

Background: Angiotensin AT1 receptors use G protein-independent signals to stimulate protein synthesis.
Results: Arrestin-dependent activation of ERK1/2 and Akt signaling regulates mTOR-p70/85S6K and p90RSK, leading to increased protein translation in HEK293 and primary vascular smooth muscle.
Conclusion: Arrestin-dependent ERK1/2 and Akt signaling cooperatively regulate cell growth.
Significance: Arrestin pathway-selective AT1 receptor agonists stimulate Akt-mTOR signaling and protein translation.
Keywords: Akt PKB, Angiotensin II, Arrestin, G Protein-coupled Receptor (GPCR), Mammalian Target of Rapamycin (mTOR), Vascular Smooth Muscle Cells, Biased Agonism
Abstract
Control of protein synthesis is critical to both cell growth and proliferation. The mammalian target of rapamycin (mTOR) integrates upstream growth, proliferation, and survival signals, including those transmitted via ERK1/2 and Akt, to regulate the rate of protein translation. The angiotensin AT1 receptor has been shown to activate both ERK1/2 and Akt in arrestin-based signalsomes. Here, we examine the role of arrestin-dependent regulation of ERK1/2 and Akt in the stimulation of mTOR-dependent protein translation by the AT1 receptor using HEK293 and primary vascular smooth muscle cell models. Nascent protein synthesis stimulated by both the canonical AT1 receptor agonist angiotensin II (AngII), and the arrestin pathway-selective agonist [Sar1-Ile4-Ile8]AngII (SII), is blocked by shRNA silencing of βarrestin1/2 or pharmacological inhibition of Akt, ERK1/2, or mTORC1. In HEK293 cells, SII activates a discrete arrestin-bound pool of Akt and promotes Akt-dependent phosphorylation of mTOR and its downstream effector p70/p85 ribosomal S6 kinase (p70/85S6K). In parallel, SII-activated ERK1/2 helps promote mTOR and p70/85S6K phosphorylation, and is required for phosphorylation of the known ERK1/2 substrate p90 ribosomal S6 kinase (p90RSK). Thus, arrestins coordinate AT1 receptor regulation of ERK1/2 and Akt activity and stimulate protein translation via both Akt-mTOR-p70/85S6K and ERK1/2-p90RSK pathways. These results suggest that in vivo, arrestin pathway-selective AT1 receptor agonists may promote cell growth or hypertrophy through arrestin-mediated mechanisms despite their antagonism of G protein signaling.
Introduction
Protein synthesis is essential for cell growth and is integral to both cellular hypertrophy and proliferation. The rate of protein translation is tightly regulated due to its high energy cost and, in proliferating cells, the necessity of integrating cell growth and cell cycle progression. Two main signal transduction pathways, the Akt-mammalian target of rapamycin (mTOR)2 and extracellular signal-regulated kinase (ERK1/2) mitogen-activated protein kinase (MAPK) cascades, connect insulin, growth factor, and G protein-coupled receptor (GPCR) activation to the modulation of protein synthesis (Fig. 1A). The Ser/Thr protein kinase, mTOR, acts as a master regulator that integrates the availability of metabolic energy and substrate amino acids with growth, proliferation and survival signals transmitted by membrane receptors. mTOR forms two distinct complexes, termed mTOR Complexes 1 and 2 (mTORC1/2), of which mTORC1 controls protein translation (1). Akt activates mTORC1 by at least three distinct mechanisms. First, Akt promotes the phosphorylation, either directly or indirectly, of mTOR Ser-2448. Second, Akt directly phosphorylates and inhibits the tuberous sclerosis complex proteins 1 and 2 (TSC1/TSC2) that restrain the mTORC1 activator, RHEB. Third, Akt phosphorylates and inactivates the kinase, PRAS40, relieving its inhibition of mTORC1. In turn, mTORC1 induces protein synthesis by activating p70S6K, and by inhibiting the eI4E-binding protein, 4E-BP1. The ERK1/2 MAPK cascade, Ras-cRaf1-MEK1/2-ERK1/2, stimulates translation by activating at least two subgroups of the MAPK-activated protein kinase (MAPKAPK) family of proteins: the ribosomal S6 kinases, p90RSK1–4, and the MAPK-interacting kinases, MNK1/2.
FIGURE 1.

General model for the arrestin-dependent regulation of protein synthesis by GPCRs. A, reported roles of the ERK1/2 and Akt pathways in protein translation (1–7). Signal transducer proteins are shown in ovals. Translation factors are shown at bottom of either cascade in boxes. B, cartoon depicting angiotensin AT1A receptor regulation of ERK1/2-p90RSK and Akt-mTORC1-p70S6K signaling via arrestin-based signalsome complexes. It is unknown if arrestins scaffold ERK1/2 and PP2A-Akt on the same arrestin or in parallel complexes. The locus of action of the MEK1/2 inhibitor, PD98059, and the PDK1 inhibitor, OSU03012, are shown.
The Akt and ERK1/2 pathways engage in cross talk at multiple levels. For example, p90RSK can also directly phosphorylate and inhibit TSC2 (2, 3). ERK1/2 can directly phosphorylate and activate p70S6K in vitro (4); however p70S6K may be independent of ERK1/2 in intact cells (5). The two pathways ultimately converge on an array of translation factors that interact with ribosomes to regulate protein translation. All three steps of protein synthesis require a unique set of translation factors: the eukaryotic initiation, termination, and release factors; eIFs, eEFs, and eRFs, respectively. The Akt and ERK pathways ultimately converge on these translation factors; both p70S6K and p90RSK phosphorylate eIF4B at Ser-422 (6) and eEF2 kinase at Ser-366 (7).
GPCRs, like the angiotensin AT1 receptor, have been shown to regulate both ERK1/2 and Akt through non-canonical G protein-independent signaling mechanisms involving arrestins. Arrestin-dependent activation of ERK1/2 is one of the prototypic arrestin-mediated signals (8). Ligand-dependent recruitment of arrestin to the receptor promotes assembly of a stable cRaf1-MEK-ERK1/2 signalsome complex, leading to activation of a discrete cytosolic pool of activated ERK1/2 (8, 9). Signalsome-bound ERK1/2 cannot translocate to the nucleus and is transcriptionally silent (10, 11), but has been reported to promote phosphorylation of cytosolic ERK1/2 substrates, including p90RSK and MNK-1, and to stimulate protein synthesis (12, 13).
Arrestins also scaffold a protein phosphatase 2A (PP2A)/Akt/glycogen synthase kinase 3β (GSK3β) complex, and in the central nervous system, βarrestin2 has been shown to provide tonic restraint of GSK3β signaling in dopaminergic neurons (14–16). Arrestin-dependent activation of Akt in response to GPCR stimulation has also been reported in HEK293 and vascular smooth muscle cells (VSMCs) (17, 18). In HEK293 cells, AT1 receptor-mediated Akt activation results from ligand-induced phosphorylation of the PP2A inhibitor, I2PP2A, which transiently inhibits PP2A bound to the signalsome complex, permitting phosphorylation of Akt on the activating phosphoinositide-dependent protein kinase 1 (PDK1) site, Thr-308, to rise (18).
Given the capacity of arrestins to regulate both the ERK1/2 and Akt pathways via GPCR-arrestin signalsomes, we hypothesized that they may function in the control of protein translation by integrating the activity of the two principal pathways controlling mTORC1 (Fig. 1B). We find that both angiotensin II (AngII) and the arrestin pathway-selective AT1 receptor agonist, [Sar1-Ile4-Ile8]AngII (SII) (7, 19), stimulate mTORC1-dependent protein translation in HEK293 and primary vascular smooth muscle cells. The responses are arrestin-dependent, and require both ERK1/2 and Akt activity. Both Akt and ERK1/2 contribute to activation of the mTORC1-p70S6K axis, while ERK1/2 is the principal activator of the MAPKAPK, p90RSK. These data underscore the pivotal role of arrestins in G protein-independent AT1 receptor signaling and identify novel properties of arrestin pathway-selective agonist ligands.
