


Abstract
Effective splice site selection is critically controlled by flanking splicing regulatory elements (SREs) that can enhance or repress splice site use. Although several computational algorithms currently identify a multitude of potential SRE motifs, their predictive power with respect to mutation effects is limited. Following a RESCUE-type approach, we defined a hexamer-based ‘HEXplorer score’ as average Z-score of all six hexamers overlapping with a given nucleotide in an arbitrary genomic sequence. Plotted along genomic regions, HEXplorer score profiles varied slowly in the vicinity of splice sites. They reflected the respective splice enhancing and silencing properties of splice site neighborhoods beyond the identification of single dedicated SRE motifs. In particular, HEXplorer score differences between mutant and reference sequences faithfully represented exonic mutation effects on splice site usage. Using the HIV-1 pre-mRNA as a model system highly dependent on SREs, we found an excellent correlation in 29 mutations between splicing activity and HEXplorer score. We successfully predicted and confirmed five novel SREs and optimized mutations inactivating a known silencer. The HEXplorer score allowed landscaping of splicing regulatory regions, provided a quantitative measure of mutation effects on splice enhancing and silencing properties and permitted calculation of the mutationally most effective nucleotide.
INTRODUCTION
A wide spectrum of functionally different mRNAs and protein isoforms can be obtained from a single primary transcript by way of alternative pre-mRNA splicing, an active process in the majority of human genes (1). Intron excision is performed by the spliceosome (2), which precisely recognizes the exon–intron borders guided by a multitude of cis-acting sequence motifs and trans-acting factors like regulatory proteins—altogether termed the ‘splicing code’ (3,4). In particular, splice site selection is equally controlled both by the proper 5′ and 3′ splice sites (5′ss and 3′ss) and by nearby activating or repressing splicing regulatory elements (SREs) (5,6).
Disease-associated aberrant splicing can e.g. be caused by mutations as well in splice sites as in SREs, which often are 6–8-nucleotide long binding sites for SR-proteins (exonic splicing enhancers) or hnRNP-proteins (intronic splicing enhancers). These SREs have signature positional distributions (7) and their enhancing or silencing effects on splice site usage depend on their localization with respect to the splice site (8). In addition, weaker splice sites can be compensated by stronger neighboring splicing enhancers, thus avoiding aberrant splicing (8).
Computational identification of SREs is highly important e.g. in human genetics, and several current bioinformatics algorithms for SRE identification provide sets of hexamers (9–15) as binding sites for splicing regulatory proteins. Aggregating SREs across available algorithms currently yields 979 exonic splicing enhancers (ESE) motifs and 496 exonic splicing silencer (ESS) motifs (16), as well as recently described position-dependent enrichment of multivalent tetramer motifs (17). In a systematic experimental evaluation, Ke et al. inserted all 4096 hexamers at five positions in two different internal exons of a 3-exon minigene, obtaining 1182 ESE and 1090 ESS motifs (15). Thus, in genomic sequences SRE searches frequently detect entire arrays of motif ‘hits’, and most mutations in the vicinity of a splice site alter at least one putative SRE, which renders mutation assessment difficult (18). Recently, a machine learning approach using a random forest algorithm yielded a mutation classifier using a variety of SNP-, exon- and gene-based features, which takes into account mutational changes both in splice site and SRE sequences (16). While this approach attempts to unify the effects of intrinsic splice site properties and of neighboring regulatory elements and thus presents a step toward a ‘functional splice site score’, it also includes highly non-local information like evolutionary conservation and properties of the entire gene. In contrast, it is our goal to derive a scoring for SREs that only uses properties of the splice site neighborhood.
We selected the HIV-1 genome as a model system for several reasons: (i) it is ∼10 000 nt small and its splicing patterns during early and late phases of the replication cycle are well characterized, (ii) it contains highly regulated splice sites dependent on many known SREs (19–39) and (iii) HIV-1 splicing is easily experimentally accessible both in subgenomic splicing reporter constructs and in replication competent virus.
Within HIV-1-infected cells, more than 40 different viral mRNAs are spliced from a single primary RNA transcript. Depending on introns retained, these RNAs can be separated into three distinct classes: intronless (2 kb), intron-containing (4 kb) and unspliced (9 kb) viral mRNAs (40,41). The sophisticated splicing pattern is derived from alternatively used subsets of at least four viral 5′ss and eight 3′ss. Splice site selection is controlled by SREs, which can either activate or repress functional recognition of a nearby splice site (40). Disruption of only one of these viral SREs can severely interfere with the viral splicing balance, which has to be maintained for proper replication (19,20). For instance, exon 3 splicing is repressed by the vpr exonic splicing silencer ESSV (19,21–22), and inactivation of vpr exonic splicing silencer (ESSV) results in dramatically increased levels of exon 3 inclusion, abolishing unspliced viral mRNAs and thus suppressing virus particle production (19,21).
In this study, we defined and validated a ‘HEXplorer score’ for every nucleotide in a genomic sequence, based on all overlapping hexamers rather than only on dedicated SRE motifs. We hypothesize this HEXplorer score to capture the splice enhancing and silencing properties of genomic regions in the vicinity of splice sites. Using the HIV-1 pre-mRNA as a model system highly dependent on SREs, we found an excellent correlation in 29 mutations between splicing activity and HEXplorer score. We successfully predicted and confirmed five novel ESEs and optimized mutations inactivating the known silencer ESSV. The HEXplorer score (www.uni-duesseldorf.de/rna) allows landscaping of splicing regulatory regions, provides a quantitative measure of mutation effects on splice enhancing and silencing properties, and permits calculation of the mutationally most effective nucleotide.
MATERIALS AND METHODS
Oligonucleotides
Oligonucleotides were obtained from Metabion GmbH (Martinsried, Germany).
Primers used for site-directed mutagenesis (see Supplementary Tables S1 and S2).
Primers used for semi-quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) analyses (see Supplementary Table S3).
HIV-1-based subgenomic splicing reporter
The LTR ex2 ex3 minigenes were generated as described previously (20,23). Exon 3 mutants were constructed by PCR mutagenesis. For construction, the AlwNI/SpeI fragment of LTR ex2 ex3 was replaced by the respective PCR products using the appropriate forward PCR primer (see Supplementary Table S1) and #2588 as a reverse PCR primer containing AlwNI and SpeI restriction sites. After cloning, all PCR amplicons were validated by sequencing.
SV-env (24) derived splicing reporters (see Supplementary Table S2) were constructed by replacing the EcoRI/SacI fragment with respective PCR products: exon 3 and exon 3 ESSV− (#1906/#1907) or linkers: part I (#1958/#1959), part II (#1960/#1961), part II ESSV− (#1962/#1963) and part III (#1964/#1965).
SV-env/eGFP reporters (42) were generated by substitution of the EcoRI/NdeI fragment with respective PCR products using the appropriate forward PCR primer (see Supplementary Table S2) and #640 as a reverse primer. SV-SD4-env/eGFP reporters (corresponding to SV-env/eGFP with HIV-1 D4 sequence instead of D1) were cloned by replacing the EcoRI/SacI fragment with the indicated linker sequences.
Proviral HIV-1 plasmids
pNL4-3 mutants were constructed by replacing the region between PflMI and EcoRI with mutated LTR ex2 ex3 fragments as described previously (20).
Cell culture and RT-PCR analysis
HeLa and HEK 293T cells were maintained in Dulbecco's high glucose-modified Eagle's medium (Invitrogen) supplemented with 10% fetal calf serum and 50 μg/ml of each penicillin and streptomycin (Invitrogen). Transfections were done in 6-well plates with 2.5 × 105 cells per plate using TransIT®-LT1 reagent (Mirus) according to the manufacturer's instructions. Total RNA samples were collected 48 h after transfection from either HeLa or HEK 293T cells transfected with subgenomic or proviral constructs and pXGH1 to control transfection efficiency. For reverse transcription, 4 μg of RNA were subjected to DNA digestion with 10 U of DNase I (Roche). DNase I was heat-inactivated at 70°C for 5 min and cDNA synthesis occurred for 1 h at 50°C and 15 min at 72°C by using 200 U Superscript III RNAse H− Reverse Transcriptase (Invitrogen), 7.5-pmol oligo(dT)12–18 (Invitrogen) as primer, 20 U of RNAsin (Promega) and 10 mM of each deoxynucleoside triphosphate (Qiagen). For semi-quantitative analysis of LTR ex2 ex3 minigene mRNAs, cDNA was used as a template for a PCR reaction with forward primer #1544 and reverse primer #2588 (see Supplementary Table S3). For semi-quantitative analysis of SV-env/eGFP-derived reporter mRNAs, cDNA was used as a template for a PCR reaction with forward primer #3210 and reverse primer #3211. For transfection control, PCR was performed with primers #1224 and #1225 to specifically detect GH1 mRNA. For analysis of exon 3 inclusion in viral tat mRNAs and vpr mRNA splicing, a PCR reaction was carried out using primers #1544 (E1) and #3632 (E4). For the analysis of intronless 1.8-kb HIV-1 mRNAs, PCR reaction was carried out with forward primer #1544 (E1) and reverse primer #3392 (E7). Finally, intron containing 4.0-kb HIV-1 mRNAs were detected with primers #1544 (E1) and #640 (I4). PCR products were separated on 8% non-denaturing polyacrylamide gels and stained with ethidium bromide for visualization.
Antibodies
The following primary antibodies were used for immunoblot analysis: mouse antibody against α-actin (A2228) was obtained from Sigma-Aldrich. Sheep antibody against HIV-1 p24 was purchased from Biochrom AG. For detection, we used a horseradish peroxidase (HRP)-conjugated anti-mouse antibody (NA931) from GE Healthcare and an HRP-conjugated anti-sheep antibody from Jackson Immunoresearch Laboratories Inc.
Protein analysis
Transfected cells were lysed in radio immunoprecipitation assay (RIPA) buffer (25-mM Tris·HCl pH 7.6, 150-mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), protease inhibitor cocktail (Roche)). Proteins were separated by SDS polyacrylamide gel electrophoresis, transferred on a nitrocellulose membrane and subjected to immunoblotting procedure. Membranes were probed with the respective primary and secondary antibodies and developed with ECL chemiluminescence reagents (GE Healthcare).
