

Background: The EGF receptor has multiple ligands that can induce different biological effects.
Results: EGF and TGFα, but not BTC or AREG, bias the EGF receptor toward heterodimer formation with ErbB2. AREG induces dimerization with different kinetics than the other ligands.
Conclusion: EGFR ligands differentially induce receptor dimerization.
Significance: This contributes to the biological differences elicited by different ligands.
Keywords: Epidermal Growth Factor (EGF), Growth Factor, Receptor Structure-Function, Receptor Tyrosine Kinase, Signal Transduction, Transforming Growth Factor α (TGF-α), Protein-Tyrosine Kinase (Tyrosine Kinase), Amphiregulin, Betacellulin
Abstract
The EGF receptor has seven different cognate ligands. Previous work has shown that these different ligands are capable of inducing different biological effects, even in the same cell. To begin to understand the molecular basis for this variation, we used luciferase fragment complementation to measure ligand-induced dimer formation and radioligand binding to study the effect of the ligands on subunit-subunit interactions in EGF receptor (EGFR) homodimers and EGFR/ErbB2 heterodimers. In luciferase fragment complementation imaging studies, amphiregulin (AREG) functioned as a partial agonist, inducing only about half as much total dimerization as the other three ligands. However, unlike the other ligands, AREG showed biphasic kinetics for dimer formation, suggesting that its path for EGF receptor activation involves binding to both monomers and preformed dimers. EGF, TGFα, and betacellulin (BTC) appear to mainly stimulate receptor activation through binding to and dimerization of receptor monomers. In radioligand binding assays, EGF and TGFα exhibited increased affinity for EGFR/ErbB2 heterodimers compared with EGFR homodimers. By contrast, BTC and AREG showed a similar affinity for both dimers. Thus, EGF and TGFα are biased agonists, whereas BTC and AREG are balanced agonists with respect to selectivity of dimer formation. These data suggest that the differences in biological response to different EGF receptor ligands may result from partial agonism for dimer formation, differences in the kinetic pathway utilized to generate activated receptor dimers, and biases in the formation of heterodimers versus homodimers.
Introduction
The EGF receptor tyrosine kinase is a classic receptor tyrosine kinase that mediates cell proliferation in response to a variety of different ligands. Structurally, the receptor possesses an extracellular ligand binding domain and an intracellular tyrosine kinase domain (1). In its inactive state, the receptor appears to exist primarily as a monomer (2). However, upon binding ligand, the receptor undergoes a substantial conformational change that allows it to dimerize, in a back-to-back orientation, with a second EGF receptor (3, 4).
Dimerization of the extracellular domains leads to dimerization and activation of the intracellular kinase domain (5). The active kinase phosphorylates a number of different tyrosine residues in the C-terminal tail of its partner subunit (6–8). This permits the binding of SH2 and PTB domain-containing proteins to the phosphorylated receptor and initiates intracellular signaling events (9–11).
Although this basic pattern of EGF receptor activation is relatively straightforward, a number of variations serve to make the system significantly more complex. The EGF receptor is a member of the ErbB family of homologous receptors that also includes ErbB2, ErbB3, and ErbB4 (12). The ErbB receptors are structurally similar and exhibit comparable mechanisms of activation (5, 13, 14). As a result of these similarities, the ErbB receptors can interact with each other to form heterodimers. Although most combinations of ErbB receptors can form, in general, it appears that ErbB2 is the preferred heterodimerization partner (15–17). This is noteworthy because ErbB2 is the only ErbB receptor that does not appear to bind a ligand (18, 19).
In addition to the variation in dimerization partners, the EGF receptor also possesses seven different cognate ligands (for a review, see Ref. 20), including EGF, TGFα, betacellulin (BTC),2 heparin-binding EGF, amphiregulin (AREG), epiregulin, and epigen. Of these EGFs, TGFα, AREG, and epigen bind only to the EGF receptor, whereas BTC, heparin-binding EGF, and epiregulin also bind to ErbB4 (20). These seven ligands can also be subdivided on the basis of their affinity for the EGF receptor. EGF, TGFα, BTC, and heparin-binding EGF exhibit affinities of 0.1–1 nm for the EGF receptor, whereas AREG, epiregulin, and epigen exhibit affinities 10- to 100-fold lower than this (20, 21). Therefore, there is variation in both receptor specificity and receptor affinity.
Despite the fact that the seven EGF receptor ligands all bind to and activate the same EGF receptor, they are capable of inducing different biological effects, even within the same cell (for a review, see Ref. 20). For example, in 32D cells expressing the EGF receptor, TGFα and AREG stimulate higher levels of cell proliferation than EGF and heparin-binding EGF (22). In human fibroblasts, EGF stimulates cell migration through a mechanism involving p70S6K, whereas TGFα stimulates migration via phospholipase C (23). In β-HC9 cells, EGF stimulates MAP kinase activation via ras, whereas BTC-stimulated MAP kinase activation occurs independently of ras (24), and AREG, but not EGF, activates NF-κB in SUM149 cells (25). In some cases, these differences in biological effects have been correlated with differences in the phosphorylation of specific sites on the C-terminal tail of the EGF receptor (22, 24). However, the molecular basis for these differences remains largely unexplored.
In this work, we use luciferase fragment complementation imaging and radioligand binding to probe the interaction of the EGF receptor and ErbB2 subunits after stimulation with EGF, TGFα, BTC, and AREG. The results suggest that differences in ligand-induced receptor-receptor interactions, along with variations in dimerization kinetics, likely contribute to the different biological effects induced by the binding of different growth factors to the EGF receptor.
EXPERIMENTAL PROCEDURES
Materials
EGF was purchased from Biomedical Technologies. TGFα was from Leinco. Human BTC was from Prospec, and AREG was from Leinco. Antibodies to the EGF receptor, pTyr-845, pTyr-992, pTyr-1045, pTyr-1068, and pTyr-1221 were from Cell Signaling Technology. The antibody against pTyr-1173 was from Thermo Scientific. Antibodies against ErbB2 were from Millipore. FetalPlex was from Gemini Bio-Products. Na125I was from PerkinElmer Life Sciences. 125I-EGF was synthesized using the method of Doran and Spar (26).
