

Background: The basic mechanisms of sulfotransferase (SULT) catalysis are not yet well understood.
Results: A complete, quantitative model of human SULT2A1 catalysis is constructed.
Conclusion: The model resolves extant ambiguities and establishes a catalytic paradigm for the SULT family.
Significance: SULTs regulate the functions of thousands of signaling small molecules. Understanding their mechanisms is fundamental to understanding their biological functions.
Keywords: Enzyme Mechanism, Ligand-binding protein, Metabolic Regulation, Metabolism, Sulfotransferase, DHEA, Pre-steady State, SULT2A1, Rate Constant, Substrate Inhibition
Abstract
Human cytosolic sulfotransferases (SULTs) regulate the activities of thousands of signaling small molecules via transfer of the sulfuryl moiety (-SO3) from 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to the hydroxyls and primary amines of acceptors. Sulfonation controls the affinities of ligands for their targets, and thereby regulates numerous receptors, which, in turn, regulate complex cellular responses. Despite their biological and medical relevance, basic SULT mechanism issues remain unresolved. To settle these issues, and to create an in-depth model of SULT catalysis, the complete kinetic mechanism of a representative member of the human SULT family, SULT2A1, was determined. The mechanism is composed of eight enzyme forms that interconvert via 22 rate constants, each of which was determined independently. The result is a complete quantitative description of the mechanism that accurately predicts complex enzymatic behavior. This is the first description of a SULT mechanism at this resolution, and it reveals numerous principles of SULT catalysis and resolves previously ambiguous issues. The structures and catalytic behaviors SULTs are highly conserved; hence, the mechanism presented here should prove paradigmatic for the family.
Introduction
Human cytosolic sulfotransferases (SULTs)2 are extensively involved in regulating metabolism, and their activities are tightly, causally linked to disease (1–5). These broad specificity enzymes transfer the sulfuryl moiety (-SO3) from a universal donor, 3′-phosphoadenosine 5′-phosphosulfate (PAPS), to the hydroxyls and primary amines of thousands of acceptors: endogenous metabolites, signaling small molecules, drugs, and xenobiotics (6–10). Sulfonation regulates the activities of compounds by altering, often dramatically, their affinities for their targets (5, 10, 11), and controls their half-lives by increasing their plasma solubility and targeting them for degradation (12–14). Sulfatases hydrolytically remove the sulfuryl moiety and the in situ balance of SULT, and sulfatase activities determine the activity of a compound in the cell.
Although much is known of the ways in which sulfonation regulates metabolism, the biology of sulfuryl transfer is far from fully understood. Receptors whose ligands are regulated by sulfonation include steroid (4, 15, 16), thyroid (5), peptide (17), pheromone (18), and dopamine receptors (7). Lymph circulation (6), aspects of the immune system (19), hemostasis (20), and growth factor recognition (21) depend critically on precise positioning of the sulfuryl group. Human diseases linked to atypical sulfonation include Parkinson disease (22), cystic fibrosis (23), hemophilia (24), heart disease (25), and obesity (15). These lists, which are far from complete, emphasize the ubiquity and importance of the modification.
Fundamental to understanding the functions of an enzyme is an understanding of its mechanism. Enzyme mechanisms evolve to satisfy the requirements of the cells and organisms that depend on them. As such, they often provide deep insights into the inner workings of biology. SULT mechanisms have been studied for decades, yet a clear consensus has not been reached on several basic mechanistic issues; the order of binding remains unresolved, and numerous mechanisms have been offered to explain substrate inhibition (26–34). The rate-determining step in the mechanism has not been determined for any SULT, and the molecular basis of half-site reactivity in these systems remains unknown. To address these issues, the complete kinetic mechanism of the human SULT2A1 was determined. This isozyme regulates the binding of steroids to their receptors and detoxifies steroid-like xenobiotics. Using a series of experimental approaches (stopped- and quenched-flow, initial rate studies, advanced modeling, and isotope trapping), microscopic rate constants were determined for each of the 11 steps in the mechanism. This set of constants accurately predicts complex behavior of the enzyme, initial rate parameters, substrate inhibition, burst rates, and amplitudes, and identifies the rate-determining steps in both the substrate-inhibited and the uninhibited mechanisms. This complete rate constant level description of the mechanism is the first of its kind in the SULT field. It resolves existing mechanistic disparities and offers a robust, definitive model that can serve as a mechanistic paradigm for the SULT family.
EXPERIMENTAL PROCEDURES
Materials
The materials and their sources are as follows: dehydroepiandrosterone (DHEA), DTT, EDTA, imidazole, isopropyl-thio-β-d-galactopyranoside, LB medium, lysozyme, β-mercaptoethanol, pepstatin A, and sodium phosphate were obtained from Sigma. Ampicillin, HEPES, KOH, MgCl2, NaCl, KCl, and PMSF were purchased from Fisher Scientific. Glutathione- and nickel-chelating resins were obtained from GE Healthcare. Competent Escherichia coli (BL21(DE3)) was purchased from Novagen. Dehydroepiandrosterone sulfate (DHEAS) was obtained from United States Biological Life Sciences. PAP and PAPS were synthesized in-house as described previously (16) and were ≥98% pure as assessed by anion-exchange HPLC.
