

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive and ultimately fatal neurodegenerative disease. Pyrazolone containing small molecules have shown significant disease attenuating efficacy in cellular and murine models of ALS. Pyrazolone based affinity probes were synthesized to identify high affinity binding partners and ascertain a potential biological mode of action. Probes were confirmed to be neuroprotective in PC12-SOD1G93A cells. PC12-SOD1G93A cell lysates were used for protein pull-down, affinity purification, and subsequent proteomic analysis using LC-MS/MS. Proteomics identified the 26S proteasome regulatory subunit 4 (PSMC1), 26S proteasome regulatory subunit 6B (PSMC4), and T-complex protein 1 (TCP-1) as putative protein targets. Coincubation with appropriate competitors confirmed the authenticity of the proteomics results. Activation of the proteasome by pyrazolones was demonstrated in the absence of exogenous proteasome inhibitor and by restoration of cellular protein degradation of a fluorogenic proteasome substrate in PC12-SOD1G93A cells. Importantly, supplementary studies indicated that these molecules do not induce a heat shock response. We propose that pyrazolones represent a rare class of molecules that enhance proteasomal activation in the absence of a heat shock response and may have therapeutic potential in ALS.
Keywords: Amyotrophic lateral sclerosis, Target identification, Pyrazolone, Proteasome activator, Neurodegeneration; Drug Discovery
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease in the United States and as motor neurone disease in the United Kingdom, is a neurodegenerative disease affecting the upper and lower motor neurons controlling voluntary muscles that produce actions such as walking and respiration. The disease afflicts approximately 2 per 100 000 people worldwide, is invariably fatal, and has no known cure.
The phenotype and pathology of sporadic ALS (SALS), which accounts for 90% of patient cases, are indistinguishable from those of familial ALS (FALS) patients,1 20% of which are caused by missense mutations in the gene encoding for Cu/Zn superoxide dismutase type 1 (SOD1).2 Because SOD1-containing astrocytes have been identified as being common between both forms of ALS, the ALS mouse expressing mutant SOD1 is a widely used model of both FALS and SALS.3 Furthermore, evidence shows that under conditions of cellular stress wild-type SOD1 plays a role in a significant fraction of sporadic ALS cases, supporting the use of SOD1-based models in the search for treatments of the sporadic form of the disease.4
While the underlying pathophysiology of the disease remains unknown, there is mounting evidence that toxic protein misfolding and/or aggregation may be a primary trigger for motor neuron dysfunction and loss.5 The underlying pathological mechanism that produces ALS has been the subject of extensive inquiry in studies of patients with familial forms of the disease. Many of the mutant proteins that cause FALS are misfolded and aggregated in these patients, including SOD1,6 ubiquilin 2 (UBQLN2),7 TAR DNA binding protein (TDP-43)8 (also seen in motor neurons of sporadic ALS patients9), and fused in sarcoma/translated in liposarcoma (FUS/TLS).10 Recently, there is evidence that cytosolic mislocalization of FUS or TDP-43 in vitro and ALS in vivo kindle the misfolding of wtSOD1 in non-SOD1 FALS and SALS.11 ALS shares the presence of prominent misfolded proteins as with many other neurodegenerative diseases.12
We have previously reported the identification and optimization of molecular scaffolds for the treatment of ALS.13 Included among these are the pyrazolones, represented by hit structure 1 and lead compound 2 (Figure 1).14,15 The lead pyrazolone 2 is neuroprotective in a cellular model of ALS using PC12-SOD1G93A cells and increases median survival time in an ALS transgenic mouse model by 13%, confirming its potential as a therapeutic candidate for ALS. Here we report mechanism of action studies and the use of a novel biotinylated probe for affinity purification and proteomic identification of high affinity binding proteins. The data generated support a mechanism of action involving proteasome activation by pyrazolones and provides insight into the potential of this chemical class in ALS therapy.
Figure 1.

Structures of initial hit pyrazolone 1 and optimized lead 2.
Results and Discussion
As a result of the use of mutant SOD1 models during evaluations of pyrazolone efficacy, a natural starting point was to assess the binding of a variety of pyrazolone analogues to SOD1. Utilizing a 96-well plate colorimetric assay (Cayman Chemical), SOD1 (Sigma) was treated with varying concentrations of pyrazolones (1 μM to 10 mM) and dismutation of superoxide radicals generated by xanthine was measured. None of the compounds exhibited any SOD1 inhibition at concentrations in excess of their determined EC50 values (Supporting Information Chart S1).
To further probe the target specificity of pyrazolone 2 across a range of CNS receptors, ion channels, and transporters the compound was analyzed in the NIMH Psychoactive Drug Screening Program at the University of North Carolina. The only protein antagonized by 2 to a significant level (>50%) was the G protein-coupled receptor metabotrophic glutamate receptor 5 (mGluR5), showing 65% antagonism at 10 μM concentration of pyrazolone 2. Antagonists of the mGluR5 receptor have been reported to be therapeutic in ALS.16 If mGluR5 represented the target of action of the pyrazolones, known mGluR5 antagonists should prove active in our cell-based assay. However, when we screened seven known mGluR5 receptor antagonists (including MPEP and fenobam) and antagonists of other mGluR receptor subtypes (LY 456236, specific for mGluR1, and LY 341495, specific for mGluR2/mGluR3), none demonstrated antiaggregation activity (Supporting Information Table S1). On the basis of these results, we conclude that antagonists of the mGluR5 receptor are inactive in our assay, and it is, therefore, unlikely that mGluR5 antagonism is a significant mode of action for these compounds.