DNA Constructs
The c'698-EGF receptor fused to the N-terminal half of firefly luciferase (NLuc) or the C-terminal half of luciferase (CLuc) was generated by introducing a BsiWI site into the EGF receptor after Ala-698 in pcDNA3.1 Zeo. The NheI to BsiWI fragment was then isolated and ligated into the pBI Tet vector or pcDNA3.1 Zeo vector cut with BsiWi and NheI and encoding the NLuc or CLuc fragments, respectively. The c'709-ErbB2-NLuc construct was generated in a similar fashion by introducing a BsiWI site into ErbB2 after Ala-709 and ligating an NheI to BsiWI fragment into the pBI Tet vector containing the NLuc fragment.
Cell Lines and Cell Culture
The generation of CHO cells constitutively expressing C-terminally truncated forms of the EGF receptor (after residue 645) or ErbB2 (truncated after residue 678) and fused to the NLuc or CLuc fragments on a Tet-inducible plasmid has been described previously (27). The construction of the kinase-dead K721A-EGFR-NLuc and K721A-EGFR-CLuc cell lines has also been described previously (28). Cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% FetalPlex and maintained in an incubator at 37 °C in 5% CO2.
Luciferase Assays
CHO cells were plated into 96-well black-walled dishes 2 days prior to use. Immediately before the assay, cells were transferred to Dulbecco's modified Eagle's medium without phenol red but with 1 mg/ml bovine serum albumin and 50 mm Hepes (pH 7.4). Cells were then incubated with 0.6 mg/ml D-luciferin for 20 min at 37 °C prior to the addition of growth factor and the start of imaging. Cell radiance (photons/second/centimeter squared/Steradian) was measured every 30 s for 25 min using a cooled charge-coupled device camera and the IVIS50 imaging system. Assays were performed in quintuplicate. Data were fit to single or double exponential model curves using GraphPad Prism 6.
Ligand Binding Studies
Cells constitutively expressing ∼300,000 EGF receptors/cell with ErbB2 on a Tet-inducible promoter were plated onto 6-well dishes 48 h prior to use (27). Cells were grown in Dulbecco's modified Eagle's medium in the absence or presence of 1 μg/ml doxycycline to induce the expression of ErbB2. This concentration of doxycycline induced the expression of approximately ∼1.5 × 106 ErbB2 molecules/cell. Before use, cells were washed in chilled phosphate-buffered saline and cooled to 4 °C prior to the addition of 30 pm 125I-EGF in Ham's F12 medium containing 50 mm Hepes (pH 7.2) and 5 mg/ml bovine serum albumin. Increasing concentrations of unlabeled EGF, TGFα, BTC, or AREG were added to the wells in triplicate. After incubation overnight at 4 °C, plates were washed three times in cold phosphate-buffered saline. Monolayers were dissolved in 1 ml 1 N NaOH and counted in a Beckman γ counter. Assays were done in triplicate. Data were fit to the equation for log(inhibitor) versus response (variable slope) using GraphPad Prism 6. The significance of the differences between the EC50 values in the absence and presence of ErbB2 was based on the p value assigned to those differences by Prism 6.
Receptor Phosphorylation
CHO cells constitutively expressing the EGF receptor and stably transfected with ErbB2 on a Tet-inducible plasmid were plated in 6-well dishes and grown for 2 days before use. When desired, 50 ng/ml doxycycline was added to the growth medium. Immediately before the assay, cells were transferred into warmed Ham's F12 medium containing 25 mm Hepes (pH 7.2) and 1 mg/ml bovine serum albumin and stimulated with growth factor for the indicated time. Plates were incubated at 37 °C, and the assay was stopped by washing in ice-cold phosphate-buffered saline followed by the addition of radioimmune precipitation assay buffer. Monolayers were scraped into the radioimmunoprecipitation assay buffer, and cells were solubilized by passage through a fine-gauge needle. After pelleting unsolubilized material, equal amounts of protein were analyzed on SDS-polyacrylamide gels, and proteins were identified by Western blotting. Results were quantitated using ImageJ software.
RESULTS
Receptor Phosphorylation Studies
We first compared the biological effects of EGF, TGFα, BTC, and AREG in CHO cells that constitutively expressed ∼300,000 EGF receptors/cell and contained ErbB2 on a tetracycline-inducible promoter. This allowed us to determine the effect of the four different growth factors in cells containing only EGF receptors or in cells containing both the EGF receptor and ErbB2.
To compare the biological effects of these four growth factors, CHO cells grown in the absence or presence of doxycycline were stimulated with a saturating concentration of EGF, TGFα, BTC, or AREG and assayed by Western blotting for phosphorylation of the EGF receptor and ErbB2. The Western blot analyses are shown in Fig. 1, A and B, left panels, and are quantitated in the right panels. As can be seen in Fig. 1A, in cells expressing only the EGF receptor, all four ligands stimulated maximal tyrosine phosphorylation of the EGF receptor within 2 min. However, EGF, TGFα, and BTC each stimulated about twice as much phosphorylation as AREG. This phosphorylation was transient because it had declined by 5 min after growth factor treatment. The effects of AREG appeared to decline somewhat more slowly than those of the other three growth factors.
FIGURE 1.

Ligand-stimulated phosphorylation of the EGF receptor and ErbB2. CHO cells expressing EGF receptors alone (A) or EGF receptors plus ErbB2 (B) were treated with 10 nm EGF, 10 nm TGFα, 10 nm BTC, or 300 nm AREG for the indicated times. RIPA lysates were prepared and analyzed by Western blotting with antibodies against the EGF receptor, ErbB2, and phosphotyrosine. Quantitation of the blots is shown in the right panels. C, control; E, EGF; T, TGFα; B, BTC; A, AREG. C, samples of the lysates from the 2-min time point were blotted with site-specific antibodies for the EGF receptor (pTyr-845, pTyr-992, pTyr-1045, pTyr 1068, and pTyr-1173) or ErbB2 (pTyr-1221).
A similar phenomenon was seen in cells expressing both the EGF receptor and ErbB2 (Fig. 1B). Again, phosphorylation was maximal by 2 min, and EGF, TGFα, and BTC stimulated about twice as much phosphorylation of both the EGF receptor and ErbB2 as AREG. Therefore, AREG is a partial agonist for the phosphorylation of both EGFR homodimers and EGFR/ErbB2 heterodimers.