Hexamer score calculation
Following the RESCUE concept (9), hexamer frequencies were determined in the four data sets of up to 100-nt long exonic and intronic sequences up- and downstream of 10 359 weak and 10 407 strong constitutive canonical 5′ splice sites, respectively. All 11 nucleotides (three exonic, eight intronic) of the 5′ splice site consensus sequence were excluded from the analysis to avoid hexamer count bias from splice site motif conservation. Weak and strong 5′ss were selected as lower (HBS ≤ 13.5) and upper (HBS > 17.0) quartiles of the HBond score (HBS) distribution (43).
To compare the relative occurrences in both data sets of a given hexamer that occurred fE times in the exonic and fI times in the intronic sequence data set, the normal distributed z-score ZEI was calculated as 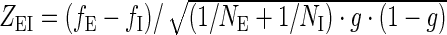 .
.
Here, NE and NI denote the total numbers of exonic and intronic positions in the respective data sets, and 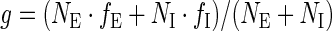 is the weighted average frequency. The ‘weak–strong’ Z-score ZWS was calculated accordingly, using hexamer frequencies fW and fS in exons upstream weak and strong 5′ splice sites, respectively.
is the weighted average frequency. The ‘weak–strong’ Z-score ZWS was calculated accordingly, using hexamer frequencies fW and fS in exons upstream weak and strong 5′ splice sites, respectively.
Detection of SREs by other available algorithms
The comparison of the novel HEXplorer score with other available algorithms for the identification of SREs was performed on 50 nucleotides upstream of each 5′ss in the HIV-1 genome. These sequences were screened for enhancer and silencer motifs, using web resources for the following available algorithms (9–15): ESEfinder 3.0 (http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home), RESCUE-ESE (http://genes.mit.edu/burgelab/rescue-ese/), FAS-ESS-hex3 (http://genes.mit.edu/fas-ess/), PESX (http://cubweb.biology.columbia.edu/pesx/) and ESRsearch (http://esrsearch.tau.ac.il/). Where appropriate, the respective default settings for SRE detection were applied.
From all these algorithms, motifs were identified in reference and mutated sequences, and a weight of +1 or −1 was assigned to a sequence for each predicted enhancer or silencer motif that overlapped with a mutated nucleotide. From the change in the number of ESE- and ESS-motifs induced by these mutations, we calculated an overall ‘exonic splicing motif difference’ (ESMD) similar to Ke et al. (44), capturing the summary effect of ESR gain and loss predicted by the various algorithms.
Systematic comparison of HEXplorer score and ESRseq score was performed by extracting hexamer weights for 1182 ESS and 1090 ESS hexamer motifs provided by Ke et al. (15). Using these hexamer weights instead of the weights obtained from our data set, we calculated the average ESRseq score of all hexamers overlapping with any index nucleotide in a genomic sequence. In this way, we obtained a metric based on the ESRseq score that permitted a direct comparison to the HEXplorer score.
RESULTS
Derivation of HEXplorer score—an in silico approach for SREs
The HEXplorer score presented in this work was based on hexamer weights calculated by a RESCUE-type approach (9). Splice site recognition depends more strongly on the presence of SREs for weak than for strong 5′ss (8). Therefore, hexamers associated with ESEs are generally assumed to be over-represented in exonic sequences upstream of weak 5′ss compared to sequences upstream of strong 5′ss, as well as in exonic versus intronic sequences. Here, RESCUE-type hexamer weights were derived from hexamer frequencies in four sets of exonic and intronic sequences taken from the human 5′ splice site data set described in (45). This data set contained 43 464 constitutively spliced internal human exons with canonical 5′ss collected from the ENSEMBL database (chromosomes 6, 7, 9, 10, 13, 14, 20, 22, X). Intrinsic strength of 5′ splice sites was assessed by the HBS describing 5′ss complementarity with all 11 nucleotides of the free 5′ end of U1 snRNA (43). In the whole set of 43 464 constitutive canonical 5′ splice sites, the HBS was approximately Gaussian distributed with mean 15.0 and standard deviation (SD) 2.59 (range 1.80–23.8; Figure 1A). Weak and strong 5′ss were selected as upper and lower quartiles of the HBS distribution, resulting in 10 359 weak 5′ss with HBS ≤ 13.5 and 10 407 strong 5′ss with HBS > 17.0. Subsequently, hexamer frequencies were counted in 100-nt long exonic and intronic sequences up- and downstream of these weak and strong 5′ss, respectively. Each 5′ss could maximally contribute 179 different hexamer positions, and a total of 1 728 912 exonic and 1 835 932 intronic hexamer occurrences were counted in these four sequence data sets.
Figure 1.
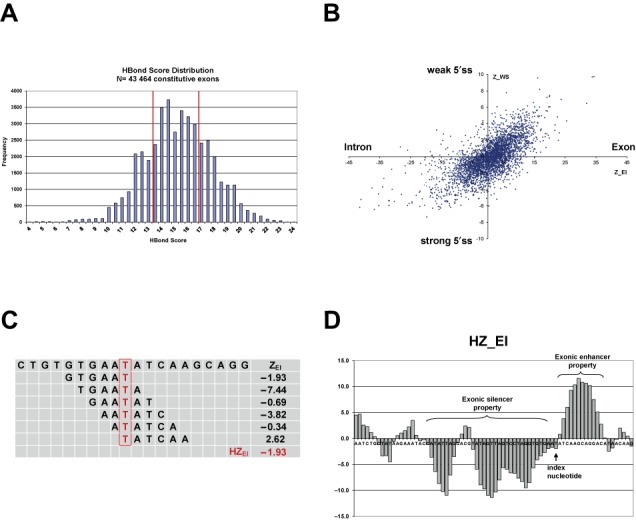
(A, B) HBond score distribution and relative hexamer abundance in human 5′ss neighborhoods. (A) HBond score distribution in 43 464 canonical 5′ss of constitutively spliced internal human exons. Vertical red lines denote lower and upper quartile boundaries at HBS = 13.5 and HBS = 17.0, respectively. (B) Scatterplot of hexamer overabundance Z-scores comparing exons with introns (ZEI) and exons of weak (10 359 5′ss with HBS ≤ 13.5) with strong (10 407 5′ss with HBS > 17.0) 5′ss (ZWS). Each dot represents one of the 4096 hexamers at coordinates (ZEI, ZWS). Hexamers e.g. in the top right hand corner were significantly over-represented in exons versus introns and in exons of weak versus strong 5′ss, and were potential RESCUE-ESE candidates. (C, D) HEXplorer score: sliding window average of ZEI-score. (C) Schematic of HEXplorer score calculation for an exemplary region from HIV-1 exon 3. For the index nucleotide T (denoted in red), the HEXplorer score HZEI was calculated as average ZEI score of all six overlapping hexamers. These six individual hexamers and their respective ZEI scores are given in the rows below. The last row contains the HEXplorer score as average of the six ZEI. (D) Exemplary HZEI plot of HIV-1 exon 3. The arrow indicates the position of the index nucleotide from (A). Prominent HZEI positive and negative regions with exonic splice enhancing or silencing properties are indicated by curly braces.
Following the RESCUE concept, two normalized Z-scores were subsequently determined for all 4096 hexamers (9). Each score reflects the difference in hexamer occurrence between two sets of 5′ss neighborhoods: for each hexamer, its exon–intron Z-score ZEI is the scaled difference of hexamer frequency between 100-nt long sequences up- and downstream of 5′ss, while its weak–strong Z-score ZWS measures the difference in hexamer occurrence between exons upstream of weak and strong 5′ss, respectively. Figure 1 B shows the scatterplot of ZWS versus ZEI for all individual 4096 hexamers. Hexamers plotted e.g. in the upper right hand corner were found more frequently in exons compared to introns, and more often in exons of weak than of strong splice sites. Within the RESCUE framework, such hexamers are considered as candidate sequences for exonic splicing enhancers. Both hexamer scores ZEI and ZWS were significantly correlated, having Pearson's r = 0.70 (P < 0.0001), as suggested by the elongated shape of the scatterplot cloud. However, hexamer frequencies differed more widely between exons and introns than between exons of weak and strong 5′ss. This was quantitatively reflected in the different hexamer score ranges: across all 4096 hexamers, ZEI had a range of −73.3–34.4 (mean of 0.746, SD 8.76, median 1.92), while ZWS had a six times smaller range of −7.34–10.5 (mean of −0.167, SD 2.50, median −0.25).
Distinct sets of splicing regulatory proteins have been found to support U1 snRNP binding in a strictly position-dependent manner. In particular, exonic or intronic enhancer sequences frequently act as silencers in their respective position opposite the 5′ss—they have a signature positional distribution around the splice site (7,8). Therefore, sequences acting as exonic splicing silencers that are statistically depleted upstream of functional 5′ss are at the same time enriched downstream as intronic enhancers. Thus, we assumed the exon–intron hexamer score ZEI to also capture enhancer/silencer sequence properties beyond general exon–intron differences, and selected ZEI for our further studies. Furthermore, we expected ZEI to have higher discriminatory power due to its larger range of values across hexamers.
Aiming at developing a RESCUE-concept-based score for each nucleotide position in a given sequence, we next calculated the HEXplorer score HZEI for every index nucleotide as the average hexamer score of all six hexamers overlapping with the index nucleotide. Beyond the index nucleotide, this HEXplorer score HZEI depends on 5 nt to both sides (up- and downstream). In the exemplary sequence GTGAATATCAA, e.g. the index nucleotide T (HZEI = −1.93) is located at the last position in hexamer GTGAAT (ZEI = −1.93), at the fourth position in hexamer GAATAT (ZEI = −0.69) and at the first position in TATCAA (ZEI = 2.62). A schematic representation of HZEI calculation from all six hexamers containing the index nucleotide T in this exemplary sequence is depicted in Figure 1C. By its construction as a moving average, the HEXplorer score varies more slowly along the sequence than the individual contributing hexamer scores ZEI, similar to the effect of a low-pass filter.
