

Background: Current understanding of mitochondrial PDH inhibition involves Ser-293 phosphorylation that impedes active site accessibility.
Results: Tyr-301 phosphorylation also inhibits PDHA1, likely by blocking pyruvate binding, which is important for the glycolytic switch and tumor growth.
Conclusion: Tyrosine phosphorylation may function to regulate PDH activity.
Significance: These data provide novel insights into the molecular mechanisms underlying PDC regulation and the Warburg effect.
Keywords: Cell Proliferation, Phosphotyrosine Signaling, Pyruvate Dehydrogenase Complex (PDC), Tumor Metabolism, Warburg Effect
Abstract
The mitochondrial pyruvate dehydrogenase complex (PDC) plays a crucial role in regulation of glucose homoeostasis in mammalian cells. PDC flux depends on catalytic activity of the most important enzyme component pyruvate dehydrogenase (PDH). PDH kinase inactivates PDC by phosphorylating PDH at specific serine residues, including Ser-293, whereas dephosphorylation of PDH by PDH phosphatase restores PDC activity. The current understanding suggests that Ser-293 phosphorylation of PDH impedes active site accessibility to its substrate pyruvate. Here, we report that phosphorylation of a tyrosine residue Tyr-301 also inhibits PDH α 1 (PDHA1) by blocking pyruvate binding through a novel mechanism in addition to Ser-293 phosphorylation. In addition, we found that multiple oncogenic tyrosine kinases directly phosphorylate PDHA1 at Tyr-301, and Tyr-301 phosphorylation of PDHA1 is common in EGF-stimulated cells as well as diverse human cancer cells and primary leukemia cells from human patients. Moreover, expression of a phosphorylation-deficient PDHA1 Y301F mutant in cancer cells resulted in increased oxidative phosphorylation, decreased cell proliferation under hypoxia, and reduced tumor growth in mice. Together, our findings suggest that phosphorylation at distinct serine and tyrosine residues inhibits PDHA1 through distinct mechanisms to impact active site accessibility, which act in concert to regulate PDC activity and promote the Warburg effect.
Introduction
In mammalian cells, mitochondrial PDC6 converts pyruvate to acetyl-CoA (pyruvate decarboxylation). Thus, PDC is at the center of aerobic metabolism of carbohydrate, which regulates the flow of energy in mammalian cells by determining when pyruvate generated in glycolysis should be used for oxidative phosphorylation or, under hypoxic conditions, converted to lactate to sustain glycolysis (1, 2). PDC is a large complex that is organized around a 60-meric dodecahedral core formed by acetyltransferase E2 protein (E2p) and E3-binding protein (3), which binds pyruvate dehydrogenase (PDH, also known as E1p), PDH upstream pyruvate dehydrogenase kinase (PDK) and phosphatase (PDP), and dihydrolipoamide dehydrogenase (E3) (4). PDH is the most important enzyme component of PDC and transforms pyruvate into acetyl-CoA, which, along with the acetyl-CoA from the fatty acid β-oxidation, enters the Krebs cycle to produce ATP and electron donors, including NADH (5).
The activity of PDH is regulated by reversible phosphorylation of three serine residues on the E1α subunit. The phosphorylation of these sites is catalyzed by PDK, which is a Ser/Thr kinase that inactivates PDC by phosphorylating PDH at least at one of three specific serine residues (sites 1, 2, and 3 are Ser-293, Ser-300, and Ser-232, respectively), whereas dephosphorylation of PDH by PDP restores PDC activity (6). Among these three sites, phosphorylation of Ser-293 was suggested to impede active site accessibility of PDH to its substrate pyruvate (7). However, it is not clear whether other types of post-translational modifications such as tyrosine phosphorylation and lysine acetylation are also involved in PDH regulation in mammalian cells.
Accumulating evidence suggests that aerobic glycolysis appears to be a key metabolic factor in human tumors (2) and leukemia (8, 9); however, the detailed mechanisms underlying the glycolytic switch in tumor/leukemia cells remain unclear. It was suggested that the metabolic switch allowing cancer cells to rely more on glycolysis is, in part, due to functional attenuation of mitochondria (10). We recently reported that acetylation at Lys-321 and Lys-202 inhibits PDHA1 and PDP1, respectively, which is common in EGF-stimulated cells and diverse human cancer cells, and is important for tumor growth (11). Moreover, we found that oncogenic tyrosine kinases promote the Warburg effect in cancer cells by attenuating mitochondria function via tyrosine phosphorylation and activation of PDK1 (12) as well as tyrosine phosphorylation and inhibition of PDP1 (11, 13). Here, we report that phosphorylation of PDHA1 at tyrosine residue Tyr-301 is also common in EGF-stimulated cells as well as diverse human cancer cells, which inhibits PDHA1 by blocking pyruvate binding through a novel molecular mechanism in addition to Ser-293 phosphorylation, and promotes the Warburg effect.