MG-132 is a well-established proteasome inhibitor that causes the accumulation of misfolded proteins into large toxic protein aggregates in mutant PC12-SOD1G93A cells. Hence, the ability of pyrazolones to attenuate MG-132 induced cell death was anticipated to involve increased degradation and clearance of misfolded proteins. We used a reporter assay to monitor degradation of polyubiquitinated proteins in living cells (Figure 2A, B).17 PC12 cells expressing a degradation-tagged (polyubiquitinated) yellow fluorescent protein (Ubi-YFP) generated basal fluorescence emission of 0.12 AU, which increased to 0.35 AU when the proteasome was inhibited upon incubation with MG-132 (10 nM). By comparison, the internal coexpressed cyan fluorescence protein (CFP) control reporter was unaffected. Upon treatment of MG-132-inhibited cells with initial hit pyrazolone compounds 1 (EC50 = 0.7 μM) or 3 (EC50 = 0.55 μM) (Figure 2C), the relative fluorescence was reduced to approximately control levels with a 25 μM dose of compound. This demonstrates that pyrazolones can overcome MG-132-mediated proteasome inhibition to increase intracellular protein degradation albeit at a 36–45-fold higher concentration than cellular EC50 values. On its own, this result may suggest direct interaction between pyrazolones and MG-132. However, our in vivo studies have clearly demonstrated that pyrazolones possess disease-modifying efficacy in a disease model that does not involve MG-132.14 Also, there was no reaction of 1 with MG-132, as seen by NMR spectroscopy. Furthermore, our in vitro neuroprotection assay was validated with reversal of bortezomib (a proteasome inhibitor)-induced cytotoxicity in PC12-SOD1G93A cells during initial assay development.13 These results implicate proteasome activation as a mechanism of action for these early hit compounds.
Figure 2.
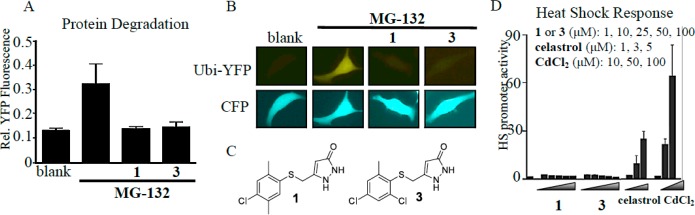
Pyrazolone mode of action studies; impact on protein degradation and heat shock response induction. (A, B) Ability of initial hit pyrazolones 1 and 3 (25 μM) to enhance protein degradation in PC-12 cells transiently transfected with a construct encoding a ubiquitin-tagged yellow fluorescent protein (Ubi-YFP) in the presence of the proteasome inhibitor MG-132 (10 nM) visualized by confocal microscopy and phase contrast microscopy (DIC). Single cell is representative of larger field. Panel (A) shows the quantitation of panel (B) represented as the mean intensity ± SEM. Coexpression of a cyan fluorescence protein (CFP) control reporter was unaffected. (C) Structures of pyrazolone analogues 1 and 3. (D) Pyrazolones do not induce a heat shock response in a HeLa cell based assay that monitors a Hsp70 promoter-luciferase reporter. Positive controls (celastrol and CdCl2) resulted in a significant increase in heat shock promoter activity.
Another possible mode of action for pyrazolones could relate to effects on protein stability or nascent chain folding, thereby shifting the equilibrium to misfolding and activating the heat shock response; activation of the heat shock response (HSR) has been well established as an effective therapeutic target and there are several examples in the literature of proteasomal activators acting via a mechanism that relies on heat shock induction.18,19 Several pyrazolones were tested for their ability to induce a HSR using a HeLa cell based assay monitoring a human Hsp70 promoter-luciferase reporter; representative examples are initial hits 1 and 3 (Figure 2D). Pyrazolones at concentrations equal to those used in the proteasome activation assay above did not induce a heat shock response relative to the known positive control HSR activators celastrol and CdCl2. Indeed, the pyrazolones did not induce a HSR even at concentrations ∼100 times greater than their established neuroprotective EC50 values. These results establish that the pyrazolones do not cause cellular stress resulting in HSR induction and are a unique chemical class that is capable of enhancing proteasomal activity in the absence of a heat shock response.
Lead compound 2 has demonstrated excellent in vitro and in vivo efficacy, and was amenable to biological probe synthesis via biotinylation to provide a water-soluble biotinylated probe,20 BP [Figure 3A (see the Supporting Information or published work for synthetic details)]. The inclusion of a tetraethylene glycol linker was necessary to afford sufficient solubility for biochemical target identification studies.21 We have previously demonstrated that the BP retains activity in our cellular ALS model, with an EC50 = 0.67 μM.22 Hence, the BP is ideally suited for use in a target identification study using affinity-bait chromatography.23
Figure 3.