Methods
Protein Purification
The SULT2A1 expression plasmid (35) contains a triple affinity tag (His/GST/MBP, where MBP is maltose-binding protein) at the 5′ terminus of the 2A1 coding region. The enzyme was expressed in E. coli BL21(DE3) and purified according to a published protocol (36). Briefly, enzyme expression was induced with isopropyl-thio-β-d-galactopyranoside (0.50 mm) in LB medium at 16 °C for 18 h. The cells were pelleted, resuspended in lysis buffer, sonicated, and centrifuged. The supernatant was loaded onto a chelating Sepharose Fast Flow column charged with Ni2+. The enzyme was eluted with imidazole (10 mm) onto a glutathione-Sepharose column and then eluted with glutathione (10 mm). The tag was removed from the enzyme by PreScission protease cleavage and separated from the enzyme using a glutathione resin. Finally, the protein was concentrated using a Millipore ultrafiltration disc (Ultracel, 10 KDa). Protein concentration was determined by the Bradford method (37), and enzyme was flash-frozen stored at −80 °C.
Pre-steady State Binding Study
Pre-steady state binding experiments were performed using an Applied Photophysics SX20 stopped-flow spectrometer. The rate constants for binding of ligands to enzyme were determined by monitoring changes in the intrinsic fluorescence of the enzyme (35). SULT2A1 fluorescence was excited at 285 nm and detected above 320 nm using a cutoff filter. A solution containing enzyme, MgCl2 (5.0 mm), and NaPO4 (25 mm), pH 7.2, was rapidly mixed (1:1) with a solution that was identical except that it lacked enzyme and contained ligand. Solutions were thermally equilibrated at 25 ± 2 °C prior to mixing. All reactions were pseudo-first order with respect to ligand. Independent progress curves were averaged to obtain the data sets used to derived apparent rate constants (kobs) by fitting to single-exponential functions using the Applied Photophysics Pro-Data analysis software (Marquardt fitting algorithm). kobs values were determined at four ligand concentrations, and the on- and off-rate constants were obtained by linear least squares analysis of kobs versus [ligand] plots.
Isotopic Trapping of [3H]DHEAS
A pulse solution (20 μl) containing SULT 2A1 (30 μm, 27 × Kd) and [3H]DHEA (1.0 μm, 0.9 × Kd, specific activity 95 Ci/mmol) was mixed with a chase solution (980 μl) containing PAPS (0.10 mm) and unlabeled DHEA (25 μm). The solutions contained MgCl2 (5.0 mm) and NaPO4 (50 mm), pH 7.2, and were equilibrated at 25 ± 2 °C prior to mixing. 3 s after mixing, the reaction was quenched by the addition of NaOH (0.20 m, final). NaPO4 (50 mm, pH 7.2) was added to neutralize the base. Chloroform extraction was performed twice to remove the nonsulfated acceptors. Sulfated acceptor in the aqueous phase was quantified using a PerkinElmer W450624 scintillation spectrometer. Total counts were corrected by subtraction of residual aqueous [3H]DHEA counts (≤1%) that were determined in control experiments in which enzyme was absent.
Quenched-flow Studies
Reactions were initiated and quenched using a KinTek quenched-flow RQF-3 instrument. A solution containing SULT2A1 (24 μm) was rapidly mixed (1:1) with [35S]PAPS (40 μm, 130 × Kd), and the mixture was then mixed with an equal volume of solution containing DHEA (50 μm, 50 × Kd, 0.5% v/v dimethyl sulfoxide (DMSO)). Reactions were quenched by the addition of NaOH (0.20 m, final). Quenched solutions were neutralized, boiled for 2 min, and centrifuged to remove denatured enzyme, and [35S]PAPS and [35S]DHEAS were separated using PEI-F TLC (36) and quantitated by STORM imaging (Nikon Instruments).
DHEA Substrate Inhibition
The conditions were as follows: SULT2A1 (0.50 nm, dimer), PAPS (30 μm, 100 × Kd), DHEA (0.30–25 μm), MgCl2 (5.0 mm), NaPO4 (50 mm), pH 7.2, 25 ± 2 °C. Reactions were initiated by the addition of SULT2A1 and quenched by the addition of NaOH (0.20 m, final). 170 μl of NaPO4 (50 mm, pH 7.2) was added to neutralize the base and increase the solution volume to 200 μl. Nonsulfated acceptors were removed by extraction with 1.0 ml of chloroform. The aqueous phase was carefully removed and extracted again with 1.0 ml of chloroform. Sulfated acceptor in the aqueous phase was quantified using a PerkinElmer W450624 scintillation spectrometer. DHEA was the concentration limiting substrate in all cases, and its consumption was ≤5% of that consumed at the end point of the reaction. The experiments were performed duplicate.
Kinetic Modeling
Data fitting was performed with Gepasi (38). Simulations using the constants in Table 1 were performed with ENZO (39).
TABLE 1.
