

Abstract
Discovery of new inhibitors of the plasmalemmal monoamine transporters (MATs) continues to provide pharmacotherapeutic options for depression, addiction, attention deficit disorders, psychosis, narcolepsy, and Parkinson’s disease. The windfall of high-resolution MAT structural information afforded by X-ray crystallography has enabled the construction of credible computational models. Elucidation of lead compounds, creation of compound structure–activity series, and pharmacologic testing are staggering expenses that could be reduced by using a MAT computational model for virtual screening (VS) of structural libraries containing millions of compounds. Here, VS of the PubChem small molecule structural database using the S1 (primary substrate) ligand pocket of a serotonin transporter homology model yielded 19 prominent “hit” compounds. In vitro pharmacology of these VS hits revealed four structurally unique MAT substrate uptake inhibitors with high nanomolar affinity at one or more of the three MATs. In vivo characterization of three of these hits revealed significant activity in a mouse model of acute depression at doses that did not elicit untoward locomotor effects. This constitutes the first report of MAT inhibitor discovery using exclusively the primary substrate pocket as a VS tool. Novel-scaffold MAT inhibitors offer hope of new medications that lack the many classic adverse effects of existing antidepressant drugs.
Keywords: Computational chemistry, antidepressant drug, virtual screening, neurotransmitter transporter
Current therapeutics targeting one or more of the three plasma membrane monoamine transporters (MATs), those for the substrates serotonin (SERT), norepinephrine (NET) or dopamine (DAT), carry numerous adverse effects such as gastrointestinal disorders, increased hunger, insomnia/hypersomnia, and impotence. Additionally, the therapeutic response is suboptimal for many patients.1−6 Toward rectifying these issues, new selective serotonin reuptake inhibitors (SSRIs), serotonin–norepinephrine reuptake inhibitors (SNRIs), and serotonin–norepinephrine–dopamine triple reuptake inhibitors (TRIs) are in demand. TRIs, once viewed as useless in the process of searching for inhibitors for specific transporter proteins, are under renewed study as therapeutics not only for depression but also for obsessive-compulsive disorder, anhedonia, substance abuse, chronic pain, Parkinson’s disease, attention deficit hyperactivity disorder, autism, and obesity.7−16 With the recent development of reliable 3D models of the plasmalemmal MAT proteins, rational discovery and design of novel MAT inhibitors should be achievable by focusing on the MAT ligand-binding pockets
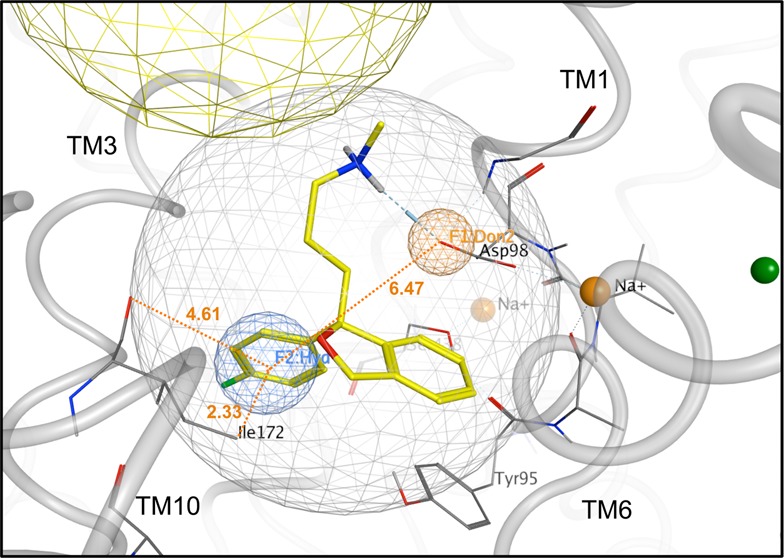
At least two ligand-binding pockets, S1 and S2, are postulated to exist for the MATs (reviewed in refs (17 and 18)). Extrapolating from the position of leucine in the crystallized bacterial leucine transporter (LeuT) protein,19 the MAT primary substrate pocket (S1) is at the approximate midpoint of the lipid bilayer and the center of the MAT transmembrane domains (Figure 1A). This positioning of the substrate is consistent with MAT mutagenesis reports.20−22 The competitive inhibition of substrate binding by certain psychostimulants, SSRIs, and tricyclic antidepressant (TCA) drugs22,23 suggested an inhibitor binding site that overlapped or coincided with the S1 site. Indeed, cocaine, benztropine, and various antidepressants including the SSRI fluoxetine (Prozac) and the TCA nortriptyline (Pamelor) have now been shown to reach the S1 pocket.24−30
Figure 1.

Preparation of the SERT S1 pocket for VS. (A) Side view (lipid bilayer cross-section) of SERT computational model (gray tubes). The S1 (light gray spherical net) and S2 (approximated by the yellow spherical net) ligand-binding pockets are indicated. Bilayer interfaces with the extracellular space and cytoplasm are delineated (upper and lower dashed red lines, respectively). (B) Zoom view of the S1 pocket. Polypeptide backbones (gray tubes) of relevant TM domains are labeled. Donor projection (F1:Don2, orange spherical net), hydrophobic (F2:Hyd, blue spherical net) and volume constraint (ligand-encompassing gray spherical net) pharmacophore features are shown. Citalopram’s docking pose (carbon atoms, yellow; oxygen atoms, red; nitrogen atoms, blue; fluorine atom, green) was used to generate the pharmacophore. Distances between the Don and Hyd features, the Hyd feature and the Ile172 terminal methyl group, and the Hyd feature and the Ile172 backbone carbonyl oxygen are shown (orange dotted lines) in angstroms (orange text). Na+ ions (solid orange spheres) serving as transport cofactors are shown.
