

Background: The androgen receptor (AR) is a transcription factor regulating progression of prostate cancer.
Results: Developed compounds inhibit AR transcriptional activity in vitro and in vivo by selective targeting of the AR-DNA-binding domain (DBD).
Conclusion: By targeting the DBD, the compounds differ from conventional anti-androgens.
Significance: Anti-androgens with a novel mechanism of action have the potential to treat recurrent prostate cancer.
Keywords: Androgen Receptor, DNA-binding Protein, Drug Discovery, Nuclear Receptor, Prostate Cancer
Abstract
The androgen receptor (AR) is a transcription factor that has a pivotal role in the occurrence and progression of prostate cancer. The AR is activated by androgens that bind to its ligand-binding domain (LBD), causing the transcription factor to enter the nucleus and interact with genes via its conserved DNA-binding domain (DBD). Treatment for prostate cancer involves reducing androgen production or using anti-androgen drugs to block the interaction of hormones with the AR-LBD. Eventually the disease changes into a castration-resistant form of PCa where LBD mutations render anti-androgens ineffective or where constitutively active AR splice variants, lacking the LBD, become overexpressed. Recently, we identified a surfaced exposed pocket on the AR-DBD as an alternative drug-target site for AR inhibition. Here, we demonstrate that small molecules designed to selectively bind the pocket effectively block transcriptional activity of full-length and splice variant AR forms at low to sub-micromolar concentrations. The inhibition is lost when residues involved in drug interactions are mutated. Furthermore, the compounds did not impede nuclear localization of the AR and blocked interactions with chromatin, indicating the interference of DNA binding with the nuclear form of the transcription factor. Finally, we demonstrate the inhibition of gene expression and tumor volume in mouse xenografts. Our results indicate that the AR-DBD has a surface site that can be targeted to inhibit all forms of the AR, including enzalutamide-resistant and constitutively active splice variants and thus may serve as a potential avenue for the treatment of recurrent and metastatic prostate cancer.
Introduction
The androgen receptor (AR)4 is a ligand-inducible transcription factor that contributes to the growth, recurrence, and metastasis of prostate cancer (PCa) tumors (1, 2). The AR is activated by binding to androgens, such as testosterone and dihydrotestosterone, which causes localization of the transcription factor into the nucleus (3) where it drives the expression of genes responsible for cell survival and proliferation (4). Early forms of prostate cancer are treatable with surgery and radiation, but recurrent or metastatic disease requires the application of alternative therapies. Thus, blockade of androgen signaling can be achieved with drugs designed to interfere with androgen production or with anti-androgens, which directly compete with hormones for binding onto the AR (5). Although these treatments are initially effective, the disease eventually progresses to castration-resistant PCa where tumor growth has become resistant to hormonal therapies (1).
The AR structural organization includes an N-terminal domain (NTD), followed by the DBD and LBD domains (6). X-ray crystal structures of the LBD (7–9) and DBD (10) have assisted in defining surface-exposed regions on the AR that facilitate ligand, DNA, and co-factor binding. Thus, it has been well characterized how androgens coordinate into a ligand binding pocket on the surface of the LBD termed the androgen-binding site. That site is the best understood target for anti-androgen compounds (such as enzalutamide) that compete with testosterone for binding. In addition, the LBD contains alternative surface-exposed pockets such the activation-function 2 (AF2) region, important for co-regulator recruitment (11), and the binding-function 3 (BF3) site of unknown function, located near the androgen-binding site (12, 13). The structure of the rat AR-DBD dimer in complex with DNA has helped us to understand the role of the DBD in AR function (10). The AR-DBD contains P-box and D-box amino acid motifs that are involved in nucleic acid binding and DBD-mediated dimerization.
Growing evidence suggests that castration-resistant PCa may be partially mediated by AR splice variants, which arise from messenger RNA lacking the ligand-binding domain coding sequence (14–18). The absence of an LBD would prevent targeting the androgen-binding site, leaving only the NTD and DBD as viable domains that are targetable by small molecules. The best characterized AR splice variants, AR-V7 and AR-v567es, are implicated in several studies to contribute to reactivation of AR signaling in castration-resistant tumors (18–22). Inhibition of splice variant transcriptional activity would be a significant breakthrough in the development of a new class of anti-AR drugs.
Here, we explore the AR-DBD as a novel target for small molecules to block full-length and splice variant AR signaling. Recently, using our established in silico drug design approach (13, 23, 24), we discovered a surface-exposed region on the AR-DBD, including residues Ser-579 to Lys-610, which was established to be targetable by small molecule inhibitors to potentially inhibit the AR by interfering with DNA binding. Computer-aided high throughput screening identified candidate molecules that were effective for abolishing AR transcriptional activity in LNCaP cells and displayed no interference or binding with the AR-LBD (25). In this study, we validate the target with point mutations at positions on the DBD that are predicted to interact with the candidates. Those compounds could also inhibit the transcriptional activity of splice variants, either when transiently expressed in PC3 cells or when encoded on the genome of a genetically modified CWR-R1 cell line. In parallel, we examine the effect of the inhibitors on DNA binding in vitro using ChIP and biophysical methods and also on AR subcellular localization using confocal microscopy. Finally, we show the leading inhibitor is effective to slow growth of tumor xenografts and to block PSA expression in castrated mice. Collectively, our data support the importance of the AR-DBD as a prospective target for the treatment of advanced metastatic PCa.
EXPERIMENTAL PROCEDURES
Compounds
VPC-14228 (4-(4-phenylthiazol-2-yl)morpholine), VPC-14449 (4-(4-(4,5-bromo-1H-imidazol-1-yl)thiazol-2-yl)morpholine), and VPC-14337 (pyrvinium) were purchased from Enamine (Monmouth Jct., NJ).
Constructs
Full-length human AR (hARWT) or splice variant (AR-V7) was encoded on a pcDNA3.1 expression plasmid. Point mutations in the DBD were generated with the QuikChange mutagenesis kit (Stratagene) using hARWT or AR-V7 templates. Mutagenic primers were generated using a primer design tool (Agilent). The glucocorticoid receptor (GR) was expressed from the pGR mammalian expression vector as described previously (26). Progesterone receptor (PR) was expressed from the pSG5-PRB vector and was obtained from Dr. X. Dong. The AR-DBD + hinge domain (amino acids 558–689) was amplified from the hARWT construct and cloned into the pTrc expression vector (N-terminal His6 tag, Invitrogen) using the polymerase incomplete primer extension method (PIPE). Briefly, the AR-DBD + hinge domain was cloned by mixing the PCR products resulting from the following templates and primers: AR(558–689) insert from hARWT template 5′-CAT CAT CAT CAT CAT CAT GGT ACC TGC CTG ATC TGT GG and 5′-CAG GCT GAA AAT CTT CTC TCA GTG TCC AGC ACA CAC TAC AC; pTrc vector lacking multiple cloning site 5′-ATC TCC ACA GAT CAG GCA GGT ACC ATG ATG ATG ATG ATG ATG and 5′-GGT GTA GTG TGT GCT GGA CAC-TGA GAG AAG ATT TTC AGC CTG; the underlined primer sections anneal to the specified template, although their 5′-extensions (italicized) are complementary to the corresponding primer sequence of the other PCR. Plasmid assembly is achieved by mixing the PCR products from each reaction (27), followed by transformation into chemically competent bacteria. A similar strategy was used to clone the AR-DBD + hinge into the Pan4 vector (avidity) expressing the N-terminal biotinylation sequence (GLNDIFEAQKIEWHE) and C-terminal His tag. YFP-AR plasmid was a gift from Dr. Jan Trapman (28) and is based on pEYFP-C1 (Clontech). YFP-V7 was constructed by polymerase incomplete primer extension method using the following primers and templates: AR-V7 insert from pcDNA3.1 AR-V7 template 5′-GGT GCT GGA GCA GGT GCT GGA ATG GAA GTG CAG TTA GGG CTG and 5′-GGA AAT AGG GTT TCC AAT GCT TCA GGG TCT GGT CAT TTT GAG; pEYFPC1 vector lacking the full-length AR 5′-CAG CCC TAA CTG CAC TTC CAT TCC AGC ACC TGC TCC AG and 5′-CTC AAA ATG ACC AGA CCC TGA AGC ATT GGA AAC CCT ATT TCC.