HEXplorer score values were graphically represented by a bar graph along a sequence (Figure 1D). In this picture, HZEI-positive areas above the horizontal axis indicated regions with exonic enhancer properties, while areas below the axis corresponded to sequences with exonic silencer properties. Upstream of 5′ss, HZEI-positive sequence stretches that contained more nucleotides and/or had higher HEXplorer scores were expected to possess stronger exonic enhancing property and more efficiently support splicing.
It is important to note that within this framework we associated ‘exonic splice enhancing’ with being a property of an entire sequence ‘region’ rather than that of a single hexamer. Correspondingly, the HEXplorer score was constructed to take into account ‘all’ hexamer frequencies in any given sequence rather than just a small subset of ‘Z-extreme’ hexamers indicating specific protein binding sites. Quantitatively, the splice enhancing property of a region in the context of a given splice site was measured by the area under the HZEI graph in this region.
HEXplorer score plots provide distinct exonic enhancer/silencer profiles for all HIV exons
In order to validate the HEXplorer score, we systematically examined HZEI in all exons of the entire HIV-1 genome, which is known to contain a large number of SREs (40). Figure 2 presents an overview of the HIV-1 genome with all known exonic splicing regulatory motifs marked by green (enhancer) or red (silencer) boxes.
Figure 2.
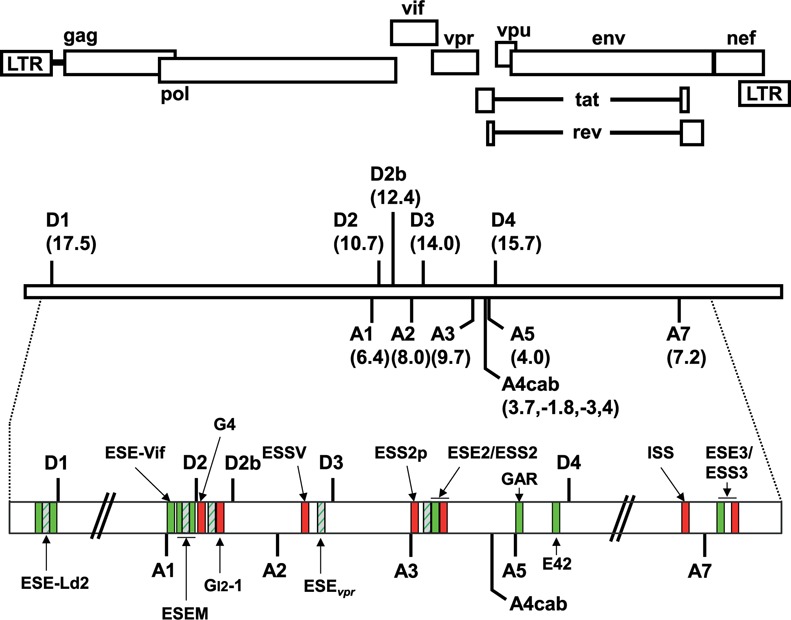
Schematic drawing of known and HEXplorer-predicted splicing regulatory elements (SREs) in the HIV-1 genome. Top: open reading frames are indicated by open boxes. The long terminal repeats (LTRs) are located at both ends of the provirus. Center: all HIV-1 proteins are encoded by a single primary RNA. Alternative splicing leads to more than 40 different mRNA transcripts enabling efficient translation of all open reading frames within the host cell. Intrinsic strength of the 5′ss (D1–D4) and 3′ss (A1–A7) is indicated in the parentheses (5′ss: HBond Score, http://www.uni-duesseldorf.de/rna; 3′ss: MaxEntScore, http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq_acc.html). Bottom: positions of the SREs within the HIV-1 pre-mRNA: known splicing enhancers (green) and silencers (red) are indicated together with HEXplorer-predicted enhancers (dashed green). [ESE-Ld2 (25); ESE-Vif (27); ESEM (26); guanosine (G)-rich silencer G4 (27); GI2–1 (23); ESSV (19,21–22); ESEvpr (20); ESS2p (28); ESE2 (29,30); ESS2 (31–33); guanosine-adenosine-rich (GAR) ESE (24,34,46); E42 fragment (34); ISS (35); ESE3 (36); ESS3 (36–38) (adapted to (34,39)].
We then calculated and plotted HZEI HEXplorer profiles for splicing relevant sequences in HIV-1 exons 1, 2, 2b, 3, 4, 5 and 7 (Figure 3). The examined exonic sequences were between 50-nucleotides (for exon 2) and 97 nucleotides (for exon 7) long. In all seven HIV-1 exons, HZEI graphs slowly varied between peak values of −11 and 33 and contained both HZEI-positive and -negative sequence stretches. In six out of seven exons, HZEI graphs were mostly dominated by large positive areas, and only exon 3 contained a long HZEI-negative sequence stretch corresponding to the known silencer ESSV. In all seven exons, known exonic enhancers and silencers correlated well with HZEI-positive and -negative sequence stretches, respectively.
Figure 3.
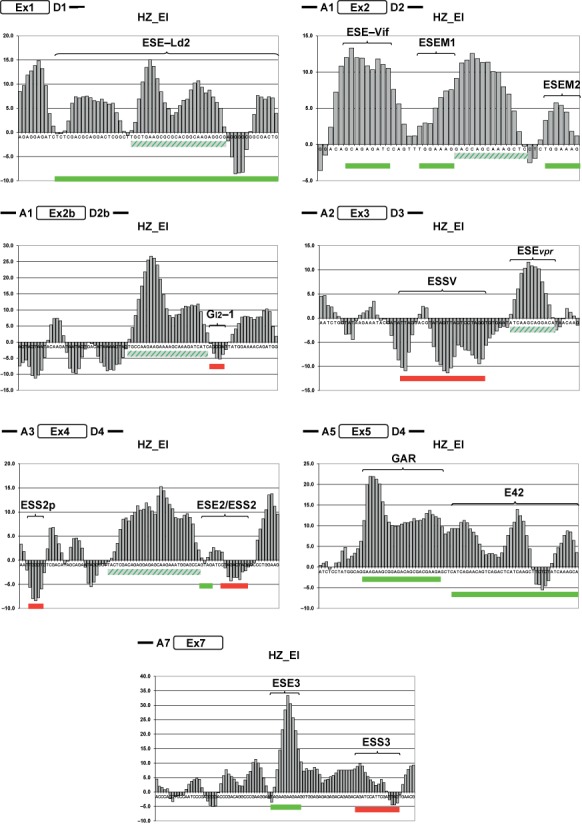
HEXplorer score profiles of HIV-1 exons. HZEI-score plots of HIV-1 exonic sequences derived from the molecular clone NL4–3. Nucleotides are plotted along the horizontal axis and HEXplorer score values HZEI on the vertical axis. Location of the plotted subgenomic region is schematically shown as an open box on the left-hand side. Known exonic SREs are indicated by curly braces and green (enhancer) or red (silencer) rectangles. Five HEXplorer-predicted enhancers are indicated by dashed green rectangles.
In particular, the exonic splicing silencers GI2–1 (exon 2b), ESSV (exon 3), ESS2p and ESS2 (exon 4) as well as ESS3 (exon 7) mostly lie in HZEI-negative regions, whereas the exonic splicing enhancers ESE-Ld2 (exon 1), ESE-Vif, ESEM1 and ESEM2 (exon 2), ESE2 (exon 4), GAR (exon 5), as well as ESE3 (exon 7) lie in HEXplorer score positive regions (Figure 3).
From a helicopter view, these findings confirmed the general qualitative association of HEXplorer score positive and negative sequence stretches with all known HIV-1 exonic enhancers and silencers, respectively. To examine this association in more detail, we studied HIV-1 exon 3 containing both the well-known splicing silencer ESSV (19) and the recently found enhancer ESEvpr (20).
Splice enhancing and silencing properties of exon 3 fragments correlate with HEXplorer score
The HZEI-plot along the pNLA1-derived HIV-1 exon 3 sequence (47) clearly depicted HZEI-positive and -negative regions (Figure 4A). For a closer examination, exon 3 was partitioned into three consecutive parts (I–III) of equal sequence length. With respect to the HEXplorer score, these three parts corresponded to (I) a HZEI heterogeneous region, (II) a predominantly HZEI-negative region and (III) a mainly HZEI-positive region. The entire exon 3 sequence and the three individual parts I–III were tested in the context of the HIV-1-based splicing reporter (SV-env; Figure 4B). SV-env is a single intron splicing reporter that contains an enhanced green fluorescent protein (eGFP) coding region downstream of the splice acceptor, permitting fluorescence monitoring and western blot analysis of HIV-1 glycoprotein expression depending on U1 snRNA binding to 5′ss D4. In addition, a proximal enhancer is required for binding of the U1 snRNP to the splicing reporter's 5′ss D4. Moreover, only in the presence of the HIV-1 regulatory protein Rev, U1 snRNP binding at the reporter's 5′ss D4 correlates with the expression of viral glycoprotein gp160 and gp120 (24,43,46).
Figure 4.