Replicate lysates at the 2-min time point in Fig. 1, A and B, were subjected to Western blotting with phosphosite-specific antibodies to determine whether there was differential phosphorylation of sites by the different growth factors. As shown in Fig. 1C, AREG stimulated less phosphorylation than EGF, TGFα, or BTC at essentially all sites on the EGF receptor, except for pTyr-1173. At this site, AREG-stimulated phosphorylation was comparable or higher in EGF receptor homodimers but less in heterodimers compared with the other three growth factors. AREG also stimulated less phosphorylation of ErbB2 at pTyr-1221. These data demonstrate that AREG ultimately induces a quantitatively different biological response than EGF, TGFα, and BTC in these cells.
It is possible that this difference in phosphorylation was due to differences in down-regulation of the EGF receptor by the four growth factors. Therefore, we examined the ability of the four different ligands to induce down-regulation of the EGF receptor. Cells were incubated for the indicated times with a saturating dose of the growth factor and then washed with low-pH buffer to remove surface-bound ligand. Residual cell surface EGF receptors were then detected via 125I-EGF binding. As shown in Fig. 2A, in cells expressing only the EGF receptor, all four ligands induced a similar, ∼65% decrease in cell surface EGF receptors over the time course of our assay.
FIGURE 2.
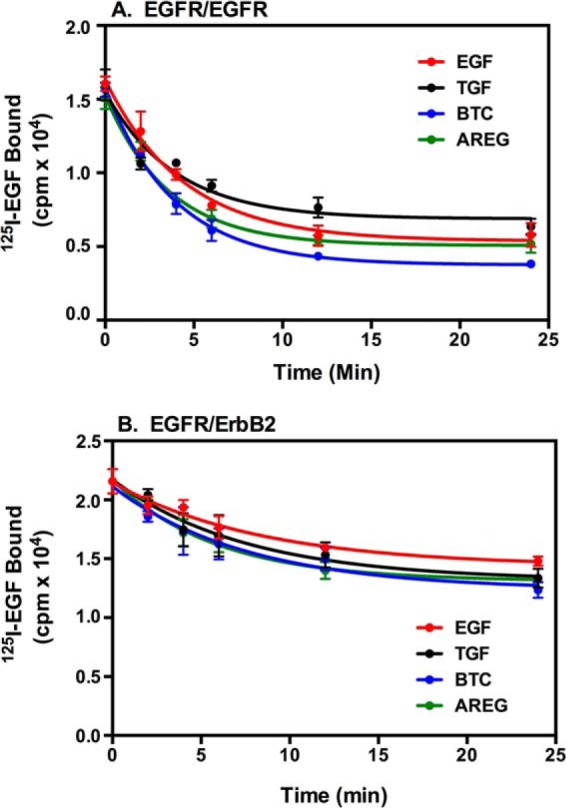
Down-regulation of cell surface EGF receptors by the four ligands. CHO cells expressing EGF receptor alone (A) or EGF receptors plus ErbB2 (B) were treated for 30 min at 37 °C with 30 nm EGF, 30 nm TGFα, 30 nm BTC, or 1 μm AREG for the indicated times. The medium was removed, and cells were washed once with cold phosphate-buffered saline and twice with an acid wash (36). Following an additional wash with cold phosphate-buffered saline, cells were incubated with 200 nm 125I-EGF, and residual cell surface EGF receptor binding was measured as described under “Experimental Procedures.”
Expression of ErbB2 is known to reduce the ligand-stimulated internalization of the EGF receptor (29, 30). This was apparent in cells expressing both the EGF receptor and ErbB2 (Fig. 2B), where each ligand induced only an ∼30% decrease in cell surface EGF receptors. Because all four ligands appear to induce the same extent of down-regulation in both cell lines, the differences observed in our phosphorylation assays do not appear to be associated with differences in ligand-induced internalization of the EGF receptor.
Luciferase Fragment Complementation Studies
One possible explanation for the partial agonism of AREG in the phosphorylation assay is that it is less effective than the other growth factors at inducing the formation of EGF receptor dimers. We have previously used luciferase fragment complementation assays to measure the formation of EGF receptor homodimers and EGFR/ErbB2 heterodimers (31, 32). Therefore, we used this method to compare dimer formation induced by the four different ligands.
Initially, we performed luciferase fragment complementation imaging using EGF receptors and ErbB2 that had been C-terminally truncated at the end of the transmembrane domain and thus lacked the entire intracellular domain (31, 32). Therefore, in these truncated constructs, dimerization was mediated only by interactions between the extracellular and/or transmembrane domains of the receptor subunits.
For these experiments, the N-terminal half (NLuc) or C-terminal half (CLuc) of firefly luciferase was fused to the C terminus of the truncated EGFR (ΔC-EGFR) or the truncated ErbB2 (ΔC-ErbB2). A flexible linker was added between the truncated receptor and the NLuc or CLuc fragments to facilitate complementation. The appropriate pairs of receptors were then stably expressed in CHO cells.
We first determined the dose response to all four ligands for complementation between ΔC-EGFR-NLuc and ΔC-EGFR-CLuc (Fig. 3). In this system, each ligand stimulated an initial rapid increase in light production, which then slowed toward a plateau after 10–15 min. A similar pattern was observed for complementation between ΔC-ErbB2-NLuc and ΔC-EGFR-CLuc (Fig. 4).
FIGURE 3.

Luciferase complementation in CHO cells stably expressing ΔC-EGFR-CLuc and inducible ΔC-EGFR-NLuc. CHO cells were plated into 96-well dishes and treated with doxycycline to induce the expression of the ΔC-EGFR-NLuc. Cells were treated with the indicated concentrations of EGF (A), TGFα (B), BTC (C), or AREG (D) and assayed for luciferase fragment complementation as described under “Experimental Procedures.”
FIGURE 4.

Luciferase complementation in CHO cells stably expressing ΔC-EGFR-CLuc and inducible ΔC-ErbB2-NLuc. CHO cells were plated into 96-well dishes and treated with doxycycline to induce the expression of the ΔC-ErbB2-NLuc. Cells were treated with the indicated concentrations of EGF (A), TGFα (B), BTC (C), or AREG (D) and assayed for luciferase fragment complementation as described under “Experimental Procedures.”