Cell Culture, Transfection, and Luciferase Assays
PC3 human PCa cells (AATC) were serum-starved in RPMI 1640 media (Invitrogen) supplemented with 5% charcoal-stripped serum (CSS) (RPMI 1640 medium with 5% CSS) for 5 days prior to transfection. For luciferase assays, PC3 cells were seeded in 96-well plates (5000 cells/well) in RPMI 1640 medium with 5% CSS for 24 h, followed by transfection with 50 ng of hAR or other nuclear receptor plasmid, 50 ng of ARR3tk-luciferase, and 0.3 μl/well TransIT20/20 transfection reagent (TT20, Mirus) for 48 h. Cells were then treated with compounds at various concentrations and 0.1 nm R1881 (in 100% ethanol) for 24 h. GR or PR activation was stimulated with 1 nm dexamethasone or progesterone, respectively. ER-α transcriptional activity was measured with a MCF-7 cell line bearing the stable transfection of an estrogen-response element-luciferase reporter, with transcriptional activity stimulated by 1 nm estradiol. Cell lysis was carried out with 60 μl of 1× passive lysis buffer/well (Promega). 20 μl of cell lysate from each well were mixed with 50 μl of luciferase assay reagent (Promega), and luminescence was recorded on a TECAN M200pro plate reader. Luciferase assays with splice variant AR were performed the same way but with only 5 ng of pcDNA3.1 AR-V7 (to limit the high level of AR-V7 expression) and no R1881. R1-AD1 and TALEN-engineered R1-D567 cell lines have been described previously (22). Assays with R1-AD1 and R1-D567 cells were performed as above but with transfection of only ARR3tk-luciferase reporter and 10,000 cells/well.
Western Blots
Cell lysates (40 μl) from luciferase assays (96-well plate) were separated on a 10% SDS-polyacrylamide mini gel. Protein was transferred to methanol-charged PVDF membranes and probed with anti-AR441 (mouse, Sigma) monoclonal primary antibody. Blots were also probed with polyclonal anti-actin (rabbit, Sigma) to show equal loading and polyclonal anti-PARP/anti-cleaved PARP (rabbit, Sigma) to test for induction of apoptosis. Lysates from CWR-R1 cells were additionally probed with polyclonal anti-FKBP5 (rabbit, Sigma) following 2 days of incubation with compounds.
PSA Measurements
LNCaP cells maintained in RPMI 1640 medium with 5% CSS were incubated in 96-wells (10,000 cells/well) for 2 days in the same culture medium and in the presence of compounds and 1 nm R1881. Following the incubation period, 150 μl of the media was taken from each well, and PSA levels were quantified using a Cobas e411 analyzer (Roche Applied Science) according to the manufacturer's instructions. The same instrument was used to analyze serum PSA from mice during the in vivo analysis.
Microarray Genetic Profile
LNCaP cells were grown for 24 h under the following four conditions: 1) DMSO without R1881; 2) DMSO with R1881 (1 nm); 3) compound 14449 at 400 nm with R1881; and 4) enzalutamide at 120 nm with R1881. Compound concentration followed approximately the IC50 concentration determined in luciferase reporter assays in Fig. 1. Each condition was repeated in triplicate. After 24 h, the total cellular mRNA was extracted from each of the 12 samples (four conditions three times), and the gene expression level of 50,737 transcripts was measured from custom Agilent microarrays. The gene expression data were quantile normalized across all the samples and transformed into a log2 scale. A two-sample t test was performed on the expression level of each transcript between condition 3 (compound VPC-14449 with R1881) and condition 2 (DMSO with R1881), and between condition 4 (enzalutamide with R1881) and condition 2 (DMSO with R1881). A gene is considered to be differentially expressed if the p value from the two-sample t test is less than 0.05. Fisher's exact test and odds ratio were used to evaluate the overlap between different sets of differentially expressed genes.
FIGURE 1.

Small molecules target the DBD of full-length hAR. A, chemical structure of VPC-14228, VPC-14449, and VPC-14337 (pyrvinium). B, multiple sequence alignment of nuclear receptor DBD domains. The amino acid sequences from human androgen, estrogen, glucocorticoid, and progesterone receptors were aligned with ClustalW, and visualized using JalView with Blossom62 color scheme. The positions that were mutated in this study, Gln-592 and Tyr-594, are highlighted in red. Numbering is according to the human AR. C–E, 50 ng of hARWT, hARY594D, or hARQ592D plasmids were co-transfected with ARR3tk-luciferase into PC3 cells. 0.1 nm R1881 and the indicated concentrations of compounds were added for 24 h. 100% refers to luminescence recorded with 0.1% DMSO only. Bottom panels in C–E present Western blots (AR-441/actin) of cell lysates incubated with DMSO or 50 μm compound. Lysates from hARWT were also probed with anti-PARP/cleaved PARP antibodies. Errors bars represent the mean ± S.D. from 4 to 6 replicates per point. IC50 values (micromolar) were calculated by fitting curves to a sigmoidal dose-response (variable slope) equation. F and G, Y594A or Q592A mutants of the full-length human AR were tested in the luciferase reporter assay and analyzed by Western blot as described in C. enz, enzalutamide.
Confocal Microscopy
Approximately 40,000 PC3 cells were seeded for 48 h on sterile coverslips placed within 12-well plates in RPMI 1640 medium with 5% CSS. Transfection of YFP-AR or YFP-V7 plasmids (100 ng per well) was performed using TT20 (3 μl) for 48 h. Cells were then treated with 10 nm R1881 and 25 μm compounds for 6 h. After aspiration of the media, cells were washed once with PBS and fixed in 4% paraformaldehyde overnight at 4 °C, followed by mounting on charged cover slides using DAPI mount (Vector Laboratories). Images were taken on a Zeiss LSM 780 confocal spinning disk microscope controlled with Zen 2012 software. YFP and DAPI were visualized with excitation wavelengths of 508 and 388 nm, respectively.
Chromatin Immunoprecipitation (ChIP)
Androgen-deprived LNCAP cells were treated for 24 h with DMSO alone, DMSO + R1881, or compounds + R1881. DNA-protein cross-linking was performed with 1% formaldehyde treatment for 10 min at room temperature and quenched with 125 mm glycine for 5 min. Cell lysates (1 × 107 cells/ml) were subjected to sonication with a Thermo Scientific 1/8-inch sonication probe and Sonic Dismembrator 550 instrument to yield DNA fragments of 200–1000 bp in size. Immunoprecipitation of lysates (3.3 × 106 cell eq) was performed with 5 μg of anti-AR-N20 antibody (Santa Cruz Biotechnology) or 1 μg of rabbit isotype control IgG (Santa Cruz Biotechnology) using a EZ-ChIP chromatin immunoprecipitation kit (Millipore). Bound DNA was quantified by quantitative PCR (SYBR Green master mix, Invitrogen) using the following primer sets: PSA enhancer, forward 5′-ATG GAG AAA GTG GCT GTG GC and reverse 5′-TGC AGT TGG TGA GTG GTC AT; FKBP5 enhancer, forward 5′-CCC CCC TAT TTT AAT CGG AGT AC and reverse 5′-TTT TGA AGA GCA CAG AAC ACC CT; GAPDH promoter, forward 5′-TAC TAG CGG TTT TAC GGG CG and reverse 5′-TCG AAC AGG AGC AGA GAG CGA. The quantitative PCR results are presented as fold enrichment of PCR amplification over control IgG antibody and normalized based on the total input (nonprecipitated chromatin). Primers for the GAPDH promoter were used as a negative control lacking any androgen-response element.