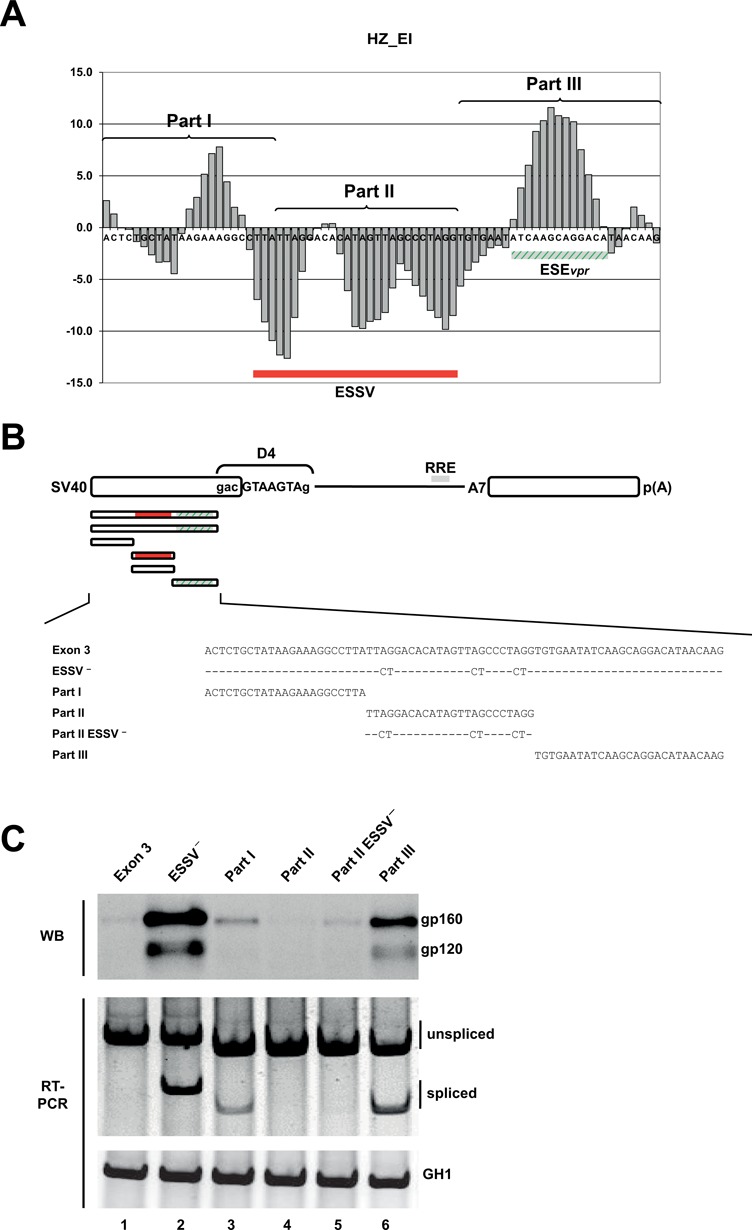
Splice enhancing and silencing properties of exon 3 fragments correlate with HEXplorer score. (A) HZEI-score plot of the HIV-1 exon 3 sequence partitioned into three consecutive fragments (parts I–III) of equal sequence length. HZEI-positive and -negative regions are indicated in green and red, respectively. (B) Schematic of the SV-env expression plasmid carrying the env open reading frame (ORF). HIV-1 exon 3-derived sequences that were inserted upstream of the splicing enhancer dependent 5′ss D4 are enlarged below. ESSV− is known to disrupt the silencer ESSV. Either single part I, part II, part II ESSV− or part III was inserted, but no combinations. (C) Western blot analysis of cell lysates from HeLa cells transiently transfected with 1 μg of each of the constructs together with 1-μg SVcrev and 1 μg of pGL3 (Promega) to control for equal transfection efficiencies. Forty-eight hours post transfection proteins were extracted and separated by 7% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Samples were normalized for equal protein and luciferase amounts as described previously (46). After transfer to a nitrocellulose membrane, samples were probed with a mouse monoclonal antibody specifically detecting splicing-dependent gp120 expression within the transfected cells (87–133/026; kindly provided by Dade Behring). (SV40: simian virus 40 early promoter; RRE: Rev responsive element.)
While the wild-type exon 3 sequence failed to enhance glycoprotein expression, inactivation of the repressing ESSV activity as described previously (21) led to considerable expression of gp160 (and the proteolytic cleavage product gp120) within the cells (Figure 4C, upper panel, cf. lanes 1 and 2). The HZEI-heterogeneous part I alone slightly enhanced glycoprotein expression (Figure 4C, upper panel, lane 3) consistent with its weak enhancing property predicted by HZEI. Similarly, both in the presence and absence of an intact ESSV, the HZEI-negative part II did not support glycoprotein expression (Figure 4C, upper panel, lanes 4 and 5). In contrast, the HZEI-positive part III expected to support U1 snRNP binding strongly increased the levels of gp160 and gp120, confirming ESEvpr activity (20). RT-PCR analysis showed the same pattern for the spliced message (Figure 4C, lower panel). However, a high amplification had to be chosen in order to detect even low levels of the spliced message (cf., e.g. Figure 4C, lower panel, lane 5), leading to an overamplification of the unspliced message in the nonlinear range.
For each part, the HEXplorer score-based prediction of exonic enhancer/silencer property correlated well with glycoprotein expression as surrogate marker of U1 snRNP binding. Therefore, the HEXplorer score may be a valuable screening tool for narrowing down regions with enhancer or silencer property as candidates for more detailed analysis by mutagenesis.
HIV-1 exon 3 inclusion correlates well with HEXplorer score change in ESEvpr mutation analysis
In order to examine HEXplorer predictions of mutation effects on splice site usage, we systematically introduced single- and double-point mutations in the recently described Tra2α-/Tra2β-dependent ESEvpr (20). We used a subgenomic pNLA1-derived four exon splicing reporter (Figure 5A) with an inactivated ESSV resulting in an intermediate exon 3 splicing phenotype (Figure 5E, cf. lanes 1 and 2). This reporter permitted determining positive and negative effects on the degree of exon 3 inclusion. All 16 mutants tested in the ESEvpr region are given in Figure 5B and their positions are denoted by arrows in the HEXplorer graph shown in Figure 5C. Semi-quantitative RT-PCR of RNA isolated from transiently transfected HeLa cells was carried out to measure the level of exon 3 splicing for each mutated minigene.
Figure 5.
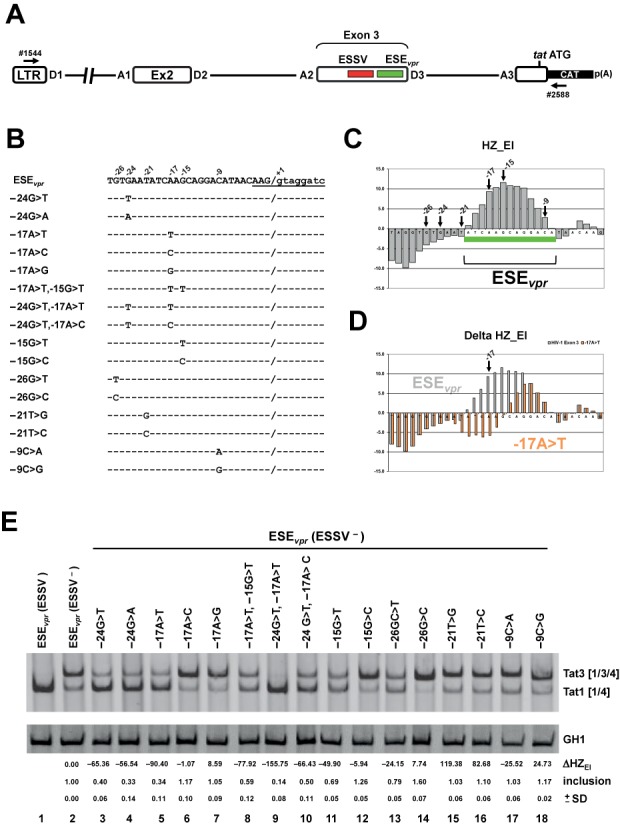
Detailed HEXplorer-guided mutational analysis of ESEvpr. (A) Schematic of the HIV-1-based LTR ex2 ex3 splicing reporter. Positions of the RT-PCR primers (#1544/#2588) are indicated by arrows. (B) ESEvpr reference (pNLA1) and mutant sequences. Nucleotide residues are denoted by their position relative to the GT-dinucleotide. Exon–intron border of exon 3 is denoted by ‘/’ and 5′ss D3 sequence is underlined. (C) HEXplorer score profile plotted along the 3’ end of exon 3 containing ESEvpr (green bar). Positions of mutated nucleotides are indicated by arrows. (D) HEXplorer score profiles for reference ESEvpr (gray) and exemplary point mutation −17A>T (red, indicated by arrow) plotted along the same sequence as in (C). (E) RT-PCR of RNA from HeLa cells transfected with mutants described in (B). HEXplorer score difference ΔHZEI between mutant and reference (ESEvpr (ESSV−) lane 2) as well as exon 3 inclusion (upper band Tat3 [1/3/4]) level is given below each lane. Mean inclusion level and standard deviation (±SD) were calculated from triplicates. GH1 (growth hormone) serves as control for transfection efficiency. 2.5 × 105 HeLa cells were transiently transfected with 1 μg of each of the constructs together with 0.2 μg of SVctat and 1 μg of pXGH5. Thirty hours after transfection RNA was isolated from the cells and subjected to RT-PCR analysis using primer pairs #1544/#2588 and #1224/#1225 (GH1). PCR products were separated by 8% non-denaturating polyacrylamide gel electrophoresis and stained with ethidium bromide.
In order to assess the point mutations’ effects on the enhancer property of the ESEvpr region, we calculated the difference between the two HEXplorer score areas of mutant and reference (intact ESEvpr with ESSV−) sequences in a sufficiently long window upstream of 5′ss D3. We provide the HEXplorer score algorithm in an Excel file as Supplementary material (cf. Supplementary XLSM-file) and for download at www.uni-duesseldorf.de/rna. Any such point mutation affected only the HEXplorer scores of 11 nucleotides centered on the point mutation itself (5 nt up- and downstream; Figure 5D). A HEXplorer score difference of zero thus corresponded to comparable enhancer property levels in mutant and reference, which in this case exhibited partial exon inclusion. Figure 5E shows the exon 3 inclusion ratio and HEXplorer score difference ΔHZEI for wild type, reference (intact ESEvpr with ESSV−) and all 16 mutants.
Observed changes both for higher and lower levels of exon 3 inclusion were mostly consistent with the HEXplorer score differences ΔHZEI mutant–reference. A large positive or slightly negative HEXplorer score difference was found, if the mutant sequence had a stronger enhancer property than the reference (intact ESEvpr with ESSV−), and such mutants were associated with a higher degree of ESEvpr-dependent exon inclusion (e.g. Figure 5E, lanes 12–18). Correspondingly, negative HEXplorer score differences were mostly found in mutant sequences with considerably weaker splice enhancing property than the reference (e.g. Figure 5E, lanes 3–5 and 9, 10). Thus, mutants with large negative ΔHZEI were prone to disrupt the enhancer ESEvpr.
These experiments showed a good semi-quantitative correlation between inclusion ratio and HEXplorer score difference in a series of mutations in the splicing enhancer ESEvpr. In the next step, we applied the HEXplorer score to ESSV mutation optimization.