On the basis of these dose-response curves, the concentration of each growth factor that gave the highest level of complementation was selected and compared directly with the others in a luciferase complementation assay in cells coexpressing ΔC-EGFR-NLuc and ΔC-EGFR-CLuc (Fig. 5A) or ΔC-ErbB2-NLuc and ΔC-EGFR-CLuc (Fig. 5B).
FIGURE 5.
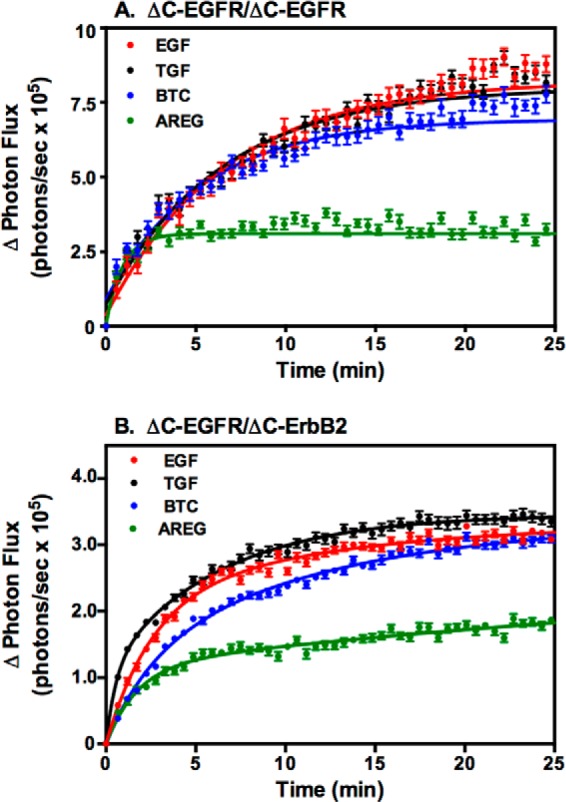
Comparison of the effect of saturating doses of the four ligands on luciferase fragment complementation in cells expressing truncated receptors. A, CHO cells expressing ΔC-EGFR-NLuc and ΔC-EGFR-CLuc were stimulated with 10 nm EGF, 10 nm TGFα, 10 nm BTC, or 100 nm AREG, and luciferase complementation was measured. B, CHO cells expressing ΔC-ErbB2-NLuc and ΔC-EGFR-CLuc were stimulated with 10 nm EGF, 10 nm TGFα, 10 nm BTC, or 100 nm AREG, and luciferase complementation was measured.
In both the homodimer (Fig. 5A) and the heterodimer (Fig. 5B), stimulation with EGF, TGFα, and BTC induced approximately twice as much complementation as the optimal dose of AREG. This is consistent with the results of the receptor autophosphorylation assays and suggests that the smaller biological effect of AREG may be due to a decreased ability to generate dimerized receptors compared with the other three growth factors.
The lower level of dimerization induced by AREG is not simply due to the truncated nature of the receptor in which these assays were done. As shown in Fig. 6, A and B, AREG also stimulated lower levels of complementation in homodimers and heterodimers composed of receptors that were truncated after the intracellular juxtamembrane domain of the receptors (c'698-EGFR and c'709-ErbB2), known to form an antiparallel helical dimer (33). Furthermore, the same pattern was observed when the full-length, kinase-dead version of the EGF receptor was used (Fig. 6, C and D). Therefore, AREG appears to function as a partial agonist with respect to the induction of dimer formation, regardless of receptor structure.
FIGURE 6.

Luciferase complementation in cells expressing mutant EGF receptors and ErbB2. CHO cells were transiently transfected with constructs encoding c'698-EGFR-NLuc and c'698-EGFR-Cluc (A), c'709-ErbB2-NLuc and c'698-EGFR-Cluc (B), K721A-EGFR-NLuc and K721A-EGFR-Cluc (C), or K721A-EGFR-CLuc and ErbB2-Nluc (D). Cultures were stimulated with 10 nm EGF, 10 nm TGFα, 10 nm BTC, or 100 nm AREG, and luciferase complementation was measured.
Kinetics of Receptor Dimerization
As is apparent from the dose-response curves of AREG in Figs. 3 and 4, at very high doses, the luciferase activity exhibits an unusual rapid rise and fall, followed by a slow rise. This suggests that receptor dimerization involves a multistep process. In fact, all of the curves for the different AREG doses (except for 1 μm, which cannot be fit) are better fit using a double exponential model than a single exponential model (p < 0.0001). This contrasts with the situation for EGF, TGFα, and BTC, in which all curves could be well fit by a single exponential. To further examine this unusual kinetic behavior, secondary plots were constructed from the data in Fig. 3, in which the observed rate constants for the fast component and the slow component were plotted against the [AREG]. The results are shown in Fig. 7.
FIGURE 7.

Secondary plots from luciferase complementation in cells expressing ΔC-EGFR-NLuc and ΔC-EGFR-CLuc. The curves for luciferase complementation for the four ligands shown in Fig. 7 were fit to either single (EGF, TGFα, and BTC) or double (AREG) exponential models (see text). The resulting kobs was plotted against the concentration of growth factor. A, kobs for the fast phase of the AREG curves plotted against [AREG]. B, kobs for the slow phase of the AREG curves plotted against [AREG]. C, fraction of the total signal attributable to the fast phase plotted against [AREG]. D, kobs for the curves for EGF, TGFα, and BTC plotted against the concentration of that growth factor. E, model for dimerization of the EGF receptor.
The plot of kobs(fast) versus [AREG] (Fig. 7A) is linear with respect to ligand concentration. This suggests that this component reflects a simple ligand binding event. By contrast, the plot of kobs(slow) versus [AREG] is saturable with respect to [AREG] (Fig. 7B), consistent with the conclusion that this component represents a change in conformation or, possibly, a dimerization event. As shown in Fig. 7C, the fraction of the luciferase signal attributable to the fast component increases with increasing [AREG] but, ultimately, plateaus at about 0.5. Therefore, at high concentrations of AREG, the two phases contribute equally to the signal. For the other three growth factors, the plots of kobs versus [growth factor] (Fig. 7D) show hyperbolic behavior, and the values are in the same range as those of the slow phase of the AREG curves. Therefore, it would appear that the four growth factors share the slow phase and that AREG exhibits an additional fast phase of complementation.