The longstanding observation that TCA drugs could also noncompetitively inhibit MAT substrate binding further implied a second, distinct MAT ligand-binding pocket.31,32 TCA drugs cocrystallized with LeuT were bound in the protein’s extracellular vestibule, in a pocket just to the extracellular side of the four “external gate” residues that control entry into the S1 pocket.33−35 Docking of substrates and inhibitors to the first LeuT-based MAT computational models indicated a similar position for this second binding pocket; inhibitors could progress to S1 only by application of external force, via molecular dynamics.36,37 This vestibular pocket was suggested to be a “staging area” for substrates prior to their relocation to the S1 pocket via a MAT conformational change.37 A similar scenario was described for binding of the MAT inhibitor cocaine.36 Evidence for the vestibular, or S2, pocket serving as the initial substrate binding site was provided with the dopamine transporter, in which two dopamine molecules were seen to be necessary for substrate translocation through the cell membrane.38 The S2 binding pocket has been equated with the MAT allosteric inhibitor binding site;28 alternatively, these sites may be nonidentical, or multiple allosteric sites may exist.39
The necessity for, and existence of, the S2 pocket is in question. S2 as a substrate site does not appear to be necessary for LeuT,40,41 although variability in experimental conditions has been proposed as a factor in detecting association of a second substrate molecule within the DAT.42 Regarding the observed TCA ligand occupation of LeuT’s extracellular vestibule, the relevance of a TCA drug binding (weakly) to a bacterial leucine transporter has itself been questioned.43 Replacement of residues in the vicinity of the S1 pocket of LeuT with their SERT counterparts (“LeuBAT”) rendered crystal structures in which the TCA clomipramine (Anafranil) occupied S1, not S2. LeuBAT crystals containing eight different SERT inhibitors covering different structural classes all localized the ligand to S1.30 The first MAT crystal structure, a Drosophila DAT protein bound to the TCA nortriptyline, also positions the drug in S1. Similar to LeuBAT, the DAT-bound TCA drug cannot progress through the substrate pore as a substrate would because its binding extends into the region of the external gate, preventing gate closure.27 Taken together, these findings suggest that MAT drug discovery efforts should include the S1 pocket.
Virtual screening (VS) has been successfully applied to a number of protein targets for the discovery of novel ligands.44,45 VS employs a computational model of the drug receptor in question and involves a rapid in silico ligand docking survey of a structural library containing thousands to millions of chemical compounds. Herein, a VS hybrid approach that included both docking and structure-based pharmacophore filtering has been applied to the SERT S1 pocket, yielding SERT ligand chemotypes that one would be unlikely to find by conventional methods.
Results
Computational Model VS of a Small Molecule Structural Database for Novel SERT Ligands
Using induced-fit docking, citalopram (Celexa) was allowed to associate with the S1 pocket of the SERT model (Figure 1B). This SSRI drug, among the most SERT-preferring, has been localized to the S1 pocket20,28,29,46 and was chosen as the template in building an S1 pocket pharmacophore. Features of the pharmacophore were based on the selected binding pose of citalopram and were added to refine the screening protocol prior to ligand VS (Figure 1C). The VS protocol was verified using an enrichment study in which 10 known non-TCA SERT ligands (Supporting Information, Figure S1) were used to seed a structural library of 1990 random compounds. (Because the evidence for TCA binding at S1 was equivocal at the time the model was optimized, TCA drugs were excluded in the 10 compound training set.) Seven of the 10 seeded compounds were among the 54 hit compounds retrieved by SERT S1 VS in screening the verification library. Following this verification step, a considerably larger structural library was screened for potential SERT ligands of novel structural scaffold.
SERT model S1 pocket screening of the PubChem database of almost half a million compounds yielded 13 378 VS hit compounds. From these, 49 were selected on the basis of visual inspection that focused on the presence of a protonatable amine, receptor placement, ligand conformation, and interactions with side chain residues. Nineteen of the 49 were found to be commercially available; these were purchased for in vitro pharmacologic characterization and labeled TN-01–TN-19 (Figure 2). All 19 VS hits contain a positively charged nitrogen atom and some aromaticity, consistent with the known SERT ligands; interestingly, only two of the 19 contain the indole ring shared with serotonin.
Figure 2.

Structures of the final 19 VS hit compounds. The randomly numbered hit compounds TN-01, TN-05, TN-06, and TN-13 (boxed in red) were selected for additional pharmacologic characterization.
In Vitro Pharmacologic Characterization of VS Hit Compounds
Using the pan-specific MAT radioligand and cocaine analogue [125I]RTI-55, initial in vitro binding assays tested the ability of a single concentration (10 μM) of each nonradioactive VS hit compound in displacing the radioligand at the three plasma membrane MATs. A similar concentration of nonradioactive citalopram, mazindol, or nisoxetine served as a positive control for SERT-, DAT- or NET-selective [125I]RTI-55 binding inhibition, respectively. Depending on the transporter protein, one-quarter to one-half of the 19 VS hits displayed 50% or better inhibition of radioligand binding. Four VS hits, TN-01, TN-05, TN-06, and TN-13, displayed higher relative affinity for at least one MAT (Figure 3).
Figure 3.

VS hit compound in vitro MAT binding screen. Compounds at 10 μM final concentration were tested for the ability to inhibit [125I]RTI-55 binding at hDAT N2A neuroblastoma cells (top panel), hNET N2A neuroblastoma cells (middle panel), or hSERT HEK293 cells (bottom panel). Nonspecific binding was assessed by the presence of 10 μM citalopram (CIT), mazindol (MAZ), and nisoxetine (NIS) for SERT, DAT, and NET, respectively. Data represent n = 3 independent experiments performed in duplicate. Data are presented as the mean ± SEM and were analyzed by one-way ANOVA with Dunnett’s multiple comparison posthoc test. *p < 0.01 vs total binding for that assay; ***p < 0.0001 vs total binding for that assay.
These four hit compounds were more rigorously characterized at each of the three MATs to determine binding affinities (Ki values) and substrate uptake inhibition potencies (IC50 values). Of the hit compounds, TN-05 displayed the strongest SERT affinity, with a Ki value of 668 nM. This compound had an even higher affinity at the NET (323 nM) and no detectable affinity at the DAT (Table 1). Of the four characterized compounds, TN-13 displayed the strongest NET (215 nM) and DAT (780 nM) affinities. Interestingly, this compound was a poor SERT ligand, registering a SERT affinity 50-fold weaker than that for the NET. TN-06, with an NRI profile, displayed the same rank order of affinities as that of TN-13, with modest NET affinity (841 nM) and selectivity over DAT and SERT. For TN-01 and TN-05, the NET was favored over the SERT by less than 2-fold. The TN-05 affinity for the NET was relatively high (323 nM) and no DAT affinity was detected, suggesting a SNRI classification for this hit compound. With only a slight bias toward the NET, TN-01 displayed high affinity for all three MATs, indicating potential as a lead TRI compound (Table 1).