HEXplorer pre-screening identifies minimal point mutations disrupting ESSV
Using the HEXplorer score-based mutation assessment, automatic screening of entire sequence regions for point mutations with maximal effects became possible. To this end, every single nucleotide in the region was systematically replaced with each of the three other possible nucleotides by the HEXplorer algorithm. From the 3·N alternative sequences obtained from an N nucleotide long sequence, point mutations with similar HEXplorer score differences were suspected to alter the sequence's enhancing/silencing property to a comparable degree.
We tested this procedure on the splicing silencer ESSV. Starting from the known mutation ESSV− (pNEU), which inactivates ESSV by substituting 7 nucleotides (19) (Figure 6A), we searched for a functionally equivalent but smaller set of point mutations. The known mutation ESSV− (pNEU) had a higher HEXplorer score compared to the reference sequence pNL4-3 (ΔHZEI = 144.0). From a thorough scrutiny of the HEXplorer score table we identified the double mutation −29G>C and −36A>C that together increased the pNL4-3 HEXplorer score in a similar order of magnitude (ΔHZEI = 157.9). From Figure 6B it is evident that this double mutation ESSV− (dm) changed the shape of the HEXplorer graph in a similar way as ESSV− (pNEU). In comparison, each single-point mutation had a smaller effect on the HEXplorer score: ΔHZEI = 92.8 for −29G>C and 65.0 for −36A>C, respectively.
Figure 6.
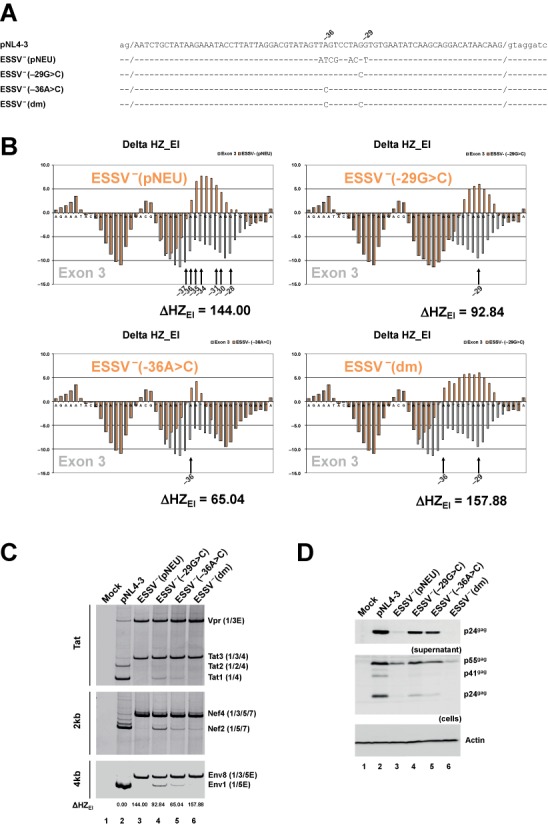
HEXplorer screening identifies minimal point mutations disrupting ESSV. (A) pNL4-3-derived wild-type and mutated HIV-1 exon 3 sequences. Nucleotide coordinates refer to the 3’ end of exon 3. (dm) refers to the double mutation −29G>C and -36A>C. (B) HZEI score profiles of HIV exon 3 reference pNL4–3 (gray) versus mutant sequences ESSV− (pNEU), ESSV− −29G>C, ESSV− −36A>C and ESSV− (dm) (red). HEXplorer score differences are given below the profile graphs, and point mutations are indicated by black arrows and corresponding positions. (C) RT-PCR analysis of splicing patterns for different classes of viral RNAs. 2.5 × 105 HEK 293T cells were transiently transfected with 1 μg of each of the proviral DNAs. Thirty hours after transfection, total RNA was isolated from the cells and subjected to RT-PCR analyses with different sets of primer pairs covering intronless and intron-containing mRNA classes described elsewhere (20). RT-PCR products were separated by 8% non-denaturing polyacrylamide gel electrophoresis and visualized using ethidium bromide staining. HIV-1 mRNA isoforms are indicated on the right and correspond to the nomenclature published previously (41). HZEI-score differences (ΔHZEI) with respect to the reference pNL4-3 are given below the gel. (D) Western blot analysis of Gag expressed by reference and mutant proviruses. 2.5 × 105 HEK 293T cells were transiently transfected with 1 μg of each of the proviral DNAs. Forty-eight hours post transfection viral supernatants were collected, layered onto 20% sucrose solution and centrifuged at 28 000 rpm for 1.30 h at 4°C to pellet the released viral particles. In addition, cells were harvested and resuspended in lysis buffer. Supernatants and cellular lysates were resolved in 12% SDS-PAGE and electroblotted on nitrocellulose membranes. To determine virus particle production and the expression of viral proteins, samples were probed with a primary antibody against HIV-1 p24 (Biochrom AG). Equal amounts of cell lysates were controlled by the detection of α-actin (Sigma-Aldrich, A2228).
These mutations were now experimentally analyzed for their ability to disrupt the ESSV activity within the infectious molecular clone NL4-3 (GenBank Accession No. M19921). Following transfection of HEK 293T cells with pNL4-3 or the ESSV mutants, the HIV-1 splicing pattern was determined by semi-quantitative RT-PCR using primer pairs spanning intronless or intron-containing HIV-1 mRNA classes (Figure 6C). In NL4-3, only low levels of exon 3-including viral mRNA species could be detected (Figure 6C, lane 2, e.g. Tat3 or Nef4) due to the silencing activity of ESSV. As expected from preceding studies (19,20) and in consistence with the higher HEXplorer score (ΔHZEI = 144.0) relative to pNL4-3, the ESSV− (pNEU) mutation led to an almost complete exon 3 inclusion (Figure 6C, cf. lanes 2 and 3, e.g. Tat3 or Nef4) obviously at the expense of the respective exon 3-lacking isoforms (Figure 6C, cf. lanes 2 and 3, e.g. Tat1 or Nef2). In agreement with their increased HEXplorer scores (ΔHZEI = 92.8 for −29G>C and 65.0 for −36A>C), each of the HEXplorer-identified single-point mutations was already capable to substantially increase the levels of exon 3 inclusion (Figure 6C, lanes 4 and 5, e.g. Tat3 or Nef4). However, both point mutations still retained some mRNAs lacking exon 3 (Figure 6C, cf. lanes 4 and 5, e.g. Tat1 or Nef2), and were thus less efficient in including exon 3 than ESSV− (pNEU). The same observations were made within the 4-kb class mRNAs (Figure 6C; 4-kb mRNAs). Both in 2-kb and 4-kb mRNA classes, the double-mutation ESSV− (dm) was as efficient in promoting exon 3 inclusion as ESSV− (pNEU) (Figure 6C, cf. lanes 3 and 6), which was consistent with a HEXplorer score difference ΔHZEI = 157.9 similar to ESSV− (pNEU), but clearly exceeding the single-point mutations’ ΔHZEI.
Transfection experiments were complemented by western blot analyses of intracellular Gag protein levels (Figure 6D). To evaluate the importance of ESSV for virus particle production, cell-free supernatants were harvested from HEK 293T cells transfected with proviral DNAs, and viral release for each sample was detected via p24 levels. Western blot results revealed that ESSV− (pNEU) led to low amounts of intracellular Gag protein (Figure 6D, cf. lanes 2 and 3), as expected from excessive exon 3 splicing causing a replication defect (19). While the single-point mutations ESSV− (−29G>C) and ESSV− (−36A>C) partially inactivated ESSV leading to slight replication defects as seen in Gag protein and virus particle production (Figure 6D, cf. lanes 2, 4 and 5), the double mutation ESSV− (dm) showed Gag protein levels not exceeding those detected in ESSV− (pNEU) both within cells and supernatants (Figure 6D, cf. lanes 3 and 6).
In these experiments, we have exemplified the HEXplorer-score-guided mutagenesis inactivating the exonic splicing silencer ESSV with just two point mutations, obtaining an efficiency comparable to the known 7-nucleotide mutation ESSV− (pNEU).
A HEXplorer-based screening of the HIV-1 genome uncovers novel SREs
In the next step, we aimed at discovering novel ESEs guided by the HEXplorer profiles of all HIV-1 exons shown in Figure 3. To this end, we first identified exonic sequences with uninterrupted positive HEXplorer scores flanked by valley regions with lower positive or negative HEXplorer scores. In these typically 20-40-nucleotide long regions with supposed exonic enhancer property we searched for point mutations that disrupted the putative enhancer sequences. The mutation positions were selected inside the HEXplorer positive regions, and the substituted nucleotide was chosen to maximize the HEXplorer score difference ΔHZEI.
Both the putative ESE containing reference and mutated sequences were tested in the context of the ESE-dependent SV-env splicing reporter ((8,46); Figure 4B). The exonic splicing enhancer SRSF7 binding site and the splicing neutral sequence were used for calibration of ESE strength (8,48). Since this analysis covers different exons, we denote mutated positions with a single coordinate counting from the start of the NL4-3 genome.
In exon 1, we narrowed down the recently published enhancer region upstream of the major 5′ss D1 (25) by identifying the double mutation 708G>T, 718C>G that effectively suppressed splice site usage in a degree comparable to the neutral sequence (Figure 7A, cf. lanes 2–4).
Figure 7.