Affinity-bait protein pull-down experiments. (A) Biotinylated probe (BP). (B) Protein pull-down experiments with BP.
To identify intracellular targets of lead compound 2 within the rat PC12 SOD1G93A cellular model, the BP was immobilized on neutravidin bound agarose beads under saturating conditions and incubated with PC12 SOD1G93A cell lysate.17 Proteins binding nonspecifically were removed by sequential washing with a prepared washing solution (4% DMSO in PBS) followed by elution of the proteins in the loading buffer and denaturation by heating (70 °C, 10 min). The eluted proteins were run on SDS-PAGE and visualized by silver staining (Figure 3B, lane 5). A blank sample of PC12 SOD1G93A lysate incubated with neutravidin beads was examined to ascertain background levels of proteins associated with the neutravidin beads (Figure 3B, lane 3). Bands present in lane 3 indicate some nonspecific background, which is a common artifact in avidin pull-down experiments.24 Comparison of lane 3 (neutravidin binding proteins) and lane 5 (BP-bound-neutravidin binding proteins) indicated the presence of new authentic protein bands. Additionally, the higher affinity lead, 2, outcompeted the BP for the target proteins (Figure 3B, lane 4). Coomassie blue staining was found to be ineffective in visualizing protein bands in this experiment, indicating that very little protein was captured.25 Figure 3B is representative of pull-down experiment results, and additional pull-down controls can be found in the Supporting Information (Figure S1).
Replicate experiments of lane 3 (background pull-down) and lane 5 (target pull-down) were carried out and subjected to proteomic analysis using LC-MS/MS.26 Each sample was run in two lanes on the same gel, which was severed in half; one half was silver stained to visualize the bands, and the other half was left untreated for band excision (see Figure S1, for stained gel). Recombination of the two halves allowed for identification of the band locations without the need to destain, a process that can cause erroneous data in the mass spectrometric analysis in the case of silver staining. The proteins in the unstained gel (20–150 kDa range) were excised in 25–35 kDa sections and submitted to in-gel tryptic digest followed by LC-MS/MS. Protein targets presented by the proteomics database were further analyzed by sequence matching probability score, mass, and known biological function. Relevance criteria were set as follows: (a) sequence probability of 99.9% was required for a protein to be considered a hit; (b) the biological function of the protein needed to have a strong relevance to the observed bioactivity; (c) the mass was required to be a close match to the enriched bands appearing on the SDS-PAGE gel; (d) the identified protein must not be present in the background neutravidin pull-down sample.
In-solution digestion is a milder technique that allows proteomics analysis of proteins from the pull-down solution without running SDS-PAGE separation. The affinity protocol was repeated with the exception that SDS-PAGE analysis was not performed. The “in-solution” BP bound to its protein targets was submitted directly to proteomics analysis. Direct comparison of the merged results from the in-gel and in-solution digestion, and subtraction of “background” proteins retained by the neutravidin/lysate solution (lane 3), provided excellent insights into potential targets (Table 1).
Table 1. Truncated List of Relevant Proteins Identified during Proteomic Analysis.
| identified proteina | accession no. | molecular weight (kDa) | total spectrum count (BP) | total spectrum count (2 and BP)b |
|---|---|---|---|---|
| 26S protease regulatory subunit 4 GN=Psmc1 PE=2 SV=1 | P62193 | 49 | 7 | 0 |
| 26S protease regulatory subunit 6B GN=Psmc4 PE=1 SV=1 | Q63570 | 47 | 4 | 0 |
| T-complex protein 1 subunit alpha GN=Tcp1 PE=1 SV=1 | P28480 | 60 | 5 | 0 |
| T-complex protein 1 subunit epsilon GN=Cct5 PE=1 SV=1 | Q68FQ0 | 59 | 4 | 0 |
| annexin A6 GN=Anxa6 PE=1 SV=2 | P48037 | 75 | 7 | 0 |
| ATP synthase subunit alpha, mitochondrial GN=Atp5a1 PE=1 | P15999 | 60 | 6 | 0 |
| ATP synthase subunit beta, mitochondrial GN=Atp5b PE=1 SV=2 | P10719 | 56 | 11 | 0 |
| coatomer subunit delta GN=Arcn1 PE=2 SV=1 | Q66H80 | 57 | 2 | 0 |
| coatomer subunit gamma-1 GN=Copg1 PE=2 SV=1 | Q4AEF8 | 98 | 4 | 0 |
| large neutral amino acids transporter small subunit 1 GN=Slc7a5 | Q63016 | 56 | 4 | 0 |
| transgelin-2 GN=Tagln2 PE=1 SV=1 | Q5XFX0 | 22 | 3 | 0 |
Proteomic analysis was carried out using SwissProt_2012_07 sequencing database using MASCOT software (selected for Rattus norgevicus species). Proteins were identified with 99.9% certainty and authenticated using control experiments. Consistent proteomic results were obtained on separate occasions by more than one researcher with blinded samples and multiple digestion methods using our affinity-bait technique. Proteins relevant to our mechanism of action that were identified in both in-gel and in-solution digests are listed above.