EXPERIMENTAL PROCEDURES
Materials
PD98059, rapamycin, and LY294002 were from Calbiochem/Millipore (Billerica, MA). OSU03012 was from Selleck Chemicals (Houston, TX). PP242 and angiotensin II were from Sigma-Aldrich. The SII peptide [Sar-Arg-Val-Ile-Ile-His-Pro-Ile] was synthesized by the Cleveland Clinic protein core facility (Cleveland, OH). 10 mm stock SII solutions were prepared in a 10 mm acetic acid, 9 mg/ml NaCl, 0.1% BSA vehicle. The pCR3.1zeo expression plasmid encoding flag epitope-tagged rat βarrestin2 has been previously described (20). Double-stranded siRNAs targeting βarrestin1/2 (18) and control scrambled siRNA were from Qiagen (Germantown, MD). Control and rat βarrestin1/2 shRNA Lentiviral vectors were from Santa Cruz Biotechnology, Inc. (Dallas, TX). Cell culture medium and cell culture additives were from Invitrogen (Grand Island, NY). GeneSilencerTM transfection reagent was from Genelantis (San Diego, CA). FuGENE 6 was from Roche Diagnostics (Indianapolis, IN). Rabbit polyclonal anti-βarrestin1/2 was a gift from Robert J. Lefkowitz (Duke University, Durham, NC). Anti-phospho-ERK1/2 IgG (Thr-202/Tyr-204; 9101), anti-ERK1/2 IgG (4695), anti-phospho-p90RSK1 IgG (Thr-359/Ser-363; 9344), anti-GAPDH IgG, anti-phospho-Akt IgG (Thr-308; 4056), anti-Akt IgG (9272), anti-phospho-p70S6K IgG (Thr-421/Ser-424; 9204), and phospho-mTOR (Ser-2448; 2971) were from Cell Signaling Technology (Beverly, MA). The anti-phospho-p70S6K antibody also detects the homologous phosphorylation events on p85S6K. EZ view red M2 anti-flag affinity resin was from (Sigma-Aldrich). Horseradish peroxidase-conjugated donkey anti-rabbit and anti-mouse IgG were from Jackson Immuno-Research Laboratories, Inc. (West Grove, PA).
Cell Culture and Transfection
HEK293 cells stably expressing the rat AT1A receptor (21) were maintained in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum, and 1% antibiotic/antimycotic solution. Prior to experimentation, cells were serum-deprived overnight in serum-free growth medium supplemented with 0.1% bovine serum albumin and 1% antibiotic/antimycotic solution. Transient transfection of HEK293 cells for flag-βarrestin2 immunoprecipitation was performed in 10 cm dishes (8 million cells/dish) using FuGENE 6 according to the manufacturer's instructions with 10 μg of plasmid DNA per dish and 3 μl of FuGENE 6 per μg of DNA.
Primary rat aortic VSMCs were isolated and maintained as described previously (22). VSMCs were cultured in minimum essential medium supplemented with 10% fetal bovine serum and 1% antibiotic/antimycotic solution. Cells were fed every 2 days and subcultured upon reaching 90% confluence. Prior to each experiment, cells were incubated for 24 to 48 h in serum-free growth medium supplemented with 0.1% bovine serum albumin and 1% antibiotic/antimycotic solution. All experiments employing primary VSMC were performed between passages four and nine.
Silencing βarrestin1/2 Expression by RNA Interference
The HEK293 FRT/TO βarrestin1/2 shRNA cell line carrying tetracycline-inducible shRNA simultaneously targeting the βarrestin1 and 2 isoforms (5′-CGTCCACGTCACCAACAAC-3′) was generated as previously described (20). Transcription of shRNA was induced by 48 h exposure to 1 μm doxycycline and down-regulation of βarrestin1/2 was documented by immunoblotting. Alternatively, transient down-regulation of βarrestin1/2 was performed using 19-nucleotide duplex siRNA with 3′ dTdT overhangs. The sequence 5′-ACCUGCGCCUUCCGCUAUGTT-3′ was used for silencing and the non-silencing RNA duplex 5′-AAUUCUCCGAACGUGUCACGU-3′ was used as the negative control. HEK293 cells at 50% confluence in 10 cm plates were transfected with 6 μg of control or βarrestin1/2-targeted siRNA using 50 μl of GeneSilencerTM according to the manufacturer's instructions. Assays were performed 72 h after gene silencing. Silencing of βarrestin1/2 expression was confirmed by immunoblotting.
For lentiviral down-regulation of βarrestin1/2 expression, VSMCs were grown to 70–80% confluence then treated at a MOI of 50 with a 1:1 mixture of βarrestin1 and βarrestin2 shRNA lentiviral particles or cop GRP control lentiviral particles for 8 h in serum-free medium. Following infection, cells were incubated in complete medium for 24 h then incubated for 48 h in serum-free growth medium containing 0.1% BSA prior to treatment with 50 μm SII for the times indicated. Silencing of βarrestin1/2 expression was confirmed by immunoblotting.
Protein Translation
The rate of net de novo protein synthesis was determined by l-[3H]leucine pulse labeling. Subconfluent cells in 6-cm dishes were given a 2 h pulse of 5 μCi of l-[3H]leucine (Perkin Elmer NEN, Waltham, MA) in the presence or absence of agonist. After labeling, cells were solubilized in 500 μl of lysis buffer (15 mm Na citrate, 150 mm NaCl, 0.25% SDS) with one liquid nitrogen freeze-thaw cycle. Total cell protein extracts were prepared by precipitating with an equal volume of 1 N perchloric acid on ice for 10 min, followed by centrifugation at 13,000 × g for 10 min at 4 °C. Protein precipitates were washed three times with ice-cold 0.5 N perchloric acid and resuspended in 0.3 N NaOH at 37 °C for 1 h, after which 25 μl of protein extract was added to 5 ml of scintillation mixture and 3H dpm incorporated into protein determined by scintillation counting. Nonspecific counts measured in parallel plates that were lysed immediately following exposure to l-[3H]leucine were background subtracted, and specific counts were normalized to total cellular protein determined by bicinchoninic acid protein assay (Pierce/Thermo Fisher Scientific).
Alternatively, protein translation rates were determined using the Click-iT® AHA Alexa Fluor® 488 Protein Synthesis High Content Screening Assay (Molecular Probes/Invitrogen, Grand Island, NY). Cells grown to subconfluence in 96-well plates were stimulated for 2 h as appropriate, and metabolically labeled for the last 30 min of incubation with the azide-containing amino acid, l-azidohomoalanine. Cells were then fixed and permeabilized, and labeled nascent protein was visualized using Click-iT chemistry that covalently attaches the fluorophore Alexa-488 to l-azidohomoalanine according to the manufacturer's protocol. Nuclei were counterstained using Hoechst 33342 dye. Cell monolayers were then imaged using an IN Cell High Content Analyzer (GE Healthcare Life Sciences, Pittsburgh, PA) to measure total levels of fluorescence in each field. Alexa-488 fluorescence was divided by the Hoechst 33342 fluorescence to normalize for cell number between fields.
Protein Immunoprecipitation and Immunoblotting
To assess the activation state of arrestin-bound Akt, HEK293-AT1A cells were transiently transfected with pCR3.1zeo-flag-βarrestin2 and stimulated as described in the figure legends. Monolayers were lysed with 0.4 ml of ice-cold glycerol lysis buffer (50 mm 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; pH 7.5, 0.5% Nonidet P-40, 250 mm NaCl, 2 mm EDTA, and 10% glycerol) containing 10 mm N-ethyl-maleimide, 1 mm Na3VO4, 10 mm NaF, and protease inhibitor mixture. Lysates were clarified by centrifugation and flag-βarrestin2 immunoprecipitated using 30 μl of a 50% slurry of M2 anti-flag affinity resin with 2 h tumbling at 4 °C. The resin was washed twice in lysis buffer, and proteins were eluted in Laemmli sample buffer for resolution by sodium dodecyl sulfate, polyacrylamide gel electrophoresis on 4–20% gradient Tris-glycine gels (Invitrogen, Grand Island, NY). Immunoblots of phospho-ERK1/2, ERK1/2, phospho-Akt, Akt, phospho-p70/p85S6K, phospho-p90RSK, βarrestin1/2, and GAPDH were performed using rabbit polyclonal IgG with HRP-conjugated goat anti-rabbit IgG as secondary antibody. Proteins were visualized using enhanced chemiluminescence (PerkinElmer, Wellesley, MA).