EXPERIMENTAL PROCEDURES
Reagents
PDHA1 cDNA image clone (Open Biosystems) was used to engineer several PDHA1 variants with a FLAG epitope tag and were subsequently subcloned into pDEST27 for GST-tagged PDHA1 expression and purification in mammalian cells and pET53 vectors (Invitrogen) for His-tagged PDHA1 expression and purification in bacteria, respectively. Point mutations were introduced using QuikChange-XL site-directed mutagenesis kit (Stratagene). [5-3H]glucose, [1-14C]pyruvate and [2-14C]pyruvate were purchased from PerkinElmer Life Sciences. Stable knockdown of endogenous PDHA1 was achieved using a lentiviral vector harboring an shRNA construct (Open Biosystems; 5′-CGAATGGAGTTGAAAGCAGAT-3′). PDHA1 rescue H1299 cell lines were generated as described previously (11). Briefly, retroviral vector pLHCX (Clontech) containing shRNA-resistant, FLAG-tagged human PDHA1 WT or mutant forms harboring silent mutations in the shRNA-targeted region were transfected into H1299 cells containing shRNA directed against endogenous PDHA1. Antibody against PDHA1 was purchased from Invitrogen. Phospho-PDHA1 (Ser-293) antibody was purchased from Calbiochem. Phosphotyrosine antibody Tyr(P)-99 was purchased from Santa Cruz Biotechnology. Anti-FLAG, β-actin, and GST antibodies were purchased from Sigma. Specific antibody against phospho-PDHA1 (Tyr(P)-301) and acetyl-PDHA1 (K321-Ac) was generated by Cell Signaling Technology specifically for the current project and is not available commercially.
Cell Culture
Human lung cancer FGFR1-expressing H1299 (14) cells and A549 (15) cells and leukemogenic tyrosine kinase expressing leukemia HEL (16), KG-1a (17), MO91 (18), EOL1 (19), Molm14 (20), and K562 (21) cells were cultured in RPMI 1640 medium with 10% bovine serum (FBS). 293T cells, human breast cancer MDA-MB231 (22), and MCF7 (23) cells, as well as normal control human proliferating cells, including human foreskin fibroblasts (24) and immortal human keratinocyte HaCaT cells (25) were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS. TKI258 (generously provided by Novartis Pharma) treatment was performed by incubating cells with 1 μm TKI258 for 4 h. For the cell proliferation assay, 125 nm oligomycin was added in the cell culture medium as described previously (26). Imatinib was purchased from Santa Cruz Biotechnology.
Purification of PDHA1 Proteins
(His)6-tagged PDHA1 protein were purified by sonication of high expressing BL21(DE3)pLysS cells obtained from a 250-ml culture subjected to isopropyl 1-thio-β-d-galactopyranoside induction for 16 h. Cell lysates were resulted by centrifugations and loaded onto a nickel-nitrilotriacetic acid column in 20 mm imidazole. After washing twice, the protein was eluted with 250 mm imidazole. Proteins were desalted on a PD-10 column, and the purification efficiency was examined by Coomassie staining and Western blotting.
PDC Activity Assay
The PDC activity was measured by conversion rate of [1-14C]pyruvate (PerkinElmer Life Sciences) to 14CO2 as described (11, 27). In brief, 14CO2 production through PDC was measured using isolated mitochondria (1 mg) in radioactive mitochondria resuspension buffer (1 ml) containing [1-14C]pyruvate (0.1 μCi/ml), 200 mm sucrose, 10 mm Hepes-HCl (pH 7.4), 1 mm pyruvate, 1 mm malate, 2 mm sodium monophosphate, and 1 mm EGTA. The incubation mixture was placed at the bottom of a vial with a rubber stopper and maintained in agitation. The 14CO2 produced during incubation was trapped by hyamine hydroxide placed in an Eppendorf tube in the vial. The reaction was terminated by adding 0.5 ml of 50% TCA to the reaction after 1 h. Twenty minutes after the TCA injection, all of the samples in hyamine hydroxide were transferred to mini-vials together with 5 ml of scintillation fluid, and radioactivity was assayed on a scintillation counter. The results were normalized based on mitochondrial protein levels assayed by Bradford assay using BSA as a standard.
In Vitro Kinase Assay
For FGFR1 kinase assay, 200 ng of purified recombinant PDHA1 WT, Y301F, or Y289F proteins were incubated with 250 ng of recombinant active FGFR1 in FGFR1 kinase buffer including 10 mm Hepes pH 7.5, 150 mm NaCl, 10 mm MnCl2, 0.01% Triton X-100, 5 mm DTT, 200 μm ATP for 1 h at 30 °C. For ABL kinase assay, purified recombinant PDHA1 proteins were incubated with 175 ng of recombinant active ABL in ABL kinase buffer including 50 mm Tris pH 7.5, 10 mm MgCl2, 0.01% Nonidet P-40, 1 mm DTT, 200 μm ATP for 1 h at 30 °C. For JAK2 kinase assay, purified recombinant PDHA1 proteins were incubated with 200 ng of recombinant active JAK2 in JAK2 kinase buffer including 25 mm Hepes pH 7.5, 10 mm MgCl2, 0.5 mm EGTA, 0.01% Triton X-100, 2.5 mm DTT, 0.5 mm sodium orthovanadate, 5 mm glycerophosphate, 200 μm ATP for 1 h at 30 °C. The samples were electrophoresed on 10% SDS-polyacrylamide gel, transferred on a nitrocellulose membrane, and then detected with the phospho-PDHA1 (Tyr(P)-301) and anti-phosphotyrosine (Tyr(P)-99) antibodies.
Pyruvate Binding Assay
Purified recombinant His-FLAG-PDHA1 proteins, including WT, Y301F, and control Y289F, that were immobilized on anti-FLAG beads were treated with or without rFGFR1 in an in vitro kinase assay as described above. The beads were incubated with 0.1 μm [2-14C]pyruvate for 2 h at room temperature. The beads were then washed twice with TBS to remove the unbound 14C-labeled pyruvate. The PDHA1 proteins were eluted and the retained [2-14C]pyruvate on PDHA1 was measured using a scintillation counter.