Purification of AR-DBD Proteins
The plasmid encoding the AR-DBD + hinge was transformed into BL21 (DE3). BL21 cells designated for expression of biotin-labeled AR-DBD + hinge were co-transformed with the Pan4 AR-DBD + hinge (ampicillin selection) and biotin ligase expression vectors (pBir-Acm, chloramphenicol selection). Single colonies were grown in 2 liters of LB media supplemented with 50 μg/ml ampicillin and 35 μg/ml chloramphenicol (where appropriate) to A600 nm = 0.6 before induction with 0.1 mm isopropyl β-d-1-thiogalactopyranoside for 3 h at 37 °C. Cultures expressing the AR-DBD with the biotinylation sequence were simultaneously supplemented with 0.150 mm biotin during the induction step. Cell pellets were resuspended in ∼20 ml of 50 mm Tris-HCl, pH 8.0, 300 mm NaCl, 5% glycerol (Buffer A) supplemented with 10 mm imidazole and incubated with 0.1 mg/ml chicken egg white lysozyme (Sigma) and 0.1 mm PMSF protease inhibitor for 30 min on ice. Cell lysis was achieved by sonication, followed by centrifugation at 13,000 × g for 30 min at 4 °C. The supernatant was rotated with 2 ml of nickel-agarose beads (GE Healthcare) for 1 h at 4 °C and then directly loaded onto a Poly-Prep 10-ml gravity chromatography column (Bio-Rad). Washing was performed with 2× 4 ml of Buffer A supplemented with 20 mm imidazole. Pure proteins were eluted in 500-μl fractions with 2 ml of Buffer A containing 250 mm imidazole.
EMSA (Gel Shift) Assays and Biolayer Interferometry Analysis
Electrophoretic mobility shift assays (EMSA) were performed using purified AR-DBD and dsDNA bearing the ARE 2 sequence. The ARE was formed by annealing the following complementary oligonucleotides in H2O: upper strand, 5′-TAC AAA TAG GTT CTT GG AGTACT TTA CTAGGC ATG GAC AAT G, and lower strand, 5′-CAT TGT CCA T GCCTAG TAA AGTACT CCA AGA ACC TAT TTG TA. Positions of hexameric AREs are underlined. Scrambled DNA was annealed from the following sequences: upper strand, 5′-TAA AAC GTG GTC CCT GGT ACT GCC TTC GTG CCA TTC GAT TTT, and lower strand, 5′-AAA ATC GAA TGG CAC GAA GGC AGT ACC AGG GAC CAC GTT TTA. Protein-DNA complexes were allowed to incubate on ice for 30 min in loading buffer (20 mm Tris, pH 8, 50 mm NaCl, 1 mm EDTA, 10 μg/ml poly(dI-dC), 5 mm MgCl2, 200 μl/ml BSA, 5% glycerol, and 1 mm DTT), followed by electrophoresis on 6% native-PAGE in 1× TBE, pH 8.0. Visualization of protein-DNA complexes was performed with SyberSafeTM DNA staining dye.
Biolayer interferometry analysis on a ForteBio Octet Red instrument was carried out using biotinylated AR-DBD + hinge in Buffer A with 5% DMSO throughout all experiments. The DBD (0.1 mg/ml) was loaded onto streptavidin sensors in 200 μl of buffer for 30 min, followed by blocking of free streptavidin sites with biocytin (10 μg/ml) for 10 min. DBD-loaded sensors were then pre-equilibrated in 50 μm compound or 5% DMSO alone for 100 s in the same buffer. The kinetics of DNA association were monitored by moving sensors into wells containing dsDNA (ARE, 3 μm) supplemented with 50 μm compound for 120 s. This was followed by dissociation in buffer + compound, but lacking DNA, for an additional 120 s. Biocytin-blocked control sensors (no AR-DBD) were subjected to the same experimental conditions, and nonspecific interactions with dsDNA were subtracted from each curve.
Assessment of Tumor Growth and PSA for Castration-resistant LNCaP Xenografts
6–8-Week-old nude mice (Harlan Sprague-Dawley) weighing 25–31 g were subcutaneously inoculated with LNCaP cells (106 cells in BD Matrigel, BD Biosciences) at the posterior dorsal site. Tumor volume, body weight, and serum PSA levels were measured weekly. When serum PSA levels reached more than 25 ng/ml, mice were castrated. When PSA recovered to pre-castration levels, mice were randomized into three treatment groups as follows: vehicle, 10 mg/kg enzalutamide, or 100 mg/kg of compound VPC-14449 and treated via intraperitoneal injection twice daily for 4 weeks. Calipers were used to measure the three perpendicular axes of each tumor to calculate the tumor volume. Mice were also weighed weekly and monitored daily for signs of toxicity, including death, lethargy, blindness, and disorientation.
RESULTS
Small Molecules Designed to Target the AR-DBD Can Inhibit AR Transcriptional Activity
A hot spot for small molecule binding was detected based on the AR DBD-DNA crystal structure at amino acids positions including Ser-579, Val-582, Phe-583, Arg-586, Gln-592, Tyr-594, and Lys-610 at the protein-DNA interface (Protein Data Bank code 1R4I (10)). The in silico workflow to identify and refine small molecule inhibitors targeting the AR-DBD, screening of virtual hits in LNCaP cells, and general characteristics with respect to AR inhibition and cross-reactivity with other AR domains was recently described by our laboratory (25). In this study, the best hit compounds (VPC-14228 and VPC-14449) were selected for characterization and comparison with known androgen pathway inhibitors such as enzalutamide and pyrvinium (VPC-14337, Ref. 29) (Fig. 1A). Subtle differences in the corresponding pockets of closely related nuclear receptors such as ER, PR, and GR also identified potential grounds for selective targeting of the AR-DBD. In particular, sequence differences between residues 590 and 594 of the androgen receptor DBD and corresponding residues from related DBDs (Fig. 1B) might allow discrimination of small molecules.
Using a luciferase reporter assay in PC3 cells driven by the ARR3tk probasin-based promoter (30), two compounds predicted to bind to the AR-DBD showed dose-dependent inhibition of the transiently expressed full-length human AR (Fig. 1C, upper panel, VPC-14228 and VPC-14449, IC50 = 2.36 and 0.340 μm, respectively) without affecting AR protein expression (Fig. 1C, lower panel). VPC-14337 (pyrvinium) was recently reported to inhibit the full-length and splice variant AR forms by targeting the DBD (29), and could also inhibit AR transcriptional activity (Fig. 1C, IC50 = 0.194 μm). Control experiments demonstrate that VPC-14449 could inhibit the AR to levels comparable with enzalutamide (Fig. 1C, IC50 = 0.314 μm). Western blots against PARP confirm that compound treatment did not affect total PARP levels nor did it generate any PARP-cleaved product, with the exception of VPC-14337 (Fig. 1C, lower panel). These results indicate that pyrvinium (VPC-14337) strongly induces apoptosis, whereas our developed DBD inhibitors (14228/14449) exhibit little or no toxicity.
To validate the site of action of AR-DBD binders, we introduced point mutations at residues that are predicted to interact with the lead compounds. Two positions (Tyr-594 and Gln-592) that were identified in the region with amino acid differences among related nuclear receptors (Fig. 1B) could bear aspartate substitutions without abolishing full-length AR activity. Whereas the Y594D and Q592D mutants could be inhibited by enzalutamide, luciferase expression was not affected by VPC-14228 and only by high concentration (≥25 μm) of VPC-14449 (Fig. 1, D and E). In contrast, pyrvinium strongly inhibited both AR mutants (Fig. 1, D and E), suggesting that the compound engages residues other than Tyr-594/Gln-592 in the surface-exposed pocket or instead binds to a different location on the DBD surface. Western blot analysis confirms that the expression of mutant AR proteins was not changed by drug inhibition (Fig. 1, D and E, lower panels). Introducing an acidic residue at these positions may prevent hydrophobic interactions necessary for supporting compound binding.