Because of its high molecular weight, samples for mTOR immunoblotting were resolved on 4–12% Bis-Tris Gels (Invitrogen) run using 3-(N-morpholino)-propanesulfonic acid buffer and transferred to Invitrolon PVDF membranes using the wet-tank method. Membranes were blocked overnight in 5% milk in Tris-buffered saline-Tween 20 buffer. Primary antibodies were diluted 1:1000 in Tris-buffered saline-Tween 20 buffer and incubated overnight at 4 °C. Membranes were washed five times and incubated with horseradish peroxidase-conjugated secondary antibody in Tris-buffered saline-Tween 20 buffer for 1 h at room temperature prior to development.
RESULTS
AT1A Receptor-stimulated Protein Translation Requires βarrestin1/2, ERK1/2, and Akt Activity
The native AT1 receptor ligand, AngII, initiates the full spectrum of AT1 signaling, engaging both heterotrimeric G protein- and arrestin-regulated effectors. In contrast, the substituted octapeptide, SII (19), is an arrestin pathway-selective AT1 agonist with greatly diminished efficacy for heterotrimeric G protein activation, but near native ability to recruit arrestins and promote receptor internalization (9, 18, 19, 23). Despite its lack of G protein efficacy, SII has been shown in global phosphoproteomic studies to elicit a robust protein phosphorylation response (18, 24, 25).
To determine the role of arrestins, ERK1/2, and Akt in AT1 receptor-stimulated protein translation, we employed both AngII and SII, since the former reflects physiological AT1 activation, while the latter pharmacologically isolates the G protein-independent aspects of AT1 signaling. As shown in Fig. 2, the two ligands provoked similar increases in protein translation rate in HEK293-AT1A cells measured either by l-[3H]leucine (Fig. 2A) or l-azidohomoalanine (Fig. 2B) pulse labeling. AT1 receptor-mediated stimulation of protein synthesis was blocked by the MEK1/2 inhibitor, PD98059, indicating that ERK1/2 is required for the response. Ligand-activated, but not basal, protein translation was also completely blocked by the PDK1 inhibitor, OSU03012. PDK1, which phosphorylates Akt on residue Thr-308, is the proximal kinase responsible for Akt activation.
FIGURE 2.
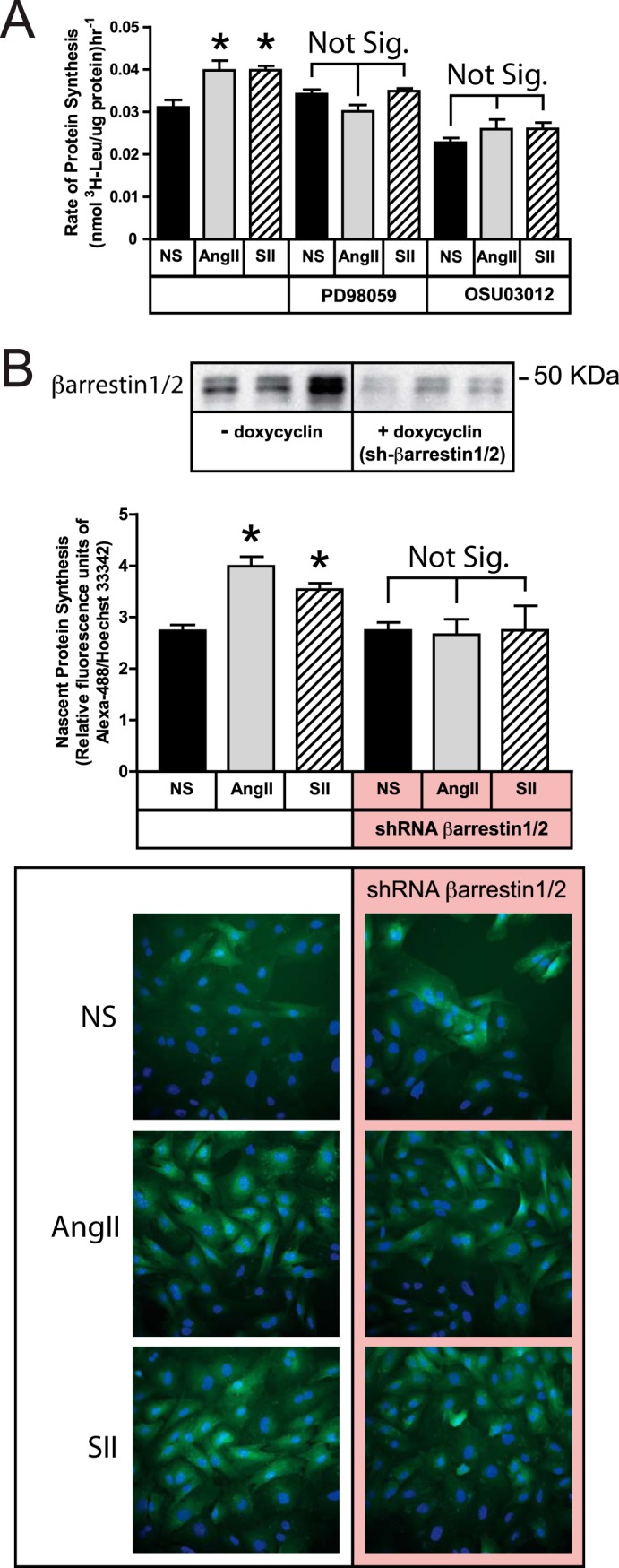
SII stimulation of protein synthesis requires ERK1/2, Akt, and arrestins. A, serum-deprived HEK293-AT1A cells were pretreated with the MEK1/2 inhibitor PD98059 (20 μm; 1 h) or the PDK1 inhibitor, OSU03012 (10 μm; 6 h), prior to pulse labeling with l-[3H]leucine for 2 h in the presence or absence of AngII (100 nm) or SII (50 μm). The bar graph depicts the mean ± S.E. of three biological replicates. B, HEK293 FRT/TO βarrestin1/2 shRNA cells, which carry a tetracycline-inducible shRNA cassette targeting βarrestin1/2, were transiently transfected with plasmid DNA encoding the rat AT1A receptor and cultured with or without doxycycline (1 μm; 48 h) prior to experimentation. After serum-deprivation, cells were stimulated with AngII or SII for 2 h. During the final 30 min of stimulation, cells were metabolically labeled with the azide-containing amino acid l-azidohomoalanine. Labeled nascent protein was visualized and quantified as described in “Experimental Procedures.” The top panel depicts an βarrestin1/2 immunoblot of cells with and without doxycycline induction. The micrographs depict representative fields of non-stimulated (NS), AngII- and SII-treated cells with and without doxycycline exposure following Click-iT® AHA Alexa Fluor® 488 labeling (green). Hoechst 33342-labeled cell nuclei are blue. The bar graph depicts the mean ± S.E. of three biological replicates. In both panels; * greater than nonstimulated (NS); p < 0.05, n = 3.
To test whether the AngII or SII effects involved arrestins, we employed HEK293 FRT/TO βarrestin1/2 shRNA cells that were transiently transfected with the rat AT1A receptor expression plasmid. Forty-eight h exposure of these cells to doxycycline results in 70–90% down-regulation of βarrestin1/2 expression (Fig. 2B). As shown, stimulation of protein translation by both AngII and SII was blocked by βarrestin1/2 silencing, suggesting that the AT1A receptor predominantly uses arrestin-mediated pathways to couple to the translational machinery.
AT1A Receptor Activation of Arrestin-bound Akt Is Independent of ERK Activation
Since the protein translation response to AngII and SII was co-dependent upon ERK1/2 and Akt activity, we next tested whether these two arrestin-regulated kinase cascades interacted proximally. As we previously described (18), basal whole cell levels of Akt phosphorylation in HEK293 cells are constitutively high, necessitating that the phosphorylation state of the arrestin-bound Akt pool be measured in βarrestin2 immunoprecipitates. Fig. 3A shows the effects of PD98059 and OSU03012 on basal Akt phosphorylation in HEK293-AT1A cells. As expected, no significant change in total cellular phospho-Akt was measurable in response to AngII or SII exposure (18). Importantly, while the PDK1 inhibitor, OSU03012, dramatically reduced global Akt phosphorylation, the MEK1/2 inhibitor PD98059 had no effect, demonstrating that it did not nonspecifically affect Akt activity at the concentration employed. To determine the effect of agonist stimulation on the arrestin-bound pool of Akt, HEK293-AT1A cells were transiently transfected with plasmid encoding flag-βarrestin2, and Akt phosphorylation was measured in flag-βarrestin2 immunoprecipitates. As shown in Fig. 3B, both SII and AngII significantly increased the level of βarrestin2-bound phospho-Akt. The response was unaffected by ERK1/2 pathway inhibition, but was completely blocked by OSU03012, suggesting that arrestin-dependent activation of Akt is independent of concomitant ERK1/2 activation.