PDHA1 Assay
PDHA1 activity was assayed by the formation of NADH after reconstitution of recombinant human protein PDHA1, E2-E3-binding protein and E3 in the ratio 1:3:3. The mixture was incubated in 37 °C for 5 min in PDHA1 buffer containing 50 mm potassium phosphate buffer, pH 7.5, containing 2 mm MgCl2, 2 mm NAD+, 156 mm CoA, 4 mm cysteine, 0.2 mm TPP. The assay was then initiated by the addition of 2 mm pyruvate (Sigma) and the formation of NADH was monitored using a spectrofluorometer (excitation, 340 nm; emission, 460 nm).
Lactate Production, Oxygen Consumption, and Intracellular ATP Assays
Cellular lactate production under normoxia was measured using a fluorescence-based lactate assay kit (MBL). Phenol red-free RPMI medium without FBS was added to a 6-well plate of subconfluent cells and was incubated for 1 h at 37 °C. After incubation, 1 μl of medium from each well was assessed using the lactate assay kit. Cell numbers were determined by cell counting using a microscope (×40). Oxygen consumption rates were measured with a Clark type electrode equipped with 782 oxygen meter (Strathkelvin Instruments). 1 × 107 cells were resuspended in RPMI 1640 medium with 10% FBS and placed into a water-jacked chamber RC300 (Strathkelvin Instruments), and recording was started immediately. Intracellular ATP concentration was measured by an ATP bioluminescent somatic cell assay kit (Sigma). Briefly, 1 × 106 cells were trypsinized and resuspended in ultrapure water. Luminescence was measured with spectrofluorometer (SpectraMax Gemini; Molecular Devices) immediately after the addition of ATP enzyme mix to cell suspension.
Glycolytic Rate Assay
Glycolytic rate was measured by monitoring the conversion of [5-3H]glucose to 3H2O. In brief, 0.5 × 106 cells were washed once in PBS prior to incubation in 1 ml of Krebs buffer without glucose for 30 min at 37 °C. The Krebs buffer was replaced with Krebs buffer containing 10 mm glucose spiked with 10 μCi of 3H-labeled glucose. After incubation for 1 h at 37 °C, triplicate 50-μl aliquots were transferred to uncapped PCR tubes containing 50 μl of 0.2 n HCl, and each tube was transferred into an Eppendorf tube containing 0.5 ml of H2O for diffusion. The tubes were sealed, and diffusion was allowed to occur for a minimum of 24 h at 34 °C. The amounts of diffused 3H2O were determined by scintillation counting.
Cell Proliferation Assays
Cell proliferation assays were performed by seeding 5 × 104 cells in a 6-well plate and culturing the cells at 37 °C in normoxia (5% CO2 and 95% air). Twenty-four hours after seeding, cells that were used for further culture under hypoxia were cultured at 37 °C in a sealed hypoxia chamber filled with 1% O2, 5% CO2, and 94% N2. Cell proliferation was determined by cell numbers recorded by TC10 Automated Cell Counter (Bio-Rad) at indicated days.
Xenograft Studies
Approval of use of mice and designed experiments was given by the Institutional Animal Care and Use Committee of Emory University. Nude mice (nu/nu, female 4–6-week-old, Harlan Laboratories) were subcutaneously injected with 20 × 106 rescue H1299 cells stably expressing hPDHA1 WT and hPDHA1 Y301F with stable knockdown of endogenous hPDHA1 on the left and right flanks, respectively. Tumor growth was recorded by measurement of two perpendicular diameters using the formula 4π/3 × (width/2)2 × (length/2). The tumors were harvested and weighed at the experimental end point, and the tumor masses were compared between tumors (g) derived from rescue cells expressing hPDHA1 WT or hPDHA1 Y301F with stable knockdown of endogenous hPDHA1. Statistical analyses were performed using a two-tailed paired Student's t test.
RESULTS
FGFR1 Inhibits PDHA1 via Phosphorylation at Tyr-301
Our phosphoproteomics studies (11, 12) and multiple proteomics-based studies performed by our collaborators at Cell Signaling Technology revealed that, in addition to its upstream kinase PDK1 (12) and phosphatase PDP1 (11, 13), PDHA1 is phosphorylated at a group of tyrosine residues in human cancer cells (Fig. 1A). Consistently, we found that co-expression of FGFR1 WT but not a kinase-dead form resulted in tyrosine phosphorylation of GST-tagged PDHA1, and such FGFR1-dependent tyrosine phosphorylation was attenuated upon treatment with FGFR1 small molecule inhibitor TKI258 (Fig. 1B). Moreover, treatment with active, recombinant FGFR1 (rFGFR1) resulted in tyrosine phosphorylation of purified His and FLAG-tagged PDHA1 in an in vitro kinase assay, leading to decreased PDHA1 catalytic activity (Fig. 1C). To examine the effect of tyrosine phosphorylation on PDHA1, we performed mutational analysis and generated diverse phosphodeficient Tyr→Phe mutants of PDHA1 to replace each of the tyrosine residues that were identified as phosphorylated. We found that treatment with FGFR1 significantly reduced PDHA1 enzyme activity (Fig. 1, C and D), whereas only substitution of PDHA1 Tyr-301 but not Tyr-242, Tyr-289, Tyr-366, or Tyr-369 abolished FGFR1-dependent inhibition of PDHA1 (Fig. 1D). These results together suggest that FGFR1 inhibits PDHA1 by phosphorylating Tyr-301.
FIGURE 1.