Total spectrum count from BP competed with compound 2. A complete list of the proteomic results and raw data can be found in the Supporting Information.
In-gel digestion and proteomics analysis identified the 49 kDa 26S proteasome regulatory subunit 4 (PSMC1) protein. Evidence suggests that inhibition of the 26S proteasome plays a role in the pathogenesis of ALS in a mouse model of the disease.27 Thus, activation of the 26S proteasome would be expected to be beneficial in ALS by increasing the rate of disposal of toxic misfolded proteins. The in-gel digestion proteomics analysis identified several relevant protein bands in the 50–60 kDa range. Cytoplasmic dynein 1 light-intermediate chain 1 is a 57 kDa protein that is the major retrograde motor, responsible for movement of freight from the synapse along the axon and back to the cell body and interacts with a large amount of signaling pathways; its many roles are only partially characterized. Mutations in the heavy chain are known to ameliorate neurodegeneration in mouse models of ALS.28 However, on the basis of control experiments and “in-solution” proteomics data, this protein was established to be nonspecific to our BP. Several low probability (<10%) hits were of particular interest in this mass region, namely, the T-complex protein 1 (TCP-1) subunits zeta (58 kDa), eta (59 kDa), gamma (61 kDa), alpha (60 kDa), theta (60 kDa), delta (58 kDa), epsilon (60 kDa), and beta (57 kDa). Detection of so many subunits seems to suggest the presence of TCP-1 that is degraded under the experimental conditions of in-gel digestion or fragmented by mass spectrometry. TCP-1 subunits alpha and epsilon (approximately 60 kDa) were identified when the milder in-solution digestion technique was employed, and remained after subtraction of the background control. A 99.9% probability, an increase of 95% from that detected in the in-gel digestion technique, was reported, indicating that the T-complex protein 1 is bound by the BP, validating the use of in-gel and in-solution methods in parallel. TCP-1 is a molecular chaperone that plays a crucial role in the folding of tubulin, actin, and a host of other cytosolic proteins, including mutant huntingtin.29,30 The 47 kDa 26S proteasome regulatory subunit 6B (PSMC4) was also identified in the in-gel digestion, further suggesting that the mode of action for these compounds involves targeting the proteasome. Three unique proteins identified from the affinity-bait pull-down experiment implicate the proteasome as an important mechanism of action for the pyrazolone compounds. We next revisited the effect of these higher potency compounds on protein degradation in PC12 SOD1G93A cells using fluorogenic proteasome substrate III, an assay method that is more sensitive at lower concentration than the previously utilized PC12 cell ubi-YFP assay. If the pyrazolone compounds do indeed activate the proteasome, they would be expected to elicit increased degradation of a substrate in the absence of the exogenous proteasome inhibitor MG-132. Pyrazolones 1, 2, and BP demonstrated proteasome activation by 50–70% above dimethyl sulfoxide (DMSO) controls level in the absence of MG-132 (Figure 4). Furthermore, all pyrazolones were able to overcome MG-132 proteasome inhibition at EC50 concentrations (Supporting Information Chart S2). This, along with the protein targets identified herein, is suggestive that the pyrazolone binding site differs from the substrate binding site occupied by MG132. These results, generated using a fluorogenic substrate, ensure that the cellular activity of these compounds is not the result of inhibition of YFP fluorescence in the PC-12 Ubi-YFP constructs or action of the compounds to antagonize MG-132. Critically, the observed activity provides biological validation of proteasome activation as a mechanism of action of these therapeutic candidates.
Figure 4.

Pyrazolones enhance proteasome activity. Compounds 1, 2, and BP significantly enhance degradation of proteasome substrate III in the absence of MG-132. Pyrazolones were assayed at EC50 concentrations (1, 700 nM; 2, 70 nM; BP, 670 nM). The two-tailed t test analysis was used to compare the statistical difference between compounds: 1, P = 2.833 × 10–8; 2, P = 2.014 × x10–7; and BP, P = 3.899 × 10–7. Bars are representative of the mean of triplicate experiments ± SD.
It is, therefore, a reasonable probability that these compounds act by stabilizing or enhancing proteasome activity. Of critical significance is that all of the identified protein targets are involved in regulating proteasome activity, and that proteasome modifications and impairment have been detected in ALS vulnerable neurons and other tissues.31
A link between ALS and the ubiquitin proteasome system (UPS) has been suggested.31 Transgenic mice expressing SOD1G93A were found to undergo a decrease in constitutive proteasome subunits during disease progression. PSMC1 and PSMC4 are proteasomal ATPases associated with diverse cellular activities (AAA)_ATPase proteins.32 The eukaryotic 26S proteasome is composed of a 20S barrel shaped catalytic core in which the enzymatic protease sites located within the barrel lumen are bound to a 19S regulatory cap structure. Access of protein substrates to the lumen is restricted and depends upon the proteasomal AAA_ATPase proteins which form the base of the 19S cap. Mechanical forces generated by cycles of ATP binding and hydrolysis unfold substrates, open the proteolytic chamber, and translocate substrate proteins into the catalytically active 20S lumen. Thus, compounds that modify the activity of these AAA_ATPase proteins would be expected to alter proteasome activity. Similarly, it has been proposed that TCP-1 interacts with the proteasome and facilitates the degradation of TCP-1 substrates that have misfolded; the TCP-1 complex functions in assisting protein degradation through interaction with the proteasome.33 It is, therefore, feasible that activation of TCP-1 could affect the conformation of misfolded substrates and, thereby, the rate of clearance by the proteasome.