In addition to introducing a charged amino acid into the DBD, we also tested Y594A and Q592A mutants, both of which could be inhibited by VPC-14228/14449 but with significantly higher IC50 values (Fig. 1, F and G, ∼3–6 μm) compared with wild type AR (Fig. 1C). It is possible that removing the Gln or Tyr side chains creates additional space in the pocket to allow compound entry but reduced ability to inhibit AR activity. The resulting increase in IC50 values further supports the importance of Tyr-594 and Gln-592 residues to compound binding that is compromised when their side chains are removed.
To determine whether our compounds cross-react with the DBDs of related nuclear receptors, we performed assays with full-length ER, GR, and PR (Fig. 2). Luciferase constructs contained the appropriate response region for the corresponding nuclear receptor with ARR3tk used for AR/GR/PR and estrogen-response element for ER. VPC-14228/14449 showed inhibition of ER transcriptional activity at concentrations higher than 5 μm (Fig. 2, B and C, black bars) but were severalfold less effective when compared with inhibition of the AR (white bars). All three tested compounds were completely ineffective on the transcriptional activity of full-length GR and PR, even when administered at 25 μm concentration (Fig. 2, dark gray and light gray bars, respectively). Remarkably, enzalutamide showed considerable cross-reactivity against the full-length ER, approaching inhibition levels comparable with that against the AR (Fig. 2A, black bars). These data suggest that the developed compounds possess potent inhibitory effects against the AR, bind to the intended target site on the AR-DBD, and show little or no cross-reactivity.
FIGURE 2.

DBD-interacting compounds are specific for AR. Enzalutamide (enz) (A), VPC-14228 (B), and VPC-14449 (C) were tested at the indicated concentrations in luciferase assays against transiently expressed AR, GR, and PR or against endogenous ER-α in MCF-7 cells. AR, GR, and PR activity was assessed with the ARR3tk-luciferase reporter. MCF-7 cells include a stably transfected estrogen-response element-luciferase gene. 100% refers to luciferase activity of each receptor with 0.1% DMSO only. Errors bars represent the mean ± S.D. six replicates. enz, enzalutamide.
DBD-interacting Compounds Down-regulate Expression of Androgen-responsive Genes in LNCaP Cells
To assess the ability of VPC-14228/14449 to block transcription of naturally occurring AR-regulated genes, LNCaP cells were treated simultaneously with R1881 and compounds, followed by measurement of secreted PSA (Fig. 3A). The results show a dose-dependent inhibition by VPC-14228/14449 and enzalutamide with corresponding IC50 values all established at sub-micromolar concentrations.
FIGURE 3.
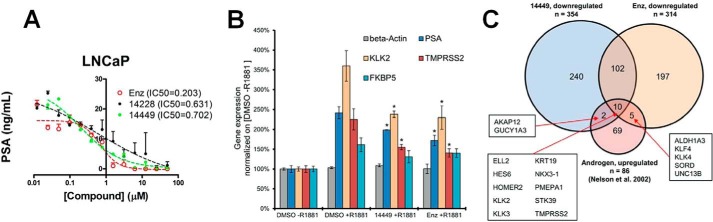
Effect of DBD-interacting compounds on expression of AR target genes. A, LNCaP cells were treated with 1 nm R1881 and compounds for 2 days at the indicated concentrations. Secreted PSA was quantified by analyzing 150 μl of cell culture media from each well from two independent experiments. B, gene expression changes of AR target genes and a non-androgen-responsive gene (β-actin, ACTB) in the presence of R1881, compound 14449, and enzalutamide (Enz). * = significant reduction in gene expressions (p value < 0.05) based on two-sample t test between 14449 + R1881 and DMSO + R1881 and between enzalutamide + R1881 and DMSO + R1881. C, overlap among three gene sets: 1) down-regulated genes by compound 14449; 2) down-regulated genes by enzalutamide; and 3) previously published androgen-up-regulated genes. Individual androgen-up-regulated genes in the overlapping regions are shown.
Using a chemo-genomic approach (31), a more extensive analysis of gene expression was conducted using the lead inhibitor VPC-14449. To determine any effect on the expression of androgen- or genotoxin-responsive genes, LNCaP cells were treated with VPC-14449 (400 nm) or enzalutamide (120 nm), in the presence of R1881, to compare transcriptional responses using Agilent gene expression microarrays (Fig. 3B). Several well known AR target genes (32, 33), which include KLK3 (PSA), KLK2, TMPRSS2, and FKBP5, increased in gene expression under the presence of R1881 (comparing DMSO + R1881 against DMSO-R1881) with fold changes of 2.42, 3.60, 2.25, and 1.61, respectively. Following treatment with VPC-14449 + R1881, gene expressions of KLK3, KLK2, and TMPRSS2 were all reduced significantly with fold changes of 0.82, 0.66, and 0.69, respectively, as compared with R1881 treatment only. The reduction of expression of these AR target genes by VPC-14449 is comparable with that by enzalutamide with fold changes of 0.71, 0.64, and 0.62. There was a decrease (fold change 0.81) of FKBP5 expression by VPC-14449; however, the p value of 0.08 did not meet the 0.05 cutoff based on the two-sample t test, likely due to the small sample size. Both VPC compounds and enzalutamide showed no effect on the expression of a non-androgen regulated gene, β-actin (ACTB).
To identify other down-regulated genes, the following criteria were applied to all the 50,737 transcripts measured on the Agilent microarrays: 1) p value <0.05 based on two-sample t test and 2) fold change ≤0.85. A total of 354 genes were down-regulated by VPC-14449, among which 112 were also down-regulated by enzalutamide (Fig. 3C). The overlap of down-regulated genes between the two compounds was significant with a p value of Fisher's exact test less than 2.20E-16 and odds ratio of 45.86. In addition, the list of down-regulated genes by either VPC-14449 and/or enzalutamide was compared with a total of 86 genes that have been previously shown to be up-regulated by androgens in LNCaP cells (32). Fig. 3C further illustrates that 12 of the 354 down-regulated genes by VPC-14449 are up-regulated by androgens (significant overlap: p value = 2.73E-08, odds ratio = 9.55), and 15 of the 314 down-regulated genes by enzalutamide are up-regulated by androgens (significant overlap: p value = 3.34E-12, odds ratio = 14.27). The genes KLK2, KLK3 (PSA), and TMPRSS2 are among 10 genes that are up-regulated by androgens, but down-regulated by both VPC-14449 and enzalutamide. The two sets of mutually exclusive down-regulated genes resulting from VPC-14449 or enzalutamide treatment indicates profoundly different mechanisms of action of each compound to inhibit AR signaling.
Finally, to identify any potential genotoxic effect from the two chemicals, up-regulated genes by either VPC-14449 or enzalutamide were compared with a list of 31 genes, the expression of which has previously been shown to increase in the presence of genotoxins (34). This set of genotoxin-responsive genes includes a number of p53 target genes and others involved in apoptosis, DNA repair, DNA damage response, and stress response. None of the genotoxin-responsive genes showed any significant change of expression by VPC-14449. Three known representative genes that were unaffected included CASP1 (caspase 1, apoptosis, fold change = 1.00, p value = 0.73), XPC (xeroderma pigmentosum complementation group C, DNA repair, fold change = 1.03, p value = 0.59), and ATF3 (activating transcription factor 3, stress response, fold change 0.99, p value = 0.89). Collectively, the results demonstrate that the developed AR DBD inhibitors can significantly down-regulate expression of known AR-regulated genes to levels comparable with that by enzalutamide, with no cytotoxicity induced.