FGFR1 inhibits PDHA1 via phosphorylation at Tyr-301. A, schematic representation of PDHA1. Five identified FGFR1 direct tyrosine phosphorylation sites are shown. B, Western blot (WB) detecting tyrosine phosphorylation of purified GST-PDHA1 from cells co-expressing FGFR1 WT or an FGFR1 kinase-dead form (KD) using a pan phosphotyrosine antibody. Cells treated with FGFR1 inhibitor TKI258 were incubated for 4 h at indicated concentrations. C, active rFGFR1 directly phosphorylates purified His-FLAG-PDHA1 at tyrosine residues in an in vitro kinase assay and attenuates PDHA1 activity. D, purified His-FLAG-PDHA1 variants were incubated with rFGFR1, followed by PDHA1 activity assay. The error bars represent mean values ± S.D. from three replicates of each sample (*, 0.01 < p < 0.05; **, 0.001 < p < 0.01; ns, not significant).
Tyr-301 Phosphorylation of PDHA1 Is Common in EGF-stimulated Human Cancer Cells
We next generated a specific phospho-PDHA1 antibody that recognizes Tyr-301-phosphorylated PDHA1. Using this antibody, we found that EGF treatment resulted in increased phosphorylation levels of PDHA1 at Tyr-301 and Ser-293 as well as previously reported Lys-321 acetylation (11) in 3T3 cells (Fig. 2A). In addition, we found that inhibition of FGFR1 by TKI258, BCR-ABL by imatinib, JAK2 by AG490, and FLT3 by TKI258 resulted in decreased Tyr-301 phosphorylation of PDHA1 in the pertinent human leukemia cell lines (Fig. 2, B–E, left panels, respectively). Consistently, purified rFGFR1, rABL, rJAK2 and rFLT3 (Fig. 2, B–E, right panels, respectively) directly phosphorylated purified rPDHA1 WT and control Y289F mutant at Tyr-301 in vitro, whereas substitution at Tyr-301 of PDHA1 abolished such phosphorylation.
FIGURE 2.

EGF stimulation and diverse oncogenic tyrosine kinases lead to Tyr-301 phosphorylation of PDHA1. A, 3T3 cells treated with EGF for increasing time were examined for phospho-PDHA1 (Tyr(P)-301 (pTyr301)) leve1 using immunoblotting. B–E, left panels: immunoblotting shows phosphorylation levels of PDHA1 Tyr-301 in FGFR1-expressing human lung cancer H1299 cells treated with FGFR1 inhibitor TKI258 (B), BCR-ABL-expressing leukemia K562 cells treated with ABL inhibitor imatinib (C), JAK2 V617F-expressing leukemia HEL cells treated with JAK2 inhibitor AG490 (D), or FLT3-ITD-expressing leukemia Molm 14 cells treated with FLT3 inhibitor TKI258 (E). Right panels: purified His-FLAG-PDHA1 WT, Y301F, or Y289F were incubated with rFGFR1 (B), rABL (C), rJAK2 (D), or rFLT3 (E), followed by immunoblotting to detect Tyr-301 phosphorylation of PDHA1. WB, Western blot.
In addition, we found that Tyr-301 phosphorylation of PDHA1 is common in diverse human tumor cells, including MCF-7 and MDA-MB-231 breast cancer cells, A549 and H1299 lung cancer cells, as well as a group of leukemia cells associated with distinct leukemogenic tyrosine kinases, including EOL1 (HIP1L1-PDGFRA), HEL (JAK2 V617F), K562 (BCR-ABL), KG-1a (FOP2-FGFR1), Molm14 (FLT3-ITD), and Mo91 (TEL-TrkC) cells (Fig. 3A), but not in normal proliferating human foreskin fibroblasts and HaCaT keratinocyte cells (Fig. 3A). Furthermore, we observed that Tyr-301 phosphorylation levels of PDHA1 were increased in primary leukemia cells from three acute myeloid leukemia and three chronic myeloid leukemia patients compared with peripheral blood cells from a representative healthy donor (Fig. 3B).
FIGURE 3.

Tyr-301 phosphorylation of PDHA1 is common in human cancer cells. A and B, immunoblotting to detect phosphorylation levels of PDHA1 Tyr-301 in diverse human tumor and leukemia cells (A) as well as human primary leukemia cells isolated from peripheral blood (PB) or bone marrow (BM) samples from representative acute myeloid leukemia (AML) and chronic myeloid leukemia (CML) patients (B). Normal proliferating human foreskin fibroblasts (HFF), HaCaT keratinocyte cells, and peripheral blood cells from healthy human donors were included as controls.
Expression of PDHA1 Y301F Mutant in H1299 Cells Leads to Decreased Proliferation under Hypoxia and Increased Oxidative Phosphorylation
We next tested whether Tyr-301 phosphorylation-dependent inhibition of PDHA1 is important for glycolysis and cancer cell proliferation. We generated “rescue” H1299 cells with stable knockdown of endogenous human PDHA1 (hPDHA1), followed by rescue expression of shRNA-resistant FLAG-PDHA1 WT and Y301F that harbor silent mutations in the target regions of shRNA (Fig. 4A). As shown in Fig. 4B, left, rescue expression of PDHA1 WT did not affect H1299 cell proliferation under normoxic or hypoxic conditions until day 5. In contrast, rescue expression of phosphodeficient PDHA1 Y301F significantly attenuated cell proliferation under hypoxia but not normoxia, whereas neither normoxia nor hypoxia affected Tyr-301 phosphorylation of PDHA1 in H1299 cells (Fig. 4B, right). Consistent with these findings, PDHA1 Y301F-expressing cells demonstrated increased PDC flux rate (Fig. 4C), along with decreased lactate production and glycolytic rate (Fig. 4D) under normoxia compared with WT cells. In addition, PDHA1 Y301F cells were more sensitive to treatment with ATP synthase inhibitor oligomycin in terms of inhibition of ATP production (Fig. 4E, left) and oxygen consumption rate (Fig. 4F), compared with control PDHA1 WT rescue cells, whereas oligomycin treatment did not affect Tyr-301 phosphorylation of PDHA1 in H1299 cells (Fig. 4E, right). These data together suggest that abolishment of Tyr-301 phosphorylation of PDHA1 leads to a metabolic change to allow cells to rely more on oxidative phosphorylation with decreased glycolysis, providing a metabolic disadvantage to cell proliferation under hypoxia.