FIGURE 3.

SII-stimulated Akt activation is independent of ERK1/2 activation. Serum-deprived HEK293-AT1A cells were pretreated with PD98059 (20 μm; 1 h) or OSU03012 (10 μm; 6 h) prior to 5 min stimulation with AngII (100 nm) or SII (50 μm). A, abundance of phospho-Akt T308 in whole cell detergent lysates. Representative phospho-Akt and total Akt immunoblots are shown above a bar graph depicting the mean ± S.E. of three biological replicates. B, abundance of phospho-Akt in flag-βarrestin2 immunoprecipitates of identically treated HEK293-AT1A cells transfected with plasmid DNA encoding flag-βarrestin2. The inset panel depicts a representative immunoblot comparing phospho-Akt in the flag-βarrestin2 immunoprecipitate with whole cell lysate. The presence of endogenous phospho-Akt above a nonspecific immunoglobulin band in the anti-flag immunoprecipitate lane is indicated. Representative phospho-Akt and Flag epitope immunoblots under each condition are shown above a bar graph depicting the mean ± S.E. of three biological replicates. C, abundance of phospho-ERK1/2 in whole cell detergent lysates of identically treated HEK293-AT1A cells. Representative phospho-ERK1/2 and total ERK1/2 immunoblots are shown above a bar graph depicting the mean ± S.E. of three biological replicates. D, abundance of phospho-ERK1/2 in whole cell detergent lysates of HEK293-AT1A cells pretreated with the PI3K inhibitor, LY294002 (10 μm; 1 h). Representative phospho-ERK1/2 and total ERK1/2 immunoblots are shown above a bar graph depicting the mean ± S.E. of three biological replicates. In all panels; * greater than non-stimulated (NS), p < 0.05, n = 3; † less than in the absence of inhibitor, p < 0.05, n = 3.
A degree of co-regulation was apparent in arrestin-dependent activation of ERK1/2. While AngII activates ERK1/2 via both Gq/11-protein kinase C and arrestin signaling, ERK1/2 activation by SII is exclusively arrestin-dependent and results from scaffolding of cRaf1-MEK-ERK1/2 in receptor-bound arrestin signalsomes (8, 9). Thus, the possible involvement of PDK1-Akt in arrestin-dependent ERK1/2 activation could be determined from whole cell lysates of SII-stimulated cells. As shown in Fig. 3C, both AngII and SII increased whole cell ERK1/2 phosphorylation. Whereas SII was at least as effective as AngII at stimulating phosphorylation of the βarrestin2-bound Akt pool, the conventional ligand was more effective at activating ERK1/2 than the arrestin pathway-selective ligand. As expected, PD98059 inhibited ERK1/2 activation by both AngII and SII. SII-induced ERK1/2 phosphorylation was also partially sensitive to OSU03012 (SII + vehicle = 21.9 ± 7.7-fold versus SII + OSU03012 = 8.4 ± 4.0-fold; p = 0.03; n = 3), while the stronger AngII-mediated signal was not significantly affected. Similar results were obtained using the phosphatidylinositol 3-kinase (PI3K) inhibitor, LY294002, as an alternative strategy to block Akt activation (Fig. 3D). Consistent with prior reports (26), this suggests PI3K-PDK1-Akt signaling may facilitate arrestin-dependent ERK1/2 activation.
Arrestin-dependent ERK1/2 and Akt Signaling Converges on p70S6K
Since arrestin-dependent activation of ERK1/2 and Akt appear to be largely independent at the signalsome level, yet both are required to stimulate protein translation, we next determined their contribution to regulation of the p70/85S6K and p90RSK pathways leading to translation initiation (Fig. 1A).
The p70 and p85S6K isoforms are products of the same gene produced from alternative AUG translation start sites. Phosphorylation of p70S6K Thr-421/Ser-424, and the homologous sites on p85S6K, is required for activation, and both Akt and ERK1/2 reportedly regulate p70S6K (27). As shown in Fig. 4A, treatment of HEK293-AT1A cells with either PD98059 or OSU03012 completely blocked SII stimulated p70/85S6K phosphorylation. The SII effect was arrestin-dependent, since siRNA-mediated down-regulation of βarrestin1/2 also prevented the response (Fig. 4B). Since SII-mediated activation of arrestin-bound Akt was not sensitive to PD98059 (Fig. 3B), this suggests signals emanating from arrestin-ERK1/2 and arrestin-Akt signalsome converge downstream at or before the level of p70/85S6K. AngII also induced phosphorylation of p70/p85S6K. As with SII, the AngII response was blocked by PD98059 pretreatment; however it was only partially sensitive to OSU03012 (Fig. 4A) and βarrestin1/2 silencing (Fig. 4B), suggesting that alternative, probably G protein-mediated, pathways also contribute to the conventional agonist response.
FIGURE 4.
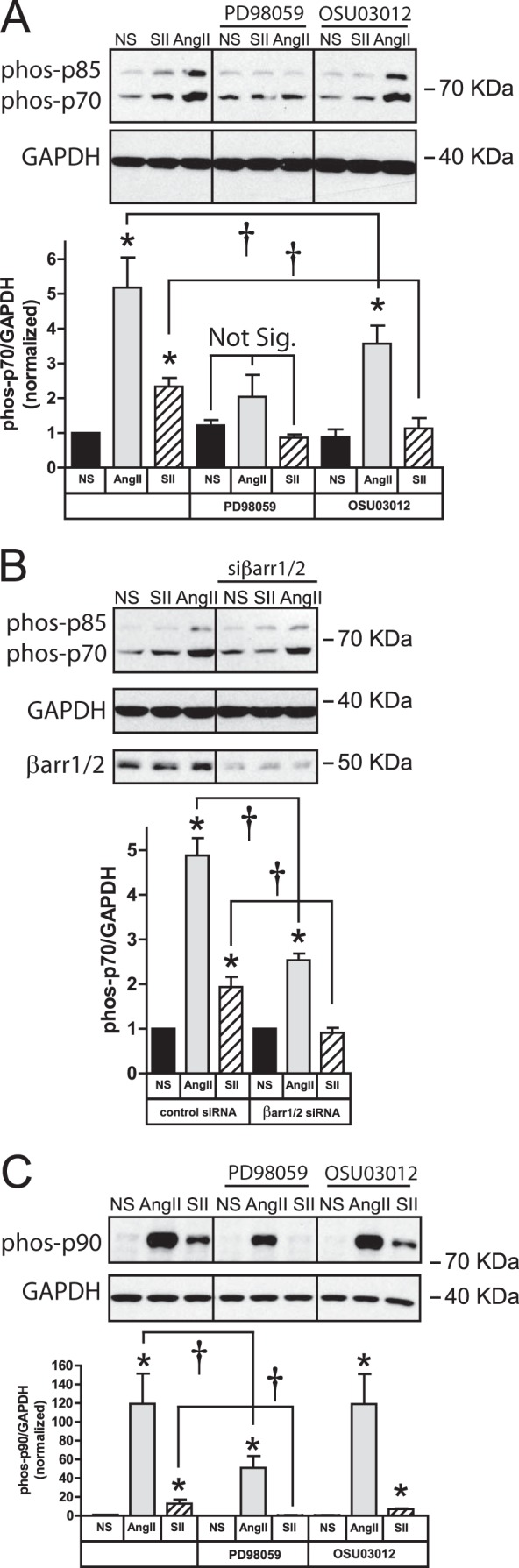
Role of ERK1/2, Akt and arrestins in SII-stimulated p70/85S6K and p90RSK activation. A, effect of ERK1/2 and Akt inhibitors on whole cell p70/85S6K phosphorylation. Serum-deprived HEK293-AT1A cells were pretreated with PD98059 (20 μm; 1 h) or OSU03012 (10 μm; 6 h) prior to 5 min stimulation with AngII (100 nm) or SII (50 μm). Representative phospho-p70/85S6K immunoblots are shown above a bar graph depicting the Mean ± S.E. of three biological replicates. Glyceraldehyde phosphate dehydrogenase (GAPDH) was immunoblotted as a protein loading control. B, effect of βarrestin1/2 down-regulation on whole cell p70/85S6K phosphorylation. HEK293-AT1A cells were transiently transfected with plasmid DNA encoding siRNA targeting βarrestin1/2 as described. Following serum-deprivation, cells were stimulated with AngII or SII for 5 min. Representative phospho-p70/85S6K and βarrestin1/2 immunoblots are shown above a bar graph depicting the mean ± S.E. of three biological replicates. GAPDH was immunoblotted as a protein loading control. C, effect of ERK1/2 and Akt inhibitors on whole cell p90RSK phosphorylation. Serum-deprived HEK293-AT1A cells were pretreated with PD98059 or OSU03012 prior to 5 min stimulation with AngII or SII. Representative phospho-p90RSK immunoblots are shown above a bar graph depicting the mean ± S.E. of three biological replicates. GAPDH was immunoblotted as a protein loading control. In all panels; * greater than non-stimulated (NS), p < 0.05, n = 3; † less than in the absence of inhibitor, p < 0.05, n = 3.