Rate constants for the SULT2A1 mechanism
RESULTS AND DISCUSSION
The Ligand Binding Rate Constants
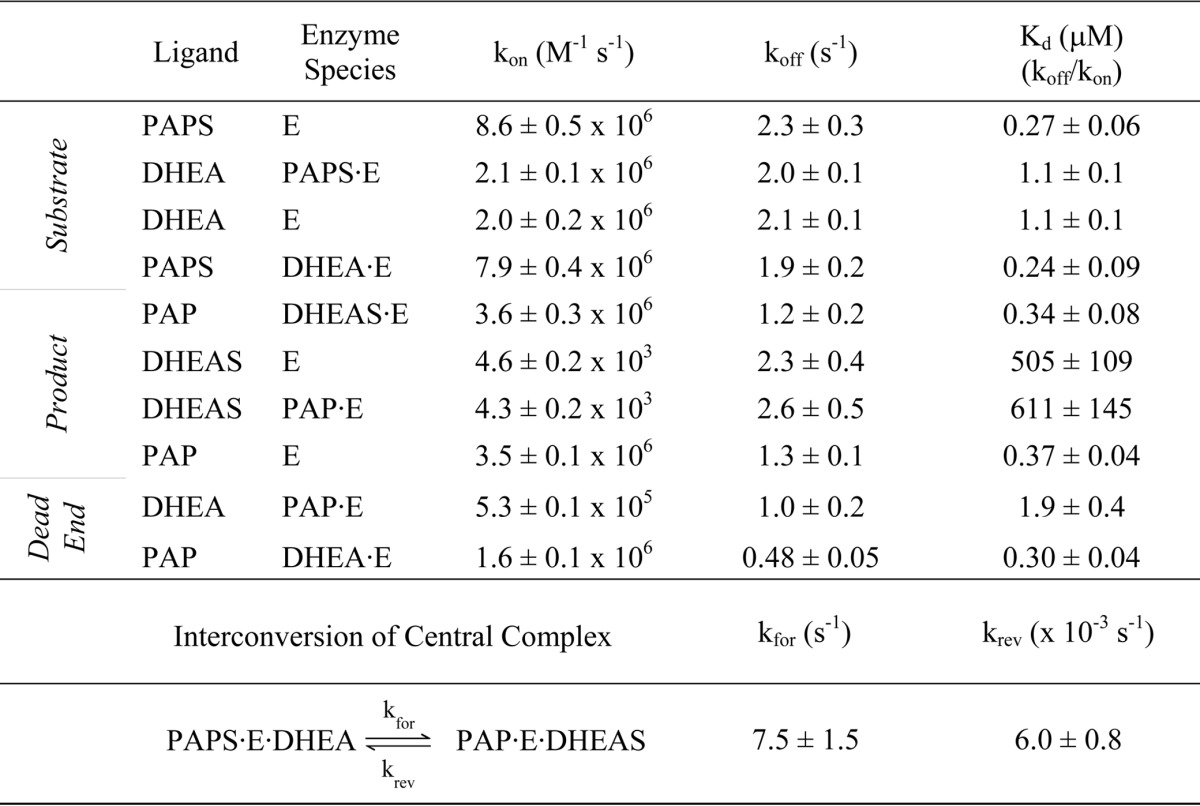
To begin to assemble a complete and quantitative mechanism of SULT2A1 catalysis, the microscopic rate constants for each ligand binding step were determined using stopped-flow fluorescence. SULT2A1, like most SULTs, undergoes significant (15–30%) changes in intrinsic fluorescence upon binding of either nucleotide or acceptor. As an example, the real-time binding of PAP to the E·DHEA complex is shown in Fig. 1A. Similar experiments were performed for every combination of ligand and complex. All binding reactions were performed under pseudo-first order conditions and were well described by a single exponential model. kobs values were obtained from single-exponential fitting of progress curve at each of four ligand concentrations, and kobs versus [ligand] plots were used to obtain kon and koff. A representative kobs versus [ligand] plot is presented in Fig. 1B.
FIGURE 1.
Pre-steady state binding of PAP to the SULT2A1·DHEA complex. A, the binding of PAP to SULT2A1·DHEA. Reactions were initiated by rapidly mixing (1:1 v/v) a solution containing PAP (0.25 μm, 1.0 × Kd [E·DHEA]) and DHEA (25 μm, 23 × Kd [E]) with a solution containing SULT2A1 (0.050 μm) and DHEA (25 μm). Binding was monitored by following changes in SULT2A fluorescence (λex = 290 nm, and λem ≥ 330 nm, where ex represents excitation and em represents emission). Fluorescence intensity is plotted relative to the intensity at time 0 (I/Io). Solutions contained MgCl2 (5.0 mm), NaPO4 (25 mm), pH 7.4, and were equilibrated at 25 ± 2 °C prior to mixing. The solid curve represents the best-fit behavior predicted by a single-exponential model. B, kobs versus [PAP]. Reactions were pseudo-first order in PAP in all cases. Each point represents the average of three independent determinations. The solid line through the points represents a linear least square fit of the data. The slope and intercept of the line provide kon and koff, respectively.
The resulting rate constants are compiled in Table 1 and shed light on several aspects of the mechanism. First, the addition and departure of each substrate is independent of the presence of its partner at the adjacent site. The ligands are “insulated” from one another. Consequently, the ligand-protein interactions that facilitate chemistry either are established prior to the addition of the second substrate and/or engage as the system moves toward the transition state. A second insight becomes apparent when the rate constants governing the addition and escape of DHEA and DHEAS are compared. The escape constants (koff) are identical within error; the ligands are equally “sticky.” The coincidence of these numbers supports that the molecular basis of ligand release is, in essence, the same for both ligands and that sulfuryl group interactions do not contribute significantly to release. In contrast, the on-rate constant for DHEAS is unusually small, 4.3 × 103 m−1 s−1, and is 2000-fold less than that for DHEA. Kd measures the relative affinity of a ligand for enzyme versus solvent. For ligands such as these, whose energetic interactions with enzyme are identical, Kd differences are due to solvent interactions. In this case, the increased solvent stability is due to the additional charge added to DHEA by the sulfonate moiety (40). A third and final insight has to do with identifying the rate-determining steps in the mechanism. The PAP off-rate constants from E and E·DHEAS are 1.3 and 1.2 s−1, respectively; kcat is 1.3 s−1 (41). The rate-determining step in the mechanism is clearly the release of nucleotide. DHEAS departs more quickly than PAP; thus, in the absence of other considerations, 64% of the enzyme is expected to be in the E·PAP complex during steady-state turnover. However, this enzyme form can be trapped by DHEA in a dead-end complex, which, as is shown below, is the basis of substrate inhibition.