While there is no evidence for structural homology between the proteasomal AAA_ATPase proteins and TCP-1 complex proteins, strong homology is seen within these protein families.32,34 National Center for Biotechnology Information protein BLAST analysis35 indicates that PSMC4 is the human protein most closely related to PSMC1, with the most closely related PSMC4 isoform showing 52% identity with PSMC1. Similarly, BLAST analysis indicates close similarity within the human genome among the TCP-1 complex proteins with the alpha subunit showing the closest homology to the eta subunit (36% identity), but also significant homology to the epsilon subunit (33% identity). Therefore, it would be reasonable to expect that chemical probes might bind similarly to highly homologous proteins within these families.
We have shown that incubation of PC12-YFP expressing cells with MG-132-induced impairment of proteasome activity with several of our pyrazolones (1, 2, and BP) increases proteasome function to statistically significant levels (Supporting Information Chart S2) at concentrations equal to the EC50 value of each compound. Furthermore, initial hit pyrazolones also demonstrate this effect at higher concentrations (Figure 2A, B). Critically, both the lead pyrazolone and biotinylated probe compounds demonstrate proteasome activation measured by the enhanced degradation of proteasome substrate III in the absence of an exogenous proteasome inhibitor (Figure 4). The similar levels of activity detected in both compound 2 and its biotinylated derivative BP validate that the probe functionality does not impede the pharmacophore of this class of molecule. This activity provides biochemical support for the proposed mechanism of action by these compounds: interaction of the pyrazolones with PSMC1, PSMC4, and/or TCP-1, which leads to proteasome activation.
Summary
When taken collectively, the data presented herein indicate that the pyrazolones act as enhancers of proteasomal activity, possibly via direct activation/stabilization. Compared to proteasome inhibitors, activators are rare and poorly understood, leading to a need for the generation of small molecules that act by this mechanism.36 In support of this concept, the proteasome activator subunit PA28γ, when overexpressed in Huntington’s disease neuronal model cells, was shown to increase the clearance of mutant aggregated huntingtin,37 and there is evidence that ALS is a protein misfolding and aggregation disease.11 Because the proteasome is the central cellular mechanism for disposing of aberrant proteins, increasing the turnover rate of the proteasome would be expected to increase the rate of disposal of these unwanted proteins and exert a therapeutic effect on a variety of neurodegenerative diseases. As the precise pathophysiology of ALS is unknown, activation of the proteasome is expected to be a valid therapeutic target. With so many proteins implicated to misfold and aggregate in ALS, targeting and activating the central disposal mechanism of the cell would be an important approach to combat misfolding and aggregation across the full range of proteins, rather than targeting only one protein in particular.
A survey of the literature reveals only one small molecule compound reported to have the effect of activating the proteasome. This compound acts by inhibiting USP14, a proteasome-associated deubiquitinating enzyme that can inhibit the processing of ubiquitin–protein complexes destined for degradation by the proteasome. Inhibition of USP14, in turn, results in proteasome activation.38
The results presented herein provide strong evidence that the mode of action by which pyrazolone-containing compounds demonstrate therapeutic activity in ALS cellular and animal models is by activation of the proteasome through direct binding to constituent proteins of the 26S proteasome. Nonbiotinylated derivative 2 is active in a SOD1G93A mouse model of ALS, extending mean average survival by 13%. The data presented posits the involvement of several proteins of the 26S proteasome, PSMC1 and PSMC4, in addition to the chaperone protein TCP-1, which is intricately involved in proteasome function. Together, these data describe a potentially multitargeted compound that activates and/or stabilizes the proteasome to provide therapeutic activity in cellular and murine models of ALS.
Methods
Proteasome Activation Assay
The proteasome protection assay was performed with PC12 cells coexpressing proteasome reporter Ubi-YFP and CFP, either treated with vehicle alone, MG-132, MG-132 + compound 1, or MG-132 + compound 3 and MG-132 + pyrazolone derivatives. Cells were incubated for 24 h, and fluorescence quantified. Experiments were performed in replicate. For full details see ref (17).