DBD-interacting Compounds Inhibit Transcriptional Activity of AR Splice Variants
Because most AR splice variants retain the DBD domain, we tested for inhibition of the transcriptional activity of AR-V7. Using the same luciferase reporter assay, the activity of transiently expressed AR-V7 was reduced with increasing concentrations of VPC-14228/14449 or pyrvinium without altering AR-V7 expression (Fig. 4A). Control experiments with enzalutamide showed no effect on AR-V7 activity (Fig. 4A) and are consistent with the absence of the LBD from this variant. Notably, the compounds did not achieve complete inhibition and were less effective against the transcriptional activity of AR-V7 (IC50 = 4–8 μm) when compared with inhibition of the full-length receptor (Fig. 1C).
FIGURE 4.

Effect of DBD-interacting compounds on AR splice variants. The luciferase reporter assay was performed as described in Fig. 1C but with 5 ng of pcDNA3.1 AR-V7 (A) or AR-V7Y594D expression plasmid (B). Western blots of cell lysates containing AR-V7 were performed as in Fig. 1C. R1-AD1 cells expressing full-length AR (C) and R1-D567 (D) cells expressing the ARv567es splice variant were tested in luciferase assays as described in Fig. 1C, but with transfection of only 50 ng of ARR3tk-luciferase reporter per well. R1-AD1 cells were stimulated with 0.1 nm R1881, whereas no androgen was administered to R1-d567 cells. E, Western blots with cell lysates before luciferase assay in C and D were performed as in Fig. 1C. F, cells were incubated with 50 μm compounds and 0.1 nm R1881 (R1-AD1 cells only; no androgen with R1-D567) for 2 days before cell lysates were probed with anti-FKBP5 antibody. enz, enzalutamide.
We reasoned that the DBDs of the full-length and splice variant ARs would share a similar protein structure. Accordingly, we introduced the Y594D mutation into the AR-V7 coding sequence to determine whether this mutation could abolish drug inhibition. The transcriptional activity of AR-V7Y594D could not be inhibited by VPC-14228/14449 (Fig. 4B), suggesting the binding location of the compounds are similar on all forms of the AR. As with the full-length AR bearing this mutation, AR-V7Y594D could still be strongly inhibited by pyrvinium (Fig. 4B).
Transient AR-V7 expression may not reflect physiological protein concentrations in cells. To investigate the effect of our compounds on endogenous expression levels, we used a pair of isogenic cell lines that express either full-length AR (R1-AD1) or the AR v567es variant (R1-D567). R1-D567 cells were derived from the R1-AD1 cell line by TALEN-mediated deletion of AR exons 5–7, reflecting an AR gene rearrangement discovered in patient-derived LuCaP 86.2 xenograft tissue (22). Following transfection of ARR3tk-luciferase plasmid into these cell lines, VPC-14228/14449 could inhibit both wild type and AR v567es transcriptional activity with increasing concentrations (Fig. 4, C and D). Western blots demonstrate no effect of the compounds on protein expression of either form of the AR (Fig. 4E). We also performed Western blots for the naturally occurring AR-regulated FK506-binding protein 5 (FKBP5). FKBP5 protein expression was reduced in R1-AD1 and R1-D567 cells following treatment with VPC-14228/14449, whereas enzalutamide treatment only affected R1-AD1 cells expressing the full-length AR (Fig. 4F). These results agree with the observed reduction in FKBP5 mRNA levels after siRNA knockdown of v567es in R1-D567 cells (22).
DBD-interacting Compounds Do Not Impede AR Nuclear Translocation
Enzalutamide and other inhibitors of the AR-LBD are thought to block nuclear localization of the AR, thereby preventing it from initiating transcription (35, 36). In contrast to this mechanism of conventional anti-androgens, we predicted that DBD-interacting compounds would exert their effect on nuclear AR. To test this idea, we transfected PC3 cells with plasmids encoding YFP-tagged full-length AR (yellow fluorescent protein, YFP-AR, see Ref. 28) and splice variant AR-V7 (YFP-V7). Both YFP-AR and YFP-V7 were able to drive luciferase expression and could be inhibited by VPC-14228/14449, demonstrating that the YFP tag did not affect AR transcriptional activity or compound inhibition (Fig. 5A). Upon treatment with R1881 and enzalutamide, confocal microscopy images revealed considerable levels of YFP-AR in the cytosol compared with control experiments (Fig. 5B). Conversely, VPC-14228/14449 did not prevent R1881-stimulated nuclear localization of YFP-AR with no fluorescence signal observed in the cytosol (Fig. 5B). Control experiments show that enzalutamide or VPC-14228/14449 could not stimulate any nuclear localization in the absence of R1881 (Fig. 5B). YFP-V7 completely localized in the nucleus under all conditions, even without R1881 (Fig. 5C), which agrees with the known property of splice variants to spontaneously undergo nuclear translocation (18). Together, these results suggest that our compounds influence the activity of the AR and its splice variants inside the cell nucleus, consistent with directly affecting DBD functions.
FIGURE 5.

Effect of compounds on AR nuclear localization. A, YFP-AR (50 ng/well) or YFP-V7 (5 ng/well) were co-transfected with ARR3tk-luciferase reporter construct as described in Fig. 1C. Where indicated, the assay was performed in the presence of 25 μm compound (or 0.1% DMSO only) and 0.1 nm R1881 (or ethanol only). The data are presented as raw luminescence values without normalization. B, PC3 cells were transfected with YFP-AR plasmid and treated with 10 nm R1881 and 25 μm of the indicated compounds. Cells were fixed, mounted, and stained with DAPI. Confocal microscopy reveals the location the cell nucleus (DAPI, 388 nm) and YFP-AR (508 nm) in the 1st two rows of images. The 3rd row depicts the merged images of DAPI and YFP-AR visualizations. Scale bars, 20 μm. C, same as B, but with a plasmid expressing YFP-V7. enz, enzalutamide.
Compounds Diminish DNA Binding by the AR at the Chromatin Level and in Vitro
Because the developed AR DBD inhibitors are predicted to bind near the protein-DNA interface on the AR-DBD, the compounds should affect AR binding to the enhancers (bearing AREs) of androgen-regulated genes. Following ChIP analysis (AR-N20 antibody) of chromatin from LNCaP cells treated with R1881 and compounds, both VPC-14428 and VPC-14449 reduced AR pulldown of the PSA and FKBP5 enhancer compared with R1881 treatment alone (Fig. 6A). Control experiments revealed no pull down of the enhancers in the absence of R1881, after enzalutamide treatment or under any condition with the GAPDH promoter negative control. Given that our compounds do not block nuclear translocation (Fig. 5), the ChIP results suggest VPC-14228/14449 blocks the interaction of the AR with androgen-response elements in the nucleus.
FIGURE 6.

Protein-DNA complexes are diminished in the presence of compounds. A, ChIP analysis of AR binding to the PSA or FKBP5 enhancers, or the GAPDH promoter, in LNCaP cells. Where indicated, nuclear translocation of the AR was stimulated with 1 nm R1881 (or ethanol only), and compounds were administered at 10 μm concentration. Sheared chromatin-protein complexes were precipitated with the AR-N20 antibody, reverse cross-linked, and analyzed by quantitative PCR. The results are normalized as fold enrichment over precipitation with a rabbit isotype control IgG antibody for each condition tested. Error bars represent the mean ± S.D. between three replicates. Fold enrichment from each tested condition (AR-N20 antibody) was statistically compared against DMSO + 1881 with a two-tailed t test (paired): *, p value < 0.05; **, p value < 0.01. B, purified AR-DBD + hinge domain at the indicated concentration was mixed with ∼2 μm ARE or scrambled 42-bp dsDNA and separated by native-PAGE. C, same as B but with 0.83 μm AR-DBD in the presence of 50 μm compound. D, biotinylated AR-DBD + hinge was loaded onto streptavidin sensors for biolayer interferometry analysis. t = 0 s. DBD was equilibrated with the indicated compound (50 μm); t = 100 s (arrow), dsDNA association was monitored with 3 μm ARE + 50 μm compound; t = 220 s, dissociation in buffer + compound. The y axis represents the nanometer shift in wavelength resulting from ligand (dsDNA) binding/dissociation. E, same as D but with the DBD bearing the Y594D mutation. enz, enzalutamide.