The Order of Binding
Whether sulfotransferase mechanisms are ordered or random remains an open question. Studies with SULT1A1, which is closely related to SULT2A1, have been interpreted in favor of an ordered mechanism with nucleotide binding first (26, 28); however, certain facts suggest a random mechanism (42). For example, nucleotide and acceptor bind independently to unliganded SULT1A1 or 2A1, and ligand affinity constants (Kd values) equal their initial rate kinetic parameters (Ki and Km) (42). Thus, the mechanism is either random with ligand binding near equilibrium during steady-state turnover or ordered with nucleotide binding first.
If the mechanism is ordered, the second substrate (the acceptor) must bind nonproductively to unliganded enzyme; otherwise, the mechanism is random. Isotope trapping was used to distinguish between these alternatives (43). [3H]DHEA (1.0 μm) was equilibrated with SULT2A1 (30 μm, 30 × Kd(DHEA)) under conditions in which 97% of DHEA is bound. The solution was rapidly mixed (1:50 v/v) with a solution containing PAPS (100 μm, 333 × Kd) and unlabeled DHEA (25 μm, 23 × Kd), and then quenched 3 s later. Given kon(PAPS) and koff(DHEA) (Table 1), one can calculate that ∼6% of DHEA dissociates from the binary complex during ternary complex formation. The remaining 94% is kinetically trapped in a ternary complex with nucleotide from which it can dissociate and, if it is bound productively, form product. The 1250-fold excess of unlabeled DHEA ensures that only a small fraction of labeled DHEA (∼4%) that dissociates into bulk solution can be converted to product during the 3-s reaction interval. The experiments were performed in triplicate, and the results show that 69 ± 2% of the [3H]DHEA was converted to [3H]DHEAS. These results definitively demonstrate that DHEA productively binds SULT2A1 in the absence of nucleotide and therefore that the binding mechanism is random.
The Interconversion of Central Complexes
The studies described above do not address the kinetics of interconversion of the central complexes. Our pre-steady state work with SULT1E1, an isozyme with high specificity for estradiol (36), demonstrated a burst of product formation whose amplitude corresponds to one-half of an active-site equivalent. These studies led to the discovery that 1E1, a native dimer, is a half-site reactive enzyme (36). If SULT 2A1 produces a similar burst, the rate constant for conversion of the substrate to the product central complex can be obtained, and evidence for half-site reactivity in another member of the SULT family will have been demonstrated.
Pre-steady state quenched-flow experiments were used to assess whether SULT2A1 yields a measurable burst of product. To circumvent the problem of PAPS hydrolysis, which occurs when nucleotide alone is incubated with SULT2A1 at high concentration (kcat = 0.003 s−1), a two-stage mixing strategy was used. In the first stage, 2A1 and PAPS are rapidly mixed, and binding is allowed to proceed for the minimum time required for binary complex formation to reach 99% completion (0.20 s, 5 × t½) before mixing with DHEA to initiate chemistry. Virtually none of the PAPS (<0.6%) is hydrolyzed at 0.20 s after mixing. The protocol was as follows: SULT2A1 (12 μm, monomer) is mixed (1:1) with [35S]PAPS (40 μm, 133 × Kd), and the solution is aged for 0.20 s and then mixed (1:1) with DHEA (50 μm, 46 × Kd [E·PAPS]). At these concentrations, the ternary complex (PAPS·E·DHEA) is essentially completely formed (>93%) after 50 ms, the earliest time point in the progress curve. The result shows a clear burst of product followed by a linear steady-state phase (Fig. 2). The burst amplitude corresponds to one-half of an active site equivalent and strongly suggests that SULT2A1 also uses a half-site mechanism. The burst is decidedly linear, indicating that the central complex equilibrium constant strongly favors product. The rate constant for conversion of the substrate complex to product, which is given by the slope of the burst, is 7.5 (± 0.4) s−1. The solid line through the data represents the behavior predicted for these conditions using the constants in Table 1.
FIGURE 2.

A burst of product. Reactions were initiated by rapidly mixing (1:1 v/v) a solution containing SULT2A1 (24 μm, dimer) with [35S]PAPS (40 μm, 4.1 Ci/μmol, 133 × Kd [E]). The solution was then mixed with an equal volume of DHEA (50 μm, 46 × Kd [E·PAPS]); all solutions contained MgCl2 (5.0 mm), NaPO4 (50 mm), pH 7.2, and were equilibrated at 25 ± 2 °C prior to mixing. Reactions were quenched with NaOH (0.20 m, final), neutralized, boiled, and centrifuged to remove the protein. [35S]DHEAS was separated from [35S]PAPS by reverse-phase TLC (see inset), and radiolabeled reactants were quantitated by two-dimensional imaging using a STORM system (Nikon Instruments). Each point is the average of two independent determinations. The solid line through the data is not a statistical fit; rather, it is the behavior predicted using the rate constants in Table 1 and the model in Fig. 5. [P]/[E] is the concentration of product divided by the concentration of enzyme.
Notably, the rate constant associated with the steady-state phase of the reaction, 0.45 (± 0.1) s−1 is within error equal to the rate constant for the release of PAP from the dead-end complex (PAP·E·DHEA), 0.48 (± 0.4) s−1. Thus, in this phase, the enzyme appears to be primarily in the dead-end complex, and PAP release from this complex appears to be rate-determining.