FIGURE 4.

Expression of PDHA1 Y301F mutant in H1299 cells leads to decreased proliferation under hypoxia and increased oxidative phosphorylation. A, generation of H1299 cells with stable knockdown of endogenous hPDHA1, followed by stable rescue expression of FLAG-tagged hPDHA1 variants, which harbor silent mutations (SM) that confer PDHA1 shRNA-resistance. B, left: PDHA1 WT and Y301F rescue H1299 cells were tested for cell proliferation rate under normoxia (17% oxygen) or hypoxia (1% oxygen). Cell proliferation was determined based on cell numbers counted daily. Right: phosphorylation level of PDHA1 Tyr-301 was examined in H1299 cells under normoxic or hypoxic conditions using immunoblotting. C and D, PDHA1 WT or Y301F rescue H1299 cells were tested for PDC flux rate (C) as well as lactate production and glycolytic rate (D; left and right, respectively) under normoxia. E and F, PDHA1 rescue cells were tested for intracellular ATP level (E) and oxygen consumption (F) in the presence and absence of ATP synthase inhibitor oligomycin under normoxia. E, right: phosphorylation level of PDHA1 Tyr-301 was examined in H1299 cells in the presence or absence of oligomycin. G, PDHA1 rescue cells were tested for PDC flux rate (left), lactate production (middle), and intracellular ATP level (right) under normoxic or hypoxic conditions for 2 h. The error bars represent mean values ± S.D. from three replicates of each sample (*, 0.01 < p < 0.05; **, 0.001 < p < 0.01; ns, not significant). WB, Western blot.
It has been reported that long term (>72 h) culture under hypoxia results in increased PDK1 (28) and LDHA1 (29) expression, leading to decreased PDC activity and increased lactate production, respectively. We found that, even although short term (2 h) hypoxic condition were insufficient to affect the increased PDC activity (Fig. 4G, left) or the decreased lactate production (Fig. 4G, middle) in PDHA1 Y301F-expressing cells, such a low oxygen condition resulted in decreased ATP levels in these cells (Fig. 4G, right), compared with control PDHA1 WT rescue cells where short term hypoxic exposure had not affected PDC activity and lactate production yet. This is consistent with our hypothesis that cells expressing PDHA1 Y301F mutant rely more on oxidative phosphorylation for ATP production compared with control WT cells, so under hypoxic conditions where oxygen is insufficient to sustain oxidative phosphorylation level, PDHA1 Y301F cells showed decreased ATP levels and subsequently reduced cell proliferation.
Tyr-301 Phosphorylation of PDHA1 Is Important for Tumor Growth
We next performed xenograft experiments and found that the growth rate (Fig. 5A) and masses of tumors (Fig. 5B) derived from PDHA1 Y301F rescue H1299 cells were significantly reduced with decreased Tyr-301 phosphorylation of PDHA1 (Fig. 5C) in tumor cells, compared with those of tumors formed by the control PDHA1 WT rescue cells. Together, these data demonstrate an important role for Tyr-301 phosphorylation of PDHA1 in tumor growth.
FIGURE 5.

Tyr-301 phosphorylation of PDHA1 is important for tumor growth. A and B, tumor growth (A) and (B) masses in xenograft nude mice injected with PDHA1 Y301F rescue cells compared with mice injected with control PDHA1 WT rescue cells are shown. p values were determined by a two-tailed paired Student's t test. C, top: dissected tumors in a representative nude mouse injected with PDHA1 WT and Y301F rescue cells on the left and right flanks, respectively, are shown. Bottom panel shows detection of Tyr-301 phosphorylation levels of PDHA1 in tumor lysates using specific phospho-PDHA1 (Tyr-301) antibody.
Tyr-301 Phosphorylation Inhibits PDHA1 by Blocking Substrate Binding through a Novel Mechanism in Addition to Ser-293 Phosphorylation
To determine whether Tyr-301 phosphorylation functions independently of Ser-293 phosphorylation, we examined whether alteration of phosphorylation level of Tyr-301 in PDHA1 would affect the level of Ser-293 phosphorylation or vice versa. We found that abolishment of Tyr-301 phosphorylation did not alter Ser-293 phosphorylation or Lys-321 acetylation levels of PDHA1 Y301F mutant, whereas altered Lys-321 acetylation attenuated phosphorylation levels of Ser-293 as reported previously (11) but not Tyr-301 in PDHA1 K321R mutant, compared with PDHA1 WT (Fig. 6A). Moreover, treatment with PDK1 inhibitor dichloroacetate resulted in decreased Ser-293 but not Tyr-301 phosphorylation levels of PDHA1 in H1299 cells (Fig. 6B).
FIGURE 6.
