 1 receptor antagonist and a histone deacetylase (HDAC) inhibitor, respectively.
EXPERIMENTAL PROCEDURES
Generation of CM Klf4 KO Mice
Animal protocols were approved by the Keio University Animal Care and Use Committee. Klf4loxP/loxP mice, provided by Dr. Klaus H. Kaestner (14), were bred with Myh6-Cre+/− mice (13) to generate Myh6-Cre+/−/Klf4loxP/+ mice. Myh6-Cre+/−/Klf4loxP/+ mice were then bred with Klf4loxP/loxP mice to generate Myh6-Cre+/−/Klf4loxP/loxP (CM Klf4 KO) mice and Myh6-Cre−/−/Klf4loxP/loxP (control) mice. Tagln promoter-dependent Klf4 knockout mice were generated by breeding Tagln-Cre mice and Klf4loxP/loxP mice as described previously (10). All mice were on the C57BL/6J background, and littermates were used for comparisons. Genotyping was performed by PCR as described previously (5, 14). The amounts of the Klf4loxP allele and the Acta2 promoter region in the genome extracted from the heart or isolated cardiomyocytes were determined by real-time PCR (5, 14), and the ratio of the Klf4loxP allele to the Acta2 promoter region was calculated to evaluate the recombination rate. Isolation of cardiomyocytes from day 1 neonates was performed with a Pierce primary cardiomyocyte isolation kit (Thermo Scientific, Rockford, IL).
Cardiac Hypertrophy Models
Mice were anesthetized with an intraperitoneal injection of pentobarbital sodium (50 mg/kg), and micro-osmotic pumps (Alzet model 1002, Durect Corp., Cupertino, CA) containing saline or ISO (Sigma-Aldrich, St. Louis, MO), the latter calibrated to release the drug at a rate of 30 mg/kg/day for 14 days, were implanted surgically and subcutaneously in the interscapular region of the mouse (15). Blood pressure and heart rate were measured by the tail cuff method (BP-98E, Softron, Tokyo, Japan) before and on day 13 after mini pump implantation. Mice were sacrificed on day 14 after the surgery. Olmesartan (Daiichi-Sankyo Co. Ltd., Tokyo, Japan) was given by gavage at a dose of 10 mg/kg/day to a subset of mice for 14 days after the surgery (16, 17). TSA (Sigma-Aldrich) at 0.6 mg/kg/day was injected intraperitoneally into a subset of mice for 14 days after the surgery (18). For the angiotensin II-induced cardiac hypertrophy model, mice received a subcutaneous infusion of angiotensin II (Sigma-Aldrich) at a rate of 2 mg/kg/day for 14 days, as described previously (19).
Tissue Harvesting, Histology, and Morphometric Analyses
Mice were weighed and euthanized. The atria were dissected away from the ventricles. The ventricles were weighed and divided into multiple pieces for histology, RNA extraction, total cellular protein extraction, and chromatin immunoprecipitation assays. For histology, tissues were fixed in 4% paraformaldehyde and embedded into OCT compound. Masson trichrome staining (Sigma-Aldrich) was performed to evaluate cardiac fibrosis. The level of fibrosis was graded as an integer number from 0–4 in a blinded manner. To quantify the individual myocyte size, sections were stained with Alexa Fluor 555-conjugated wheat germ agglutinin (Invitrogen) and DAPI. The myocyte area was assessed by Image-Pro Plus software (Media Cybernetics, Silver Spring, MD). The mean cardiomyocyte area was evaluated by measurement of 10 cells/heart (5–6 hearts/genotype).
Plasmid Constructs
Expression plasmids for Myc-KLF4, FLAG-KLF4, and FLAG-myocardin (MYOCD) have been described previously (4, 20). The rat Nppa (−633/+88) promoter-luciferase construct in the pGL3-basic vector (Promega, Madison, WI) was made by PCR amplification of the DNA fragments. Site-directed mutagenesis of the Nppa promoter-luciferase construct was performed using the QuikChange II site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). The structure was confirmed by DNA sequencing.
Cell Culture
Rat H9c2 cells (ATCC) were cultured in DMEM supplemented with 10% FBS (Invitrogen). One day after plating 10,000 cells/cm2, the medium was changed to DMEM without serum for 24 h. Cells were then transfected with the KLF4 expression plasmid or Klf4 siRNA using Superfect (Qiagen, Valencia, CA) or Lipofectamine RNAiMAX (Invitrogen), respectively (21). Cells were cultured in DMEM with 10% FBS or no serum for an additional 24 h. For luciferase assays, DNA plasmids and siRNAs were transfected into H9c2 cells, and cells were incubated for 48 h in DMEM supplemented with 10% FBS. Luciferase activity was measured as described previously (22).
Real-time RT-PCR
Total RNA prepared from the heart or cultured H9c2 cells was used for real-time RT-PCR. Primer and probe sequences for Nppa, Nppb, Myh7, Klf4, Myocd, serum response factor (Srf), and 18 S rRNA have been described previously (10, 22, 23). Expression of microRNAs (miR-1, miR-9, and miR-133a) and small nucleolar RNA 202 was measured by TaqMan microRNA assays (Invitrogen).
Western Blotting and Immunofluorescence Studies
Western blotting was performed using antibodies for NPPA (Peninsula Laboratories, San Carlos, CA) and GAPDH (6C5, Millipore, Billerica, MA), as described previously (4, 10). Immunofluorescence studies were performed by using antibodies for α-actinin (catalog no. EA-53, Sigma-Aldrich), Myc (Abcam), and FLAG (Sigma). The cell surface area where α-actinin was stained was quantified using Image-Pro Plus software by measurement of 10 cells/group.
EMSA
EMSA was performed using a LightShift chemiluminescent EMSA kit (Thermo Scientific). The 5′ biotin-labeled oligonucleotide (5′-CTCTCACACCTTTGAAG-3′) was annealed to its complementary sequence and incubated with cellular lysates from H9c2 cells transfected with the FLAG-KLF4 expression plasmid. Competition assays were performed using an unlabeled double-stranded oligonucleotide or its mutant (5′-CTCTCACTGGTTTGAAG-3′). The FLAG antibody was used for supershift assays.
Quantitative Chromatin Immunoprecipitation Assays
Quantitative chromatin immunoprecipitation assays were performed using anti-KLF4 antibody or anti-SRF antibody, as described previously (4, 5, 10). Real-time PCR was performed to amplify the promoter region of the Nppa gene. Primer sequences were as follows: Nppa-proF, 5′-GGCCAGAGGTCCACCCACGA-3′; Nppa-proR, 5′-CCAGACCCTCAGCTGCAAGA-3′.