To directly probe DNA interactions, we explored the binding of recombinant AR-DBD with an oligonucleotide containing two hexameric AREs. Purified human AR-DBD and hinge region (residues 558–689, AR-DBD + hinge) were incubated with ARE 42-bp double-stranded DNAs (dsDNA) and analyzed by native-PAGE. The protein was able discriminate between the ARE and a scrambled control (Fig. 6B), but VPC-14228/14449 could not prevent protein-DNA complex formation (Fig. 6C). We speculated that gel shift may be insufficient to detect small but significant changes in DNA binding because the acrylamide matrix might dissociate the hydrophobic compound from the protein surface. To circumvent this limitation, we used a biotinylated AR-DBD linked to streptavidin-coated sensors for use in biolayer interferometry analysis. The biotinylated DBD was exposed to the ARE oligonucleotide in the presence of DMSO or compounds. The observed association kinetics revealed a significantly slower rate of dsDNA binding in the presence of VPC-14228/14449 as compared with enzalutamide and DMSO controls, although dissociation remained relatively unchanged (Fig. 6D). The same experiment performed with the biotinylated AR-DBD bearing the Y594D mutation revealed the inability of VPC-14228/14449 to affect either association or dissociation of dsDNA (Fig. 6E). Thus, our compounds weaken protein-DNA interactions in vitro, and this reduction in binding can be neutralized by mutagenesis of the surface-exposed pocket on the AR-DBD.
VPC-14449 Reduces Tumor Volume and Blocks PSA Production in Mice
The effect of compound VPC-14449 was evaluated in mice by monitoring the growth of androgen-sensitive LNCaP xenografts. Initial experiments demonstrated no systemic toxicity and doses of up to 100 mg/kg administered i.p. twice daily could be tolerated by the mice with no decrease in body weight over a total duration 4 weeks. The in vivo screening for tumor growth inhibition was initially performed using our previously established castration-resistant tumor xenograft model (37–40) in castrated hosts (41, 42). Post-castration, animals were monitored for regrowth of LNCaP tumors and pre-castration levels of serum PSA, at which point the mice were treated with compound VPC-14449 (100 mg/kg) or enzalutamide (10 mg/kg) twice daily. Tumor volume and serum PSA were effectively suppressed by VPC-14449 in this model comparable with enzalutamide treatment (Fig. 7, A and B). The results demonstrate that compound VPC-14449 is as effective as enzalutamide in blocking androgen signaling in vivo. Thus, DBD inhibitor prototypes may yield useful AR targeting drugs that could provide benefit in treating castration-resistant prostate cancer patients.
FIGURE 7.

VPC-14449 reduces tumor volume and abolishes PSA production in LNCaP xenograft model. Castrated mice were dosed twice daily with VPC-14449 (100 mg/kg) or enzalutamide (Enz) (10 mg/kg) for 4 weeks and assessed for LNCaP xenograft tumor volume (A) and serum PSA (B). Data are presented as mean ± S.E., n = 4. p value <0.05 was considered significant (*) compared with vehicle control; p value < 0.001 was considered extremely significant (**) compared with vehicle control.
DISCUSSION
There has been little development of inhibitors that specifically target the NTD or DBD of the AR (2, 43). Sadar and co-workers (44) reported on the high throughput screening-based discovery of EPI-001, a small molecule that can interact with AR-NTD to inhibit transcriptional activity and reduce tumor volume of LNCaP xenografts. However, given that no crystal structure of the AR-NTD has been solved, further development of this drug cannot be aided by rational design. Similarly, reported strategies to inhibit AR-DBD function did not utilize its available crystal structure, and instead it relied upon targeting of androgen-response elements with polyamide mimics that can anneal to specific DNA sequences and block AR recruitment (45). Here, we capitalize on the known structural information on the AR-DBD to target the androgen receptor directly and demonstrate specific inhibition of transcriptional activity despite high sequence conservation with other receptors. Related DBD domains from ER, GR, and PR may have enough structural differences with the AR-DBD such that the surface-exposed pocket is either changed or absent. Remarkably, Gln-592 of the AR is not conserved with any other related nuclear receptor (Fig. 1B). This residue, along with Tyr-594, might contribute to the unique shape and chemistry of the surface-exposed pocket on the DBD, and consequently, when mutated, counteracts drug inhibition.
The properties of a surface-exposed or buried pocket may predictably be altered through mutagenesis. For example, a single amino acid substitution of T877A converts AR antagonists into agonists (46), and this conversion can be rationalized by crystal structures of AR-LBD in complex with drugs (2). Here, substitution of aspartic acid or alanine residues at Tyr-594 and Gln-592 of the AR had a dramatic effect on the ability of two compounds to inhibit AR transcriptional activity, providing compelling evidence for action upon the DBD domain (Fig. 1). Possible interactions between the compounds and other domains of the AR are not excluded, but the fact that the transcriptional activity of splice variants could be affected (Fig. 4) strongly argues for some preference toward the DBD. Notably, VPC-14449 displayed inhibition of Y594D and Q592D mutants at ≥25 μm concentration (Fig. 1, D and E), suggesting that either the mutations shift the binding equilibrium of the compound or indeed that VPC-14449 is able to engage the LBD or NTD domains as secondary targets.
The inhibitory effect of VPC-14228/14449 was weaker against the splice variants when compared with the full-length AR. A maximum of 70–90% inhibition for transiently expressed V7 (Fig. 4A) and 50–70% for endogenously produced V567es (Fig. 4D) could be achieved. These levels of inhibition are similar to that of EPI-001, which could achieve ∼80% inhibition of the AR(1–653) truncation mutant, lacking the LBD, when tested at 25 μm concentration on PC3 cells co-transfected with ARR3tk-luciferase reporter (44).
We demonstrate a dramatic difference between the nuclear localization profile of YFP-AR in the presence of a known anti-androgen (enzalutamide) and VPC-14228/14449 (Fig. 5B). Although unimpeded nuclear localization does not strictly rule out VPC-14228/14449 action upon the LBD, it clearly illustrates a different mechanism than enzalutamide activity, which promoted significant retention of the YFP-AR in the cytosol, likely by displacing R1881 and altering the AR protein structure (18, 47). It also suggests that VPC-14228/14449 action must occur in the nucleus, a requirement to disrupt protein-DNA interactions. The fact that YFP-V7 completely localized to the nucleus in the presence or absence of drugs (Fig. 5C) also agrees with a previous report indicating that enzalutamide could not cause AR-V7 to re-enter the cytosol (18).
The observed nuclear localization dynamics agree with the ability of the compounds to affect AR binding to chromatin or to alter association of dsDNA with the purified AR-DBD (Fig. 6). VPC-14228/14449 may not totally abolish DNA binding, but rather weakens or modulates the binding in such a way as to prevent transcriptional activation by nuclear AR. Clarifying the exact mode of interference of AR binding to androgen-response elements by VPC-14228/14449 will be an important area of investigation.
Finally, we showed that compound VPC-14449 has favorable therapeutic characteristics in vivo (Fig. 7). In addition to having no observable toxic effect on animals, both tumor volume and PSA expression were inhibited to levels comparable with enzalutamide treatment. Thus, targeting DNA binding by the AR can be as effective in vivo as preventing nuclear translocation by enzalutamide (47) or by blocking co-factor recruitment at the AR-NTD by EPI-001 (44). The ability for VPC-14449 to affect tumor xenografts from a variety of other cell lines, both androgen-sensitive and -independent, is currently underway.