The other arm of translational regulation is the ERK1/2-p90RSK cascade. ERK1/2 is known to activate p90RSK by direct phosphorylation of Thr-359/Ser-363. As shown in Fig. 4C, AngII- and SII-mediated p90RSK phosphorylation was inhibited by PD98059, consistent with previous reports that arrestin-dependent ERK1/2 signaling regulated the MAPKAPKs, p90RSK and MNK-1 (12, 13). Neither AngII- nor SII-dependent p90RSK phosphorylation was significantly affected by OSU03012, although the PDK1 inhibitor appeared to blunt the SII response (SII + vehicle = 12.9 ± 5.3-fold versus SII + OSU03012 = 7.1 ± 0.9), perhaps reflecting the reduced efficiency of SII-stimulated ERK1/2 activation observed in the presence of OSU03012 (Fig. 3C).
Arrestin-dependent ERK1/2 and Akt Signaling Regulates mTORC1 Activation and Function
Although arrestin-dependent ERK1/2 signaling appeared to be sufficient to activate p90RSK, SII-mediated activation of p70/85S6K required both ERK1/2 and Akt. Since mTORC1 is the critical upstream regulator of p70S6K, and is itself regulated by both ERK1/2 and Akt (Fig. 1A), we next tested whether the two arrestin signaling pathways converged at the level of mTORC1.
The mTORC1 complex is regulated directly by Akt phosphorylation of mTOR Ser-2448 (28), and indirectly by Akt and ERK1/2-p90RSK-mediated inhibition of TSC1/2 (1–3). As shown in Fig. 5A, both AngII and SII stimulated mTOR Ser-2448 phosphorylation. Both responses were abolished in the presence of OSU03012, which blocks ligand-dependent Akt activation (Fig. 3B). mTOR Ser-2448 phosphorylation was also significantly reduced, but not blocked, by PD98059, consistent with the known modulatory role of ERK1/2-p90RSK.
FIGURE 5.

SII stimulates mTORC1 phosphorylation and activation via ERK1/2 and Akt. A, effect of ERK1/2 and Akt inhibitors on mTOR S2448 phosphorylation. Serum-deprived HEK293-AT1A cells were pretreated with PD98059 (20 μm; 1 h) or OSU03012 (10 μm; 6 h) prior to 5 min stimulation with AngII (100 nm) or SII (50 μm). Representative phospho-mTOR immunoblots are shown above a bar graph depicting the mean ± S.E. of three biological replicates. GAPDH was immunoblotted as a protein loading control. B, SII-stimulated p70/85S6K phosphorylation is mTORC1 dependent. Serum-deprived HEK293-AT1A cells were pretreated with the mTOR inhibitors, rapamycin (2 nm; 30 min) or PP242 (100 nm; 30 min) prior to 5 min stimulation with AngII or SII. Representative phospho-p70/85S6K immunoblots are shown above a bar graph depicting the Mean ± S.E. of three biological replicates. GAPDH was immunoblotted as a protein loading control. C, SII-stimulated protein translation requires mTORC1. Serum-deprived HEK293-AT1A cells were pretreated rapamycin or PP242, prior to pulse labeling with l-[3H]leucine for 2 h in the presence or absence of AngII or SII. The bar graph depicts the Mean ± S.E. of three biological replicates. D, results of identical experiments to those shown in panel C performed using l-azidohomoalanine pulse labeling. The bar graph depicted the mean ± S.E. of three biological replicates. In all panels; * greater than non-stimulated (NS), p < 0.05, n = 3; † less than in the absence of inhibitor, p < 0.05, n = 3.
Since p70/85S6K is a direct mTOR substrate, we tested whether AT1A receptor-mediated phosphorylation p70/85S6K was mTOR-dependent. As shown in Fig. 5B, SII-induced p70/85S6K phosphorylation was blocked by pretreatment with either rapamycin, an inhibitor of the raptor-containing mTORC1, or with PP242, a dual inhibitor of mTORC1 and the rictor-containing mTORC2, suggesting that the G protein-independent co-regulation of p70/85S6K by ERK1/2 and Akt occurs primarily at the level of mTOR. In contrast, AngII-mediated p70/85S6K phosphorylation was not significantly affected by rapamycin and while significantly reduced, was not abolished, by PP242, suggesting that the AngII response is less dependent on mTORC1 than the more arrestin-dependent SII response. This ability to bypass mTOR may reflect the enhanced efficacy of AngII for ERK1/2 activation (Fig. 3C), since ERK1/2 can reportedly phosphorylate p70S6K directly (27), and AngII-stimulated p70/85S6K phosphorylation is markedly inhibited by PD98059 in HEK293-AT1A cells (Fig. 4A).
We next tested whether AT1A receptor-stimulated protein translation required mTORC1. As shown in Fig. 5C, treatment of HEK293 AT1A cells with either rapamycin or PP242 completely inhibited SII- and AngII-stimulated of l-[3H]leucine incorporation. Identical results were obtained using the l-azidohomoalanine incorporation method (Fig. 5D).
AT1 Receptor-mediated Protein Translation in Primary VSMCs Requires ERK1/2, Akt, mTORC1, and βarrestin1/2
As shown in Fig. 6A, both AngII and SII activate ERK1/2 in primary rat aortic VSMC (17). Unlike HEK293 cells, primary aortic VSMC exhibit low basal levels of Akt phosphorylation, thus permitting detection of both AngII- and SII-induced Akt phosphorylation in whole cell lysates. As in HEK293 cells, SII-stimulated Akt phosphorylation was inhibited by down-regulating βarrestin1/2 expression (Fig. 6B).
FIGURE 6.

SII independently stimulates ERK1/2 and Akt activation in primary rat aortic VSMCs. A, effect of AngII or SII on ERK1/2 and Akt phosphorylation. Serum-deprived VSMCs were stimulated for 5 min with AngII (100 nm) or SII (50 μm) prior to performing phospho-Akt and phospho-ERK1/2 immunoblots on whole cell lysates. In each panel, representative immunoblots are shown above a bar graph depicting the Mean ± S.E. of three biological replicates. B, down-regulating βarrestin1/2 expression attenuates SII-mediated Akt phosphorylation. Rat aortic VSMCs were infected with lentiviral vectors encoding shRNA targeting βarrestin1/2 or control lentivirus. After serum deprivation, cells were stimulated for 5 min with SII prior to performing phospho-Akt immunoblots on whole cell lysates. A representative phospho-Akt mmunoblot is shown above a bar graph depicting the Mean ± S.E. of three biological replicates. GAPDH was immunoblotted as a protein loading control. A representative βarrestin immunoblot of control and βarrestin1/2 shRNA lentivirus treated VSMCs is shown to the right. C, time course of AngII- and SII-stimulated Akt and ERK1/2 phosphorylation. Serum-deprived rat aortic VSMCs were stimulated for the indicated times with AngII (100 nm) or SII (50 μm) prior to performing phospho-Akt and phospho-ERK1/2 immunoblots on whole cell lysates. In each panel, representative immunoblots are shown above a bar graph depicting the mean ± S.E. of three biological replicates. D, dose dependence of AngII- and SII-stimulated Akt and ERK1/2 phosphorylation. Serum-deprived rat aortic VSMCs were stimulated for 5 min with the indicated concentrations of AngII or SII prior to performing phospho-Akt and phospho-ERK1/2 immunoblots on whole cell lysates. In each panel, representative immunoblots are shown above a bar graph depicting the mean ± S.E. of three biological replicates. EC50 values were derived by curve fitting using GraphPad Prism software. E, effect of MEK and PI3K inhibitors on AngII and SII-stimulated Akt and ERK1/2 phosphorylation. Serum-deprived rat aortic VSMCs were pretreated with PD98059 (20 μm; 1 h) or LY294002 (10 μm; 1 h) prior to 5 min stimulation with AngII or SII. Levels of whole cell phospho-Akt and -ERK1/2 were determined by immunoblotting whole cell lysates. Representative immunoblots are shown above a bar graph depicting the mean ± S.E. of three biological replicates. In all panels; * greater than non-stimulated (NS), p < 0.05, n = 3; † less than in the absence of inhibitor, p < 0.05, n = 3.