Statistical Analyses
Data are presented as mean ± S.E. Statistical analyses were done by SigmaPlot/SigmaStat9 (Systat Software Inc., San Jose, CA). After confirming that the data passed the normality test for parametric analyses, unpaired Student's t test (see Fig. 1, C and D), one-way factorial analysis of variance (Fig. 7B), two-way factorial analysis of variance (see Figs. 2, A, B, and E–G; 2I; 3, A–F; 4, A–C, E, and G; 5, A–C; 6, A–D; and 8C), and three-way factorial analysis of variance (Fig. 9, A–H) were performed with a post hoc Fisher protected least significant difference test. A Kruskal-Wallis non-parametric test was performed for Fig. 2C because the data did not pass the normality test. p < 0.05 was considered significant.
FIGURE 1.
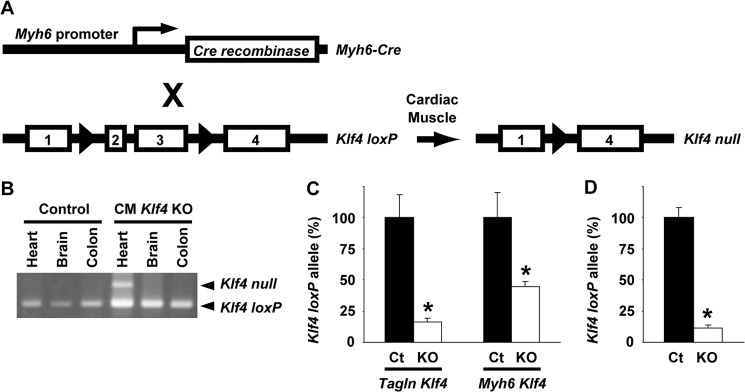
Generation of CM Klf4 KO mice. A, schematic of the cardiomyocyte-specific deletion of the Klf4 gene. The numbers represent Klf4 exons. Triangles represent the loxP sites. X, breeding. B, recombination of the Klf4loxP allele was examined by PCR in multiple tissues, including the heart, brain, and colon of CM Klf4 KO mice and control mice. A representative picture is shown (n = 4/genotype). C and D, the recombination rates of the Klf4loxP allele were determined by real-time PCR in the hearts from day 1 neonates in Tagln promoter-dependent Klf4 knockout mice and CM Klf4 KO mice (Myh6 Klf4) (C, n = 10/group) or in cardiomyocytes isolated from day 1 neonates in CM Klf4 KO mice (D, n = 3/group). The ratio of the Klf4loxP allele to the Acta2 promoter region, a nonspecific genomic region, was calculated. *, p < 0.05 compared with control (Ct) neonatal hearts (C) or control isolated cardiomyocytes (D).
FIGURE 7.
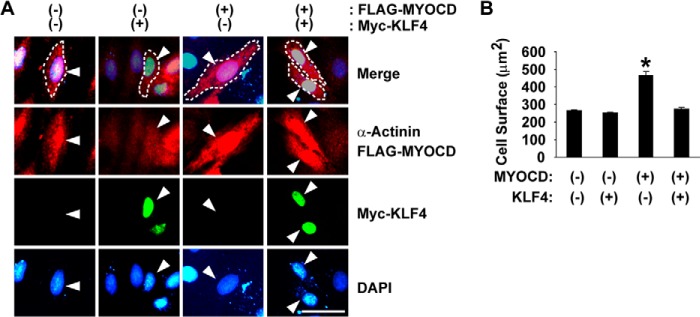
KLF4 attenuated MYOCD-induced cellular enlargement. H9c2 cells were transfected with the expression plasmids for FLAG-tagged MYOCD and/or Myc-tagged KLF4. Cells were stained with anti-FLAG antibody (red), anti-α-Actinin antibody (red), anti-Myc antibody (green), and DAPI (blue). A, representative pictures. Although costaining of FLAG and α-Actinin could not distinguish the subcellular localization of MYOCD from α-Actinin, preliminary experiments using each of these antibodies showed that FLAG-MYOCD was exclusively localized in the nucleus, whereas α-Actinin was stained in the cytoplasm (data not shown). Cells of interest are indicated by arrowheads. Dotted lines indicate the margin of the cells. Scale bar = 50 μm. B, cell surface areas were quantified in 10 cells/group. *, p < 0.05 compared with control.
FIGURE 2.

ISO-induced cardiac hypertrophy was accelerated in CM Klf4 KO mice. CM Klf4 KO mice and control (Ct) mice received a continuous subcutaneous infusion of ISO for 14 days. A–G, body weight (A), systolic and diastolic blood pressure (B), and heart rate (C) were measured on day 13, and heart weight (E), the HW/BW ratio (F), and the HW/TL ratio (G) were measured at the time of sacrifice in CM Klf4 KO mice and control mice (n = 5–6/group). The tops and bottoms of the squares in B indicate systolic and diastolic blood pressure, respectively. Cross-sections of the hearts were subjected to hematoxylin and eosin staining, and representative pictures are shown in D. Scale bar = 2 mm. H and I, wheat germ agglutinin staining was performed in the hearts of ISO-treated CM Klf4 KO mice and control mice. H, representative pictures from 5–6 analyzed mice/group. DAPI was used for nuclear staining. Scale bar = 100 μm. I, myocyte areas were evaluated by measurement of 10 cells/heart (5–6 hearts/group). *, p < 0.05 compared with ISO-untreated mice; #, p < 0.05 compared with ISO-treated control mice.
FIGURE 3.