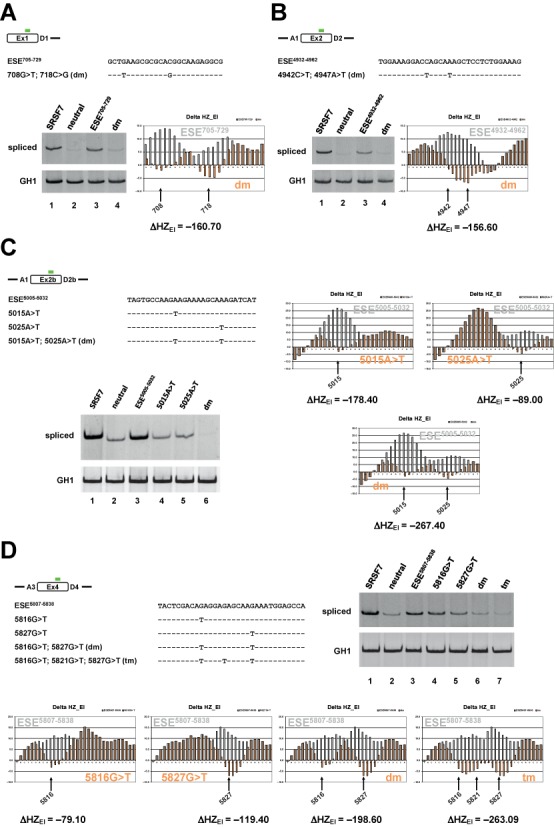
HEXplorer screening identifies novel SREs within the HIV-1 genome. Novel exonic splicing enhancers were identified by HEXplorer screening of HIV-1 exons 1 (A), 2 (B), 2b (C), 4 (D), and were experimentally confirmed by HEXplorer-predicted mutations within the SV-env reporter construct described in Figure 4B. For each HIV-1 exon, experimentally tested reference and mutated sequences are given below a schematic representation of their location (green bars). All positions in HEXplorer-predicted enhancer and mutant sequences are counted from the start of the HIV-1 NL4-3 genome. HEXplorer profiles span the entire experimentally tested sequences. HEXplorer score differences are given below the profile graphs, and mutated nucleotides are indicated by arrows and corresponding positions (dm: double mutation; tm: triple mutation). 2.5 × 105 HeLa cells were transiently transfected with 1 μg of each of the splicing reporters and 1 μg of pXHG5 for normalization. Thirty hours post transfection, RNA was extracted and reverse transcribed using the resultant cDNA in a PCR reaction with primer pair #3210/#3211. To control for equal transfection efficiency, we also performed a separate PCR reaction with primer pair #1224/#1225 detecting constitutively spliced GH1 mRNA. RT-PCR products were resolved on 8% non-denaturing polyacrylamide gels and stained with ethidium bromide.
In exon 2, we discovered a novel enhancer between ESEM1 and ESEM2 (26) that was inactivated by the double mutation 4942C>T, 4947A>T. This novel enhancer was weaker than the SRSF7 binding site, but was still functional as part of an array of consecutive enhancer motifs possibly compensating for its lack of strength (ESE-Vif, ESEM1, ESEM2; Figure 7B, cf. lanes 1–4).
By mutation of the G-run splicing silencer GI2–1, usage of alternative 5′ss D2b can be significantly increased (23). In this exon 2b, we discovered the novel enhancer ESE5005–5032 upstream of GI2–1, which was almost completely inactivated by the point mutation 5015A>T, and fully disabled by the double mutation 5015A>T, 5025A>T (Figure 7C, cf. lanes 1–6). The different degree of ESE5005–5032 inactivation was reflected by a larger HEXplorer score difference ΔHZEI = −267.4 for the double mutation compared to ΔHZEI = −178.4 for 5015A>T or ΔHZEI = −89.0 for 5025A>T.
A particularly interesting example was discovered in exon 4: the HEXplorer score profile indicated an exonic region with strong enhancing property wedged between the known silencer ESS2p and the known enhancer ESE2 (Figure 3). The enhancing property of this region was confirmed by the two single-point mutations 5816G>T (ΔHZEI = −79.1) and 5827G>T (ΔHZEI = −119.4), and the corresponding double mutation (ΔHZEI = −198.6) that reduced D4 splice site usage to a similar degree as the neutral sequence (Figure 7D, cf. lanes 2 and 6). The single-point mutations achieved only an intermediate reduction of D4 usage in line with the respective HEXplorer score differences (Figure 7D, cf. lanes 3–5). Furthermore, the additional third mutation 5821G>T (ΔHZEI = −263.1) slightly reduced both D4 usage and HEXplorer score below the levels of the double mutation. By analyzing the same RNA samples with primer pairs also detecting the unspliced messages, we additionally confirmed that the reduced amount of spliced messages was indeed due to mutation of an SRE (Supplementary Figure S1).
Taken together, HEXplorer score profiling permitted us to firstly identify novel putative splice enhancing regions in HIV-1 exons 1, 2, 2b and 4 and to secondly find specific enhancer inactivating mutations inside these regions, which can serve to identify enhancer binding proteins. In all cases, splicing reporter experiments qualitatively confirmed the HEXplorer predicted effects of enhancer mutations.
HEXplorer score difference quantitatively correlates with HIV-1 splicing activity
In order to quantitatively compare HEXplorer score differences with changes in exon recognition or splice site usage even across different reporter systems, we plotted splicing activity versus HEXplorer score difference ΔHZEI between pairs of mutated and reference sequences. In experiments using the 4-exon splicing reporter we measured splicing activity by exon inclusion rate defined as inclusion/(inclusion + exclusion), while in the single-intron splicing reporter experiments we used the ratio of 5′ splice site usage/GH1 transfection control instead. To account for different reference splicing activity levels across different exons and reporter systems, we separately normalized reference splicing activity to 100% in each reporter. In Figure 8, all 16 point mutations examined in the 4-exon splicing reporter (Figure 5) are represented by red squares, while data from four point mutations within ESSV− (pNEU) (Figure 6, and one additional mutation not shown) and nine point mutations obtained from exons 1, 2, 2b and 4 (Figure 7) are denoted by individual symbols. Across all these experiments, we obtained an excellent correlation of r = 0.85 (P < 0.001) between splicing activity and HEXplorer score difference. These promising results suggest that the HEXplorer score can be effectively used to (i) determine the splice enhancing or silencing property of an entire region and (ii) predict the effects of point mutations on splice site usage.
Figure 8.
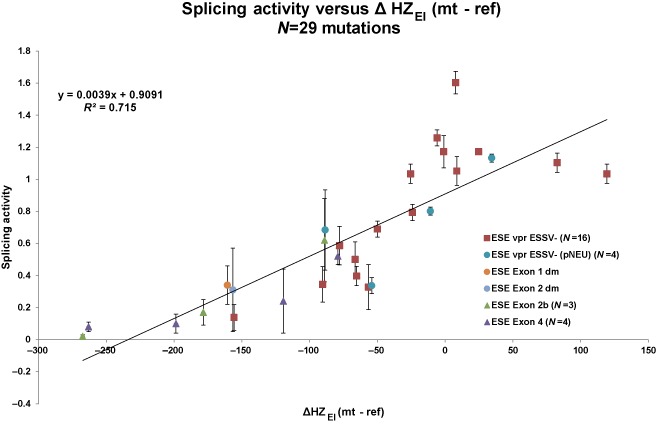
HEXplorer score difference quantitatively correlates with HIV-1 splicing activity. In the calibration experiment (Figure 5), exon 3 inclusion ratio was determined in triplicate for 16 single or double mutations in the ESEvpr region upstream 5‘ss D3. The exon 3 inclusion ratio showed a linear correlation of r = 0.75 with changes in hexamer score HZEI (red squares). Adding 13 mutations from a series of control experiments involving HIV exons 1–4 (colored circles and triangles denote exons; cf. Figure 7) even improved the correlation for the entire data set (r = 0.85, 16 + 13 mutations; error bars denote standard deviations from triplicates; N denotes the number of mutations per exon).
HEXplorer score difference correlates well with various available SRE algorithms
We additionally applied two different approaches to compare the HEXplorer score with other available algorithms for the identification of SREs, using the data set of 29 experimentally tested mutations.
First, for each of the 29 mutations shown in Figure 8, we determined all ESR motifs predicted to be overlapping with the mutated positions by the ESEfinder (10,12), RESCUE-ESE (9,49), FAS-ESS-hex3 (13), PESX (11) or ESRsearch (14) algorithms. We aggregated the results of the different ESR-identifying algorithms by assigning +1 for each ESE and −1 for each ESS motif, similar to the approach chosen in (44). Totaling these ±1-weights for every pair of reference and mutant sequences, we obtained the ‘mutant–reference’ ESMD, an integer number measuring the overall net gain and loss of ESE and ESS. For all 29 mutations, we obtained an excellent correlation of r = 0.93 (P < 0.001) between ESMD and HEXplorer score difference (see Supplementary Figure S2).
Second, we compared the individual exon–intron and weak–strong hexamer scores (ZEI and ZWS) to the ESRseq score available for all 1182 ESE and 1090 ESS hexamers identified as exonic SREs in (15). From the scatterplots in Supplementary Figure S3 we obtained a good correlation of r = 0.72 with both ZEI and ZWS. Finally, for all 29 mutations shown in Figure 8, we calculated the ESRseq difference between mutant and reference sequences, using the same averaging approach as in the HEXplorer score definition, but with the ESRscore hexamer weights instead of ZEI. We again found an excellent correlation of r = 0.93 between the ESRseq difference and HEXplorer score difference (mutant–reference) as shown in Supplementary Figure S4.
Although predictions of individual currently available ESR-identifying algorithms are not necessarily consistent with each other, our results indicate that there is a considerable agreement between the HEXplorer score prediction of SREs—in particular mutation effects—and the average of all other algorithms for ESR motif identification examined in this context.
DISCUSSION
Hexamer over-representation between different data sets of exonic and intronic sequences has been successfully used as a basis for the identification of exonic SREs (9,11). In the present study, we extended this approach by incorporating all hexamer frequencies of an entire sequence region into a novel HEXplorer score HZEI that was calculated as the average exon–intron hexamer ZEI-score of all six hexamers overlapping with any given index nucleotide. Capturing the overall hexamer content of an entire sequence region, HEXplorer score profiles plotted along exonic sequences exhibited a continuous spectrum of exonic enhancing and silencing properties in the context of a given splice site, rather than the presence or absence of individual predicted SRE hexamer binding sites.
Genome wide analyses (7,17,50–53) as well as systematic splicing reporter experiments (8) have confirmed that SREs act in a position-dependent manner: classical splice enhancing SR-proteins bind upstream of a 5′ss, but inhibit splicing from an intronic position. In a similar way, the typical exonic splice silencing hnRNP H/F proteins act as enhancers from intronic positions. Based upon the respective different exonic and intronic hexamer distributions, the HEXplorer score HZEI presumably captures these position-dependent splicing regulatory sequence properties in the context of any given splice site.