Fig. 7E shows the current model for dimerization of the EGF receptor in which there is a pre-existing equilibrium between unoccupied EGF receptor monomers and dimers (34). Ligand can bind to either the monomer, which subsequently dimerizes, or to the dimer. Either pathway leads to activation of the tyrosine kinase activity of the receptor.
On the basis of the kinetic data, we hypothesize that the complementation we observed between EGF receptor subunits can be described by a combination of two kinetic pathways. In the first, ligand binds to the pre-existing, unoccupied EGF receptor dimers. This ligand binding event occurs rapidly and induces an intramolecular conformational change that produces enhanced complementation in the luciferase assay,
where R and R* represent the basal and activated forms of the receptor and L is the ligand. This represents the fast phase of the kinetics seen in AREG-treated cells.
In a second pathway, ligand binds to the receptor monomer. This binding event would be silent in our assay because it does not involve the interaction of two EGF receptor subunits. Subsequently, however, this liganded receptor slowly dimerizes with another receptor, producing a homodimer and generating a signal in the luciferase assay.
This represents the slow phase of complementation in AREG-treated cells as well as the single phase in cells treated with the other three growth factors.
For the two kinetic pathways, we have assigned the fast and slow phases on the basis of the likely relative rates of an intramolecular conformational change (fast) versus an intermolecular dimerization reaction (slow). If these assignments are correct, our data would suggest that EGF, TGFα, and BTC almost exclusively utilize the pathway in which the growth factor first binds to a monomer that subsequently dimerizes (Fig. 7E, bottom pathway). At low doses, AREG also follows the monomer binding pathway. However, at high doses of AREG, binding to pre-existing dimers increases (Fig. 7E, top pathway) and, ultimately, accounts for half of the signal generated.
This interpretation of our data requires the presence of predimers of the truncated receptor constructs. To assess this possibility, cells expressing ΔC-EGFR-NLuc and ΔC-EGFR-CLuc or ΔC-ErbB2-NLuc and ΔC-EGFR-CLuc were pretreated with either cetuximab or pertuzumab or both and then stimulated with AREG. Cetuximab is an antibody directed against the EGF receptor that inhibits ligand binding and, hence, agonist-stimulated events. Pertuzumab targets the dimerization arm of ErbB2, blocking the formation of back-to-back dimers of ErbB2 and other ErbB receptor family members. The results of this experiment are shown in Fig. 8.
FIGURE 8.
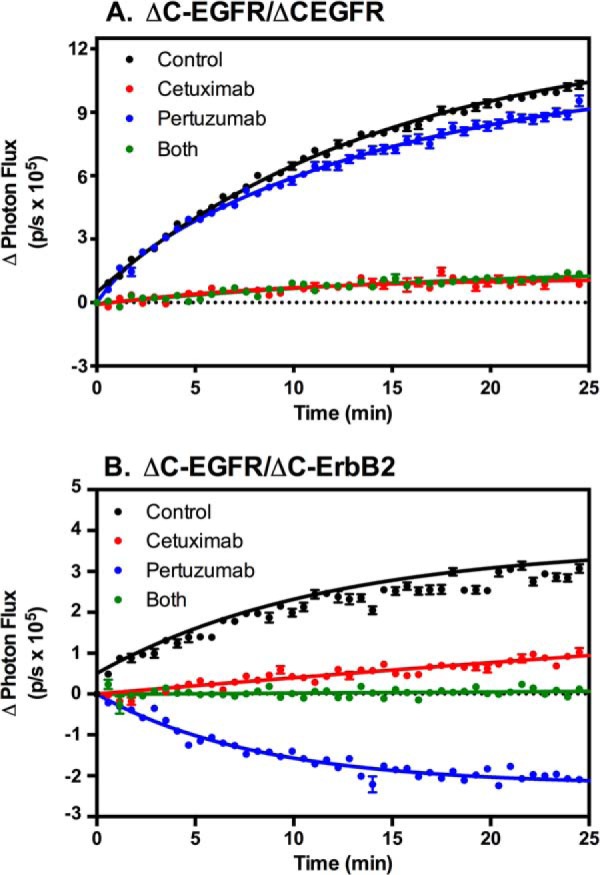
Effects of therapeutic antibodies on complementation in cells expressing truncated EGF receptors and ErbB2. A, CHO cells expressing ΔC-EGFR-NLuc and ΔC-EGFR-CLuc were treated with 5 μg/ml of cetuximab, pertuzumab, or both for 20 min. Cultures were stimulated with 10 nm EGF, and luciferase complementation was measured. B, CHO cells expressing ΔC-EGFR-CLuc and ΔC-ErbB2-NLuc were treated with 5 μg/ml of cetuximab, pertuzumab, or both for 20 min. Cultures were stimulated with 10 nm EGF, and luciferase complementation was measured.
Stimulation of either ΔC-EGFR homodimers (Fig. 8A) or ΔC-EGFR/ΔC-ErbB2 heterodimers (Fig. 8B) with EGF resulted in the expected exponential increase in luciferase activity. Pretreatment of either cell line with cetuximab resulted in a nearly complete inhibition of this effect. As expected, pretreatment of the line expressing only ΔC-EGF receptors with pertuzumab did not significantly alter the response to EGF because there are no ErbB2 subunits in these cells. However, pretreatment of the cells expressing ΔC-ErbB2-NLuc and ΔC-EGFR-CLuc heterodimers with pertuzumab resulted in an EGF-stimulated decrease in luciferase complementation. Treatment with both cetuximab and pertuzumab blocked the ability of EGF to promote dimerization and also blocked the decrease in luciferase activity following stimulation with this growth factor. This latter finding indicates that it is not simply the binding of pertuzumab that leads to the loss of the complementation signal but, rather, the binding of EGF that promotes the disruption of the predimers.
These data suggest that there are indeed predimers of ΔC-ErbB2-NLuc and ΔC-EGFR-CLuc. The addition of EGF apparently disrupts these predimers, leading to a loss of signal when stable back-to-back dimers cannot form because of the presence of pertuzumab. Presumably, ΔC-EGF receptors can also form predimers with other ΔC-EGF receptors. The fact that predimers form in the absence of the intracellular domains suggests that the extracellular domain and/or the transmembrane domains mediate the formation of these predimers.