As shown in Fig. 6C, AngII- and SII-mediated Akt phosphorylation was detectable within 1 min of stimulation, peaked at 5 min, and returned nearly to baseline by 30 min. Ang II-stimulated ERK1/2 phosphorylation followed a similar time course, while SII-stimulated ERK1/2 phosphorylation was slightly delayed in onset, consistent with prior reports (29). AngII stimulated ERK1/2 and Akt phosphorylation with similar potency (EC50 ∼0.4 nm; Fig. 6D). Consistent with its known 200-fold lower affinity for the AT1A receptor (19, 30), SII was much less potent than AngII. In addition, the EC50 of SII for ERK1/2 activation was about 5-fold higher than for Akt activation (ERK1/2 EC50 = 470 nm versus Akt EC50 = 85 nm), suggesting differences in the efficiency of coupling to the two pathways.
We next tested whether the SII-stimulated Akt and ERK1/2 responses were sensitive to PD98059 and LY294002 in VSMCs. As in HEK293-AT1A cells, SII-stimulated Akt phosphorylation was unaffected by the MEK1 inhibitor, but abolished by PI3K inhibition (Fig. 6E). Conversely, SII-stimulated ERK1/2 activation was sensitive to PD98059, but not LY294002. The attenuation of SII-stimulated ERK1/2 activation by LY294002 seen in HEK293-AT1A cells (Fig. 3D) did not occur in VSMC, underscoring that arrestin-dependent regulation of ERK1/2 and Akt signaling are largely independent. AngII-stimulated Akt and ERK1/2 phosphorylation exhibited a similar pattern of inhibitor sensitivity.
As shown in Fig. 7A, 2 h stimulation of VSMCs with SII or AngII produced a significant increase in nascent protein abundance assayed by l-azidohomo-alanine incorporation. The response was blocked by cycloheximide, demonstrating that it was due to increased de novo protein translation. To test whether the response was arrestin-dependent in a native cell background, we employed lentiviral vectors to deliver shRNA targeting βarrestin1/2 (Fig. 7B). Down-regulating βarrestin1/2 expression by 60–70% led to an increase in basal protein translation compared with VSMC treated with control lentivirus. As in HEK293 FRT/TO βarrestin1/2 shRNA cells, neither AngII nor SII stimulated protein translation following βarrestin1/2 down-regulation. In contrast, epidermal growth factor, which acts via a classical receptor tyrosine kinase, provoked similar net increases in protein translation in VSMC treated with either control or βarrestin1/2 shRNA lentivirus. Since βarrestin expression has been shown to tonically inhibit Akt-GSK3β signaling in dopaminergic neurons (14–16), the increase in basal protein translation seen upon βarrestin1/2 down-regulation may reflect de-repression of basal Akt activity. At the same time, loss of βarrestin scaffolds appears to uncouple AT1, but not epidermal growth factor, receptors from the translational machinery.
FIGURE 7.

SII stimulated protein synthesis in primary rat VSMCs requires ERK1/2, Akt, mTORC1, and βarrestin1/2. A, SII stimulates nascent protein synthesis in primary VSMCs. Serum-deprived rat VSMCs were pretreated with the translation inhibitor, cycloheximide (2 μg/ml; 6 h), prior to stimulation with AngII or SII for 2 h, and protein synthesis was measured by l-azidohomoalanine pulse labeling. The bar graph depicts the mean ± S.E. of three biological replicates. The micrographs depict representative fields of non-stimulated (NS), AngII- and SII-treated cells with and without cycloheximide exposure following Click-iT® AHA Alexa Fluor® 488 labeling (green). Hoechst 33342-labeled cell nuclei are blue. In both panels; * greater than nonstimulated (NS), p < 0.05, n = 3. B, nascent protein synthesis in VSMCs after down-regulation of βarrestin1/2 using lentiviral vectors. The top panel depicts a representative βarrestin1/2 immunoblot of VSMCs following treatment with either βarrestin1/2-targeted or control lentivirus. The bar graph depicts the mean ± S.E. of three biological replicates. C, effect of ERK1/2, Akt and mTORC1 inhibitors on SII-stimulated protein synthesis. Serum-deprived rat VSMCs were pretreated with PD98059 (20 μm; 1 h), LY294002 (10 μm; 1 h), rapamycin (2 nm; 30 min), or PP242 (100 nm; 30 min), prior to stimulation 2 h with AngII or SII, and protein synthesis was measured by l-azidohomoalanine pulse labeling. The bar graph depicts the mean ± S.E. of three biological replicates. In all panels; * greater than nonstimulated (NS), p < 0.05, n = 3.
To determine the contribution of G protein-independent signaling to endogenous AT1 receptor-stimulated protein translation, we tested whether the SII response was sensitive to ERK1/2, Akt, and mTORC1 inhibition. Consistent with our findings in HEK293-AT1A cells, exposing rat VSMCs to PD98059, the phosphatidylinositol 3-kinase inhibitor LY294002, rapamycin, or PP242 completely blocked SII-stimulated l-azidohomo-alanine incorporation (Fig. 7C). The native agonist, AngII, also demonstrated co-dependence on ERK1/2, Akt, and mTORC1.
DISCUSSION
Regulation of the protein translational machinery by extracellular signals involves complex interplay between the ERK1/2-p90RSK and Akt-mTORC1-p70S6K pathways that regulate ribosomal eIFs, eEFs, and eRFs (Fig. 1A). Our data demonstrate that arrestin-dependent signals regulating both ERK1/2 and Akt downstream of the angiotensin AT1A receptor are critical mediators of GPCR-regulated protein translation.
The co-regulation of protein translation by arrestin-dependent signaling is best demonstrated using SII, since this arrestin pathway-selective AT1A receptor agonist is devoid of significant G protein efficacy (18, 19, 23). SII elicits a protein translation response that is comparable in magnitude to that produced by AngII, and completely blocked by shRNA-mediated down-regulation of βarrestin1/2. As previously reported (12, 13), activation of p90RSK by SII is ERK1/2 dependent, in that it is abolished by MEK1/2-ERK1/2 inhibition and not significantly affected by PDK1-Akt inhibition. In contrast, both ERK1/2 and Akt signaling are involved in activation of the mTORC1-p70/85S6K pathway. The two signals converge at the level of mTORC1, since inhibiting either is sufficient to impair mTORC1 Ser-2448 phosphorylation, p70/85S6K Thr-421/Ser-424 phosphorylation, and mTORC1-dependent protein translation.
As with SII, stimulation of protein translation by AngII is both arrestin- and mTORC1-dependent. In HEK293-AT1A cells, AngII and SII produce comparable activation of arrestin-bound Akt, while AngII, which can activate both hetero-trimeric G protein and arrestin signaling, produces more robust ERK1/2 activation. While this enhanced ERK1/2 activation may render AngII-stimulated p70/85S6K phosphorylation less sensitive to Akt and mTORC1 inhibition, AngII-induced protein translation in both HEK293-AT1A cells and primary VSMCs requires Akt and mTORC1 activity, indicating that the Akt-mTORC1-p70/85S6K pathway is as important for the native ligand as for SII.
The non-visual arrestins, βarrestin1/2, perform dual roles in modulating GPCR signaling; serving both as the principal mediators of GPCR desensitization and internalization, and as signal transducers that confer additional G protein-independent signaling capability (31). Many arrestin-dependent signals result from scaffolding protein kinase and/or phosphatase cascades within receptor-arrestin signalsomes (32). Of these, scaffolding of cRaf1-MEK-ERK1/2 and PP2A-Akt-GSK3β complexes is among the best characterized. Arrestins recruit a cRaf1-MEK-ERK1/2 complex to agonist-occupied GPCRs, leading to sustained activation of a spatially, temporally and functionally discrete ERK1/2 pool (8–11, 33). Our present data support the hypothesis that one important function of this ERK1/2 pool is to regulate protein translation by activating the MAPKAPKs, p90RSK, and MNK-1, and serving as a co-activator of the mTORC1-p70/85S6K pathway.