Table 1. MAT Binding Affinities of Top VS Hit Compounds.
|
Ki (nM) |
||||
|---|---|---|---|---|
| SERT | DAT | NET | selectivity ratio SERT/DAT/NET | |
| TN-01 | 1029 ± 81 | 3058 ± 403a,d | 613 ± 162b,e | 2:5:1 |
| TN-05 | 668 ± 41 | >20 000 | 323 ± 53c,d | 2:>62:1 |
| TN-06 | >20 000 | 15 740 ± 2787 | 841 ± 225c,e | 24:19:1 |
| TN-13 | 12 600 ± 2122 | 780 ± 78a,e | 215 ± 55c,e | 58:4:1 |
SERT vs DAT.
DAT vs NET.
SERT vs NET.
p < 0.005.
p < 0.0005.
In terms of substrate uptake inhibition, the rank order of potencies for these compounds (Table 2) largely mirrored the selectivity ratios from the binding assays (Table 1). For the DAT, IC50 values for substrate uptake inhibition agreed well with binding affinity Ki values, in part because the two assays employed intact N2A cells and almost identical conditions. For the SERT and NET, binding and uptake inhibition constants did not correlate nearly as well, typically because these binding assays employed membrane preparations (a requirement for adequate radioligand binding signal-to-noise ratio).
Table 2. MAT Substrate Uptake Inhibition Potencies of Top VS Hit Compounds.
| IC50 (nM) |
||||
|---|---|---|---|---|
| SERT | DAT | NET | potency ratio SERT/DAT/NET | |
| TN-01 | 5025 ± 1894 | 2562 ± 450 | 2092 ± 291 | 2:1:1 |
| TN-05 | 3845 ± 257 | >20 000 | 635 ± 99b,d | 6:>31:1 |
| TN-06 | >20 000 | 13 880 ± 4424 | 5574 ± 1607 | >3:2:1 |
| TN-13 | 18 870 ± 5566 | 720 ± 174a,c | 615 ± 24b,c | 30:1:1 |
SERT vs DAT.
SERT vs NET.
p < 0.05.
p < 0.005.
Antidepressant-Like Effects of VS Hit Compounds in Mice
Following VS identification and in vitro pharmacology, the most promising hit compounds were evaluated via the tail suspension test (TST) for the ability to induce antidepressant-like effects in mice.47 The TST assay was first validated using citalopram and fluvoxamine (Luvox, another SSRI) as positive controls. Compared to saline-treated animals, both citalopram (10 mg/kg) and fluvoxamine (10 mg/kg) induced a significant decrease in immobility, indicative of a reduction in despair-like behavior (saline, 143.7 ± 3.3 s; citalopram, 29.7 ± 14.5 s; fluvoxamine, 59.0 ± 12.5 s; Dunnett’s multiple comparison, p < 0.001 for citalopram and fluvoxamine vs saline). Naïve mice were next acutely treated with TN-01 (0.5 mg/kg or 5 mg/kg), TN-06 (20 mg/kg), or TN-13 (10 mg/kg or 20 mg/kg). (TN-05 was not tested in vivo, as the compound was no longer commercially available at this point.) At one or more doses, all three VS hit compounds induced a statistically significant decrease in immobility compared to that of saline-treated mice (Figure 4A–C), suggesting antidepressant-like activity.
Figure 4.

In vivo characterization of three VS hit compounds. (A) Naïve C57Bl/6J mice treated with TN-01 (0.5 mg/kg, n = 6 or 5 mg/kg, n = 5) showed significant decreases in immobility in the tail suspension test compared to saline-treated mice (n = 6). (p = 0.016; Dunnett’s test *p < 0.05 compared to saline.) (B) Naïve mice treated with TN-06 (1 mg/kg, n = 6; 10 mg/kg, n = 7; 20 mg/kg, n = 6) showed a significant decrease in immobility at the highest dose compared to saline (n = 6). (p = 0.018; Dunnett’s test *p < 0.05 compared to saline.) (C) Naïve mice treated with TN-13 (1 mg/kg, n = 6; 10 mg/kg, n = 6; 20 mg/kg, n = 6) showed a significant decrease in immobility with the two highest doses compared to saline (n = 6). (p = 0.0064; Dunnett’s test *p < 0.05, **p < 0.01 compared to saline.) The distance traveled in an open field compared to saline-treated mice (n = 5) is shown for mice treated with (D) TN-01 (5 mg/kg, n = 6), (E) TN-06 (20 mg/kg, n = 5), or (F) TN-13 (1 mg/kg, n = 3; 10 mg/kg, n = 3).
To test for possible nonspecific motor effects, an open field test was conducted following VS compound injection. The open field test allows for analysis of total locomotion as well as measures of anxiety (time in the center compared to the outside edge of the open field). Neither TN-01 (5 mg/kg), TN-06 (20 mg/kg), nor TN-13 (10 mg/kg) induced statistically significant changes in the total distance traveled compared to that of saline-treated mice (Figure 4D–F), nor was there a statistically significant change in center time between vehicle groups and any of the three VS hit compounds (Supporting Information, Table S2). These data indicate that nonspecific locomotor effects of the compounds cannot account for the TST results.
Discussion
Previously, this research group created a computational model of the human SERT based on the X-ray crystal structure of the LeuT “outward-occluded” conformation (PDB: 2a65),19 within which the putative S2 substrate binding pocket in the SERT extracellular vestibule was used to find new MAT ligands via VS.49 Here, the S1 primary substrate binding pocket of the same SERT homology model was employed to screen the PubChem small molecule structural library for SERT inhibitor candidates of atypical scaffold. Access of these inhibitors to the S1 site had not been permitted using MAT computational models unless the protein was afforded enough flexibility to accommodate induced-fit ligand docking.50 Only recently have substrate- or inhibitor-free LeuT crystal structures become available in which the protein presents an S1-accessible conformation.
The VS approach was primarily employed to uncover potential MAT ligands of novel scaffold (i.e., those that are not clearly structure–activity relatives of established MAT ligands). All hits contained two conserved features: a protonated amine and a hydrophobic moiety ∼6.5 Å away. Three of the four characterized hit compounds, TN-01, TN-05, and TN-06, have scaffolds differing from other reported MAT ligands. While TN-13 is structurally related to a class of compounds recently reported as SERT imaging agents,51−56 the structural differences confer a unique selectivity profile. Interestingly, TN-13 preferred NET and DAT, whereas the imaging agents were selective for SERT, suggesting that TN-13’s extended amine linker and lack of substituents are important for NET/DAT activity (Figure 5). The lack of selectivity for SERT was not unexpected given the high sequence similarity among the three transporters at the S1 binding site.