Proteasome Substrate Assay
Compounds were dissolved to a 10 mM concentration stock solution in DMSO. Compounds were assayed at their respective EC50 concentrations. Two types of control wells were included on every plate: negative control wells containing DMSO (with and without MG-132). After 4 h incubation with the compounds, MG-132 was added to a final concentration of 700 nM to all wells except the negative control containing DMSO only and the compound assay plate with no MG-132. Cells were incubated in a 37 °C 5% CO2 incubator for 48 h. Media was removed by aspiration, and cells were washed with 500 μL of warm 1× PBS before the addition of 300 μL of cell lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1% Triton X-100) to each well. Plates were incubated at 37 °C in a 5% CO2 incubator for 30 min. Proteasome substrate III (Calbiochem 539142) was dissolved to 10 mM concentration in DMSO; 450 μL of this was diluted to 0.5 mM in 8.55 mL of cell media. A 60 μL aliquot was added to each well, which contained 300 μL of cell lysis buffer, bringing the final concentration of substrate to 100 μM in each well. The plates were incubated at 37 °C in a 5% CO2 incubator for 30 min. Fluorescence was read at 380 nm using a Biotek Synergy 4 microplate reader.
Heat Shock Response
The heat shock response was measured in a stable HeLa cell line containing a heat shock-inducible reporter construct that consists of the human Hsp70.1 promoter sequence fused to a luciferase reporter gene (HeLa-luc) that was maintained in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA) with phenol red buffered with HEPES and supplemented with 10% v/v fetal bovine serum (FBS), 1% l-glutamine, 100 mg/mL penicillin/streptomycin, and 100 mg/mL of G418. Cells were maintained at 37 °C with 5% CO2 atmosphere until they were ready for passage or harvest. The HeLa-luc cells were treated with compounds for a 24 h incubation, the assay plates were equilibrated to room temperature, and luciferase was measured using the Bright-Glo Luciferase Assay System (Promega, Madison, WI) with the luminescence signal read with an Envision multilabel plate reader (PerkinElmer, Waltham, MA). Dose–response experiments were then performed in triplicate using a serial dilution of the relevant pyrazolone compounds or positive controls as indicated.
Affinity Chromatography
Two plates of PC12-SOD1G93A cells, grown to ∼80% confluence,17 were harvested at 4 °C in PBS and centrifuged at 14 000 rpm. The resultant pellet was mixed with 400 μL of lysis buffer [20 mM HEPES, pH 7.6, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X, supplemented with “Sigmafast” protease inhibitor cocktail (Sigma-Aldrich, St Louis, MO)], and incubated on ice for 1 h. After centrifugation at 14 000 rpm, the lysate was divided into equal quantities (approximately 150 μL each) and incubated with the selected sample compound (10 mM).
Affinity beads were prepared as follows: Nutravidin-functionalized beads (1 mL per sample) were washed in cold PBS, gently shaken for 10 min, and centrifuged at 2000 rpm for 1 min; the process was repeated three times. Equal amounts (150 μL) of the slurry were transferred to separate Eppendorf tubes and nutated with 100 μL of a 10 mM solution of BP in water with 2% DMSO, for 1 h at 4 °C. The treated beads were centrifuged and washed three times with cold washing solution (4% DMSO in PBS) to remove excess compound.
Following the incubation, lystate or preincubated lysate portions as required were loaded onto the functionalized neutravidin beads, and incubation was continued for 1 h at 4 °C with gentle shaking. Upon completion of incubation, samples were centrifuged at 2000 rpm for 1 min, the supernatant was removed, and the beads were washed three times with 150 μL of washing solution. In those cases where post-treatment was described, the test compound was added to the sample (10 mM) and incubated at 4 °C with gentle shaking for 1 h. Upon completion of incubation, samples were centrifuged at 2000 rpm for 1 min, the supernatant was removed, and the beads were washed three times with 150 μL of washing solution. Samples (24 μL with 6 μL of 5× loading buffer) were boiled and loaded on a polyacrylamide gel and run at 200 V. Polyacrylamide gels were stained with silver stain dye.
Acknowledgments
We thank Dr. Cynthia Voisine for the culture and supply of PC12-SOD1G93A cells. Proteomics were performed at the Proteomics Center of Excellence at Northwestern University by Ms. Emma Doud and Dr. Paul Thomas. Receptor binding profiles and antagonist functional data for mGluR5 inhibition determination were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2008-00025-C (NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth M.D., Ph.D. at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. We thank Dr. Kristin Jansen Labby for providing technical assistance and helpful comments.
Supporting Information Available
Chemical probe synthesis and characterization data, cell culture conditions, proteomics raw data, mGluR5 assay results, and SOD1 inhibition data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ P.C.T.: Department of Pharmaceutical Sciences, School of Pharmacy, Texas Tech University Health Sciences Center, Amarillo, TX and Center for Chemical Biology, Department of Chemistry and Biochemistry, Texas Tech University, Lubbock, TX.
Author Contributions
P.C.T. performed synthetic chemistry, affinity experiments, and SOD1 inhibition assays, interpreted data, and wrote the manuscript. K.T.Z. and S.G.F. performed the proteasome activation assays and interpreted data. I.T.S. replicated affinity assays, interpreted data, and contributed to the manuscript. R.B. conducted activity assays in PC12-SOD1G93A cells. J.M. conducted protein degradation and heat shock response assays. R.B.S. wrote the manuscript. D.R.K, R.I.M, and R.B.S. conceived the project and oversaw the research.