Recently, the compound pyrvinium pamoate (VPC-14337) was reported to inhibit the transcriptional activity of full-length/splice variant ARs (29). Modeling of the AR-DBD-DNA interface (Protein Data Bank code 1R4I) was used to rationalize that pyrvinium interacts with the same surface-exposed pocket that is proposed here but in the conserved area of Lys-610 to Pro-613 (see Fig. 1B) possibly explaining the cross-reactivity of this compound with ER and GR (29). Direct evidence for an interaction with this pocket was not given, but replacing the DBD on the full-length AR with the LexA protein prevented drug inhibition, suggesting that pyrvinium binds somewhere on the DBD (29). Here, we revealed the general toxicity of pyrvinium (Fig. 1C) and the inability to inhibit the AR bearing the Q592D and Y594D mutations. Although pyrvinium strongly inhibits the androgen signaling pathway, its cross-reactivity with other nuclear receptors and disruption of Wnt/β-cat signaling by binding at nanomolar concentrations to casein kinase family members (48) makes the suitability for specific AR inhibition unclear, at least until less promiscuous derivatives of the compound are developed.
The identification of specific AR-DBD inhibitors with activity toward AR splice variants has excellent potential for treatment of enzalutamide-resistant or AR-variant driven castration-resistant PCa. This study will facilitate the development of DBD inhibitors with greater potency and stability for testing with in vitro and in vivo models of prostate cancer.
This work was supported, in whole or in part, by National Institutes of Health Grant R01CA174777-01 (to the S. D. laboratory). This work was also supported by Canadian Institutes of Health Research, Genome BC, Prostate Cancer Canada with generous support from Canada Safeway Grant SP2013-02, and American Cancer Society Research Scholar Grant RSG-031-01-TBE (to the S. D. laboratory).
- AR
- androgen receptor
- LBD
- ligand-binding domain
- DBD
- DNA-binding domain
- PCa
- prostate cancer
- NTD
- N-terminal domain
- ARE
- androgen-response element
- PSA
- prostate-specific antigen
- GR
- glucocorticoid receptor
- PR
- progesterone receptor
- ER
- estrogen response
- CSS
- charcoal-stripped serum
- PARP
- poly(ADP-ribose) protein
- hAR
- human AR.
REFERENCES
- 1. Green S. M., Mostaghel E. A., Nelson P. S. (2012) Androgen action and metabolism in prostate cancer. Mol. Cell. Endocrinol. 360, 3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lallous N., Dalal K., Cherkasov A., Rennie P. S. (2013) Targeting alternative sites on the androgen receptor to treat castration-resistant prostate cancer. Int. J. Mol. Sci. 14, 12496–12519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cutress M. L., Whitaker H. C., Mills I. G., Stewart M., Neal D. E. (2008) Structural basis for the nuclear import of the human androgen receptor. J. Cell Sci. 121, 957–968 [DOI] [PubMed] [Google Scholar]
- 4. Lamont K. R., Tindall D. J. (2010) Androgen regulation of gene expression. Adv. Cancer Res. 107, 137–162 [DOI] [PubMed] [Google Scholar]
- 5. Saad F., Miller K. (2014) Treatment options in castration-resistant prostate cancer: current therapies and emerging docetaxel-based regimens. Urol. Oncol. 32, 70–79 [DOI] [PubMed] [Google Scholar]
- 6. Jenster G., van der Korput H. A., van Vroonhoven C., van der Kwast T. H., Trapman J., Brinkmann A. O. (1991) Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol. Endocrinol. 5, 1396–1404 [DOI] [PubMed] [Google Scholar]
- 7. Bohl C. E., Gao W., Miller D. D., Bell C. E., Dalton J. T. (2005) Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 102, 6201–6206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Salvati M. E., Balog A., Wei D. D., Pickering D., Attar R. M., Geng J., Rizzo C. A., Hunt J. T., Gottardis M. M., Weinmann R., Martinez R. (2005) Identification of a novel class of androgen receptor antagonists based on the bicyclic-1H-isoindole-1,3(2H)-dione nucleus. Bioorg. Med. Chem. Lett. 15, 389–393 [DOI] [PubMed] [Google Scholar]
- 9. Duke C. B., Jones A., Bohl C. E., Dalton J. T., Miller D. D. (2011) Unexpected binding orientation of bulky-B-ring anti-androgens and implications for future drug targets. J. Med. Chem. 54, 3973–3976 [DOI] [PubMed] [Google Scholar]
- 10. Shaffer P. L., Jivan A., Dollins D. E., Claessens F., Gewirth D. T. (2004) Structural basis of androgen receptor binding to selective androgen response elements. Proc. Natl. Acad. Sci. U.S.A. 101, 4758–4763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. He B., Gampe R. T., Jr., Kole A. J., Hnat A. T., Stanley T. B., An G., Stewart E. L., Kalman R. I., Minges J. T., Wilson E. M. (2004) Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Mol. Cell 16, 425–438 [DOI] [PubMed] [Google Scholar]
- 12. Estébanez-Perpiñá E., Arnold L. A., Arnold A. A., Nguyen P., Rodrigues E. D., Mar E., Bateman R., Pallai P., Shokat K. M., Baxter J. D., Guy R. K., Webb P., Fletterick R. J. (2007) A surface on the androgen receptor that allosterically regulates coactivator binding. Proc. Natl. Acad. Sci. U.S.A. 104, 16074–16079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lack N. A., Axerio-Cilies P., Tavassoli P., Han F. Q., Chan K. H., Feau C., LeBlanc E., Guns E. T., Guy R. K., Rennie P. S., Cherkasov A. (2011) Targeting the binding function 3 (BF3) site of the human androgen receptor through virtual screening. J. Med. Chem. 54, 8563–8573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dehm S. M., Schmidt L. J., Heemers H. V., Vessella R. L., Tindall D. J. (2008) Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 68, 5469–5477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu R., Dunn T. A., Wei S., Isharwal S., Veltri R. W., Humphreys E., Han M., Partin A. W., Vessella R. L., Isaacs W. B., Bova G. S., Luo J. (2009) Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 69, 16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo Z., Yang X., Sun F., Jiang R., Linn D. E., Chen H., Chen H., Kong X., Melamed J., Tepper C. G., Kung H. J., Brodie A. M., Edwards J., Qiu Y. (2009) A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 69, 2305–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun S., Sprenger C. C., Vessella R. L., Haugk K., Soriano K., Mostaghel E. A., Page S. T., Coleman I. M., Nguyen H. M., Sun H., Nelson P. S., Plymate S. R. (2010) Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Invest. 120, 2715–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Watson P. A., Chen Y. F., Balbas M. D., Wongvipat J., Socci N. D., Viale A., Kim K., Sawyers C. L. (2010) Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. U.S.A. 107, 16759–16765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chan S. C., Li Y., Dehm S. M. (2012) Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J. Biol. Chem. 287, 19736–19749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li Y., Chan S. C., Brand L. J., Hwang T. H., Silverstein K. A., Dehm S. M. (2013) Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 73, 483–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nyquist M. D., Dehm S. M. (2013) Interplay between genomic alterations and androgen receptor signaling during prostate cancer development and progression. Horm. Cancer 4, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nyquist M. D., Li Y., Hwang T. H., Manlove L. S., Vessella R. L., Silverstein K. A., Voytas D. F., Dehm S. M. (2013) TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 110, 17492–17497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Munuganti R. S., Leblanc E., Axerio-Cilies P., Labriere C., Frewin K., Singh K., Hassona M. D., Lack N. A., Li H., Ban F., Tomlinson Guns E., Young R., Rennie P. S., Cherkasov A. (2013) Targeting the binding function 3 (BF3) site of the androgen receptor through virtual screening. 2. Development of 2-((2-phenoxyethyl) thio)-1H-benzimidazole derivatives. J. Med. Chem. 56, 1136–1148 [DOI] [PubMed] [Google Scholar]