Figure 5.

Docking placement of the top four VS hits (in yellow) in the S1 SERT pocket: (A) TN-01, (B) TN-05, (C) TN-06, and (D) TN-13. Polar hydrogen atoms are shown. Ligand atoms are color-coded (carbon atoms, yellow; oxygen atoms, red; nitrogen atoms, blue; fluoride atoms, light green; chloride atoms, dark green; sulfur atoms, pink). Relevant SERT side chains are annotated (carbon atoms, gray; oxygen atoms, red; nitrogen atoms, blue). Na+ ions serving as transport cofactors (solid orange spheres) are positioned consistent with the LeuT crystal structure, with the Na+ in the background hidden for clarity.
Ultimately, the goal is to use VS techniques to identify lead compounds with in vivo efficacy in treating depression and other monoamine-related disorders. Depression is associated with a variety of behavioral changes including loss of energy, sleep alterations, learning and memory impairments, anhedonia, and helplessness (despair). Of the four VS hits characterized in vitro for binding affinity and uptake inhibition potency, TN-01, TN-06, and TN-13 were used with naïve mice in the tail suspension test (TST). Reduced immobility (or increased movement) in the TST is associated with a decrease in despair-like behavior. Behavioral effects in the TST are often seen after acute treatment with SSRIs, even though chronic treatment is often necessary for efficacy in human patients.57 This apparent disconnect may be resolved by evidence of acute58 and subchronic59 effects of SSRIs in human subjects. In fact, the TST has shown strong predictive validity in mice as a screen for efficacious human antidepressants.57 The TST and locomotor data suggest that TN-01, TN-06, and TN-13 have antidepressant activity but not anxiolytic activity and that the antidepressant action is not confounded by nonspecific changes in locomotor output after treatment. The lack of an anxiolytic phenotype despite the presence of antidepressant-like activity is similar to that of established SSRIs such as fluoxetine.60 Future analysis of these VS hit compounds will include tests for chronic antidepressant efficacy, effects on learning and memory, and additional assays for anxiety-like behavior.
Until recently, only the extracellular vestibule region of MAT computational models was employed for VS.49,61 The first report of pharmacology-supported MAT structure-based VS exclusive to the S1 pocket was a drug-repurposing effort using a NET model. The VS yielded five novel NET inhibitors with moderate micromolar affinity; notably, all five compounds were structurally similar to that of the norepinephrine substrate.65 More recently, the SERT S1 pocket was used as a VS tool to identify two novel compounds proposed to possess better SERT affinity than that of paroxetine (Paxil), a classic SSRI with subnanomolar SERT affinity. The affinity values, however, were extrapolated from computational modeling predictions as opposed to being pharmacologically verified.62 Very recently, SERT structure-based VS has been employed utilizing an outward-facing (extracellular-facing) SERT model conformation63 that allowed simultaneous access to the S1 and S2 pockets; thus, both pockets and the extracellular vestibule served as potential hit compound binding sites. Several VS hits were obtained that displayed nanomolar to low-micromolar Ki values and a degree of structural uniqueness; SERT selectivity was not addressed.64
Here, the discovery of novel MAT ligands through a hybrid VS approach has been described. Specifically, the screening of a large small molecule structural database using exclusively the S1 binding site of a SERT homology model has afforded four submicromolar affinity hits with varying MAT selectivity profiles. The hit compounds were confirmed to have true MAT activity, as measured by inhibition of substrate uptake, as opposed to merely having MAT binding affinity. Three of these compounds show antidepressant-like activity in a rodent model. This represents the first report in which antidepressant candidate compounds have been identified in silico based solely on the primary ligand-binding (S1) pocket of the SERT and validated using in vitro and in vivo pharmacology. These compounds may now serve both as leads for the discovery of new MAT therapeutics via SAR guided by the computational SERT model as well as new tools in investigating MAT mechanism of action. With such reliable computational models in hand, the VS approach to drug discovery is accessible to research universities as well as to the pharmaceutical industry, which lacks the cost-prohibitive high-throughput in vitro screening steps characteristic of classic pharmaceutical development.
Methods
Materials
Molecular modeling was performed using MOE v2010 and v2011.10 software from Chemical Computing Group (Montreal, Quebec, CA). The radioligands [3H]serotonin (∼28 Ci/mmol), [3H]dopamine (∼26 Ci/mmol), [3H]norepinephrine (∼26 Ci/mmol), and [125I]RTI-55 (∼2200 Ci/mmol) were obtained from PerkinElmer Life and Analytical Sciences (Foster City, CA). Nonradioactive citalopram, mazindol, nisoxetine, and fluvoxamine were obtained from Tocris Bioscience (Ellisville, MO). Virtual screening hit compounds were purchased from Ambinter (Orleans, FR). C57BL/6J mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Data analysis was performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA).
Molecular Modeling
Database Generation
The PubChem database, consisting of 473 965 compounds, was downloaded from www.ncbi.nlm.nih.gov. Three-dimensional structures were generated, and partial charges were determined using the MMFF94x force field in MOE. The database was then “washed” to remove salt fragments and metals, followed by generation of a maximum of 30 tautomers for each compound. Protonation states of each compound were considered, protonating strong bases and deprotonating strong acids. Using the stochastic search method, a conformational search was carried out on the modified database, now containing 1 091 982 entries, to generate a maximum of 30 conformations for each compound. During this stage, several filters were employed to eliminate undesirable compounds and to retain compounds with the following characteristics: MW ≤ 600, donor/acceptor atoms ≤ 12, chiral centers ≤ 4, rotatable bonds < 7, and a LogP range of −4 to 5.
Docking Site Preparation
A previously described SERT model was modified for use in this study.49 An initial docking study of known SSRI and TCA compound SERT ligands revealed the inability of either class to comfortably dock into the S1 site of the occluded model (LeuT PDB: 2a65;19). On the basis of the literature, citalopram was assumed to bind in the S1 site and was therefore used as the reference compound for an induced-fit docking in an effort to expand the S1 pocket (Figure 1B).20,46 Protonate 3D was used to prepare the protein for docking. Alpha spheres selected using SiteFinder defined the S1 site for VS docking. This site corresponds to the position of serotonin and citalopram binding. Structure-based pharmacophore features were introduced (Figure 1C) in order to retain only hits capable of matching three features: a volume constraint (radius = 6 Å, centered on citalopram docked into S1), a hydrogen bond donor projection (radius = 1 Å, centered on a carboxylate oxygen of Asp-98 in TM 1), and a hydrophobic element (radius = 1.4 Å, placed within 2 Å of Ile-172 in TM 3; measurements in angstroms shown in Figure 1C).