A native βarrestin2-PP2A-Akt-GSK3β complex has been isolated from striatum (15), where it is involved in the regulation of dopamine-dependent behavior (14–16). Arrestins have been implicated in Akt regulation by several GPCRs, including the angiotensin AT1A (18), ghrelin (34), and α-thrombin (35) receptors. We have previously described a mechanism for rapid AT1A receptor-mediated activation of arrestin-bound Akt resulting from SII- or AngII-stimulated phosphorylation of I2PP2A, inhibition of PP2A, and relief of tonic PP2A-mediated inhibition of Akt in the signalsome complex (18). Our present data suggest that an important function of this arrestin-bound Akt pool is to activate the mTORC1-p70/85S6K pathway, which in conjunction with ERK1/2, leads to increased mTORC1-dependent protein translation.
Arrestin-dependent signaling has been shown to exert pleiotropic effects in cardiovascular tissues (36). Arrestin pathway-selective AT1 receptor agonists have been reported to improve contractile parameters in isolated murine cardiomyocytes (37), to reduce blood pressure and improve cardiac performance in vivo (38), and to promote cardiomyocyte survival during acute cardiac injury (39). Indeed, an arrestin-biased angiotensin analog, TRV120023 (Sar-Arg-Val-Tyr-Lys-His-Pro-Ala-OH), that possesses higher affinity for the AT1 receptor than SII, has been advanced as a potential therapeutic in cardiogenic shock (38–40). On the other hand, βarrestin2-null mice exhibit reduced neointimal VSMC hyperplasia following endothelial injury, and SII stimulates VSMC proliferation, migration, and anti-apoptotic signaling in vitro (17, 41, 42), suggesting that arrestin-dependent AT1 receptor signaling may promote atherosclerosis. Moreover, in the adrenal, βarrestin1 signaling underlies AngII-induced aldosterone biosynthesis, and inhibition of adrenal βarrestin1 signaling in vivo attenuates post-myocardial infarction heart failure and adverse remodeling by inhibiting aldosterone-dependent salt retention (43, 44). Our present data demonstrating a central role for arrestin-dependent ERK1/2 and Akt signaling in AT1 receptor regulation of the mTOCR1 complex further suggests that arrestin-dependent signaling may promote adverse effects in the cardiovascular system by stimulating cellular growth, hyperplasia, and proliferation.
This work was supported by National Institutes of Health Grants 5K12GM081265 (to R. T. K.) and 5R01DK055524 (to L. M. L.), and the Research Service of the Ralph H. Johnson Veterans Affairs Medical Center.
- mTOR
- mammalian target of rapamycin
- AngII
- angiotensin II
- ERK1/2
- extracellular signal-regulated kinases 1 and 2
- GPCR
- G protein-coupled receptor
- GAPDH
- glyceraldehyde phosphate dehydrogenase
- GSK3β
- glycogen synthase kinase 3β
- MAPK
- mitogen-activated protein kinase
- MAPKAPK
- MAPK-activated protein kinase
- mTORC1/2
- mTOR Complexes 1 and 2
- p70/85S6K
- p70/p85 ribosomal S6 kinase
- p90RSK
- p90 ribosomal S6 kinase
- PDK1
- phosphoinositide-dependent protein kinase 1
- PI3K
- phosphatidylinositol 3-kinase
- PP2A
- protein phosphatase 2A
- SII
- [Sar1-Ile4-Ile8]AngII
- TSC1/TSC2
- tuberous sclerosis complex proteins 1 and 2
- VSMC
- vascular smooth muscle cells.
REFERENCES
- 1. Proud C. G. (2007) Signalling to translation: how signal transduction pathways control the protein synthetic machinery. Biochem. J. 403, 217–234 [DOI] [PubMed] [Google Scholar]
- 2. Roux P. P., Ballif B. A., Anjum R., Gygi S. P., Blenis J. (2004) Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. U.S.A. 101, 13489–13494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rolfe M., McLeod L. E., Pratt P. F., Proud C. G. (2005) Activation of protein synthesis in cardiomyocytes by the hypertrophic agent phenylephrine requires the activation of ERK and involves phosphorylation of tuberous sclerosis complex 2 (TSC2). Biochem. J. 388, 973–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mukhopadhyay N. K., Price D. J., Kyriakis J. M., Pelech S., Sanghera J., Avruch J. (1992) An array of insulin-activated, proline-directed serine/threonine protein kinases phosphorylate the p70 S6 kinase. J. Biol. Chem. 267, 3325–3335 [PubMed] [Google Scholar]
- 5. Lenormand P., McMahon M., Pouysségur J. (1996) Oncogenic Raf-1 activates p70 S6 kinase via a mitogen-activated protein kinase-independent pathway. J. Biol. Chem. 271, 15762–15768 [DOI] [PubMed] [Google Scholar]
- 6. Shahbazian D., Roux P. P., Mieulet V., Cohen M. S., Raught B., Taunton J., Hershey J. W., Blenis J., Pende M., Sonenberg N. (2006) The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 25, 2781–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang X., Li W., Williams M., Terada N., Alessi D. R., Proud C. G. (2001) Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 20, 4370–4379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luttrell L. M., Roudabush F. L., Choy E. W., Miller W. E., Field M. E., Pierce K. L., Lefkowitz R. J. (2001) Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc. Natl. Acad. Sci. U.S.A. 98, 2449–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wei H., Ahn S., Shenoy S. K., Karnik S. S., Hunyady L., Luttrell L. M., Lefkowitz R. J. (2003) Independent β-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc. Natl. Acad. Sci. U.S.A. 100, 10782–10787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tohgo A., Pierce K. L., Choy E. W., Lefkowitz R. J., Luttrell L. M. (2002) β-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J. Biol. Chem. 277, 9429–9436 [DOI] [PubMed] [Google Scholar]
- 11. Lee M. H., El-Shewy H. M., Luttrell D. K., Luttrell L. M. (2008) Role of β-arrestin-mediated desensitization and signaling in the control of angiotensin AT1a receptor-stimulated transcription. J. Biol. Chem. 283, 2088–2097 [DOI] [PubMed] [Google Scholar]
- 12. DeWire S. M., Kim J., Whalen E. J., Ahn S., Chen M., Lefkowitz R. J. (2008) β-arrestin-mediated signaling regulates protein synthesis. J. Biol. Chem. 283, 10611–10620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aplin M., Christensen G. L., Schneider M., Heydorn A., Gammeltoft S., Kjølbye A. L., Sheikh S. P., Hansen J. L. (2007) Differential extracellular signal-regulated kinases 1 and 2 activation by the angiotensin type 1 receptor supports distinct phenotypes of cardiac myocytes. Basic Clin. Pharmacol. Toxicol. 100, 296–301 [DOI] [PubMed] [Google Scholar]
- 14. Beaulieu J. M., Sotnikova T. D., Yao W. D., Kockeritz L., Woodgett J. R., Gainetdinov R. R., Caron M. G. (2004) Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc. Natl. Acad. Sci. U.S.A. 101, 5099–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beaulieu J. M., Sotnikova T. D., Marion S., Lefkowitz R. J., Gainetdinov R. R., Caron M. G. (2005) An Akt/β-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273 [DOI] [PubMed] [Google Scholar]
- 16. Beaulieu J. M., Marion S., Rodriguiz R. M., Medvedev I. O., Sotnikova T. D., Ghisi V., Wetsel W. C., Lefkowitz R. J., Gainetdinov R. R., Caron M. G. (2008) A β-arrestin 2 signaling complex mediates lithium action on behavior. Cell 132, 125–136 [DOI] [PubMed] [Google Scholar]