Thus far, 21 of the 22 rate constants needed for a complete description of the mechanism have been determined. The final constant, which governs the rate at which the central product complex moves back into substrate, was obtained by fitting progress curves. Accurate determination of the constant requires that the reaction proceed solely due to an approach to equilibrium, rather than, for example, loss of activity. Furthermore, it is important that the initial conditions be such that the desired constant contributes to the rate of the reaction. The t0 condition was as follows: DHEA (1.1 μm) and DHEAS (0.50 mm) concentrations were equal to their Kd values; [35]PAPS and PAP were 1.0 μm (3.3 × Kd) and 10 μm (33 × Kd), respectively. The progress curve for [35S]DHEAS synthesis is plotted along with the associated fit in Fig. 3. The fit yielded a rate constant of 6 (± 0.8) × 10−3 s−1. To ensure that the first plateau corresponds to the equilibrium point of the reaction and that the enzyme activity had not significantly deteriorated, the DHEA concentration was increased to 0.50 μm and the response of the system was analyzed. The reaction moved to a new plateau that corresponds to an equilibrium constant identical to that associated with the first plateau, 3.8 (± 0.7) × 104. Moreover, the fit of the second phase yielded the same reverse rate constant.
FIGURE 3.

Progress curves and simulations of DHEAS formation. The initial reaction conditions were as follows: DHEA (1.1 μm, 1.0 × Kd [E]), DHEAS (0.50 mm, 1.0 × Kd [E]), [35S]PAPS (1.0 μm, 3.7 × Kd [E]), PAP (10 μm, 27 × Kd [E]), MgCl2 (5.0 mm), and NaPO4 (50 mm), pH 7.2, T = 25 ± 2 °C. The reaction was allowed to reach what appeared to be equilibrium (7.5 h), at which point the DHEA concentration was increased to 0.50 μm and the reaction was again allowed to plateau. The rate constant for conversion of the product to the substrate central complex was obtained by fitting the data using the 21 other constants (Table 1) associated with the mechanism (Fig. 5). Both phases yielded the same rate constant, 6 (± 0.8) × 10−3 s−1, and overall equilibrium constant, 3.8 (± 0.7) × 104.
DHEA Inhibition
Most SULTs show partial substrate inhibition at physiologically relevant concentrations, and the kcat effects and Ki values vary substantially with each inhibitor/substrate (10, 16, 28, 29). Numerous mechanisms have been used to explain this inhibition, including dead-end complex formation (31, 33, 36), allosteric regulation (16), gating (32), and the binding of multiple substrates in the active-site cavity (34).
The SULT2A1 mechanism (Fig. 4) only allows substrate inhibition via the dead-end complex. If this mechanism and its rate constants quantitatively predict SULT2A1 inhibition, there is no need to invoke an alternative mechanism. Furthermore, any such alternative would have to supplant the dead-end inhibition model and precisely mimic its effects. To test the ability of the 2A1 mechanism to predict substrate inhibition, a typical substrate inhibition dataset was constructed for modeling purposes. To do so, the initial rate of DHEAS synthesis was determined as a function of DHEA concentration (Fig. 5). In all cases, DHEA was the concentration-limiting substrate, PAPS was fixed and saturating (30 μm, 125 × Kd [E·DHEA]), and DHEA consumption was <5% of that. The velocities are normalized to enzyme active-site concentration and plotted as kcatapp versus [DHEA]. The solid curve represents the behavior predicted by the mechanism in Fig. 4 using the microscopic rate constants in Table 1. The close agreement between the predicted and experimental behavior offers very strong support for the dead-end inhibition model. Thus, inhibition occurs because increasing [DHEA] causes it to add more quickly to the E·PAP complex, trapping a greater fraction of PAP in the dead-end complex. At saturation, essentially all of the enzyme-bound PAP has been titrated into the dead-end complex. Consequently, as the DHEA concentration increases, the rate-determining step in the catalytic cycle shifts from PAP release from E·PAP, koff = 1.2 s−1, to its release from the dead-end complex, koff = 0.48 s−1.
FIGURE 4.

The mechanism of SULT2A1-catalyzed DHEA sulfation.
FIGURE 5.

Partial substrate inhibition by DHEA. The initial rate of product formation was determined as a function of DHEA concentration. The reaction conditions were as follows: SULT2A1 (0.50 nm), [3H]DHEA (0.30–25 μm), PAPS (30 μm, 100 × Kd [E·DHEA]), MgCl2 (5.0 mm), and NaPO4 (50 mm), pH 7.2, T = 25 ± 2 °C. Reactions were initiated by the addition of enzyme. Reactions were quenched with (0.20 m, final). [3H]DHEA was extracted with chloroform, and [3H]DHEAS in the aqueous phase was quantified. Data are plotted as kcatapp versus [DHEA], where kcatapp = initial rate/[enzyme active sites]. Each point is the average of two independent determinations. The solid line through the data does not represent a fit of the data; rather, it is the behavior predicted using the 22 constants listed in Table 1 and the model shown in Fig. 5.
SULTs harbor a conserved 30-residue active site cap that “sits above” both the nucleotide and the acceptor (35). Cap closure encapsulates the nucleotide, and cap dynamics govern its binding and departure (42). It is plausible that DHEA slows PAP release by shifting cap isomerization toward the closed state, thus decreasing the concentration of the open form of the cap, which is the species that allows nucleotide to add and depart. Such a mechanism predicts that DHEA will have identical effect on kon and koff, which is what is observed (Table 1).
It is notable that partial inhibition (10, 34) is inconsistent with an ordered mechanism in which nucleotide binds first. In such a mechanism, nucleotide cannot escape from the dead-end complex until DHEA departs, and thus total, rather than partial, inhibition is predicted as the DHEA concentration approaches infinity.
How General Is the Mechanism?