Radioligand Binding Studies
Another way to probe subunit-subunit interactions within a receptor dimer is through direct radioligand binding experiments. Therefore, we compared the ability of these four growth factors to interact with EGFR homodimers and EGFR/ErbB2 heterodimers in competition binding studies using 125I-EGF as the radioligand and EGF, TGFα, BTC, or AREG as the unlabeled competitor. In these studies, we used CHO cells that stably express the wild-type EGF receptor but express ErbB2 from a Tet-inducible promoter. Therefore, by growing the cells in the absence and presence of doxycycline, we could compare the binding of these hormones to cells containing only EGFR homodimers with their binding to cells containing EGFR homodimers and EGFR/ErbB2 heterodimers. The results are shown in Fig. 9.
FIGURE 9.

Competition binding dose-response curves in CHO cells expressing EGF receptors alone or in the presence of ErbB2. CHO cells were plated in the absence (dashed line) or presence (solid line) of 1 μg/ml doxycycline to induce the expression of ErbB2. Cells were cooled to 4 °C, and 125I-EGF was added to each well along with increasing concentrations of EGF (A), TGFα (B), BTC (C), or AREG (D). Plates were processed as described under “Experimental Procedures.” Points represent the mean ± S.D. of triplicate determinations.
The open circles and dashed lines show the competition binding curves for cells containing only EGFR homodimers. The filled circles and solid lines denote the competition binding curves for cells containing both the EGF receptor and ErbB2. As can be seen from the dashed lines in Fig. 9, in cells that expressed only the EGF receptor, BTC showed the highest affinity for the EGF receptor, with an IC50 of 1.3 × 10−10. EGF was the next most potent, followed by TGFα and, finally, AREG, which exhibited an IC50 of 2.4 × 10−8, two orders of magnitude lower than that exhibited by BTC.
We have shown previously that expression of ErbB2 induces a significant increase in the affinity of the EGF receptor for EGF. This appears to be due to EGF having a higher affinity for the EGFR/ErbB2 heterodimer than for the EGFR/EGFR homodimer (27). To determine whether expression of ErbB2 also affects the affinity of the EGF receptor for its other ligands, we repeated the competition binding experiments in the same CHO cell line after treatment with 1 μg/ml doxycycline to induce the expression of ErbB2.
As expected, the expression of ErbB2 enhanced the affinity of the EGF receptor for EGF (p < 0.001), as evidenced by a 3-fold shift to the left in the competition binding curve for EGF (Fig. 9A). Similarly, expression of ErbB2 increased the affinity of the EGF receptor for TGFα (π < 0.001), indicated by the ∼2-fold shift in the competition binding curve (Fig. 9B). By contrast, neither BTC nor AREG showed a substantial increase in binding affinity upon expression of ErbB2. These data suggest that EGF and TGFα have a higher affinity for EGFR/ErbB2 heterodimers than EGFR homodimers and are, therefore, biased agonists with respect to heterodimer formation. BTC and AREG, which show little preference for binding to homodimers versus heterodimers, would be considered to be balanced agonists with respect to dimer formation.
DISCUSSION
The EGF receptor binds seven different ligands, of which roughly half are high-affinity binders (∼ 1 nm Kd) and half are low-affinity binders (∼ 30 nm Kd). A number of studies have suggested that, although they bind to the same receptor, the different ligands induce different biological effects, even in the same cell type. The experiments we report here begin to explain the molecular basis for these differences.
There were distinct differences among the four growth factors when they were assayed for their ability to stimulate receptor tyrosine phosphorylation in our CHO cells. Specifically, AREG stimulated only half as much phosphorylation of either the EGF receptor or ErbB2 as EGF, TGFα, or BTC. Therefore, AREG appears to be a partial agonist in this system.
In parallel with its reduced ability to stimulate receptor phosphorylation, AREG also showed a decreased capacity to induce the formation of EGFR homodimers or EGFR/ErbB2 heterodimers, as assessed in our luciferase complementation assay. The most straightforward interpretation of these data is that AREG is a partial agonist in the phosphorylation assay because it induces only partial dimerization of the EGF receptor.
It is possible that the partial agonist behavior of AREG is related to its low affinity for the EGF receptor. The EGF receptor exhibits negative cooperativity so that EGF binds with 10-fold lower affinity to the second site on a singly occupied dimer than to the first site on the dimer (34). Because AREG already exhibits a ∼100-fold lower affinity for the EGF receptor than EGF, binding to the second site on the dimer may be exceedingly difficult. Therefore, AREG may be largely restricted to forming only occupied monomers and singly occupied dimers (see the model in Fig. 7E). As a result, it would form fewer dimeric receptors overall. However, if singly occupied dimers are active, this would be sufficient to induce at least a partial phosphorylation response. Some biological responses, such as the activation of MAP kinase, can be fully induced at very low concentrations EGF, suggesting that they are mediated largely through the formation of singly occupied dimers. Therefore, it is possible that AREG may be a full agonist for such responses and a partial agonist for others. This would clearly lead to a different overall cellular response to AREG compared with EGF, TGFα, and BTC.
In addition to being a partial agonist, AREG exhibited unusual behavior at high doses of the growth factor in the luciferase assays that used the C-terminally truncated forms of the receptors (ΔC-EGFR and ΔC-ErbB2). At the higher doses, there was rapid stimulation of complementation, followed by a loss of signal and then a slow rise. Unlike the family of curves generated for the dose responses to EGF, TGFα, or BTC, the AREG curves were best fit by a double exponential model, indicating the presence of at least two separable kinetic pathways. We hypothesize that these two pathways may be: (i) binding to monomer with subsequent slow dimerization; and (ii) rapid binding to predimers followed by an intramolecular conformational change that leads to enhanced complementation. All four growth factors utilize the first pathway, but, at high concentrations, AREG also increasingly uses the second pathway.
In this scenario, high doses of AREG rapidly induce the formation of a stabilized, ligand-occupied dimer by binding to predimerized receptors. However, the level of activated dimers generated during this rapid binding reaction is higher than the level of this species that can be maintained at equilibrium at the given ligand and receptor concentrations. Therefore, there is dissociation of the dimer and loss of signal as the system relaxes down to equilibrium.