For accurate splicing, nearby splice regulatory elements often modulate proper splice site motif recognition, and they may be evolutionarily adapted to particular splice site properties (e.g. intrinsic strength). Therefore, splice enhancing or silencing sequence properties must always be rated in the context of the actual splice site, and this rating can be different in the vicinity of weak splice sites than near strong ones. Thus, while HEXplorer score ‘differences’ upstream of a given 5′ss have proven quantitatively valid in mutation analyses, the comparative validity of ‘absolute’ HEXplorer area size was limited to its 5′ss context, and care must be taken in comparing different splice sites. It is therefore natural that the absolute HEXplorer score area size should be most meaningful together with the intrinsic 5′ss strength, measured e.g. by its maxent or HBS. A systematic examination of the interplay between intrinsic splice site strength and neighboring HEXplorer score profile and their merging into a ‘functional splice site score’ could further the understanding of normal and pathological splice site usage.
Using the HIV-1 genome heavily relying on SREs (40) as a model system, we experimentally examined whether the HEXplorer score faithfully represented the respective enhancing or silencing properties of genomic regions. In a helicopter view, we first generated HEXplorer score profiles of each HIV-1 exon exhibiting specific HEXplorer score positive and negative regions that generally coincided with known exonic enhancers or silencers.
In a set of systematic point mutations of the exemplarily chosen exonic enhancer ESEvpr, the HEXplorer score correlated well with exon inclusion in a four-exon splicing reporter. Outside this splicing reporter, ESEvpr was previously examined in a proviral context. In the absence of the repressing ESSV, strong exon 3 splice site activation led to reduced levels of unspliced viral mRNA and a deficiency in virus particle production (19), while the release of virus particles into the supernatant could be rescued by an ESEvpr double mutation that was originally suggested by the HEXplorer algorithm (20). These experiments confirmed that the four-exon splicing reporter reliably captured HIV-1 splice site usage.
Based on HEXplorer profile prediction, we discovered five novel splicing enhancers in HIV-1 exons and experimentally confirmed all of them by mutagenesis. Furthermore, we were able to design a novel, reduced set of point mutations disrupting the splicing silencer ESSV to a similar degree as the established ESSV− (pNEU). Based on the relative occurrences of all hexamers, the HEXplorer score permitted optimizing mutagenesis by a priori determining the point mutation(s) with the largest effects on the exonic enhancing or silencing property of a given sequence. Moreover, using HEXplorer score predictions of point mutation effects on splicing now opens the perspective to design silent mutations interfering with splice site usage while not altering the amino acid sequence of the encoded protein.
The HEXplorer score definition was derived from a specific set of exonic and intronic sequences extracted from the ENSEMBL database, and it is therefore based on pooled sequences expressed in different human tissues. Hexamer weights derived in this way cannot take into account tissue-specific protein expression and possibly fail to detect SRE motifs bound by splicing regulatory proteins that are not ubiquitously expressed (54–57). On the contrary, HEXplorer score predictions of regulatory properties may be invalid for a specific tissue not expressing a required splicing regulatory protein. This principal deficiency, however, holds for all algorithms identifying SREs from pooled sequence data. Future applications may overcome these limitations by deriving hexamer weights from cell-type-specific transcriptome data sets obtained from RNA deep sequencing.
In the HEXplorer score definition, the choice of exonic/intronic neighborhoods entering into the RESCUE-like hexamer weight calculation was somewhat arbitrary. Since the median size of a middle exon in human genes is 123 nucleotides (58), we chose 100-nt wide regions (or the entire exon if it was shorter). These sets of exonic and intronic sequences were sufficiently large that all hexamers were multiply represented, but shorter than the sequences used in the original work (9). Experimentally, most splice enhancers and silencers can be expected to lie within a 100-nucleotide splice site neighborhood. We comparatively evaluated an even smaller neighborhood size of only 30 nucleotides (data not shown) and found an excellent correlation of r = 0.96 between the respective exon–intron scores of all 4096 hexamers. Thus, within the range of values covered, the size of the splice site neighborhoods used for the HEXplorer score definition has only limited impact on the HEXplorer score values obtained.
We chose to derive the HEXplorer score from exon–intron hexamer scores ZEI based on heuristic arguments that splicing enhancing or silencing properties differ more widely between exonic and intronic splice site neighborhoods than between exons of strong and weak splice sites. To examine the validity of this choice, we also calculated analogous ZWS-based HEXplorer score differences for all 29 mutations shown in Figure 8. From these ZWS-based HEXplorer scores, we obtained a weaker correlation of r = 0.75 with experimentally determined splicing activity than from ΔHZEI, which was consistent with our assumption.
The novel computational HEXplorer score profiles provide a global landscaping of splicing regulatory regions, as well as a quantitative measure of mutation effects on their splice enhancing and silencing properties in the vicinity of a given splice site. In particular, HEXplorer score calculation may significantly alleviate mutational analyses by reliably predicting possibly silent point mutations most effectively disrupting or even creating SREs. This is especially helpful for the identification of proteins binding to regulatory elements. HEXplorer score analyses may also further the computational prediction of pathogenic mutation effects in human genetics.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Björn Wefers, Sarah Otten and Sebastian Wittich for excellent technical assistance.
FUNDING
Deutsche Forschungsgemeinschaft (DFG) [SCHA 909/3-1]; Stiftung für AIDS-Forschung, Düsseldorf [to H.S.]; Jürgen Manchot Stiftung [to M.W., J.O.P. and H.S.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Pan Q., Shai O., Lee L.J., Frey B.J., Blencowe B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 2.Wahl M.C., Will C.L., Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Barash Y., Calarco J.A., Gao W., Pan Q., Wang X., Shai O., Blencowe B.J., Frey B.J. Deciphering the splicing code. Nature. 2010;465:53–59. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- 4.Wang Z., Burge C.B. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blencowe B.J. Alternative splicing: new insights from global analyses. Cell. 2006;126:37–47. doi: 10.1016/j.cell.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 6.Nilsen T.W., Graveley B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim K.H., Ferraris L., Filloux M.E., Raphael B.J., Fairbrother W.G. Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc. Natl Acad. Sci. U.S.A. 2011;108:11093–11098. doi: 10.1073/pnas.1101135108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Erkelenz S., Mueller W.F., Evans M.S., Busch A., Schoneweis K., Hertel K.J., Schaal H. Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms. RNA. 2013;19:96–102. doi: 10.1261/rna.037044.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fairbrother W.G., Yeh R.F., Sharp P.A., Burge C.B. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297:1007–1013. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- 10.Cartegni L., Wang J., Zhu Z., Zhang M.Q., Krainer A.R. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X.H., Chasin L.A. Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev. 2004;18:1241–1250. doi: 10.1101/gad.1195304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith P.J., Zhang C., Wang J., Chew S.L., Zhang M.Q., Krainer A.R. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum. Mol. Genet. 2006;15:2490–2508. doi: 10.1093/hmg/ddl171. [DOI] [PubMed] [Google Scholar]
- 13.Wang Z., Rolish M.E., Yeo G., Tung V., Mawson M., Burge C.B. Systematic identification and analysis of exonic splicing silencers. Cell. 2004;119:831–845. doi: 10.1016/j.cell.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 14.Goren A., Ram O., Amit M., Keren H., Lev-Maor G., Vig I., Pupko T., Ast G. Comparative analysis identifies exonic splicing regulatory sequences—the complex definition of enhancers and silencers. Mol. Cell. 2006;22:769–781. doi: 10.1016/j.molcel.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 15.Ke S., Shang S., Kalachikov S.M., Morozova I., Yu L., Russo J.J., Ju J., Chasin L.A. Quantitative evaluation of all hexamers as exonic splicing elements. Genome Res. 2011;21:1360–1374. doi: 10.1101/gr.119628.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mort M., Sterne-Weiler T., Li B., Ball E.V., Cooper D.N., Radivojac P., Sanford J.R., Mooney S.D. MutPred Splice: machine learning-based prediction of exonic variants that disrupt splicing. Genome Biol. 2014;15:R19. doi: 10.1186/gb-2014-15-1-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cereda M., Pozzoli U., Rot G., Juvan P., Schweitzer A., Clark T., Ule J. RNAmotifs: prediction of multivalent RNA motifs that control alternative splicing. Genome Biol. 2014;15:R20. doi: 10.1186/gb-2014-15-1-r20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Betz B., Theiss S., Aktas M., Konermann C., Goecke T.O., Moslein G., Schaal H., Royer-Pokora B. Comparative in silico analyses and experimental validation of novel splice site and missense mutations in the genes MLH1 and MSH2. J. Cancer Res. Clin. Oncol. 2010;136:123–134. doi: 10.1007/s00432-009-0643-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Madsen J.M., Stoltzfus C.M. An exonic splicing silencer downstream of the 3’ splice site A2 is required for efficient human immunodeficiency virus type 1 replication. J. Virol. 2005;79:10478–10486. doi: 10.1128/JVI.79.16.10478-10486.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erkelenz S., Poschmann G., Theiss S., Stefanski A., Hillebrand F., Otte M., Stuhler K., Schaal H. Tra2-mediated recognition of HIV-1 5’ splice site D3 as a key factor in the processing of vpr mRNA. J. Virol. 