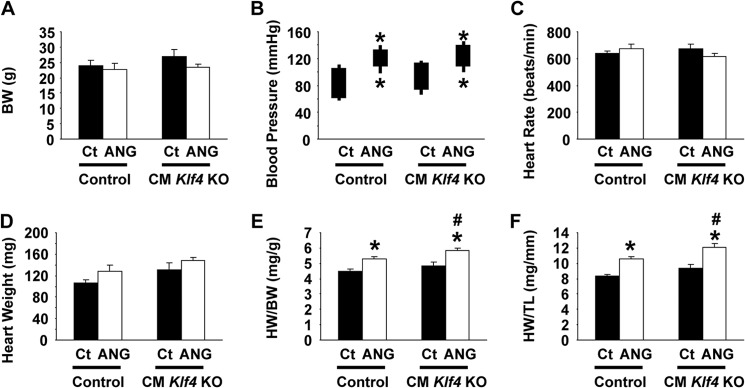
Angiotensin II-induced cardiac hypertrophy was accelerated in CM Klf4 KO mice. CM Klf4 KO mice and control (Ct) mice received a continuous subcutaneous infusion of angiotensin II (ANG) for 14 days. Body weight (A), systolic and diastolic blood pressure (B), and heart rate (C) were measured on day 13, and heart weight (D), the HW/BW ratio (E), and the HW/TL ratio (F) were measured at the time of sacrifice in CM Klf4 KO mice and control mice (n = 5–8/group). The tops and bottoms of the squares in B indicate systolic and diastolic blood pressure, respectively. *, p < 0.05 compared with angiotensin II-untreated mice; #, p < 0.05 compared with angiotensin II-treated control mice.
FIGURE 4.

ISO-induced expression of fetal cardiac markers was augmented in CM Klf4 KO mice. CM Klf4 KO mice and control (Ct) mice received a continuous subcutaneous infusion of ISO for 14 days. A–C, expression of Nppa (A), Nppb (B), and Myh7 (C) was determined by real-time RT-PCR in the hearts of ISO-treated CM Klf4 KO mice and control mice (n = 5–6/group). D and E, expression of NPPA and GAPDH in the heart was examined by Western blotting. D, representative pictures (n = 5/group). E, expression levels of NPPA and GAPDH were measured by densitometry, and the NPPA/GAPDH ratio was calculated. F and G, cardiac fibrosis was evaluated by Masson trichrome staining. F, representative pictures (n = 5–6/group). Scale bar = 200 μm. G, the levels of cardiac fibrosis were graded. *, p < 0.05 compared with ISO-untreated mice; #, p < 0.05 compared with ISO-treated control mice.
FIGURE 5.
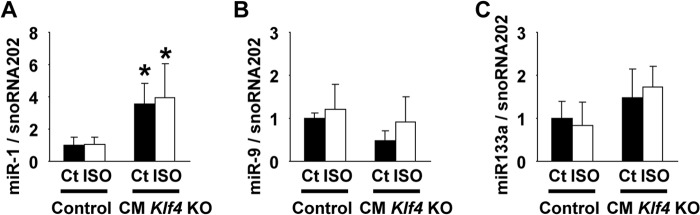
Expression of miR-1 was increased in CM Klf4 KO mice. CM Klf4 KO mice and control (Ct) mice received a continuous subcutaneous infusion of ISO for 14 days. Expression of miR-1 (A), miR-9 (B), and miR-133a (C) was determined by real-time RT-PCR in the hearts from ISO-treated CM Klf4 KO mice and control mice (n = 5–6/group). *, p < 0.05 compared with control mice.
FIGURE 6.

Induction of Myocd expression in ISO-treated hearts was enhanced in CM Klf4 KO mice. A–C, CM Klf4 KO mice and control (Ct) mice received a continuous subcutaneous infusion of ISO for 14 days. Expression of Myocd (A), Srf (B), and Klf4 (C) in the hearts was determined by real-time RT-PCR (n = 5–6/group). *, p < 0.05 compared with ISO-untreated mice; #, p < 0.05 compared with ISO-treated control mice; $, p < 0.05 compared with ISO-untreated control mice. D, H9c2 cells were transfected with KLF4 expression plasmid, empty plasmid, or siRNAs for Klf4 or scrambled sequence and incubated in DMEM supplemented with 10% FBS or serum-free medium (SFM). Expression of Nppa, Myocd, and Srf was determined by real-time RT-PCR (n = 4). *, p < 0.05 compared with cells incubated with serum-free medium.
FIGURE 8.

KLF4 attenuated MYOCD-induced Nppa expression. A, H9c2 cells were transfected with the Nppa promoter-luciferase construct, its mutant of the KLF4 binding site, or the pGL3-basic plasmid with the expression plasmid for MYOCD and increasing amounts of KLF4 expression plasmid. Luciferase (Luc) activity was measured and normalized to protein content (n = 4). Ct, control. B, EMSA was performed using cellular lysates from FLAG-KLF4-transfected H9c2 cells and a 5′ biotin-labeled, double-stranded oligonucleotide probe containing the putative KLF4 binding site. Competition assays were performed using WT or mutated (Mut) cold competitors. Supershift assays were performed using anti-FLAG antibody or control IgG. C, association of KLF4 and SRF with the Nppa promoter was examined by in vivo chromatin immunoprecipitation assays in the hearts of ISO-treated CM Klf4 KO mice and control mice (n = 4/group). *, p < 0.05 compared with ISO-untreated mice.
FIGURE 9.

ISO-induced cardiac hypertrophy in CM Klf4 KO mice was blunted by olmesartan but not by TSA. CM Klf4 KO mice and control (Ct) mice received a continuous subcutaneous infusion of ISO for 14 days. A subset of mice received olmesartan (Olme) orally for 14 days or intraperitoneal injections of TSA for 14 days after mini pump implantation. A–F, body weight (A), systolic and diastolic blood pressure (B), and heart rate (C) were measured on day 13, and heart weight (D), the HW/BW ratio (E), and the HW/TL ratio (F) were measured at the time of sacrifice in CM Klf4 KO mice and control mice (n = 5–6/group). G, wheat germ agglutinin staining was performed in the hearts, and myocyte areas were evaluated by measurement of 10 cells/heart (5–6 hearts/group). H, expression of Nppa in the hearts was determined by real-time RT-PCR (n = 5–6/group). Data for mice that were not treated with olmesartan or TSA are reproduced from Figs. 2, A–C, E, G, and I, and 4A. *, p < 0.05 compared with ISO-untreated mice; #, p < 0.05 compared with ISO-treated control mice; $, p < 0.05 compared with mice that were not treated with olmesartan or TSA.