Enrichment Docking Study
An enrichment database containing 1990 unknown compounds seeded with 10 known SERT ligands including SSRIs and SNRIs (Table S1, Supporting Information) was generated to evaluate the ability of the employed screening parameters to retrieve known ligands from a database. Several trials of docking in MOE were carried out in order to fine tune the parameters to be used for the actual screening. Additionally, the volume constraint was adjusted in order to limit the number of hit compounds while still allowing known compounds to be selected. A final protocol consisted of the Proxy Triangle placement, the Affinity dG scoring function, and a volume constraint with a 6 Å radius, all features of MOE software. Compounds capable of docking into the S1 site and matching the above pharmacophore features were considered to be hits.
While the score proved to be useful for setting a cutoff limit for hit compounds, it was not capable of rank-ordering known ligands with respect to actual experimental binding affinities. The final enrichment docking retrieved 6 of the 10 known SERT ligands out of a total of 253 hits.
PubChem Database Screening
Using the above docking protocol, the 10 subsets of the PubChem database were screened, resulting in 13 378 hit compounds. As mentioned, the scoring function was used only as a cutoff limit to select compounds for visual inspection, not for ranking. Compounds with S < 0 and MW = 200–450 were inspected both for their fit with the pharmacophore as well as for the interactions formed with the protein. Chemical complexity included the number of stereocenters; synthetic feasibility was taken into consideration. On the basis of these criteria, 49 compounds were selected, of which 19 were commercially available.
In Vitro Pharmacology
In Vitro Pharmacologic Screening
The 19 hit compounds yielded by the S1 SERT virtual screening were purchased and initially tested at a single concentration (10 μM) with cells expressing one of the three MATs, toward detecting specific binding. hDAT or hNET N2A whole cells or hSERT HEK membranes were coincubated with VS hit compound and [125I]RTI-55 (∼0.1 nM concentration). Four hit compounds capable of inhibiting >50% of the radioligand binding were examined further.
SERT Membrane Binding
Membranes were prepared using SERT HEK stable cells grown in a 5% CO2 environment. Cell monolayers were washed twice with 10 mL cold phosphate-buffered saline (DPBS). An additional 10 mL cold DPBS was added, and cells were scraped from the plate, transferred to 15 mL tubes, and centrifuged at low speed (700g). Supernatant was removed followed by resuspension of the cell pellet in 500 μL cold TE buffer (50 mM Tris, pH 7.5, 1 mM EDTA). Homogenate was transferred to cold 1.5 mL microcentrifuge tubes and centrifuged at 100 000g at 4 °C for 30 min (Sorvall Discovery M150 centrifuge). Supernatant was discarded, and the pellet was frozen for later use or resuspended in cold binding buffer (50 mM Tris, pH 7.5, 100 mM NaCl) for immediate use in a membrane-binding assay. Each sample was analyzed for protein content using the micro-Bradford protein assay (Bio-Rad). For competition binding, membrane fractions were incubated with [125I]RTI-55 (∼0.1 nM concentration) radioligand and increasing concentrations of nonradioactive competitor (1 fM to 1 μM concentration) or 10 μM citalopram for nonspecific binding. Reactions were carried out in 12 × 75 mm borosilicate glass tubes at 22 °C for 1 h and terminated by rapid filtration through GF/B filters (Schleicher and Schuell, Keene, NH) presoaked in 0.5% polyethylenimine solution (v/v). Filters were washed twice with 5 mL of cold 50 mM Tris buffer and transferred to counting vials. Radioactivity was determined using a Beckman gamma counter.
DAT and NET Competition Binding
Competition binding assays were performed using hDAT- or hNET-expressing N2A cells grown on 24-well plates in a 5% CO2 environment. Cell monolayers were initially washed twice with 1 mL of KRH buffer (25 mM HEPES, pH 7.3, 125 mM NaCl, 4.8 mM KCl, 1.3 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 5.6 mM glucose) supplemented with 50 μM ascorbic acid (KRH/AA). Cells were then incubated for 15 min with [125I]RTI-55 (0.1 nM concentration) and either increasing concentrations of nonradioactive competitor (1 fM to 1 μM concentration) or 10 μM of mazindol or nisoxetine for assessing nonspecific binding of DAT or NET, respectively. Cell monolayers were washed twice with 1 mL of KRH/AA buffer and were then treated with 1 mL of 1% SDS with gentle shaking at room temperature for 1 h. Cell lysates were transferred into 5 mL scintillation fluid for radioactivity analysis using a liquid scintillation analyzer.
DAT, NET, and SERT Uptake Inhibition
[3H]substrate uptake assays were performed using hDAT- or hNET-expressing N2A cells or hSERT HEK cells grown on 24-well plates in a 5% CO2 environment. hSERT HEK cells were grown on poly l-lysine coated plates to enhance cell adhesion. Cell monolayers were washed twice with 1 mL of KRH/AA buffer, followed by preincubation for 10 min with either increasing concentrations of the drug of interest or 10 μM mazindol, nisoxetine, or citalopram for assessment of nonspecific uptake by DAT, NET or SERT, respectively. Cells were then treated with 10 nM [3H]substrate (dopamine, norepinephrine, or serotonin) supplemented with 99 μM tropolone (total volume of 500 μL) for 5 min. Reactions were terminated by washing twice with 1 mL of KRH/AA buffer to remove any remaining substrate radioligand from the extracellular milieu. Cells were then lysed with 1 mL of 1% SDS under gentle shaking at 22 °C for 1 h, after which cell lysates were transferred into 5 mL scintillation fluid tubes for radioactivity analysis using a liquid scintillation analyzer.
Data Analysis
Experimental data, expressed as counts per minute (cpm), were analyzed with GraphPad Prism. Nonspecific binding was subtracted, and data were transformed to percentages with respect to baseline levels (“no drug”). Nonlinear regression and one-site analysis were used to determine binding (Ki) and uptake (IC50) values.
Behavioral Testing
Animals
All mouse protocols were in accordance with National Institutes of Health guidelines and were approved by the Animal Care and Use Committee of Duquesne University (Pittsburgh, PA). Male C57Bl/6J mice were group-housed on a 12 h/12 h light/dark cycle with ad libitum access to rodent chow and water.