- 24. Li H., Ren X., Leblanc E., Frewin K., Rennie P. S., Cherkasov A. (2013) Identification of novel androgen receptor antagonists using structure- and ligand-based methods. J. Chem. Inf. Model. 53, 123–130 [DOI] [PubMed] [Google Scholar]
- 25. Li H., Ban F., Dalal K., Leblanc E., Frewin K., Ma D., Adomat H., Rennie P. S., Cherkasov A. (2014) Discovery of small-molecule inhibitors selectively targeting the DNA-binding domain of the human androgen receptor. J. Med. Chem. 10.1021/jm500802j [DOI] [PubMed] [Google Scholar]
- 26. Miesfeld R., Rusconi S., Godowski P. J., Maler B. A., Okret S., Wikström A. C., Gustafsson J. A., Yamamoto K. R. (1986) Genetic complementation of a glucocorticoid receptor deficiency by expression of cloned receptor cDNA. Cell 46, 389–399 [DOI] [PubMed] [Google Scholar]
- 27. Klock H. E., Koesema E. J., Knuth M. W., Lesley S. A. (2008) Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins 71, 982–994 [DOI] [PubMed] [Google Scholar]
- 28. van Royen M. E., van Cappellen W. A., de Vos C., Houtsmuller A. B., Trapman J. (2012) Stepwise androgen receptor dimerization. J. Cell Sci. 125, 1970–1979 [DOI] [PubMed] [Google Scholar]
- 29. Lim M., Otto-Duessel M., He M., Su L., Nguyen D., Chin E., Alliston T., Jones J. O. (2014) Ligand-independent and tissue-selective androgen receptor inhibition by pyrvinium. ACS Chem. Biol. 9, 692–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Snoek R., Bruchovsky N., Kasper S., Matusik R. J., Gleave M., Sato N., Mawji N. R., Rennie P. S. (1998) Differential transactivation by the androgen receptor in prostate cancer cells. Prostate 36, 256–263 [DOI] [PubMed] [Google Scholar]
- 31. Bredel M., Jacoby E. (2004) Chemogenomics: an emerging strategy for rapid target and drug discovery. Nat. Rev. Genet. 5, 262–275 [DOI] [PubMed] [Google Scholar]
- 32. Nelson P. S., Clegg N., Arnold H., Ferguson C., Bonham M., White J., Hood L., Lin B. (2002) The program of androgen-responsive genes in neoplastic prostate epithelium. Proc. Natl. Acad. Sci. U.S.A. 99, 11890–11895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Magee J. A., Chang L. W., Stormo G. D., Milbrandt J. (2006) Direct, androgen receptor-mediated regulation of the FKBP5 gene via a distal enhancer element. Endocrinology 147, 590–598 [DOI] [PubMed] [Google Scholar]
- 34. Ellinger-Ziegelbauer H., Fostel J. M., Aruga C., Bauer D., Boitier E., Deng S., Dickinson D., Le Fevre A. C., Fornace A. J., Jr., Grenet O., Gu Y., Hoflack J. C., Shiiyama M., Smith R., Snyder R. D., Spire C., Tanaka G., Aubrecht J. (2009) Characterization and interlaboratory comparison of a gene expression signature for differentiating genotoxic mechanisms. Toxicol. Sci. 110, 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ferraldeschi R., Pezaro C., Karavasilis V., de Bono J. (2013) Abiraterone and novel antiandrogens: overcoming castration resistance in prostate cancer. Annu. Rev. Med. 64, 1–13 [DOI] [PubMed] [Google Scholar]
- 36. Rathkopf D., Scher H. I. (2013) Androgen receptor antagonists in castration-resistant prostate cancer. Cancer J. 19, 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miyake H., Nelson C., Rennie P. S., Gleave M. E. (2000) Acquisition of chemoresistant phenotype by overexpression of the antiapoptotic gene testosterone-repressed prostate message-2 in prostate cancer xenograft models. Cancer Res. 60, 2547–2554 [PubMed] [Google Scholar]
- 38. Sato N., Gleave M. E., Bruchovsky N., Rennie P. S., Goldenberg S. L., Lange P. H., Sullivan L. D. (1996) Intermittent androgen suppression delays progression to androgen-independent regulation of prostate-specific antigen gene in the LNCaP prostate tumour model. J. Steroid Biochem. Mol. Biol. 58, 139–146 [DOI] [PubMed] [Google Scholar]
- 39. Cheng H., Snoek R., Ghaidi F., Cox M. E., Rennie P. S. (2006) Short hairpin RNA knockdown of the androgen receptor attenuates ligand-independent activation and delays tumor progression. Cancer Res. 66, 10613–10620 [DOI] [PubMed] [Google Scholar]
- 40. Snoek R., Cheng H., Margiotti K., Wafa L. A., Wong C. A., Wong E. C., Fazli L., Nelson C. C., Gleave M. E., Rennie P. S. (2009) In vivo knockdown of the androgen receptor results in growth inhibition and regression of well established, castration-resistant prostate tumors. Clin. Cancer Res. 15, 39–47 [DOI] [PubMed] [Google Scholar]
- 41. Kuruma H., Matsumoto H., Shiota M., Bishop J., Lamoureux F., Thomas C., Briere D., Los G., Gleave M., Fanjul A., Zoubeidi A. (2013) A novel antiandrogen, compound 30, suppresses castration-resistant and MDV3100-resistant prostate cancer growth in vitro and in vivo. Mol. Cancer Ther. 12, 567–576 [DOI] [PubMed] [Google Scholar]
- 42. Zhang K. X., Moussavi M., Kim C., Chow E., Chen I. S., Fazli L., Jia W., Rennie P. S. (2009) Lentiviruses with trastuzumab bound to their envelopes can target and kill prostate cancer cells. Cancer Gene Ther. 16, 820–831 [DOI] [PubMed] [Google Scholar]
- 43. Caboni L., Lloyd D. G. (2012) Beyond the ligand-binding pocket: targeting alternate sites in nuclear receptors. Med. Res. Rev. 33, 1081–1118 [DOI] [PubMed] [Google Scholar]
- 44. Andersen R. J., Mawji N. R., Wang J., Wang G., Haile S., Myung J. K., Watt K., Tam T., Yang Y. C., Bañuelos C. A., Williams D. E., McEwan I. J., Wang Y., Sadar M. D. (2010) Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 17, 535–546 [DOI] [PubMed] [Google Scholar]
- 45. Nickols N. G., Dervan P. B. (2007) Suppression of androgen receptor-mediated gene expression by a sequence-specific DNA-binding polyamide. Proc. Natl. Acad. Sci. U.S.A. 104, 10418–10423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Taplin M. E., Bubley G. J., Shuster T. D., Frantz M. E., Spooner A. E., Ogata G. K., Keer H. N., Balk S. P. (1995) Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N. Engl. J. Med. 332, 1393–1398 [DOI] [PubMed] [Google Scholar]
- 47. Tran C., Ouk S., Clegg N. J., Chen Y., Watson P. A., Arora V., Wongvipat J., Smith-Jones P. M., Yoo D., Kwon A., Wasielewska T., Welsbie D., Chen C. D., Higano C. S., Beer T. M., Hung D. T., Scher H. I., Jung M. E., Sawyers C. L. (2009) Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 324, 787–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thorne C. A., Hanson A. J., Schneider J., Tahinci E., Orton D., Cselenyi C. S., Jernigan K. K., Meyers K. C., Hang B. I., Waterson A. G., Kim K., Melancon B., Ghidu V. P., Sulikowski G. A., LaFleur B., Salic A., Lee L. A., Miller D. M., 3rd, Lee E. (2010) Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat. Chem. Biol. 6, 829–836 [DOI] [PMC free article] [PubMed] [Google Scholar]