RESULTS
CM Klf4 KO Mice Grew Normally to Adulthood
To determine the role of KLF4 in cardiac hypertrophy, CM Klf4 KO mice were generated by breeding Myh6-Cre mice and Klf4loxP mice (Fig. 1A). CM Klf4 KO mice were born at the expected Mendelian ratio and were grown to adulthood without any differences in visible appearance compared with controls. Body weight (24.3 ± 1.3 g in CM Klf4 KO mice versus 23.6 ± 0.9 g in control mice, n = 10/genotype), systolic blood pressure (103.2 ± 2.8 mm Hg in CM Klf4 KO mice versus 100.1 ± 1.5 mm Hg in control mice, n = 10/genotype), diastolic blood pressure (67.9 ± 3.5 mm Hg in CM Klf4 KO mice versus 64.4 ± 2.0 mm Hg in control mice, n = 10/genotype), and heart rate (642 ± 15 beats/min in CM Klf4 KO mice versus 659 ± 30 beats/min in control mice, n = 10/genotype) at 11 weeks of age were similar in CM Klf4 KO mice and control mice. Selective recombination of the Klf4loxP allele was seen in the heart, but not in the brain and colon, in CM Klf4 KO mice (Fig. 1B). Results of our previous studies showed that smooth and cardiac muscle-selective knockout of the Klf4 gene in mice under the control of the Tagln promoter resulted in postnatal death and growth retardation, probably because of lack of Klf4 in the heart (10). To clarify the reasons for the differences in growth and survival between CM Klf4 KO mice and Tagln promoter-dependent Klf4 knockout mice, the recombination rate of the Klf4loxP allele was determined in hearts from day 1 neonates in both mice. Although 84% of the Klf4loxP allele was recombinated in the hearts of Tagln promoter-dependent Klf4 knockout neonates, the recombination rate was 55% in CM Klf4 KO neonates (Fig. 1C). Because the proportion of cardiomyocytes in the heart is about 50–60% (24), it is likely that the recombination in Tagln promoter-dependent Klf4 knockout mice occurred in multiple cardiac cell types in addition to cardiomyocytes, whereas the recombination occurred specifically in cardiomyocytes in CM Klf4 KO mice. In support of this, the recombination rate of the Klf4loxP allele in isolated cardiomyocytes derived from day 1 neonates of CM Klf4 KO mice was much higher than in the hearts of these mice (Fig. 1D). These results may explain why CM Klf4 KO mice grew up normally even though KLF4 plays a key role in late fetal and/or postnatal cardiac development in Tagln-dependent, Klf4-deficient mice (10).
ISO-induced Cardiac Hypertrophy Was Accelerated in CM Klf4 KO Mice
CM Klf4 KO mice and control mice 11–12 weeks old received a continuous subcutaneous infusion of ISO for 14 days via osmotic mini pumps. After 2 weeks of ISO infusion, body weight, systolic and diastolic blood pressure, and heart rate were unaltered in CM Klf4 KO mice and in control mice (Fig. 2, A–C). However, ISO infusion caused increases in heart weight, the heart weight to body weight (HW/BW) ratio, as well as the heart weight to tibia length (HW/TL) ratio in both mice (Fig. 2, D–G). Of particular interest, ISO-induced increases in these parameters were accelerated in CM Klf4 KO mice compared with control mice. Indeed, ISO induced a 31% increase in the HW/BW ratio in CM Klf4 KO mice, whereas it only increased 20% in control mice. Consistent with these results, microscopic analysis of cardiomyocytes stained with a fluorescently labeled wheat germ agglutinin revealed that the areas of cardiomyocytes were significantly larger in ISO-treated hearts of CM Klf4 KO mice than those of control mice, although both of them were larger than those of ISO-untreated counterparts (Fig. 2, H and I). Accelerated cardiac hypertrophy in CM Klf4 KO mice was also seen in an angiotensin II infusion model (Fig. 3).
Expression of fetal cardiac markers was examined by real-time RT-PCR. Results showed that, although the expression of Nppa, Nppb, and Myh7 in ISO-untreated hearts did not differ between CM Klf4 KO mice and control mice, the ISO-induced augmentation of these genes was more remarkable in CM Klf4 KO mice than in control mice (Fig. 4, A–C). That is, ISO-induced increases in expression of Nppa, Nppb, and Myh7 were 2.6-fold, 2.7-fold, and 4.2-fold in CM Klf4 KO mice, respectively, whereas they were 1.7-fold, 2.1-fold, and 3.1-fold in control mice. Moreover, ISO-mediated induction of NPPA expression at the protein level was more exaggerated in CM Klf4 KO mice than in control mice (Fig. 4, D and E). In addition to the enhanced induction of fetal cardiac markers, cardiac fibrosis was more significant in CM Klf4 KO mice than in control mice, as determined by Masson trichrome staining and the semiquantitative scoring system (Fig. 4, F and G). Taken together, these results suggest that ISO-induced cardiac hypertrophy is accelerated in CM Klf4 KO mice compared with control mice.
We examined the apoptotic rate and the expression of microRNAs in the hearts of ISO-treated CM Klf4 KO mice. First, the apoptotic rate in ISO-treated hearts did not differ between CM Klf4 KO mice and control mice, as determined by TUNEL staining (data not shown). Second, we hypothesized that microRNAs might play a role in the dissociation between Nppa mRNA expression (2.6-fold versus controls, Fig. 4A) and NPPA protein expression (8.4-fold versus controls, Fig. 4E) in ISO-treated CM Klf4 KO mice. Expression of miR-1, miR-9, and miR-133a, which are known to be involved in cardiac hypertrophy (25), was measured by real-time RT-PCR. The results showed that expression of miR-1 was increased in both ISO-treated and ISO-untreated hearts of CM Klf4 KO mice, although ISO did not induce miR-1 expression in CM Klf4 KO mice and control mice (Fig. 5A). Expression of miR-9 and miR-133a in the hearts was unaltered by ISO treatment or Klf4 deletion (Fig. 5, B and C). These results suggest that apoptosis and microRNAs do not play a critical role in accelerated cardiac hypertrophy in CM Klf4 KO mice.
KLF4 Attenuated Hypertrophy-related Nppa Induction by Suppressing MYOCD Expression and Activity
To clarify the molecular mechanisms by which KLF4 controls ISO-induced cardiac hypertrophy, we sought to determine whether KLF4 regulates MYOCD-induced Nppa expression. MYOCD is a cardiac and smooth muscle-specific cofactor of SRF that potently activates the transcription of CArG element-containing muscle-selective genes, including Nppa, Nppb, and Myh7 (20, 26). The results of previous studies showed that MYOCD induced cardiac hypertrophy (26) and that it was up-regulated in the hearts of piglets with doxorubicin-induced cardiomyopathy and in patients with dilated cardiomyopathy (27). As shown in Fig. 6A, quantitative analysis using real-time RT-PCR showed that ISO-induced Myocd expression was more abundant in the hearts of CM Klf4 KO mice (2.2-fold) than in control mice (1.5-fold), although Myocd expression levels in ISO-untreated hearts did not differ between CM Klf4 KO mice and control mice. By contrast, Srf expression was unaltered by either ISO infusion or Klf4 deletion (Fig. 6B). Klf4 expression in the hearts was induced by chronic ISO infusion in control mice, whereas it was diminished in CM Klf4 KO mice (Fig. 6C). The effects of KLF4 on the expression of Nppa and Myocd were then examined in cultured H9c2 cardiac cells. Consistent with the results seen in mice in vivo, siRNA-mediated knockdown of Klf4 augmented the serum-induced activation of Nppa and Myocd, whereas overexpression of KLF4 inhibited the serum-induced activation of these genes in H9c2 cells (Fig. 6D). These results suggest that the repressive effect of KLF4 on Nppa expression is mediated, at least in part, by the down-regulation of Myocd expression in the heart.