We are grateful to the National Institutes of Health (Grant 1R43NSO57849), the ALS Association (TREAT program), and the Department of Defense (Grant AL093052) for financial support of this research.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bruijn L. I.; Houseweart M. K.; Kato S.; Anderson K. L.; Anderson S. D.; Ohama E.; Reaume A. G.; Scott R. W.; Cleveland D. W. (1998) Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281, 1851–1854. [DOI] [PubMed] [Google Scholar]
- Pasinelli P.; Brown R. H. (2006) Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 7, 710–723. [DOI] [PubMed] [Google Scholar]
- Haidet-Phillips A. M.; Hester M. E.; Miranda C. J.; Meyer K.; Braun L.; Frakes A.; Song S.; Likhite S.; Murtha M. J.; Foust K. D.; Rao M.; Eagle A.; Kammesheidt A.; Christensen A.; Mendell J. R.; Burghes A. H.; Kaspar B. K. (2011) Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 29, 824–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagliardi S.; Cova E.; Davin A.; Guareschi S.; Abel K.; Alvisi E.; Laforenza U.; Ghidoni R.; Cashman J. R.; Ceroni M.; Cereda C. (2010) SOD1 mRNA expression in sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 39, 198–203. [DOI] [PubMed] [Google Scholar]
- Rothstein J. D. (2009) Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann. Neurol. 65(Suppl 1), S3–9. [DOI] [PubMed] [Google Scholar]
- Kerman A.; Liu H. N.; Croul S.; Bilbao J.; Rogaeva E.; Zinman L.; Robertson J.; Chakrabartty A. (2010) Amyotrophic lateral sclerosis is a non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial form. Acta Neuropathol. 119, 335–344. [DOI] [PubMed] [Google Scholar]
- Deng H. X.; Chen W.; Hong S. T.; Boycott K. M.; Gorrie G. H.; Siddique N.; Yang Y.; Fecto F.; Shi Y.; Zhai H.; Jiang H.; Hirano M.; Rampersaud E.; Jansen G. H.; Donkervoort S.; Bigio E. H.; Brooks B. R.; Ajroud K.; Sufit R. L.; Haines J. L.; Mugnaini E.; Pericak-Vance M. A.; Siddique T. (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477, 211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J.; Blair I. P.; Tripathi V. B.; Hu X.; Vance C.; Rogelj B.; Ackerley S.; Durnall J. C.; Williams K. L.; Buratti E.; Baralle F.; de Belleroche J.; Mitchell J. D.; Leigh P. N.; Al-Chalabi A.; Miller C. C.; Nicholson G.; Shaw C. E. (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Plotkin A. S.; Lee V. M.; Trojanowski J. Q. (2010) TAR DNA-binding protein 43 in neurodegenerative disease. Nat. Rev. Neurol. 6, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski T. J. Jr.; Bosco D. A.; Leclerc A. L.; Tamrazian E.; Vanderburg C. R.; Russ C.; Davis A.; Gilchrist J.; Kasarskis E. J.; Munsat T.; Valdmanis P.; Rouleau G. A.; Hosler B. A.; Cortelli P.; de Jong P. J.; Yoshinaga Y.; Haines J. L.; Pericak-Vance M. A.; Yan J.; Ticozzi N.; Siddique T.; McKenna-Yasek D.; Sapp P. C.; Horvitz H. R.; Landers J. E.; Brown R. H. Jr. (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. [DOI] [PubMed] [Google Scholar]
- Pokrishevsky E.; Grad L. I.; Yousefi M.; Wang J.; Mackenzie I. R.; Cashman N. R. (2012) Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PloS One 7, e35050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C.; Estrada L. D. (2008) Protein misfolding and neurodegeneration. Arch. Neurol. 65, 184–189. [DOI] [PubMed] [Google Scholar]
- Benmohamed R.; Arvanites A. C.; Kim J.; Ferrante R. J.; Silverman R. B.; Morimoto R. I.; Kirsch D. R. (2011) Identification of compounds protective against G93A-SOD1 toxicity for the treatment of amyotrophic lateral sclerosis. Amyotrophic Lateral Scler. 12, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T.; Benmohamed R.; Kim J.; Smith K.; Amante D.; Morimoto R. I.; Kirsch D. R.; Ferrante R. J.; Silverman R. B. (2012) ADME-guided design and synthesis of aryloxanyl pyrazolone derivatives to block mutant superoxide dismutase 1 (SOD1) cytotoxicity and protein aggregation: potential application for the treatment of amyotrophic lateral sclerosis. J. Med. Chem. 55, 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T.; Benmohamed R.; Arvanites A. C.; Ralay Ranaivo H.; Morimoto R. I.; Ferrante R. J.; Watterson D. M.; Kirsch D. R.; Silverman R. B. (2011) Arylsulfanyl pyrazolones block mutant SOD1-G93A aggregation. Potential application for the treatment of amyotrophic lateral sclerosis. Bioorg. Med. Chem. 19, 613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeiren C.; Hemptinne I.; Vanhoutte N.; Tilleux S.; Maloteaux J. M.; Hermans E. (2006) Loss of metabotropic glutamate receptor-mediated regulation of glutamate transport in chemically activated astrocytes in a rat model of amyotrophic lateral sclerosis. J. Neurochem 96, 719–731. [DOI] [PubMed] [Google Scholar]