- 17. Ahn S., Kim J., Hara M. R., Ren X. R., Lefkowitz R. J. (2009) β-Arrestin-2 mediates anti-apoptotic signaling through regulation of BAD phosphorylation. J. Biol. Chem. 284, 8855–8865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kendall R. T., Strungs E. G., Rachidi S. M., Lee M. H., El-Shewy H. M., Luttrell D. K., Janech M. G., Luttrell L. M. (2011) The β-arrestin pathway-selective type 1A angiotensin receptor (AT1A) agonist [Sar1,Ile4,Ile8]angiotensin II regulates a robust G protein-independent signaling network. J. Biol. Chem. 286, 19880–19891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Holloway A. C., Qian H., Pipolo L., Ziogas J., Miura S., Karnik S., Southwell B. R., Lew M. J., Thomas W. G. (2002) Side-chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol. Pharmacol. 61, 768–777 [DOI] [PubMed] [Google Scholar]
- 20. Zimmerman B., Simaan M., Lee M. H., Luttrell L. M., Laporte S. A. (2009) c-Src-mediated phosphorylation of AP-2 reveals a general mechanism for receptors internalizing through the clathrin pathway. Cell Signal. 21, 103–110 [DOI] [PubMed] [Google Scholar]
- 21. Wilson P. C., Lee M. H., Appleton K. M., El-Shewy H. M., Morinelli T. A., Peterson Y. K., Luttrell L. M., Jaffa A. A. (2013) The Arrestin-selective Angiotensin AT1 Receptor Agonist [Sar1,Ile4,Ile8]-AngII Negatively Regulates Bradykinin B2 Receptor Signaling via AT1-B2 Receptor Heterodimers. J. Biol. Chem. 288, 18872–18884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tan Y., Hutchison F. N., Jaffa A. A. (2004) Mechanisms of angiotensin II-induced expression of B2 kinin receptors. Am. J. Physiol. Heart Circ. Physiol. 286, H926–H932 [DOI] [PubMed] [Google Scholar]
- 23. Saulière A., Bellot M., Paris H., Denis C., Finana F., Hansen J. T., Altié M. F., Seguelas M. H., Pathak A., Hansen J. L., Sénard J. M., Galés C. (2012) Deciphering biased-agonism complexity reveals a new active AT(1) receptor entity. Nat. Chem. Biol. 8, 622–630 [DOI] [PubMed] [Google Scholar]
- 24. Christensen G. L., Kelstrup C. D., Lyngsø C., Sarwar U., Bogebo R., Sheikh S. P., Gammeltoft S., Olsen J. V., Hansen J. L. (2010) Quantitative phosphoproteomics dissection of seven-transmembrane receptor signaling using full and biased agonists. Mol. Cell Proteomics. 9, 1540–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xiao K., Sun J., Kim J., Rajagopal S., Zhai B., Villén J., Haas W., Kovacs J. J., Shukla A. K., Hara M. R., Hernandez M., Lachmann A., Zhao S., Lin Y., Cheng Y., Mizuno K., Ma'ayan A., Gygi S. P., Lefkowitz R. J. (2010) Global phosphorylation analysis of β-arrestin-mediated signaling downstream of a seven transmembrane receptor (7TMR). Proc. Natl. Acad. Sci. U.S.A. 107, 15299–15304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hawes B. E., Luttrell L. M., van Biesen T., Lefkowitz R. J. (1996) Phosphatidylinositol 3-kinase is an early intermediate in the G βγ-mediated mitogen-activated protein kinase signaling pathway. J. Biol. Chem. 271, 12133–12136 [DOI] [PubMed] [Google Scholar]
- 27. Pullen N., Thomas G. (1997) The modular phosphorylation and activation of p70s6k. FEBS Lett. 410, 78–82 [DOI] [PubMed] [Google Scholar]
- 28. Navé B. T., Ouwens M., Withers D. J., Alessi D. R., Shepherd P. R. (1999) Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem. J. 344, 427–431 [PMC free article] [PubMed] [Google Scholar]
- 29. Ahn S., Shenoy S. K., Wei H., Lefkowitz R. J. (2004) Differential kinetic and spatial patterns of β-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J. Biol. Chem. 279, 35518–35525 [DOI] [PubMed] [Google Scholar]
- 30. Zimmerman B., Beautrait A., Aguila B., Charles R., Escher E., Claing A., Bouvier M., Laporte S. A. (2012) Differential β-arrestin-dependent conformational signaling and cellular responses revealed by angiotensin analogs. Sci. Signal. 5, ra33. [DOI] [PubMed] [Google Scholar]
- 31. Kendall R. T., Luttrell L. M. (2009) Diversity in arrestin function. Cell Mol. Life Sci. 66, 2953–2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Luttrell L. M., Miller W. E. (2013) Arrestins as regulators of kinases and phosphatases. Prog. Mol. Biol. Transl. Sci. 118, 115–147 [DOI] [PubMed] [Google Scholar]
- 33. Coffa S., Breitman M., Hanson S. M., Callaway K., Kook S., Dalby K. N., Gurevich V. V. (2011) The effect of arrestin conformation on the recruitment of c-Raf1, MEK1, and ERK1/2 activation. PLoS One. 6, e28723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goel R., Phillips-Mason P. J., Raben D. M., Baldassare J. J. (2002) alpha-Thrombin induces rapid and sustained Akt phosphorylation by β-arrestin1-dependent and -independent mechanisms, and only the sustained Akt phosphorylation is essential for G1 phase progression. J. Biol. Chem. 277, 18640–18648 [DOI] [PubMed] [Google Scholar]
- 35. Alvarez C. J., Lodeiro M., Theodoropoulou M., Camiña J. P., Casanueva F. F., Pazos Y. (2009) Obestatin stimulates Akt signalling in gastric cancer cells through β-arrestin-mediated epidermal growth factor receptor transactivation. Endocr. Relat. Cancer 16, 599–611 [DOI] [PubMed] [Google Scholar]
- 36. Lymperopoulos A., Bathgate A. (2013) Arrestins in the cardiovascular system. Prog. Mol. Biol. Transl. Sci. 118, 297–334 [DOI] [PubMed] [Google Scholar]
- 37. Rajagopal K., Whalen E. J., Violin J. D., Stiber J. A., Rosenberg P. B., Premont R. T., Coffman T. M., Rockman H. A., Lefkowitz R. J. (2006) β-Arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc. Natl. Acad. Sci. U.S.A. 103, 16284–16289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Violin J. D., DeWire S. M., Yamashita D., Rominger D. H., Nguyen L., Schiller K., Whalen E. J., Gowen M., Lark M. W. (2010) Selectively engaging β-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J. Pharmacol. Exp. Ther. 335, 572–579 [DOI] [PubMed] [Google Scholar]
- 39. Kim K. S., Abraham D., Williams B., Violin J. D., Mao L., Rockman H. A. (2012) β-Arrestin-biased AT1R stimulation promotes cell survival during acute cardiac injury. Am. J. Physiol. Heart Circ. Physiol. 303, H1001–H1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Monasky M. M., Taglieri D. M., Henze M., Warren C. M., Utter M. S., Soergel D. G., Violin J. D., Solaro R. J. (2013) The β-arrestin-Biased Ligand TRV120023 Inhibits Angiotensin II-Induced Cardiac Hypertrophy While Preserving Enhanced Myofilament Response to Calcium. Am. J. Physiol. Heart Circ Physiol. 10.1152/ajpheart.00327.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim J., Zhang L., Peppel K., Wu J. H., Zidar D. A., Brian L., DeWire S. M., Exum S. T., Lefkowitz R. J., Freedman N. J. (2008) β-Arrestins regulate atherosclerosis and neointimal hyperplasia by controlling smooth muscle cell proliferation and migration. Circ. Res. 103, 70–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim J., Ahn S., Rajagopal K., Lefkowitz R. J. (2009) Independent β-arrestin2 and Gq/protein kinase Cζ pathways for ERK stimulated by angiotensin type 1A receptors in vascular smooth muscle cells converge on transactivation of the epidermal growth factor receptor. J. Biol. Chem. 284, 11953–11962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lymperopoulos A., Rengo G., Zincarelli C., Kim J., Soltys S., Koch W. J. (2009) An adrenal β-arrestin 1-mediated signaling pathway underlies angiotensin II-induced aldosterone production in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 106, 5825–5830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lymperopoulos A., Rengo G., Zincarelli C., Kim J., Koch W. J. (2011) Adrenal β-arrestin 1 inhibition in vivo attenuates post-myocardial infarction progression to heart failure and adverse remodeling via reduction of circulating aldosterone levels. J. Am. Coll. Cardiol. 57, 356–365 [DOI] [PMC free article] [PubMed] [Google Scholar]