In addition to the repressive effect of KLF4 on Myocd expression, we found that KLF4 also suppressed MYOCD activity, on the basis of the following observations. First, although overexpression of MYOCD enlarged cell surface areas in H9c2 cells, coexpression of KLF4 abolished MYOCD-induced cellular enlargement (Figs. 7, A and B). Second, the results of transfection/luciferase assays showed that MYOCD induced the transcriptional activity of the Nppa (−633/+88) luciferase construct and that the effect of MYOCD was attenuated by KLF4 in H9c2 cells (Fig. 8A). These results suggest that KLF4 inhibits the effect of MYOCD on cellular enlargement as well as Nppa expression through the posttranslational mechanisms in cardiac cells. Of interest, a consensus KLF4 binding site, 5′-(G/A)(G/A)GG(C/T)G(C/T)-3′, was found at −250/−244 bp between two CArG elements (−409/−400 bp and −117/−108 bp) within the Nppa (−633/+88) promoter. Therefore, we examined the effects of mutation of the KLF4 binding site by site-directed mutagenesis. The results showed that mutation of the KLF4 binding site within the Nppa promoter attenuated the inhibitory effect of KLF4 on MYOCD-induced Nppa activation (Fig. 8A). In addition, KLF4 binding to the KLF4 binding site was detectable by EMSA (Fig. 8B). Indeed, FLAG-tagged KLF4 bound with the biotin-labeled double-stranded oligonucleotide probe containing the KLF4 binding site, and the binding disappeared in the presence of an excess of unlabeled probe or anti-FLAG antibody. Moreover, in vivo chromatin immunoprecipitation assays showed that KLF4 binding with the Nppa promoter was increased by ISO infusion in the hearts of control mice but that the binding was undetectable in CM Klf4 KO mouse hearts (Fig. 8C). On the other hand, SRF binding with the Nppa promoter was increased by ISO infusion in control mice, and it was enhanced further in the hearts of ISO-treated CM Klf4 KO mice. Taken together, these results suggest that KLF4 regulates Nppa expression by at least two mechanisms: KLF4 suppresses Myocd expression, and KLF4 represses the activity of MYOCD by binding to the KLF4 binding site between CArG elements within the Nppa promoter.
Olmesartan, but Not TSA, Attenuated ISO-induced Cardiac Hypertrophy in CM Klf4 KO Mice
Both angiotensin II type 1 receptor antagonists (16, 17) and histone deacetylase inhibitors (18, 28) have been shown to attenuate the progression of cardiac hypertrophy. We determined whether the antihypertrophic effects of olmesartan and TSA were mediated by KLF4 using CM Klf4 KO mice. CM Klf4 KO mice and control mice received a daily oral dose of olmesartan or a daily intraperitoneal injection of TSA during the continuous subcutaneous ISO infusion for 14 days. The doses and the routes of administration for these antihypertrophic reagents were as those employed in previous studies (16, 18). Results showed that body weight, systolic blood pressure, and heart rate were unaltered by treatment with olmesartan or TSA, although the diastolic blood pressure was decreased by olmesartan treatment in ISO-untreated CM Klf4 KO mice and control mice (Fig. 9, A–C). Consistent with the results of previous studies (16–18, 28), treatment with olmesartan and TSA attenuated ISO-induced cardiac hypertrophy in control mice, as assessed by heart weight, the HW/BW ratio, and the HW/TL ratio (Fig. 9, D–F). However, significantly, olmesartan, but not TSA, blunted ISO-induced cardiac hypertrophy in CM Klf4 KO mice. Indeed, olmesartan treatment decreased the HW/BW ratio by 81% in ISO-treated CM Klf4 KO mice, but TSA only decreased it by 95%. Similar trends were observed in the HW/TL ratio, the areas of cardiomyocytes, as well as Nppa expression, although TSA slightly decreased ISO-induced Nppa expression in CM Klf4 KO mice (Fig. 9, F–H). Consistently, the results of transfection/luciferase assays showed that serum-induced transcriptional activation of the Nppa gene was blocked by TSA and that this effect was abolished by siRNA-mediated knockdown of Klf4 in H9c2 cells (Fig. 10). As such, these results suggest that the antihypertrophic effect of TSA, but not olmesartan, was mediated by KLF4 in the heart.
FIGURE 10.

The repressive effect of TSA on serum-induced Nppa activation was abolished by knockdown of Klf4. H9c2 cells were transfected with the Nppa promoter-luciferase construct and siRNAs for Klf4 or scrambled sequence, treated with 100 nmol/l TSA or control (Ct), and incubated in DMEM supplemented with 10% FBS or serum-free medium (SFM). Luciferase activity was measured and normalized to protein content (n = 4).
DISCUSSION
In this study, we showed that ISO-induced cardiac hypertrophy was enhanced in CM Klf4 KO mice compared with control mice. Indeed, the HW/BW ratio and the HW/TL ratio, established measures of cardiac hypertrophy, were both increased in ISO-treated CM Klf4 KO mice compared with ISO-treated control mice. Accelerated cardiac hypertrophy in CM Klf4 KO mice was accompanied by the augmented cellular enlargement of cardiomyocytes as well as the exaggerated expression of fetal cardiac marker genes, including Nppa, Nppb, and Myh7. Mechanistic analyses demonstrated that KLF4 suppressed the expression and the activity of MYOCD, a potent transcriptional cofactor for SRF. Because MYOCD has been shown to be an inducer of cardiac hypertrophy (26), KLF4-mediated alterations in the expression and the activity of MYOCD are likely to be the underlying mechanisms for accelerated cardiac hypertrophy in ISO-treated CM Klf4 KO mice. Moreover, we showed that olmesartan, but not TSA, attenuated ISO-induced cardiac hypertrophy in CM Klf4 KO mice, although both reagents efficiently inhibited it in control mice. As such, the results of this study provide evidence that KLF4 regulates ISO-induced cardiac hypertrophy by modulating MYOCD expression and activity and that KLF4 mediates the antihypertrophic effect of the HDAC inhibitor TSA in mice in vivo.