This difference between the kinetics of dimer formation and the equilibrium level of dimers maintained could again yield a distinctive biological response to AREG compared with the three other ligands. The ability of AREG to transiently generate high levels of activated receptor dimers could allow it to rapidly and strongly trigger early biological responses. The drop to lower equilibrium levels of dimer formation over time could limit the activation of pathways that require longer term receptor activation and, in particular, those that negatively regulate EGF receptor function. Indeed, AREG induces lower levels of ubiquitination and recruitment of Cbl to the EGF receptor than EGF (35). Therefore, a different response would be elicited by each growth factor because there would be a different ratio of early versus late responses.
We have shown previously through ligand binding studies that, in cells, there is a pre-existing equilibrium of EGF receptor monomers and dimers. EGF can bind to the monomer, the first site on the dimer, or the second site on the dimer. Each of these species exhibits a different affinity for EGF. Consequently, the binding curves for EGF reflect this heterogeneity in binding affinities, and the binding isotherms shift right or left, depending on the concentration of EGF receptors in the cell (34). We have also shown that EGF binds with higher affinity to the EGFR/ErbB2 heterodimer than to the EGF receptor homodimer (27). As a result, EGF binding isotherms shift leftward when ErbB2 is expressed in the presence of the EGF receptor (27). The size of this shift is a rough indication of the extent of the preference of EGF for binding to the heterodimer as opposed to the homodimer.
Our radioligand binding experiments on full-length EGF receptors reported here demonstrate that the competition binding curves for EGF and TGFα both shift significantly leftward upon expression of ErbB2. This suggests that EGF and TGFα induce a different conformation of the EGF receptor than BTC and AREG. The conformation induced by EGF or TGFα is better stabilized when the EGFR is part of an EGFR/ErbB2 heterodimer than when it is in an EGFR homodimer. By contrast, the conformation induced by BTC or AREG is equally stable in the homodimer and the heterodimer.
This difference in conformation is not simply due to affinity differences among these ligands because BTC is a high-affinity ligand, like EGF and TGFα, whereas AREG is a low-affinity ligand. More likely, it reflects a difference in the way in which BTC and AREG interact with the EGF receptor. It is possible that, because BTC binds to both the EGF receptor and ErbB4, its broader specificity limits the nature of the contacts it is able to make with either receptor. Likewise, the low affinity of AREG implies a relatively limited suite of interactions with the EGF receptor that may be insufficient to induce a conformation of the receptor that can distinguish between dimerization partners.
CONCLUSIONS
Our findings suggest several molecular mechanisms that may contribute to the different biological responses induced by the binding of different growth factors to the EGF receptor. First, different growth factors may exhibit differences in their ability to induce dimer formation. AREG is clearly a partial agonist in this regard, and this is reflected in the lower level of receptor phosphorylation induced by this growth factor compared with EGF, TGFα, and BTC. The decreased signaling potential would clearly affect the net output signal induced by AREG.
Second, the ligands may differ with respect to the kinetic pathways through which they induce dimer formation. EGF, TGFα, and BTC induced a slower rate of dimerization than AREG, but, ultimately, they maintained a higher level of dimers than AREG. AREG generated high levels of dimer formation initially but could only maintain a lower level of dimers at equilibrium. Therefore, the relative ratio of rapid versus persistent signaling will vary among the growth factors, again yielding an overall biological signal that is unique to each growth factor.
Finally, different ligands appear to bias the system toward one type of dimer over another (e.g. EGFR/ErbB2 heterodimers versus EGFR homodimers). Given the differences in the sites of phosphorylation available on these two ErbB subunits as well as the differences in their internalization and trafficking (29, 30), such a preference could easily alter the signal produced.
This work was supported, in whole or in part, by National Institutes of Health Grant R01GM099695 (to L. J. P.).
- BTC
- betacellulin
- AREG
- amphiregulin.
REFERENCES
- 1. Ullrich A., Coussens L., Hayflick J. S., Dull T. J., Gray A., Tam A. W., Lee J., Yarden Y., Libermann T. A., Schlessinger J. (1984) Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 309, 418–425 [DOI] [PubMed] [Google Scholar]
- 2. Ferguson K. M., Berger M. B., Mendrola J. M., Cho H.-S., Leahy D. J., Lemmon M. A. (2003) EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell 11, 507–517 [DOI] [PubMed] [Google Scholar]
- 3. Garrett T. P., McKern N. M., Lou M., Elleman T. C., Adams T. E., Lovrecz G. O., Zhu H.-J., Walker F., Frenkel M. J., Hoyne P. A., Jorissen R. N., Nice E. C., Burgess A. W., Ward C. W. (2002) Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor a. Cell 110, 763–773 [DOI] [PubMed] [Google Scholar]
- 4. Ogiso H., Ishitani R., Nureki O., Fukai S., Yamanaka M., Kim J.-H., Saito K., Sakamoto A., Inoue M., Shirouzu M., Yokoyama S. (2002) Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 110, 775–787 [DOI] [PubMed] [Google Scholar]
- 5. Zhang X., Gureasko J., Shen K., Cole P. A., Kuriyan J. (2006) An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149 [DOI] [PubMed] [Google Scholar]
- 6. Hsuan J. J., Totty N., Waterfield M. D. (1989) Identification of a novel autophosphorylation site (P4) on the epidermal growth factor receptor. Biochem. J. 262, 659–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Margolis B. L., Lax I., Kris R., Dombalagian M., Honegger A. M., Howk R., Givol D., Ullrich A., Schlessinger J. (1989) All autophosphorylation sites of epidermal growth factor (EGF) receptor and HER2/neu are located in their carboxyl-terminal tails. J. Biol. Chem. 264, 10667–10671 [PubMed] [Google Scholar]
- 8. Downward J., Parker P., Waterfield M. D. (1984) Autophosphorylation sites on the epidermal growth factor receptor. Nature 311, 483–485 [DOI] [PubMed] [Google Scholar]
- 9. Burgess A. W. (2008) EGFR family: structure, physiology, signalling and therapeutic targets. Growth Factors 26, 263–274 [DOI] [PubMed] [Google Scholar]