2013;87:2721–2734. doi: 10.1128/JVI.02756-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bilodeau P.S., Domsic J.K., Mayeda A., Krainer A.R., Stoltzfus C.M. RNA splicing at human immunodeficiency virus type 1 3’ splice site A2 is regulated by binding of hnRNP A/B proteins to an exonic splicing silencer element. J. Virol. 2001;75:8487–8497. doi: 10.1128/JVI.75.18.8487-8497.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Domsic J.K., Wang Y., Mayeda A., Krainer A.R., Stoltzfus C.M. Human immunodeficiency virus type 1 hnRNP A/B-dependent exonic splicing silencer ESSV antagonizes binding of U2AF65 to viral polypyrimidine tracts. Mol. Cell. Biol. 2003;23:8762–8772. doi: 10.1128/MCB.23.23.8762-8772.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Widera M., Erkelenz S., Hillebrand F., Krikoni A., Widera D., Kaisers W., Deenen R., Gombert M., Dellen R., Pfeiffer T., et al. An intronic G run within HIV-1 intron 2 is critical for splicing regulation of vif mRNA. J. Virol. 2013;87:2707–2720. doi: 10.1128/JVI.02755-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kammler S., Leurs C., Freund M., Krummheuer J., Seidel K., Tange T.O., Lund M.K., Kjems J., Scheid A., Schaal H. The sequence complementarity between HIV-1 5’ splice site SD4 and U1 snRNA determines the steady-state level of an unstable env pre-mRNA. RNA. 2001;7:421–434. doi: 10.1017/s1355838201001212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Asang C., Erkelenz S., Schaal H. The HIV-1 major splice donor D1 is activated by splicing enhancer elements within the leader region and the p17-inhibitory sequence. Virology. 2012;432:133–145. doi: 10.1016/j.virol.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 26.Kammler S., Otte M., Hauber I., Kjems J., Hauber J., Schaal H. The strength of the HIV-1 3’ splice sites affects Rev function. Retrovirology. 2006;3:89. doi: 10.1186/1742-4690-3-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Exline C.M., Feng Z., Stoltzfus C.M. Negative and positive mRNA splicing elements act competitively to regulate human immunodeficiency virus type 1 vif gene expression. J. Virol. 2008;82:3921–3931. doi: 10.1128/JVI.01558-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacquenet S., Mereau A., Bilodeau P.S., Damier L., Stoltzfus C.M., Branlant C. A second exon splicing silencer within human immunodeficiency virus type 1 tat exon 2 represses splicing of Tat mRNA and binds protein hnRNP H. J. Biol. Chem. 2001;276:40464–40475. doi: 10.1074/jbc.M104070200. [DOI] [PubMed] [Google Scholar]
- 29.Zahler A.M., Damgaard C.K., Kjems J., Caputi M. SC35 and heterogeneous nuclear ribonucleoprotein A/B proteins bind to a juxtaposed exonic splicing enhancer/exonic splicing silencer element to regulate HIV-1 tat exon 2 splicing. J. Biol. Chem. 2004;279:10077–10084. doi: 10.1074/jbc.M312743200. [DOI] [PubMed] [Google Scholar]
- 30.Hallay H., Locker N., Ayadi L., Ropers D., Guittet E., Branlant C. Biochemical and NMR study on the competition between proteins SC35, SRp40, and heterogeneous nuclear ribonucleoprotein A1 at the HIV-1 Tat exon 2 splicing site. J. Biol. Chem. 2006;281:37159–37174. doi: 10.1074/jbc.M603864200. [DOI] [PubMed] [Google Scholar]
- 31.Si Z., Amendt B.A., Stoltzfus C.M. Splicing efficiency of human immunodeficiency virus type 1 tat RNA is determined by both a suboptimal 3’ splice site and a 10 nucleotide exon splicing silencer element located within tat exon 2. Nucleic Acids Res. 1997;25:861–867. doi: 10.1093/nar/25.4.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amendt B.A., Hesslein D., Chang L.J., Stoltzfus C.M. Presence of negative and positive cis-acting RNA splicing elements within and flanking the first tat coding exon of human immunodeficiency virus type 1. Mol. Cell. Biol. 1994;14:3960–3970. doi: 10.1128/mcb.14.6.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caputi M., Mayeda A., Krainer A.R., Zahler A.M. hnRNP A/B proteins are required for inhibition of HIV-1 pre-mRNA splicing. EMBO J. 1999;18:4060–4067. doi: 10.1093/emboj/18.14.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Asang C., Hauber I., Schaal H. Insights into the selective activation of alternatively used splice acceptors by the human immunodeficiency virus type-1 bidirectional splicing enhancer. Nucleic Acids Res. 2008;36:1450–1463. doi: 10.1093/nar/gkm1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tange T.O., Damgaard C.K., Guth S., Valcarcel J., Kjems J. The hnRNP A1 protein regulates HIV-1 tat splicing via a novel intron silencer element. EMBO J. 2001;20:5748–5758. doi: 10.1093/emboj/20.20.5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Staffa A., Cochrane A. Identification of positive and negative splicing regulatory elements within the terminal tat-rev exon of human immunodeficiency virus type 1. Mol. Cell. Biol. 1995;15:4597–4605. doi: 10.1128/mcb.15.8.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amendt B.A., Si Z.H., Stoltzfus C.M. Presence of exon splicing silencers within human immunodeficiency virus type 1 tat exon 2 and tat-rev exon 3: evidence for inhibition mediated by cellular factors. Mol. Cell. Biol. 1995;15:4606–4615. doi: 10.1128/mcb.15.8.4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Si Z.H., Rauch D., Stoltzfus C.M. The exon splicing silencer in human immunodeficiency virus type 1 Tat exon 3 is bipartite and acts early in spliceosome assembly. Mol. Cell. Biol. 1998;18:5404–5413. doi: 10.1128/mcb.18.9.5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stoltzfus C.M., Madsen J.M. Role of viral splicing elements and cellular RNA binding proteins in regulation of HIV-1 alternative RNA splicing. Curr. HIV Res. 2006;4:43–55. doi: 10.2174/157016206775197655. [DOI] [PubMed] [Google Scholar]
- 40.Stoltzfus C.M. Chapter 1. Regulation of HIV-1 alternative RNA splicing and its role in virus replication. Adv. Virus Res. 2009;74:1–40. doi: 10.1016/S0065-3527(09)74001-1. [DOI] [PubMed] [Google Scholar]
- 41.Purcell D.F., Martin M.A. Alternative splicing of human immunodeficiency virus type 1 mRNA modulates viral protein expression, replication, and infectivity. J. Virol. 1993;67:6365–6378. doi: 10.1128/jvi.67.11.6365-6378.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Voelker R.B., Erkelenz S., Reynoso V., Schaal H., Berglund J.A. Frequent gain and loss of intronic splicing regulatory elements during the evolution of vertebrates. Genome Biol. Evol. 2012;4:659–674. doi: 10.1093/gbe/evs051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Freund M., Asang C., Kammler S., Konermann C., Krummheuer J., Hipp M., Meyer I., Gierling W., Theiss S., Preuss T., et al. A novel approach to describe a U1 snRNA binding site. Nucleic Acids Res. 2003;31:6963–6975. doi: 10.1093/nar/gkg901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ke S., Zhang X.H., Chasin L.A. Positive selection acting on splicing motifs reflects compensatory evolution. Genome Res. 2008;18:533–543. doi: 10.1101/gr.070268.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hartmann L., Theiss S., Niederacher D., Schaal H. Diagnostics of pathogenic splicing mutations: does bioinformatics cover all bases. Front. Biosci. 2008;13:3252–3272. doi: 10.2741/2924. [DOI] [PubMed] [Google Scholar]
- 46.Caputi M., Freund M., Kammler S., Asang C., Schaal H. A bidirectional SF2/ASF- and SRp40-dependent splicing enhancer regulates human immunodeficiency virus type 1 rev, env, vpu, and nef gene expression. J. Virol. 2004;78:6517–6526. doi: 10.1128/JVI.78.12.6517-6526.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strebel K., Daugherty D., Clouse K., Cohen D., Folks T., Martin M.A. The HIV ‘A’ (sor) gene product is essential for virus infectivity. Nature. 1987;328:728–730. doi: 10.1038/328728a0. [DOI] [PubMed] [Google Scholar]
- 48.Zhang X.H., Arias M.A., Ke S., Chasin L.A. Splicing of designer exons reveals unexpected complexity in pre-mRNA splicing. RNA. 2009;15:367–376. doi: 10.1261/rna.1498509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fairbrother W.G., Yeo G.W., Yeh R., Goldstein P., Mawson M., Sharp P.A., Burge C.B. RESCUE-ESE identifies candidate exonic splicing enhancers in vertebrate exons. Nucleic Acids Res. 2004;32:W187–W190. doi: 10.1093/nar/gkh393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ule J., Stefani G., Mele A., Ruggiu M., Wang X., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–586. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- 51.Llorian M., Schwartz S., Clark T.A., Hollander D., Tan L.Y., Spellman R., Gordon A., Schweitzer A.C., de la Grange P., Ast G., et al. Position-dependent alternative splicing activity revealed by global profiling of alternative splicing events regulated by PTB. Nat. Struct. Mol. Biol. 2010;17:1114–1123. doi: 10.1038/nsmb.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huelga S.C., Vu A.Q., Arnold J.D., Liang T.Y., Liu P.P., Yan B.Y., Donohue J.P., Shiue L., Hoon S., Brenner S., et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 2012;1:167–178. doi: 10.1016/j.celrep.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rossbach O., Hung L.H., Khrameeva E., Schreiner S., Konig J., Curk T., Zupan B., Ule J., Gelfand M.S., Bindereif A. Crosslinking-immunoprecipitation (iCLIP) analysis reveals global regulatory roles of hnRNP L. RNA Biol. 2014;11:146–155. doi: 10.4161/rna.27991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Castle J.C., Zhang C., Shah J.K., Kulkarni A.V., Kalsotra A., Cooper T.A., Johnson J.M. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat. Genet. 2008;40:1416–1425. doi: 10.1038/ng.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Norris A.D., Gao S., Norris M.L., Ray D., Ramani A.K., Fraser A.G., Morris Q., Hughes T.R., Zhen M., Calarco J.A. A pair of RNA-binding proteins controls networks of splicing events contributing to specialization of neural cell types. Mol. Cell. 2014;54:946–959. doi: 10.1016/j.molcel.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grosso A.R., Gomes A.Q., Barbosa-Morais N.L., Caldeira S., Thorne N.P., Grech G., von Lindern M., Carmo-Fonseca M. Tissue-specific splicing factor gene expression signatures. Nucleic Acids Res. 2008;36:4823–4832. doi: 10.1093/nar/gkn463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Venables J.P., Brosseau J.P., Gadea G., Klinck R., Prinos P., Beaulieu J.F., Lapointe E., Durand M., Thibault P., Tremblay K., et al. RBFOX2 is an important regulator of mesenchymal tissue-specific splicing in both normal and cancer tissues. Mol. Cell. Biol. 2013;33:396–405. doi: 10.1128/MCB.01174-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scherer S. A Short Guide to the Human Genome. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.