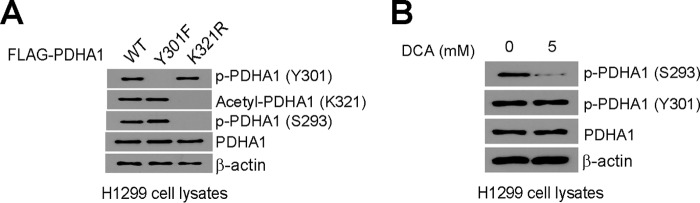
Tyr-301 phosphorylation provides a novel and distinct mechanism to inhibit PDHA1. A, distinct PDHA1 rescue cells were tested in immunoblotting to detect Tyr-301 phosphorylation, Lys-321 acetylation and Ser-293 phosphorylation levels. B, immunoblotting results detecting phosphorylation levels of Ser-293 and Tyr-301 in H1299 cells treated with PDK inhibitor dichloroacetate (DCA).
Tyr-301 Phosphorylation Inhibits PDHA1 by Blocking Substrate Pyruvate Binding
Tyr-301 of PDHA1 is evolutionarily conserved (Table 1). Structural analysis revealed that Tyr-301 is close to the active site (<10Å away), a similar proximity to the active site as Ser-293, whereas, for example, the nonfunctional phosphorylation site Tyr-289 is more distal (Fig. 7A). Because phosphorylation of Ser-293 impedes active site accessibility (7), we hypothesized that Tyr-301 phosphorylation could also impact active site accessibility. We tested this hypothesis by incubating rPDHA1 with 14C-labeled pyruvate (substrate) in the presence and absence of active, rFGFR1. Phosphorylation of rPDHA1 by rFGFR1 resulted in decreased 14C-labeled pyruvate binding to PDHA1 WT and control Y289F mutant proteins, whereas substitution of Tyr-301 abolishes FGFR1-dependent inhibition of substrate binding (Fig. 7B).
TABLE 1.
Phylogenetic analysis of PDHA1 in different species
| Species | PDHA1 sequence (Tyr-301) |
|---|---|
| Homo sapiens (human) | MSDPGVSYRTREEIQa |
| Pan troglodytes (chimpanzee) | MSDPGVSYRTREEIQ |
| Macaca mulatta (Rhesus monkey) | MSDPGVSYRTREEIQ |
| Canis lupus familiaris (dog) | MSDPGVSYRTREEIQ |
| Bos taurus (cattle) | MSDPGVSYRTREEIQ |
| Mus musculus (house mouse) | MSDPGVSYRTREEIQ |
| Rattus norvegicus (Norway rat) | MSDPGVSYRTREEIQ |
| Gallus gallus (chicken) | MSDPGISYRTREEIQ |
| Danio rerio (zebrafish) | MSDPGVSYRTREEIQ |
| Caenorhabditis elegans (C. elegans) | MSDPGTSYRTREEIQ |
| Saccharomyces cerevisiae | MSDPGTTYRTRDEIQ |
a Underlined residue indicates Tyr-301.
FIGURE 7.

Tyr-301 phosphorylation inhibits PDHA1 by blocking substrate pyruvate binding. A, schematic representation of PDHA1 structure (Protein Data Bank code 1NI4 (32)). Tyr-301 is proximal to the PDHA1 catalytic site where substrate (pyruvate) binds (<10 Å), a distance similar to Ser-293 pyruvate. B, purified His-FLAG-PDHA1 variants were incubated with rFGFR1, followed by incubation with 2-14C-labeled pyruvate. PDHA1-bound [2-14C]pyruvate was assessed by scintillation counting. The error bars represent mean values ± S.D. from three replicates of each sample (*, 0.01 < p < 0.05; **, 0.001 < p < 0.01; ns, not significant). C, proposed model shows that phosphorylation at Tyr-301 inhibits PDHA1 and blocks substrate pyruvate binding through a novel and distinct mechanism in addition to Ser-293 phosphorylation in EGF-stimulated cells and cancer cells where tyrosine kinase signaling is commonly up-regulated.
DISCUSSION
Our findings shed new insight into the molecular mechanisms by which growth factors and oncogenic tyrosine kinase signaling attenuate mitochondrial function by inhibiting PDHA1 through direct phosphorylation at Tyr-301. The consequently decreased PDC activity in part contributes to the metabolic switch to allow proliferating and cancer cells to rely more on glycolysis instead of mitochondrial oxidative phosphorylation, providing a metabolic advantage to proliferating cells and cancer cell proliferation/tumor growth, respectively. Interestingly, Tyr-301 phosphorylation also inhibits PDHA1 by impacting substrate (pyruvate) binding, similar as the well known inhibitory Ser-293 phosphorylation of PDHA1 regulated by its upstream kinase PDK1 and phosphatase PDP1. However, we demonstrated that Tyr-301 phosphorylation represents a novel and distinct molecular mechanism underlying regulation of PDHA1 involving active site accessibility. These findings, in addition to our previous reports (11–13), together showcase the beauty of precise and organized signal transduction-based regulation of cellular processes, in which growth factors as well as oncogenic tyrosine kinases regulate PDHA1 activity directly through Tyr-301 phosphorylation and indirectly through tyrosine phosphorylation of PDK1 (12) and PDP1 (11, 13) to control Ser-293 phosphorylation (Fig. 7C). Thus, these distinct mechanisms act in concert to provide parallel regulation of PDHA1 that ensures appropriate control of PDC in normal proliferating cells and cancer cells.
Our findings also suggest that tyrosine phosphorylation of PDHA1 is common in diverse human tumor and leukemia cells, as well as primary leukemia cells from human patients, which represents an acute mechanism to mediate upstream oncogenic tyrosine kinase signaling-dependent regulation to mitochondrial PDC. These findings add another example to the emerging concept that supports the importance of tyrosine phosphorylation of metabolic enzymes, including PGAM1 (30), PKM2 (31), LDH-A (26), PDP1 (11, 13), and PDK1 (12), in cancer cell metabolism and proliferation as well as tumor growth. Future studies are warranted to explore how tyrosine kinase signaling coordinates phosphorylation and regulation of these enzymes to provide an ultimately optimized metabolic advantage to normal cell proliferation, which is, unfortunately, “hijacked” by the Warburg effect in cancer cells and tumor growth.