Behavioral Analysis
All behavioral analyses were performed by an observer blinded to treatment. Behavioral tests were conducted with adult male mice 8–20 weeks of age. Mice were treated with citalopram (10 mg/kg in 0.9% normal saline), fluvoxamine (10 mg/kg in 0.9% normal saline), TN-01 (0.5 or 5 mg/kg in 0.9% normal saline), TN-06 (1, 10, or 20 mg/kg in 0.9% normal saline), TN-13 (1, 10, or 20 mg/kg in 0.9% normal saline), or vehicle (0.9% normal saline, pH 7.5). Treatment was given 30 min prior to testing via intraperitoneal injection (0.3 mL). The highest safe dose of each compound showing efficacy in the tail suspension test was chosen for use in the open field test.
Tail Suspension Test (TST)
The TST apparatus consisted of a cubicle made of 1.2 cm Plexiglas with inside dimensions of 40 (w) × 40 (l) × 35 (d) cm3. Mice were suspended by the distal 1.5 cm of their tails with tape. Activity was continuously scored for immobility behavior during the entire 6 min trial. Immobility was defined as the lack of all motion except respiration.
Open Field Test (OFT)
Lighting was provided by a single 100 W incandescent light bulb placed 2 m above the Plexiglas box. Each mouse was placed in a corner of the box and subjected to one 10 min trial. Between sessions, the box was rinsed with 70% ethanol and dried with paper towels. Total distance traveled and time in center square (31 × 31 cm2) were analyzed using Any-Maze software (Stoelting).
Data Analysis
Experimental data, expressed as mean ± SEM, were analyzed with GraphPad Prism. All behavior was analyzed with Student’s t test or one-way ANOVA followed by Dunnett’s multiple comparison tests. p < 0.05 was considered statistically significant.
Acknowledgments
We thank Bernadie Jean for helpful discussions and for providing ChemDraw structures in Supporting Information Figure S1.
Supporting Information Available
Table S1: Structures of the 10 MAT inhibitors used to seed the 1990-compound database in the enrichment docking study. Figure S1: Center time test for nonspecific motor effects of three VS hit compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
T.L.N., B.J.K., J.D.M., and C.K.S. designed the research; T.L.N. and L.M.G. performed the research; T.L.N., L.M.G., B.J.K., J.D.M., and C.K.S. analyzed the data and wrote the manuscript.
This work was supported by NIH grants DA026530 (C.K.S.) and DA027806 (J.D.M.), the Pittsburgh Foundation Hunkele Dreaded Disease Fund (B.J.K., C.K.S.), and a Sharon S. Keller Chronic Pain Research Grant from the American Pain Society (B.J.K.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Ferguson J. M. (2001) SSRI antidepressant medications: adverse effects and tolerability. Prim Care Companion J. Clin. Psychiatry 3, 22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preskorn S. H. (1995) Comparison of the tolerability of buproprion, fluoxetine, imipramine, nefazodone, paroxetine, sertraline, and venlafaxine. J. Clin. Psychiatry 56, 12–21. [PubMed] [Google Scholar]
- Artigas F. (2013) Serotonin receptors involved in antidepressant effects. Pharmacol. Ther. 137, 119–131. [DOI] [PubMed] [Google Scholar]
- Stahl S. M.; Lee-Zimmerman C.; Cartwright S.; Morrissette D. A. (2013) Serotonergic drugs for depression and beyond. Curr. Drug Targets 14, 578–585. [DOI] [PubMed] [Google Scholar]
- Blier P.; Mansari M. E. (2013) Serotonin and beyond: therapeutics for major depression. Philos. Trans. R. Soc., B 368, 20120536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immadisetty K.; Geffert L.; Surratt C.; Madura J. D. (2013) New design strategies for antidepressant drugs. Expert Opin. Drug Discovery 8, 1399–1414. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Skolnick P. (2007) Triple uptake inhibitors: therapeutic potential in depression and beyond. Expert Opin. Invest. Drugs 16, 1365–1377. [DOI] [PubMed] [Google Scholar]
- Marks M. M.; Pae C.-U.; Patkar A. A. (2008) Triple reuptake inhibitors: a premise and a promise. Psychiatry Invest. 5, 142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascol O.; Poewe W.; Lees A.; Aristin M.; Salin L.; Juhel N.; Waldhauser L.; Schindler T. (2008) Tesofensine (NS 2330), a monoamine reuptake inhibitor, in patients with advanced Parkinson disease and motor fluctuations: the ADVANS study. Arch. Neurol. 65, 577–583. [DOI] [PubMed] [Google Scholar]
- Schoedel K. A.; Meier D.; Chakraborty B.; Manniche P. M.; Sellers E. M. (2010) Subjective and objective effects of the novel triple reuptake inhibitor tesofensine in recreational stimulant users. Clin. Pharmacol. Ther. 88, 69–78. [DOI] [PubMed] [Google Scholar]
- Sjodin A.; Gasteyger C.; Nielsen A. L.; Raben A.; Mikkelsen J. D.; Jensen J. K.; Meier D.; Astrup A. (2010) The effect of the triple monoamine reuptake inhibitor tesofensine on energy metabolism and appetite in overweight and moderately obese men. Int. J. Obes. 34, 1634–1643. [DOI] [PubMed] [Google Scholar]
- Tran P.; Skolnick P.; Czobor P.; Huang N. Y.; Bradshaw M.; McKinney A.; Fava M. (2012) Efficacy and tolerability of the novel trriple reuptake inhibitor amitifadine in the treatment of patients with major depressive disorder: a randomized, double-blind, placebo-controlled trial. J. Psychiatr. Res. 46, 64–71. [DOI] [PubMed] [Google Scholar]
- Chen R.; Tilley M. R.; Wei H.; Zhou F.; Zhou F. M.; Ching S.; Quan N.; Stephens R. L.; Hill E. R.; Nottoli T.; Han D. D.; Gu H. H. (2006) Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter. Proc. Natl. Acad. Sci. U.S.A. 103, 9333–9338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn M. K.; Blakely R. D. (2002) Monoamine transporter gene structure and polymorphisms in relation to psychiatric and other complex disorders. Pharmacogenomics J. 2, 217–235. [DOI] [PubMed] [Google Scholar]
- Murphy D. L.; Lerner A.; Rudnick G.; Lesch K. P. (2004) Serotonin transporter: gene, genetic disorders, and pharmacogenetics. Mol. Interventions 4, 109–123. [DOI] [PubMed] [Google Scholar]