Although the results of our previous studies showed that Tagln promoter-dependent, Klf4-deficient mice exhibited severe growth retardation and postnatal death (10), CM Klf4 KO mice grew normally to adulthood. The differences in phenotype between these mice are caused by the differences in the promoter driving Cre recombinase, i.e. the Tagln promoter versus the Myh6 promoter. The Tagln promoter is active in embryonic and adult SMCs as well as in hearts between embryonic days (E) 8.0 and E12.5 (11). By contrast, MYH6 is a marker of matured cardiomyocytes, but results of several previous studies showed that Cre recombination in Myh6-Cre mice started at E9.5 and occurred in nearly all cardiomyocytes by E11.5 (29, 30). Because KLF4 expression in the heart is first detectable at E18.5 (10), it is unlikely that the difference in timing of the recombination affected the distinct phenotype between these mice. Alternatively, it is highly possible that the cell specificity of Cre recombination contributed to the distinct phenotype. Indeed, although the recombination rate of the Klf4loxP allele was 55% in neonatal hearts of CM Klf4 KO mice, it was over 80% in the hearts of Tagln promoter-dependent, Klf4-deficient mouse neonates. Because 50–60% of cells in the heart are cardiomyocytes (24), our results suggest that the recombination occurs specifically in cardiomyocytes in CM Klf4 KO mice, whereas it occurs in multiple cell types, including SMCs, cardiomyocytes, and other cell types, in Tagln promoter-dependent, Klf4-deficient mice. In support of this, the results of previous studies using Tagln-Cre mice and indicator mice containing a Cre-activated LacZ gene showed that Cre-dependent LacZ expression was observed in almost all cells in the left and right ventricles (31). Further studies are needed to determine the cellular fate of temporary TAGLN-positive cells in the heart.
The results of this study showed that KLF4 regulated Myocd expression in cardiac cells. Indeed, siRNA-mediated repression of Klf4 augmented serum-activated Myocd expression, whereas overexpression of KLF4 inhibited it in H9c2 cells. Moreover, induction of Myocd expression was enhanced more in the hearts of ISO-treated CM Klf4 KO mice than in control mice. Consistent with these results, KLF4 has been shown to repress Myocd expression in cultured aortic SMCs (32, 33). Oxidized phospholipids induced an increase in KLF4 expression and a decreased expression of Myocd in cultured SMCs, and the reduction in Myocd expression was abolished by the presence of Klf4 siRNA (32). Likewise, interleukin-1β increased KLF4 expression, but it repressed expression of Myocd and SMC differentiation markers in cultured SMCs (33). The repressive effect of interleukin 1β on Myocd expression is considered to be mediated through KLF4 because interleukin 1β-induced repression of Myocd expression was abolished in Klf4-deficient SMCs (33). KLF4 repressed Myocd expression by binding to the consensus KLF4 binding site within the Myocd promoter in concert with p65, a component of NF-κB in SMCs (33). Mutation of either the KLF4 binding site or the NF-κB binding site attenuated interleukin 1β-induced transcriptional repression of the Myocd gene in SMCs. In addition, both KLF4 and p65 were enriched within the Myocd promoter in SMCs after interleukin 1β treatment, as determined by chromatin immunoprecipitation assays. Moreover, KLF4 has been shown to interact with p65 by coimmunoprecipitation assays (34). As such, the preceding studies provide evidence that KLF4 represses Myocd expression in concert with p65 in SMCs. However, inhibition of the NF-κB pathway has been shown to reduce ISO-induced cardiac hypertrophy in mice (19), suggesting that KLF4 and NF-κB counteract each other in the heart. It is of interest to further determine the relationship between KLF4, NF-κB, and other transcription factors during the progression of cardiac disease.
In this study, we showed that KLF4 not only decreased Myocd expression but that it also suppressed the activity of MYOCD in cardiac cells. Overexpression of KLF4 inhibited MYOCD-induced cellular enlargement. In addition, KLF4 attenuated MYOCD-induced Nppa expression by binding to the KLF4 binding site located between CArG elements within the Nppa promoter. MYOCD is a non-DNA-binding transcriptional cofactor of SRF, and SRF is a binding factor for CArG elements (20, 35). Many muscle-specific genes, such as Nppa, contain multiple CArG elements within their promoter, and homodimerization of MYOCD activates the transcription of these genes by eliciting the interaction between multiple SRF-CArG complexes (35). Because the KLF4 binding site within the Nppa promoter is located between two CArG elements, KLF4 would efficaciously interfere with MYOCD, which brings two SRF-CArG complexes closer to each other. In fact, the binding affinity of SRF to CArG elements was increased greatly in the hearts of ISO-treated CM Klf4 KO mice compared with ISO-treated control mice. Of importance, the KLF4 binding site within the Nppa promoter is completely conserved among humans, mice, and rats. Moreover, it also appears at −432/−426 bp within the NPPB promoter, which is located near the proximal CArG element (−181/−172 bp). As such, the results of this study demonstrate that KLF4 regulates cardiac hypertrophy through multiple mechanisms, including the modulation of MYOCD expression and activity.
Recently, multiple HDAC inhibitors, such as TSA, valproic acid, SK-7041, and Scriptaid, have been shown to be useful for preventing cardiac hypertrophy in vivo (18, 28). The antihypertrophic effect of these HDAC inhibitors is thought to be mediated by blocking HDAC2 because inactivation of HDAC2 resulted in constitutive activation of glycogen synthase kinase-3β, an important negative regulator of cardiac hypertrophy, and because Hdac2 deficiency in mice attenuated cardiac hypertrophy in response to hypertrophic stimuli, whereas Hdac2 transgenic mice exhibited augmented cardiac hypertrophy (36). In this study, we showed that KLF4 was a factor mediating the repressive effect of TSA on ISO-induced cardiac hypertrophy in mice. Of interest, KLF4 has been shown to physically interact with HDAC2, as determined by GST pulldown assays (37). Moreover, the results of previous studies in cultured cardiomyocytes showed that KLF4 was a downstream target of HDAC2 (38). As such, KLF4 is likely to be a factor mediating the antihypertrophic effect of HDAC inhibitors by associating with HDAC2. Further studies are needed to examine the physical and/or functional relationship between KLF4 and glycogen synthase kinase 3β in hypertrophic hearts.