Lastly, our findings suggest that functional activation of PDHA1 may be explored as a therapeutic strategy in treatment of human cancers that heavily depend on glycolysis. Future studies are warranted to develop PDHA1 activators as novel anti-cancer agents.
This work was supported in part by National Institutes of Health Grants CA140515, CA183594, CA174786 (to J. C.), CA175316 (to S. K.), the Career Development Fellow Award (to J. F.) from National Institutes of Health SPORE in Head and Neck Cancer P50CA128613 (to D. M. S.) and the Pharmacological Sciences Training Grant T32 GM008602 (to S. E.), Department of Defense Grant W81XWH-12-1-0217 (to J. C.), Charles Harris Run For Leukemia, Inc. (to H. J. K.) and the Hematology Tissue Bank of the Emory University School of Medicine (to H. J. K.). J. X., T.-L. G., M. A., and S. L. are employees of Cell Signaling Technology, Inc.
- PDC
- pyruvate dehydrogenase complex
- PDH
- pyruvate dehydrogenase
- PDK
- PDH kinase
- PDP
- PDH phosphatase
- PDHA1
- PDH α 1
- FGFR1
- fibroblast growth factor receptor 1
- rFGFR1
- recombinant FGFR1
- hPDHA1
- human PDHA1.
REFERENCES
- 1. Cairns R. A., Harris I. S., Mak T. W. (2011) Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95 [DOI] [PubMed] [Google Scholar]
- 2. Kroemer G., Pouyssegur J. (2008) Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell 13, 472–482 [DOI] [PubMed] [Google Scholar]
- 3. Hiromasa Y., Fujisawa T., Aso Y., Roche T. E. (2004) Organization of the cores of the mammalian pyruvate dehydrogenase complex formed by E2 and E2 plus the E3-binding protein and their capacities to bind the E1 and E3 components. J. Biol. Chem. 279, 6921–6933 [DOI] [PubMed] [Google Scholar]
- 4. Read R. J. (2001) Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr. D 57, 1373–1382 [DOI] [PubMed] [Google Scholar]
- 5. Harris R. A., Bowker-Kinley M. M., Huang B., Wu P. (2002) Regulation of the activity of the pyruvate dehydrogenase complex. Adv. Enzyme Regul. 42, 249–259 [DOI] [PubMed] [Google Scholar]
- 6. Roche T. E., Baker J. C., Yan X., Hiromasa Y., Gong X., Peng T., Dong J., Turkan A., Kasten S. A. (2001) Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog. Nucleic Acid Res. Mol. Biol. 70, 33–75 [DOI] [PubMed] [Google Scholar]
- 7. Seifert F., Ciszak E., Korotchkina L., Golbik R., Spinka M., Dominiak P., Sidhu S., Brauer J., Patel M. S., Tittmann K. (2007) Phosphorylation of serine 264 impedes active site accessibility in the E1 component of the human pyruvate dehydrogenase multienzyme complex. Biochemistry 46, 6277–6287 [DOI] [PubMed] [Google Scholar]
- 8. Elstrom R. L., Bauer D. E., Buzzai M., Karnauskas R., Harris M. H., Plas D. R., Zhuang H., Cinalli R. M., Alavi A., Rudin C. M., Thompson C. B. (2004) Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 64, 3892–3899 [DOI] [PubMed] [Google Scholar]
- 9. Gottschalk S., Anderson N., Hainz C., Eckhardt S. G., Serkova N. J. (2004) Imatinib (STI571)-mediated changes in glucose metabolism in human leukemia BCR-ABL-positive cells. Clin. Cancer Res. 10, 6661–6668 [DOI] [PubMed] [Google Scholar]
- 10. Kim J. W., Dang C. V. (2006) Cancer's molecular sweet tooth and the Warburg effect. Cancer Res. 66, 8927–8930 [DOI] [PubMed] [Google Scholar]
- 11. Fan J., Shan C., Kang H. B., Elf S., Xie J., Tucker M., Gu T. L., Aguiar M., Lonning S., Chen H., Mohammadi M., Britton L. M., Garcia B. A., Alečković M., Kang Y., Kaluz S., Devi N., Van Meir E. G., Hitosugi T., Seo J. H., Lonial S., Gaddh M., Arellano M., Khoury H. J., Khuri F. R., Boggon T. J., Kang S., Chen J. (2014) Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol. Cell 53, 534–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hitosugi T., Fan J., Chung T. W., Lythgoe K., Wang X., Xie J., Ge Q., Gu T. L., Polakiewicz R. D., Roesel J. L., Chen G. Z., Boggon T. J., Lonial S., Fu H., Khuri F. R., Kang S., Chen J. (2011) Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol. Cell 44, 864–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shan C., Kang H. B., Elf S., Xie J., Gu T. L., Aguiar M., Lonning S., Hitosugi T., Chung T. W., Arellano M., Khoury H. J., Shin D. M., Khuri F. R., Boggon T. J., Fan J. (2014) Tyr-94 phosphorylation inhibits pyruvate dehydrogenase phosphatase 1 and promotes tumor growth. J. Biol. Chem. 289, 21413–21422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. (1996) NCI-Navy Medical Oncology Branch cell line supplement. J. Cell Biochem. Suppl. 24, 1–291 [PubMed] [Google Scholar]