Although the mechanisms of closely related enzymes need not be identical, they often differ only in the constants of the mechanism, which define specificities and turnover. In humans, there are two main SULT families (44), SULT1 and SULT2. Superposition of the structures of seven different members of the two families reveals that they are nearly identical; their Cα root mean square deviation values are less than 1.0 Å (Table 2). The deviations lie primarily in the active-site “caps,” which are ∼30 residues in length and confer much of the specificity differences seen among SULT isoforms (42). Structures show that members of each of the SULT1 and SULT2 subfamilies form dead-end complexes (16, 33, 45–48) (Table 2). Of the seven subfamily members listed in Table 2, six are partially inhibited by substrate (16, 28, 29, 31, 49, 50), and the seventh (SULT2B1b) has not been examined for such inhibition. The fact that the structures of these seven subfamily members are nearly identical, that all of them form dead-end complexes (a cardinal feature of the current mechanism), and that six show partial substrate inhibition (which argues against the ordered mechanism) strongly suggests that these and other related SULTs share a common mechanism, which has been defined here using a representative member of the SULT family.
TABLE 2.
SULT structures and inhibition
| SULT | Ternary complex PDBa ID | Cα r.m.s.d.b | Substrate inhibition |
|---|---|---|---|
| Å | |||
| SULT2A1 | 3F3Y | 0.00 | 31 |
| SULT2B1b | 1Q22 | 0.67 | NAc |
| SULT1A1 | 2D06 | 0.86 | 28 |
| SULT1A3 | 2A3R | 0.88 | 29 |
| SULT1B1 | 3CKL | 0.89 | 49 |
| SULT1C2 | 2GWH | 0.96 | 50 |
| SULT1E1 | 1G3M | 0.97 | 16 |
a PDB, Protein Data Bank.
b r.m.s.d., root mean square deviation.
c NA, not applicable.
Conclusions
The complete kinetic mechanism of human SULT2A1 has been determined. Forward and reverse microscopic rate constants have been determined for each of the steps of the mechanism. This level of mechanistic scrutiny has provided answers to a number of important mechanistic questions in the SULT field. The controversy over whether the mechanism is ordered or random has been resolved; the mechanism is rapid equilibrium random. The equilibrium constant for the interconversion of the central complexes was determined and strongly favors product (Keq = 1.3 × 103). The amplitude of the pre-steady state burst of product indicates that SULT2A1, like SULT1E1, is a half-site enzyme, suggesting that half-site reactivity may be the rule, rather than the exception. The coincidence of kcat and koff for nucleotide release reveals that release is the rate-limiting step in the mechanism. Numerous mechanisms have been used to explain the partial substrate inhibition observed with most SULTs. Here we show that inhibition is caused by trapping of PAP in a dead-end complex (E·PAP·substrate), which slows the release of nucleotide.
This work was supported, in whole or in part, by National Institutes of Health Grants GM38953 (to C. N. F.) and GM54469 (to T. S. L.).
- SULT
- human cytosolic sulfotransferase
- PAP
- 3′,5′-diphosphoadenosine
- PAPS
- 3′-phosphoadenosine 5′-phosphosulfate
- PAP
- 3′-phosphoadenosine 5′-phosphate
- DHEA
- dehydroepiandrosterone
- DHEAS
- dehydroepiandrosterone sulfate.
REFERENCES
- 1. Leyte A., van Schijndel H. B., Niehrs C., Huttner W. B., Verbeet M. P., Mertens K., van Mourik J. A. (1991) Sulfation of Tyr1680 of human blood coagulation factor VIII is essential for the interaction of factor VIII with von Willebrand factor. J. Biol. Chem. 266, 740–746 [PubMed] [Google Scholar]
- 2. Kotov A., Falany J. L., Wang J., Falany C. N. (1999) Regulation of estrogen activity by sulfation in human Ishikawa endometrial adenocarcinoma cells. J. Steroid Biochem. Mol. Biol. 68, 137–144 [DOI] [PubMed] [Google Scholar]
- 3. Tuddenham E. G., Cooper D. N., Gitschier J., Higuchi M., Hoyer L. W., Yoshioka A., Peake I. R., Schwaab R., Olek K., Kazazian H. H., et al. (1991) Haemophilia A: database of nucleotide substitutions, deletions, insertions and rearrangements of the factor VIII gene. Nucleic Acids Res. 19, 4821–4833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Parker C. R. (1999) Dehydroepiandrosterone and dehydroepiandrosterone sulfate production in the human adrenal during development and aging. Steroids 64, 640–647 [DOI] [PubMed] [Google Scholar]
- 5. Visser T. J. (1994) Role of sulfation in thyroid hormone metabolism. Chem. Biol. Interact. 92, 293–303 [DOI] [PubMed] [Google Scholar]
- 6. Tangemann K., Bistrup A., Hemmerich S., Rosen S. D. (1999) Sulfation of a high endothelial venule-expressed ligand for L-selectin: effects on tethering and rolling of lymphocytes. J. Exp. Med. 190, 935–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goldstein D. S., Swoboda K. J., Miles J. M., Coppack S. W., Aneman A., Holmes C., Lamensdorf I., Eisenhofer G. (1999) Sources and physiological significance of plasma dopamine sulfate. J. Clin Endocrinol. Metab. 84, 2523–2531 [DOI] [PubMed] [Google Scholar]