- Ozaki N.; Goldman D.; Kaye W. H.; Greenberg B. D.; Lappalainen J.; Rudnick G.; Murphy D. L. (2003) Serotonin transporter missense mutation associated with a complex neuropsychiatric phenotype. Mol. Psychiatry 8, 933–936. [DOI] [PubMed] [Google Scholar]
- Kristensen A. S.; Andersen J.; Jorgensen T. N.; Sorensen L.; Eriksen J.; Loland C. J.; Stromgaard K.; Gether U. (2011) SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol. Rev. 63, 585–640. [DOI] [PubMed] [Google Scholar]
- Manepalli S.; Surratt C. K.; Madura J. D.; Nolan T. L. (2012) Monoamine transporter structure, function, dynamics, and drug discovery: a computational perspective. AAPS J. 14, 820–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A.; Singh Satinder K.; Kawate T.; Jin Y.; Gouaux E. (2005) Crystal structure of a bacterial homologue of Na+/Cl–-dependent neurotransmitter transporters. Nature 437, 215–223. [DOI] [PubMed] [Google Scholar]
- Andersen J.; Olsen L.; Hansen K. B.; Taboureau O.; Jorgensen F. S.; Jorgensen A. M.; Bang-Andersen B.; Egebjerg J.; Stromgaard K.; Kristensen A. S. (2010) Mutational mapping and modeling of the binding site for (S)-citalopram in the human serotonin transporter. J. Biol. Chem. 285, 2051–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker E. L.; Perlman M. A.; Adkins E. M.; Houlihan W. J.; Pristupa Z. B.; Niznik H. B.; Blakely R. D. (1998) High affinity recognition of serotonin transporter antagonists defined by species-scanning mutagenesis. An aromatic residue in transmembrane domain I dictates species-selective recognition of citalopram and mazindol. J. Biol. Chem. 273, 19459–19468. [DOI] [PubMed] [Google Scholar]
- Henry L. K.; Field J. R.; Adkins E. M.; Parnas M. L.; Vaughan R. A.; Zou M. F.; Newman A. H.; Blakely R. D. (2006) Tyr-95 and Ile-172 in transmembrane segments 1 and 3 of human serotonin transporters interact to establish high affinity recognition of antidepressants. J. Biol. Chem. 281, 2012–2023. [DOI] [PubMed] [Google Scholar]
- Talvenheimo J.; Nelson P. J.; Rudnick G. (1979) Mechanism of imipramine inhibition of platelet 5-hydroxytryptamine transport. J. Biol. Chem. 254, 4631–4635. [PubMed] [Google Scholar]
- Beuming T.; Kniazeff J.; Bergmann M. L.; Shi L.; Gracia L.; Raniszewska K.; Newman A. H.; Javitch J. A.; Weinstein H.; Gether U.; Loland C. J. (2008) The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat. Neurosci. 11, 780–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgaard H.; Larsen M. A.; Mazier S.; Beuming T.; Newman A. H.; Weinstein H.; Shi L.; Loland C. J.; Gether U. (2011) The binding sites for benztropines and dopamine in the dopamine transporter overlap. Neuropharmacology 60, 182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinning S.; Musgaard M.; Jensen M.; Severinsen K.; Celik L.; Koldso H.; Meyer T.; Bols M.; Jensen H. H.; Schiott B.; Wiborg O. (2010) Binding and orientation of tricyclic antidepressants within the central substrate site of the human serotonin transporter. J. Biol. Chem. 285, 8363–8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penmatsa A.; Wang K. H.; Gouaux E. (2013) X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503, 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenge P.; Shi L.; Beuming T.; Te J.; Newman A. H.; Weinstein H.; Gether U.; Loland C. J. (2012) Steric hindrance mutagenesis in the conserved extracellular vestibule impedes allosteric binding of antidepressants to the serotonin transporter. J. Biol. Chem. 287, 39316–39326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen L.; Andersen J.; Thomsen M.; Hansen S. M.; Zhao X.; Sandelin A.; Stromgaard K.; Kristensen A. S. (2012) Interaction of antidepressants with the serotonin and norepinephrine transporters: mutational studies of the S1 substrate binding pocket. J. Biol. Chem. 287, 43694–43707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Goehring A.; Wang K. H.; Penmatsa A.; Ressler R.; Gouaux E. (2013) Structural basis for action by diverse antidepressants on biogenic amine transporters. Nature 503, 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenge P.; Mellerup E. T. (1997) An affinity-modulating site on neuronal monoamine transport proteins. Pharmacol. Toxicol. 80, 197–201. [DOI] [PubMed] [Google Scholar]
- Wennogle L. P.; Meyerson L. R. (1982) Serotonin modulates the dissociation of [3H]imipramine form human platelet recognition sites. Eur. J. Pharmacol. 86, 303–307. [DOI] [PubMed] [Google Scholar]
- Singh S. K.; Yamashita A.; Gouaux E. (2007) Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature 448, 952–956. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Zhen J.; Karpowich N. K.; Goetz R. M.; Law C. J.; Reith M. E. A.; Wang D. N. (2007) LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science 317, 1390–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.; Zhen J.; Karpowich N. K.; Law C. J.; Reith M. E.; Wang D. N. (2009) Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nat. Struct. Mol. Biol. 16, 652–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Gu H. H.; Zhan C. G. (2009) Mechanism for cocaine blocking the transport of dopamine: insights from molecular modeling and dynamics simulations. J. Phys. Chem. B 113, 15057–15066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indarte M.; Madura J. D.; Surratt C. K. (2008) Dopamine transporter comparative molecular modeling and binding site prediction using the LeuTAa leucine transporter as a template. Proteins: Struct., Funct., Bioinf. 70, 1033–1046. [DOI] [PubMed] [Google Scholar]
- Shi L.; Quick M.; Zhao Y.; Weinstein H.; Javitch J. A. (2008) The mechanism of a neurotransmitter:sodium symporter—inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol. Cell 30, 667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortagere S.; Fontana A. C.; Rose D. R.; Mortensen O. V. (2013) Identification of an allosteric modulator of the serotonin transporter with novel mechanism of action. Neuropharmacology 72, 282–290. [DOI] [PubMed] [Google Scholar]