- 15. Giard D. J., Aaronson S. A., Todaro G. J., Arnstein P., Kersey J. H., Dosik H., Parks W. P. (1973) In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J. Natl. Cancer Inst. 51, 1417–1423 [DOI] [PubMed] [Google Scholar]
- 16. Papayannopoulou T., Nakamoto B., Yokochi T., Chait A., Kannagi R. (1983) Human erythroleukemia cell line (HEL) undergoes a drastic macrophage-like shift with TPA. Blood 62, 832–845 [PubMed] [Google Scholar]
- 17. Gu T. L., Goss V. L., Reeves C., Popova L., Nardone J., Macneill J., Walters D. K., Wang Y., Rush J., Comb M. J., Druker B. J., Polakiewicz R. D. (2006) Phosphotyrosine profiling identifies the KG-1 cell line as a model for the study of FGFR1 fusions in acute myeloid leukemia. Blood 108, 4202–4204 [DOI] [PubMed] [Google Scholar]
- 18. Chen C. R., Kang Y., Siegel P. M., Massagué J. (2002) E2F4/5 and p107 as Smad cofactors linking the TGFβ receptor to c-myc repression. Cell 110, 19–32 [DOI] [PubMed] [Google Scholar]
- 19. Cools J., Quentmeier H., Huntly B. J., Marynen P., Griffin J. D., Drexler H. G., Gilliland D. G. (2004) The EOL-1 cell line as an in vitro model for the study of FIP1L1-PDGFRA-positive chronic eosinophilic leukemia. Blood 103, 2802–2805 [DOI] [PubMed] [Google Scholar]
- 20. Matsuo Y., MacLeod R. A., Uphoff C. C., Drexler H. G., Nishizaki C., Katayama Y., Kimura G., Fujii N., Omoto E., Harada M., Orita K. (1997) Two acute monocytic leukemia (AML-M5a) cell lines (MOLM-13 and MOLM-14) with interclonal phenotypic heterogeneity showing MLL-AF9 fusion resulting from an occult chromosome insertion, ins(11;9)(q23;p22p23). Leukemia 11, 1469–1477 [DOI] [PubMed] [Google Scholar]
- 21. Lozzio C. B., Lozzio B. B. (1975) Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood 45, 321–334 [PubMed] [Google Scholar]
- 22. Cailleau R., Young R., Olivé M., Reeves W. J., Jr. (1974) Breast tumor cell lines from pleural effusions. J. Natl. Cancer Inst. 53, 661–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Soule H. D., Vazguez J., Long A., Albert S., Brennan M. (1973) A human cell line from a pleural effusion derived from a breast carcinoma. J. Natl. Cancer Inst. 51, 1409–1416 [DOI] [PubMed] [Google Scholar]
- 24. Takahashi K., Yamanaka S. (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 [DOI] [PubMed] [Google Scholar]
- 25. Schoop V. M., Mirancea N., Fusenig N. E. (1999) Epidermal organization and differentiation of HaCaT keratinocytes in organotypic coculture with human dermal fibroblasts. J. Invest. Dermatol. 112, 343–353 [DOI] [PubMed] [Google Scholar]
- 26. Fan J., Hitosugi T., Chung T. W., Xie J., Ge Q., Gu T. L., Polakiewicz R. D., Chen G. Z., Boggon T. J., Lonial S., Khuri F. R., Kang S., Chen J. (2011) Tyrosine phosphorylation of lactate dehydrogenase A is important for NADH/NAD(+) redox homeostasis in cancer cells. Mol. Cell. Biol. 31, 4938–4950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pezzato E., Battaglia V., Brunati A. M., Agostinelli E., Toninello A. (2009) Ca2+-independent effects of spermine on pyruvate dehydrogenase complex activity in energized rat liver mitochondria incubated in the absence of exogenous Ca2+ and Mg2+. Amino Acids 36, 449–456 [DOI] [PubMed] [Google Scholar]
- 28. Kim J. W., Tchernyshyov I., Semenza G. L., Dang C. V. (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185 [DOI] [PubMed] [Google Scholar]
- 29. Semenza G. L., Roth P. H., Fang H. M., Wang G. L. (1994) Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 269, 23757–23763 [PubMed] [Google Scholar]
- 30. Hitosugi T., Zhou L., Fan J., Elf S., Zhang L., Xie J., Wang Y., Gu T. L., Alečković M., LeRoy G., Kang Y., Kang H. B., Seo J. H., Shan C., Jin P., Gong W., Lonial S., Arellano M. L., Khoury H. J., Chen G. Z., Shin D. M., Khuri F. R., Boggon T. J., Kang S., He C., Chen J. (2013) Tyr26 phosphorylation of PGAM1 provides a metabolic advantage to tumours by stabilizing the active conformation. Nat. Commun. 4, 1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hitosugi T., Kang S., Vander Heiden M. G., Chung T. W., Elf S., Lythgoe K., Dong S., Lonial S., Wang X., Chen G. Z., Xie J., Gu T. L., Polakiewicz R. D., Roesel J. L., Boggon T. J., Khuri F. R., Gilliland D. G., Cantley L. C., Kaufman J., Chen J. (2009) Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal. 2, ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ciszak E. M., Korotchkina L. G., Dominiak P. M., Sidhu S., Patel M. S. (2003) Structural basis for flip-flop action of thiamin pyrophosphate-dependent enzymes revealed by human pyruvate dehydrogenase. J. Biol. Chem. 278, 21240–21246 [DOI] [PubMed] [Google Scholar]