- 8. Lipscombe R. J., Nakhoul A. M., Sanderson C. J., Coombe D. R. (1998) Interleukin-5 binds to heparin/heparan sulfate: a model for an interaction with extracellular matrix. J. Leukoc. Biol. 63, 342–350 [DOI] [PubMed] [Google Scholar]
- 9. Poeppel P., Habetha M., Marcão A., Büssow H., Berna L., Gieselmann V. (2005) Missense mutations as a cause of metachromatic leukodystrophy: degradation of arylsulfatase A in the endoplasmic reticulum. Febs. J. 272, 1179–1188 [DOI] [PubMed] [Google Scholar]
- 10. Cook I. T., Duniec-Dmuchowski Z., Kocarek T. A., Runge-Morris M., Falany C. N. (2009) 24-Hydroxycholesterol sulfation by human cytosolic sulfotransferases: formation of monosulfates and disulfates, molecular modeling, sulfatase sensitivity and inhibition of LXR activation. Drug Metab. Dispos. 37, 2069–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kuiper G. G., Carlsson B., Grandien K., Enmark E., Häggblad J., Nilsson S., Gustafsson J. A. (1997) Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors α and β. Endocrinology 138, 863–870 [DOI] [PubMed] [Google Scholar]
- 12. Argiolas A., Hedlund H. (2001) The pharmacology and clinical pharmacokinetics of apomorphine SL. BJU Int. 88, Suppl. 3, 18–21 [DOI] [PubMed] [Google Scholar]
- 13. Robertson J. F., Harrison M. (2004) Fulvestrant: pharmacokinetics and pharmacology. Br. J. Cancer 90, Suppl. 1, S7–S10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nagar S., Walther S., Blanchard R. L. (2006) Sulfotransferase (SULT) 1A1 polymorphic variants *1, *2, and *3 are associated with altered enzymatic activity, cellular phenotype, and protein degradation. Mol. Pharmacol. 69, 2084–2092 [DOI] [PubMed] [Google Scholar]
- 15. Bai Q., Xu L., Kakiyama G., Runge-Morris M. A., Hylemon P. B., Yin L., Pandak W. M., Ren S. (2011) Sulfation of 25-hydroxycholesterol by SULT2B1b decreases cellular lipids via the LXR/SREBP-1c signaling pathway in human aortic endothelial cells. Atherosclerosis 214, 350–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang H., Varlamova O., Vargas F. M., Falany C. N., Leyh T. S. (1998) Sulfuryl transfer: the catalytic mechanism of human estrogen sulfotransferase. J. Biol. Chem. 273, 10888–10892 [DOI] [PubMed] [Google Scholar]
- 17. Matsubayashi Y., Sakagami Y. (2006) Peptide hormones in plants. Annu. Rev. Plant. Biol. 57, 649–674 [DOI] [PubMed] [Google Scholar]
- 18. Stowers L., Logan D. W. (2010) Sexual dimorphism in olfactory signaling. Curr. Opin. Neurobiol. 20, 770–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Selvan R. S., Ihrcke N. S., Platt J. L. (1996) Heparan sulfate in immune responses. Ann. N.Y. Acad. Sci. 797, 127–139 [DOI] [PubMed] [Google Scholar]
- 20. Anderson J. A., Fredenburgh J. C., Stafford A. R., Guo Y. S., Hirsh J., Ghazarossian V., Weitz J. I. (2001) Hypersulfated low molecular weight heparin with reduced affinity for antithrombin acts as an anticoagulant by inhibiting intrinsic tenase and prothrombinase. J. Biol. Chem. 276, 9755–9761 [DOI] [PubMed] [Google Scholar]
- 21. Mesiano S., Jaffe R. B. (1997) Developmental and functional biology of the primate fetal adrenal cortex. Endocr. Rev. 18, 378–403 [DOI] [PubMed] [Google Scholar]
- 22. Steventon G. B., Heafield M. T., Waring R. H., Williams A. C. (1989) Xenobiotic metabolism in Parkinson's disease. Neurology 39, 883–887 [DOI] [PubMed] [Google Scholar]
- 23. Li L., Falany C. N. (2007) Elevated hepatic SULT1E1 activity in mouse models of cystic fibrosis alters the regulation of estrogen responsive proteins. J. Cyst. Fibros. 6, 23–30 [DOI] [PubMed] [Google Scholar]
- 24. Moore K. L. (2003) The biology and enzymology of protein tyrosine O-sulfation. J. Biol. Chem. 278, 24243–24246 [DOI] [PubMed] [Google Scholar]
- 25. Bischoff E. D., Daige C. L., Petrowski M., Dedman H., Pattison J., Juliano J., Li A. C., Schulman I. G. (2010) Non-redundant roles for LXRα and LXRβ in atherosclerosis susceptibility in low density lipoprotein receptor knockout mice. J. Lipid Res. 51, 900–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tyapochkin E., Cook P. F., Chen G. (2008) Isotope exchange at equilibrium indicates a steady state ordered kinetic mechanism for human sulfotransferase. Biochemistry 47, 11894–11899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Duffel M. W., Jakoby W. B. (1981) On the mechanism of aryl sulfotransferase. J. Biol. Chem. 256, 11123–11127 [PubMed] [Google Scholar]
- 28. Whittemore R. M., Pearce L. B., Roth J. A. (1986) Purification and kinetic characterization of a phenol-sulfating form of phenol sulfotransferase from human brain. Arch. Biochem. Biophys 249, 464–471 [DOI] [PubMed] [Google Scholar]
- 29. Whittemore R. M., Pearce L. B., Roth J. A. (1985) Purification and kinetic characterization of a dopamine-sulfating form of phenol sulfotransferase from human brain. Biochemistry 24, 2477–2482 [DOI] [PubMed] [Google Scholar]
- 30. Barnes S., Waldrop R., Crenshaw J., King R. J., Taylor K. B. (1986) Evidence for an ordered reaction mechanism for bile salt: 3′phosphoadenosine-5′-phosphosulfate:sulfotransferase from rhesus monkey liver that catalyzes the sulfation of the hepatotoxin glycolithocholate. J. Lipid Res. 27, 1111–1123 [PubMed] [Google Scholar]