- Matsumoto G.; Stojanovic A.; Holmberg C. I.; Kim S.; Morimoto R. I. (2005) Structural properties and neuronal toxicity of amyotrophic lateral sclerosis-associated Cu/Zn superoxide dismutase 1 aggregates. J. Cell Biol. 171, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto R. I. (2008) Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 22, 1427–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calamini B.; Silva M. C.; Madoux F.; Hutt D. M.; Khanna S.; Chalfant M. A.; Saldanha S. A.; Hodder P.; Tait B. D.; Garza D.; Balch W. E.; Morimoto R. I. (2012) Small-molecule proteostasis regulators for protein conformational diseases. Nat. Chem. Biol. 8, 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trippier P. C. (2013) Synthetic strategies for the biotinylation of bioactive small molecules. ChemMedChem 8, 190–203. [DOI] [PubMed] [Google Scholar]
- Sato S.; Murata A.; Shirakawa T.; Uesugi M. (2010) Biochemical target isolation for novices: affinity-based strategies. Chem. Biol. 17, 616–623. [DOI] [PubMed] [Google Scholar]
- Trippier P. C.; Benmohammed R.; Kirsch D. R.; Silverman R. B. (2012) Substituted pyrazolones require N(2) hydrogen bond donating ability to protect against cytotoxicity from protein aggregation of mutant superoxide dismutase 1. Bioorg. Med. Chem. Lett. 22, 6647–6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie B. J.; Hergenrother P. J. (2008) Identification of the cellular targets of bioactive small organic molecules using affinity reagents. Chem. Soc. Rev. 37, 1347–1360. [DOI] [PubMed] [Google Scholar]
- Sreenivasan V. K.; Kelf T. A.; Grebenik E. A.; Stremovskiy O. A.; Say J. M.; Rabeau J. R.; Zvyagin A. V.; Deyev S. M. (2013) A modular design of low-background bioassays based on a high-affinity molecular pair barstar:barnase. Proteomics 13, 1437–1443. [DOI] [PubMed] [Google Scholar]
- Switzer R. C. 3rd; Merril C. R.; Shifrin S. (1979) A highly sensitive silver stain for detecting proteins and peptides in polyacrylamide gels. Anal. Biochem. 98, 231–237. [DOI] [PubMed] [Google Scholar]
- Aebersold R.; Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207. [DOI] [PubMed] [Google Scholar]
- Kabashi E.; Agar J. N.; Taylor D. M.; Minotti S.; Durham H. D. (2004) Focal dysfunction of the proteasome: a pathogenic factor in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 89, 1325–1335. [DOI] [PubMed] [Google Scholar]
- Banks G. T.; Fisher E. M. (2008) Cytoplasmic dynein could be key to understanding neurodegeneration. Genome Biol. 9, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiess C.; Meyer A. S.; Reissmann S.; Frydman J. (2004) Mechanism of the eukaryotic chaperonin: protein folding in the chamber of secrets. Trends Cell Biol. 14, 598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura A.; Kubota H.; Pack C. G.; Matsumoto G.; Hirayama S.; Takahashi Y.; Kimura H.; Kinjo M.; Morimoto R. I.; Nagata K. (2006) Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat. Cell Biol. 8, 1163–1170. [DOI] [PubMed] [Google Scholar]
- Cheroni C.; Marino M.; Tortarolo M.; Veglianese P.; De Biasi S.; Fontana E.; Zuccarello L. V.; Maynard C. J.; Dantuma N. P.; Bendotti C. (2009) Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 18, 82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Nun S.; Glickman M. H. (2012) Proteasomal AAA-ATPases: structure and function. Biochim. Biophys. Acta 1823, 67–82. [DOI] [PubMed] [Google Scholar]
- Guerrero C.; Milenkovic T.; Przulj N.; Kaiser P.; Huang L. (2008) Characterization of the proteasome interaction network using a QTAX-based tag-team strategy and protein interaction network analysis. Proc. Natl. Acad. Sci. U.S.A. 105, 13333–13338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valpuesta J. M.; Martin-Benito J.; Gomez-Puertas P.; Carrascosa J. L.; Willison K. R. (2002) Structure and function of a protein folding machine: the eukaryotic cytosolic chaperonin CCT. FEBS Lett. 529, 11–16. [DOI] [PubMed] [Google Scholar]
- Altschul S. F.; Madden T. L.; Schaffer A. A.; Zhang J.; Zhang Z.; Miller W.; Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L.; Chen C. H. (2009) Proteasome regulators: activators and inhibitors. Curr. Med. Chem. 16, 931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo H.; Sonntag K. C.; Kim W.; Cattaneo E.; Isacson O. (2007) Proteasome activator enhances survival of Huntington’s disease neuronal model cells. PloS One 2, e238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. H.; Lee M. J.; Park S.; Oh D. C.; Elsasser S.; Chen P. C.; Gartner C.; Dimova N.; Hanna J.; Gygi S. P.; Wilson S. M.; King R. W.; Finley D. (2010) Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.