In contrast to the relationship between KLF4 and HDAC signaling, we showed that KLF4 did not mediate the antihypertrophic effect of olmesartan on ISO-induced cardiac hypertrophy. In this regard, it is worth noting that angiotensin II type 1 receptor antagonists are able to interfere with β-adrenergic receptors via receptor-receptor interaction (39). The results of previous studies showed that angiotensin II type 1 receptors and β-adrenergic receptors formed constitutive complexes and that a single receptor antagonist effectively blocked downstream signaling of both receptors simultaneously (39). On the basis of these findings, it is highly possible that olmesartan blocked ISO-mediated activation of β-adrenergic receptor signaling at the cell surface level, which was positioned far upstream of KLF4. Although speculative, this may be why the antihypertrophic effect of olmesartan was not attenuated in CM Klf4 KO mice.
In summary, we provide novel evidence that KLF4 regulates cardiac hypertrophy by modulating the expression and the activity of MYOCD. The reagents enhancing KLF4 activity would be a novel candidate for treatment of cardiac hypertrophy.
This work was supported by a grant-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to T. Y. and M. H.) and by the Mochida Memorial Foundation for Medical and Pharmaceutical Research (to T. Y.).
- SMC
- smooth muscle cell
- CM Klf4 KO
- cardiomyocyte-specific Klf4 knockout
- ISO
- isoproterenol
- HDAC
- histone deacetylase
- MYOCD
- myocardin
- miR
- microRNA
- HW
- heart weight
- BW
- body weight
- TL
- tibia length
- SRF
- serum response factor
- E
- embryonic day(s).
REFERENCES
- 1. Lorell B. H., Carabello B. A. (2000) Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation 102, 470–479 [DOI] [PubMed] [Google Scholar]
- 2. Yoshida T., Hayashi M. (2014) Role of Krüppel-like factor 4 and its binding proteins in vascular disease. J. Atheroscler. Thromb. 21, 402–413 [DOI] [PubMed] [Google Scholar]
- 3. Liu Y., Sinha S., McDonald O. G., Shang Y., Hoofnagle M. H., Owens G. K. (2005) Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J. Biol. Chem. 280, 9719–9727 [DOI] [PubMed] [Google Scholar]
- 4. Yoshida T., Gan Q., Owens G. K. (2008) Krüppel-like factor 4, Elk-1, and histone deacetylases cooperatively suppress smooth muscle cell differentiation markers in response to oxidized phospholipids. Am. J. Physiol. Cell Physiol. 295, C1175–C1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoshida T., Kaestner K. H., Owens G. K. (2008) Conditional deletion of Krüppel-like factor 4 delays downregulation of smooth muscle cell differentiation markers but accelerates neointimal formation following vascular injury. Circ. Res. 102, 1548–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hamik A., Lin Z., Kumar A., Balcells M., Sinha S., Katz J., Feinberg M. W., Gerzsten R. E., Edelman E. R., Jain M. K. (2007) Kruppel-like factor 4 regulates endothelial inflammation. J. Biol. Chem. 282, 13769–13779 [DOI] [PubMed] [Google Scholar]
- 7. Zhou G., Hamik A., Nayak L., Tian H., Shi H., Lu Y., Sharma N., Liao X., Hale A., Boerboom L., Feaver R. E., Gao H., Desai A., Schmaier A., Gerson S. L., Wang Y., Atkins G. B., Blackman B. R., Simon D. I., Jain M. K. (2012) Endothelial Kruppel-like factor 4 protects against atherothrombosis in mice. J. Clin. Invest. 122, 4727–4731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yoshida T., Yamashita M., Horimai C., Hayashi M. (2014) Deletion of Krüppel-like factor 4 in endothelial and hematopoietic cells enhances neointimal formation following vascular injury. J. Am. Heart Assoc. 3, e000622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cowan C. E., Kohler E. E., Dugan T. A., Mirza M. K., Malik A. B., Wary K. K. (2010) Krüppel-like factor-4 transcriptionally regulates VE-cadherin expression and endothelial barrier function. Circ. Res. 107, 959–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yoshida T., Gan Q., Franke A. S., Ho R., Zhang J., Chen Y. E., Hayashi M., Majesky M. W., Somlyo A. V., Owens G. K. (2010) Smooth and cardiac muscle-selective knock-out of Krüppel-like factor 4 causes postnatal death and growth retardation. J. Biol. Chem. 285, 21175–21184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li L., Miano J. M., Cserjesi P., Olson E. N. (1996) SM22α, a marker of adult smooth muscle, is expressed in multiple myogenic lineages during embryogenesis. Circ. Res. 78, 188–195 [DOI] [PubMed] [Google Scholar]
- 12. Liao X., Haldar S. M., Lu Y., Jeyaraj D., Paruchuri K., Nahori M., Cui Y., Kaestner K. H., Jain M. K. (2010) Krüppel-like factor 4 regulates pressure-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 49, 334–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Agah R., Frenkel P. A., French B. A., Michael L. H., Overbeek P. A., Schneider M. D. (1997) Gene recombination in postmitotic cells: targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J. Clin. Invest. 100, 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Katz J. P., Perreault N., Goldstein B. G., Lee C. S., Labosky P. A., Yang V. W., Kaestner K. H. (2002) The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development 129, 2619–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kee H. J., Eom G. H., Joung H., Shin S., Kim J. R., Cho Y. K., Choe N., Sim B. W., Jo D., Jeong M. H., Kim K. K., Seo J. S., Kook H. (2008) Activation of histone deacetylase 2 by inducible heat shock protein 70 in cardiac hypertrophy. Circ. Res. 103, 1259–1269 [DOI] [PubMed] [Google Scholar]
- 16. Ido A., Hasebe N., Takeuchi T., Kikuchi K. (2003) Effects of Temocapril and olmesartan on myocardial sympathetic nervous activity and fatty acid metabolism in rats with chronic β-adrenergic stimulation. J. Cardiovasc. Pharmacol. 41, S133–S137 [PubMed] [Google Scholar]