- 31. Gulcan H. O., Duffel M. W. (2011) Substrate inhibition in human hydroxysteroid sulfotransferase SULT2A1: studies on the formation of catalytically non-productive enzyme complexes. Arch. Biochem. Biophys 507, 232–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lu L. Y., Hsieh Y. C., Liu M. Y., Lin Y. H., Chen C. J., Yang Y. S. (2008) Identification and characterization of two amino acids critical for the substrate inhibition of human dehydroepiandrosterone sulfotransferase (SULT2A1). Mol. Pharmacol 73, 660–668 [DOI] [PubMed] [Google Scholar]
- 33. Gamage N. U., Tsvetanov S., Duggleby R. G., McManus M. E., Martin J. L. (2005) The structure of human SULT1A1 crystallized with estradiol: an insight into active site plasticity and substrate inhibition with multi-ring substrates. J. Biol. Chem. 280, 41482–41486 [DOI] [PubMed] [Google Scholar]
- 34. Gamage N. U., Duggleby R. G., Barnett A. C., Tresillian M., Latham C. F., Liyou N. E., McManus M. E., Martin J. L. (2003) Structure of a human carcinogen-converting enzyme, SULT1A1: structural and kinetic implications of substrate inhibition. J. Biol. Chem. 278, 7655–7662 [DOI] [PubMed] [Google Scholar]
- 35. Cook I., Wang T., Falany C. N., Leyh T. S. (2012) A nucleotide-gated molecular pore selects sulfotransferase substrates. Biochemistry 51, 5674–5683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sun M., Leyh T. S. (2010) The human estrogen sulfotransferase: a half-site reactive enzyme. Biochemistry 49, 4779–4785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 38. Mendes P. (1997) Biochemistry by numbers: simulation of biochemical pathways with Gepasi 3. Trends Biochem. Sci. 22, 361–363 [DOI] [PubMed] [Google Scholar]
- 39. Bevc S., Konc J., Stojan J., Hodošček M., Penca M., Praprotnik M., Janežič D. (2011) ENZO: a web tool for derivation and evaluation of kinetic models of enzyme catalyzed reactions. PLoS One 6, e22265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pauff S. M., Miller S. C. (2013) A trifluoroacetic acid-labile sulfonate protecting group and its use in the synthesis of a near-IR fluorophore. J. Org. Chem. 78, 711–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Falany C. N., Vazquez M. E., Kalb J. M. (1989) Purification and characterization of human liver dehydroepiandrosterone sulphotransferase. Biochem. J. 260, 641–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cook I., Wang T., Almo S. C., Kim J., Falany C. N., Leyh T. S. (2013) The gate that governs sulfotransferase selectivity. Biochemistry 52, 415–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cleland W. W. (1975) Partition analysis and the concept of net rate constants as tools in enzyme kinetics. Biochemistry 14, 3220–3224 [DOI] [PubMed] [Google Scholar]
- 44. Blanchard R. L., Freimuth R. R., Buck J., Weinshilboum R. M., Coughtrie M. W. (2004) A proposed nomenclature system for the cytosolic sulfotransferase (SULT) superfamily. Pharmacogenetics 14, 199–211 [DOI] [PubMed] [Google Scholar]
- 45. Lu J. H., Li H. T., Liu M. C., Zhang J. P., Li M., An X. M., Chang W. R. (2005) Crystal structure of human sulfotransferase SULT1A3 in complex with dopamine and 3′-phosphoadenosine 5′-phosphate. Biochem. Biophys. Res. Commun. 335, 417–423 [DOI] [PubMed] [Google Scholar]
- 46. Allali-Hassani A., Pan P. W., Dombrovski L., Najmanovich R., Tempel W., Dong A., Loppnau P., Martin F., Thornton J., Edwards A. M., Bochkarev A., Plotnikov A. N., Vedadi M., Arrowsmith C. H. (2007) Structural and chemical profiling of the human cytosolic sulfotransferases. PLoS Biol 5, e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee K. A., Fuda H., Lee Y. C., Negishi M., Strott C. A., Pedersen L. C. (2003) Crystal structure of human cholesterol sulfotransferase (SULT2B1b) in the presence of pregnenolone and 3′-phosphoadenosine 5′-phosphate: rationale for specificity differences between prototypical SULT2A1 and the SULT2BG1 isoforms. J. Biol. Chem. 278, 44593–44599 [DOI] [PubMed] [Google Scholar]
- 48. Shevtsov S., Petrotchenko E. V., Pedersen L. C., Negishi M. (2003) Crystallographic analysis of a hydroxylated polychlorinated biphenyl (OH-PCB) bound to the catalytic estrogen binding site of human estrogen sulfotransferase. Environ. Health Perspect. 111, 884–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Saeki Y., Sakakibara Y., Araki Y., Yanagisawa K., Suiko M., Nakajima H., Liu M. C. (1998) Molecular cloning, expression, and characterization of a novel mouse liver SULT1B1 sulfotransferase. J. Biochem. 124, 55–64 [DOI] [PubMed] [Google Scholar]
- 50. Sakakibara Y., Yanagisawa K., Katafuchi J., Ringer D. P., Takami Y., Nakayama T., Suiko M., Liu M. C. (1998) Molecular cloning, expression, and characterization of novel human SULT1C sulfotransferases that catalyze the sulfonation of N-hydroxy-2-acetylaminofluorene. J. Biol. Chem. 273, 33929–33935 [DOI] [PubMed] [Google Scholar]