- 10. Hynes N. E., Lane H. A. (2005) ErbB receptors and cancer: the complexity of targeted inhibitors. Nat. Rev. Cancer 5, 341–354 [DOI] [PubMed] [Google Scholar]
- 11. Lemmon M. A., Schlessinger J. (2010) Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ferguson K. M. (2008) Structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 37, 353–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aertgeerts K., Skene R., Yano J., Sang B.-C., Zou H., Snell G., Jennings A., Iwamoto K., Habuka N., Hirokawa A., Ishikawa T., Tanaka T., Miki H., Ohta Y., Sogabe S. (2011) 1) Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J. Biol. Chem. 286, 18756–18765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Qiu C., Tarrant M. K., Choi S. H., Sathyamurthy A., Bose R., Banjade S., Pal A., Bornmann W. G., Lemmon M. A., Cole P. A., Leahy D. J. (2008) Mechanism of activation and inhibition of the HER4/ErbB4 kinase. Structure 16, 460–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Graus-Porta D., Beerli R. R., Daly J. M., Hynes N. E. (1997) ErbB2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 16, 1647–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Karunagaran D., Tzahar E., Beerli R. R., Chen X., Graus-Porta D., Ratzkin B. J., Seger R., Hynes N. E., Yarden Y. (1996) ErbB-2 is a common auxiliary subunit of NDF and EGF receptors: implications for breast cancer. EMBO J. 15, 254–264 [PMC free article] [PubMed] [Google Scholar]
- 17. Tzahar E., Waterman H., Chen X., Levkowitz G., Karunagaran D., Lavi S., Ratzkin B. J., Yarden Y. (1996) A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell Biol. 16, 5276–5287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qian X., LeVea C. M., Freeman J. K., Dougall W. C., Greene M. I. (1994) Heterodimerization of epidermal growth factor receptor and wild-type or kinase-deficient Neu: a mechanism of interreceptor kinase activation and transphosphorylation. Proc. Natl. Acad. Sci. U.S.A. 91, 1500–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wada T., Qian X. L., Greene M. I. (1990) Intermolecular association of the p185neu protein and EGF receptor modulates EGF receptor function. Cell 61, 1339–1347 [DOI] [PubMed] [Google Scholar]
- 20. Wilson K. J., Gilmore J. L., Foley J., Lemmon M. A., Riese D. J., 2nd (2009) Functional selectivity of EGF family peptide growth factors: implications for cancer. Pharmacol. Ther. 122, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jones J. T., Akita R. W., Sliwkowski M. X. (1999) Binding specificities and affinities of EGF domains for ErbB receptors. FEBS Lett. 447, 227–231 [DOI] [PubMed] [Google Scholar]
- 22. Wilson K. J., Mill C., Lambert S., Buchman J., Wilson T. R., Hernandez-Gordillo V., Gallo R. M., Ades L. M., Settleman J., Riese D. J., 2nd (2012) EGFR ligands exhibit functional differences in models of paracrine and autocrine signaling. Growth Factors 30, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ellis I. R., Schor A. M., Schor S. L. (2007) EGF and TGF-a motogenic activities are mediated by the EGF receptor via distinct matrix-dependent mechanisms. Exp. Cell Res. 313, 732–741 [DOI] [PubMed] [Google Scholar]
- 24. Saito T., Okada S., Ohshima K., Yamada E., Sato M., Uehara Y., Shimizu H., Pessin J. E., Mori M. (2004) Differential activation of epidermal growth factor (EGF) receptor downstream signaling pathways by βcellulin and EGF. Endocrinology 145, 4232–4243 [DOI] [PubMed] [Google Scholar]
- 25. Streicher K. L., Willmarth N. E., Garcia J., Boerner J. L., Dewey T. G., Ethier S. P. (2007) Activation of a nuclear factor-κB /interleukin-1 positive feedback loop by amphiregulin in human breast cancer cells. Mol. Cancer Res. 5, 847–861 [DOI] [PubMed] [Google Scholar]
- 26. Doran D. M., Spar I. L. (1980) Oxidative iodine monochloride iodination technique. J. Immunol. Methods 39, 155–163 [DOI] [PubMed] [Google Scholar]
- 27. Li Y., Macdonald-Obermann J., Westfall C., Piwnica-Worms D., Pike L. J. (2012) Quantitation of the effect of ErbB2 on EGF receptor binding and dimerization. J. Biol. Chem. 287, 31116–31125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Macdonald-Obermann J. L., Piwnica-Worms D., Pike L. J. (2012) The mechanics of EGF receptor/ErbB2 kinase activation revealed by luciferase fragment complementation imaging. Proc. Natl. Acad. Sci. U.S.A. 109, 137–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haslekås C., Breen K., Pedersen K. W., Johannessen L. E., Stang E., Madshus I. H. (2005) The inhibitory effect of ErbB2 on epidermal growth factor-induced formation of clathrin-coated pits correlates with retention of epidermal growth factor receptor-ErbB2 oligomeric complexes at the plasma membrane. Mol. Biol. Cell 16, 5832–5842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Worthylake R., Opresko L. K., Wiley H. S. (1999) ErbB2 amplification inhibits down-regulation and induces constitutive activation of both ErbB2 and epidermal growth factor receptors. J. Biol. Chem. 274, 8865–8874 [DOI] [PubMed] [Google Scholar]
- 31. Macdonald-Obermann J. L., Adak S., Landgraf R., Piwnica-Worms D., Pike L. J. (2013) Dynamic analysis of the epidermal growth factor (EGF) receptor-ErbB2-ErbB3 protein network by luciferase fragment complementation imaging. J. Biol. Chem. 288, 30773–30784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang K. S., Ilagan M. X., Piwnica-Worms D., Pike L. J. (2009) Luciferase fragment complementation imaging of conformational changes in the EGF receptor. J. Biol. Chem. 284, 7474–7482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jura N., Endres N. F., Engel K., Deindl S., Das R., Lamers M. H., Wemmer D. E., Zhang X., Kuriyan J. (2009) Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 137, 1293–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Macdonald J. L., Pike L. J. (2008) Heterogeneity in EGF binding affinities arises from negative cooperativity in an aggregating system. Proc. Natl. Acad. Sci. U.S.A. 105, 112–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stern K. A., Place T. L., Lill N. L. (2008) EGF and amphiregulin differentially regulate Cbl recruitment to endosomes and EGF receptor fate. Biochem. J. 410, 585–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schuh S. M., Newberry E. P., Dalton M. A., Pike L. J. (1994) Mutation of proline-1003 to glycine in the epidermal growth factor (EGF) receptor enhances responsiveness to EGF. Mol. Biol. Cell 5, 739–746 [DOI] [PMC free article] [PubMed] [Google Scholar]