- 17. Zhang G. X., Ohmori K., Nagai Y., Fujisawa Y., Nishiyama A., Abe Y., Kimura S. (2007) Role of AT1 receptor in isoproterenol-induced cardiac hypertrophy and oxidative stress in mice. J. Mol. Cell. Cardiol. 42, 804–811 [DOI] [PubMed] [Google Scholar]
- 18. Kee H. J., Sohn I. S., Nam K. I., Park J. E., Qian Y. R., Yin Z., Ahn Y., Jeong M. H., Bang Y. J., Kim N., Kim J. K., Kim K. K., Epstein J. A., Kook H. (2006) Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation 113, 51–59 [DOI] [PubMed] [Google Scholar]
- 19. Freund C., Schmidt-Ullrich R., Baurand A., Dunger S., Schneider W., Loser P., El-Jamali A., Dietz R., Scheidereit C., Bergmann M. W. (2005) Requirement of nuclear factor-κB in angiotensin II- and isoproterenol-induced cardiac hypertrophy in vivo. Circulation 111, 2319–2325 [DOI] [PubMed] [Google Scholar]
- 20. Wang D., Chang P. S., Wang Z., Sutherland L., Richardson J. A., Small E., Krieg P. A., Olson E. N. (2001) Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105, 851–862 [DOI] [PubMed] [Google Scholar]
- 21. Yoshida T., Yamashita M., Hayashi M. (2012) Krüppel-like factor 4 contributes to high phosphate-induced phenotypic switching of vascular smooth muscle cells into osteogenic cells. J. Biol. Chem. 287, 25706–25714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yoshida T., Sinha S., Dandré F., Wamhoff B. R., Hoofnagle M. H., Kremer B. E., Wang D. Z., Olson E. N., Owens G. K. (2003) Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ. Res. 92, 856–864 [DOI] [PubMed] [Google Scholar]
- 23. Yoshida T., Kawai-Kowase K., Owens G. K. (2004) Forced expression of myocardin is not sufficient for induction of smooth muscle differentiation in multipotential embryonic cells. Arterioscler. Thromb. Vasc. Biol. 24, 1596–1601 [DOI] [PubMed] [Google Scholar]
- 24. Banerjee I., Fuseler J. W., Price R. L., Borg T. K., Baudino T. A. (2007) Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am. J. Physiol. Heart Circ. Physiol. 293, H1883–H1891 [DOI] [PubMed] [Google Scholar]
- 25. Da Costa Martins P. A., De Windt L. J. (2012) MicroRNAs in control of cardiac hypertrophy. Cardiovasc. Res. 93, 563–572 [DOI] [PubMed] [Google Scholar]
- 26. Xing W., Zhang T. C., Cao D., Wang Z., Antos C. L., Li S., Wang Y., Olson E. N., Wang D. Z. (2006) Myocardin induces cardiomyocyte hypertrophy. Circ. Res. 98, 1089–1097 [DOI] [PubMed] [Google Scholar]
- 27. Torrado M., López E., Centeno A., Medrano C., Castro-Beiras A., Mikhailov A. T. (2003) Myocardin mRNA is augmented in the failing myocardium: expression profiling in the porcine model and human dilated cardiomyopathy. J. Mol. Med. 81, 566–577 [DOI] [PubMed] [Google Scholar]
- 28. Kong Y., Tannous P., Lu G., Berenji K., Rothermel B. A., Olson E. N., Hill J. A. (2006) Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 113, 2579–2588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gaussin V., Van de Putte T., Mishina Y., Hanks M. C., Zwijsen A., Huylebroeck D., Behringer R. R., Schneider M. D. (2002) Endocardial cushion and myocardial defects after cardiac myocyte-specific conditional deletion of the bone morphogenetic protein receptor ALK3. Proc. Natl. Acad. Sci. U.S.A. 99, 2878–2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zeisberg E. M., Ma Q., Juraszek A. L., Moses K., Schwartz R. J., Izumo S., Pu W. T. (2005) Morphogenesis of the right ventricle requires myocardial expression of Gata4. J. Clin. Invest. 115, 1522–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lepore J. J., Cheng L., Min Lu M., Mericko P. A., Morrisey E. E., Parmacek M. S. (2005) High-efficiency somatic mutagenesis in smooth muscle cells and cardiac myocytes in SM22α-Cre transgenic mice. Genesis 41, 179–184 [DOI] [PubMed] [Google Scholar]
- 32. Pidkovka N. A., Cherepanova O. A., Yoshida T., Alexander M. R., Deaton R. A., Thomas J. A., Leitinger N., Owens G. K. (2007) Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ. Res. 101, 792–801 [DOI] [PubMed] [Google Scholar]
- 33. Yoshida T., Yamashita M., Horimai C., Hayashi M. (2013) Smooth muscle-selective inhibition of nuclear factor-κB attenuates smooth muscle phenotypic switching and neointima formation following vascular injury. J. Am. Heart Assoc. 2, e000230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Feinberg M. W., Cao Z., Wara A. K., Lebedeva M. A., Senbanerjee S., Jain M. K. (2005) Kruppel-like factor 4 is a mediator of proinflammatory signaling in macrophages. J. Biol. Chem. 280, 38247–38258 [DOI] [PubMed] [Google Scholar]
- 35. Wang Z., Wang D. Z., Pipes G. C., Olson E. N. (2003) Myocardin is a master regulator of smooth muscle gene expression. Proc. Natl. Acad. Sci. U.S.A. 100, 7129–7134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Trivedi C. M., Luo Y., Yin Z., Zhang M., Zhu W., Wang T., Floss T., Goettlicher M., Noppinger P. R., Wurst W., Ferrari V. A., Abrams C. S., Gruber P. J., Epstein J. A. (2007) Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3β activity. Nat. Med. 13, 324–331 [DOI] [PubMed] [Google Scholar]
- 37. Noti J. D., Johnson A. K., Dillon J. D. (2005) The leukocyte integrin gene CD11d is repressed by gut-enriched Kruppel-like factor 4 in myeloid cells. J. Biol. Chem. 280, 3449–3457 [DOI] [PubMed] [Google Scholar]
- 38. Kee H. J., Kook H. (2009) Krüppel-like factor 4 mediates histone deacetylase inhibitor-induced prevention of cardiac hypertrophy. J. Mol. Cell. Cardiol. 47, 770–780 [DOI] [PubMed] [Google Scholar]
- 39. Barki-Harrington L., Luttrell L. M., Rockman H. A. (2003) Dual inhibition of β-adrenergic and angiotensin II receptors by a single antagonist: a functional role for receptor-receptor interaction in vivo. Circulation 108, 1611–1618 [DOI] [PubMed] [Google Scholar]