- Piscitelli C. L.; Krishnamurthy H.; Gouaux E. (2010) Neurotransmitter/sodium symporter orthologue LeuT has a single high-affinity substrate site. Nature 468, 1129–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Elferich J.; Gouaux E. (2012) Structures of LeuT in bicelles define conformation and substrate binding in a membrane-like context. Nat. Struct. Mol. Biol. 19, 212–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick M.; Shi L.; Zehnpfennig B.; Weinstein H.; Javitch J. A. (2012) Experimental conditions can obscure the second high-affinity site in LeuT. Nat. Struct. Mol. Biol. 19, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnick G. (2007) What is an anitdepressant binding site doing in a bacterial transporter?. ACS Chem. Biol. 2, 606–609. [DOI] [PubMed] [Google Scholar]
- Cheng T.; Li Q.; Zhou Z.; Wang Y.; Bryant S. H. (2012) Structure-based virtual screening for drug discovery: a problem-centric review. AAPS J. 14, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider G. (2010) Virtual screening: an endless staircase?. Nat. Rev. Drug Discovery 9, 273–276. [DOI] [PubMed] [Google Scholar]
- Andersen J.; Taboureau O.; Hansen K. B.; Olsen L.; Egebjerg J.; Stromgaard K.; Kristensen A. S. (2009) Location of the antidepressant binding site in the serotonin transporter: importance of Ser-438 in recognition of citalopram and tricyclic antidepressants. J. Biol. Chem. 284, 10276–10284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryan J. F.; Mombereau C.; Vassout A. (2005) The tail suspension test as a model for assessing antidepressant activity: review of pharmacological and genetic studies in mice. Neurosci. Biobehav. Rev. 29, 571–625. [DOI] [PubMed] [Google Scholar]
- Manepalli S.; Geffert L. M.; Surratt C. K.; Madura J. D. (2011) Discovery of novel selective serotonin reuptake inhibitors through development of a protein-based pharmacophore. J. Chem. Inf. Model. 51, 2417–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldso H.; Autzen H. E.; Grouleff J.; Schiott B. (2013) Ligand induced conformational changes of the human serotonin transporter revealed by molecular dynamics simulations. PLoS One 8, e63635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. L.; Parhi A. K.; Oya S.; Lieberman B.; Kung H. F. (2009) In vivo characterization of a series of 18F-diaryl sulfides (18F-2-(2′-((dimethylamino)methyl)-4′-(fluoroalkoxy)phenylthio)benzenamine) for PET imaging of the serotonin transporter. J. Nucl. Med. 50, 1509–1517. [DOI] [PubMed] [Google Scholar]
- Wang J. L.; Deutsch E. C.; Oya S.; Kung H. F. (2010) FlipADAM: a potential new SPECT imaging agent for the serotonin transporter. Nucl. Med. Biol. 37, 577–586. [DOI] [PubMed] [Google Scholar]
- Vercouillie J.; Mavel S.; Galineau L.; Ragusa T.; Innis R.; Kassiou M.; Chalon S.; Dolle F.; Besnard J.-C.; Guilloteau D.; Emond P. (2006) Synthesis and in vitro evaluation of novel derivatives of diphenylsulfide as serotonin transporter ligands. Bioorg. Med. Chem. Lett. 16, 1297–1300. [DOI] [PubMed] [Google Scholar]
- Mavel S.; Vercouillie J.; Garreau L.; Raguza T.; Ravna A. W.; Chalon S.; Guilloteau D.; Emond P. (2008) Docking study, synthesis, and in vitro evaluation of fluoro-MADAM derivatives as SERT ligands for PET imaging. Bioorg. Med. Chem. 16, 9050–9055. [DOI] [PubMed] [Google Scholar]
- Jarkas N.; Voll R. J.; Williams L.; Votaw J. R.; Owens M. J.; M.M G. (2008) Synthesis and in vivo evaluation of halogenated N,N-dimethyl-2-(2′-amino-4′-hydroxymethylphenylthio)benzylamine derivatives as PET serotonin transporter ligands. J. Med. Chem. 51, 271–281. [DOI] [PubMed] [Google Scholar]
- Chang K. W.; Chen C. C.; Lee S. Y.; Shen L. H.; Wang H. E. (2010) Development pf acute and subacute toxicity with the serotonin transporter radiopharmaceutical, ADAM. Drug Chem. Toxicol. 33, 393–402. [DOI] [PubMed] [Google Scholar]
- Cryan J. F.; Holmes A. (2005) The ascent of mouse: advances in modeling human depression and anxiety. Nat. Rev. Drug Discovery 4, 775–790. [DOI] [PubMed] [Google Scholar]
- Harmer C. J.; Bhagwagar Z.; Perrett D. I.; Vollm B. A.; Cowen P. J.; Goodwin G. M. (2003) Acute SSRI administration affects the processing of social cues in healthy volunteers. Neuropsychopharmacology 28, 148–152. [DOI] [PubMed] [Google Scholar]
- Godlewska B. R.; Norbury R.; Selvaraj S.; Cowen P. J.; Harmer C. J. (2012) Short-term SSRI treatment normalises amygdala hyperactivity in depressed patients. Psychol. Med. 42, 2609–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Garza J. C.; Bronner J.; Kim C. S.; Zhang W.; Lu X. Y. (2010) Acute administration of leptin produces anxiolytic-like effects: a comparison with fluoxetine. Psychopharmacology 207, 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indarte M.; Liu Y.; Madura J. D.; Surratt C. K. (2010) Receptor-based discovery of a plasmalemmal monoamine transporter inhibitor via high-throughput docking and pharmacophore modeling. ACS Chem. Neurosci. 1, 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger A.; Geier E.; Fan H.; Irwin J. J.; Shoichet B. K.; Giacomini K. M.; Sali A. (2011) Structure-based discovery of prescription drugs that interact with the norepinephrine transporter, NET. Proc. Natl. Acad. Sci. U.S.A. 108, 15810–15815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.-L.; Liu H.-L.; Wu J. W.; Tsao C.-W.; Chen W.-H.; Liu K.-T.; Ho Y. (2013) Combining structure-based pharmacophore and in silico approaches to discover novel selective serotonin reuptake inhibitors. Chem. Biol. Drug Des. 82, 705–717. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy H.; Gouaux E. (2012) X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 481, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielsen M.; Kurczab R.; Siwek A.; Wolak M.; Ravna A. W.; Kristiansen K.; Kufareva I.; Abagyan R.; Nowak G.; Chilmonczyk Z.; Sylte I.; Bojarski A. J. (2014) Identification of novel serotonin transporter compounds by virtual screening. J. Chem. Inf. Model. 54, 933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.