


Abstract
The c-MYC (MYC afterward) oncogene is well known for driving numerous oncogenic programs. However, MYC can also induce apoptosis and this function of MYC warrants further clarification. We report here that a clinically relevant proteasome inhibitor significantly increases MYC protein levels and that endogenous MYC is necessary for the induction of apoptosis. This kind of MYC-induced cell death is mediated by enhanced expression of the pro-apoptotic BCL2 family members NOXA and BIM. Quantitative promoter-scanning chromatin immunoprecipitations (qChIP) further revealed binding of MYC to the promoters of NOXA and BIM upon proteasome inhibition, correlating with increased transcription. Both promoters are further characterized by the presence of tri-methylated lysine 4 of histone H3, marking active chromatin. We provide evidence that in our apoptosis models cell death occurs independently of p53 or ARF. Furthermore, we demonstrate that recruitment of MYC to the NOXA as well as to the BIM gene promoters depends on MYC's interaction with the zinc finger transcription factor EGR1 and an EGR1-binding site in both promoters. Our study uncovers a novel molecular mechanism by showing that the functional cooperation of MYC with EGR1 is required for bortezomib-induced cell death. This observation may be important for novel therapeutic strategies engaging the inherent pro-death function of MYC.
INTRODUCTION
One important response of tumor cells toward current cancer therapies is the induction of the cell death program. Accordingly, resistance to apoptosis is a hallmark of cancer that contributes to treatment failure (1). A plethora of intracellular stresses can engage the mitochondrial death pathway, which is critically controlled by the B-cell lymphoma-2 (BCL-2) protein family (1). This family comprises three groups including the pro-survival BCL-2 members, the BAX subfamily multidomain death inducers and the pro-death BH3-only proteins. Different cellular stresses are primarily sensed by certain BH3-only proteins and once activated, BH3-only proteins initiate the mitochondrial cell death program (1).
The basic helix-loop-helix leucine-zipper transcription factor c-MYC (MYC afterward) heterodimerizes with MAX (MYC-associated factor X) (2). MYC binds to the consensus element CACGTG, the so-called E-box, in promoters of genes that control cellular processes such as growth, proliferation or differentiation (2). MYC is frequently overexpressed or dysregulated in cancer cells, in which it drives genetic programs that are required for the progression and maintenance of tumors. Accordingly, apoptosis in response to increased expression of MYC is an important cell-intrinsic fail-safe mechanism to restrain MYC's oncogenic properties and tumorigenesis (3,4). Moreover, pro-death functions of MYC are not limited to carcinogenesis. Endogenous MYC also sensitizes cells to undergo apoptosis in response to diverse danger signals compromising cellular integrity (5). MYC-dependent apoptosis involves p53-dependent and -independent mechanisms (6). For example, MYC induces the expression of the tumor suppressor ARF. ARF inhibits the E3 ubiquitin ligase MDM2/HDM2, which catalyzes the proteasomal degradation of p53 (7). How MYC induces p53-independent apoptosis programs is incompletely understood. This mechanism likely involves various pathways and acts in a stimulus- and context-dependent manner.
The aim of this study was to characterize direct MYC target genes in relevant models of apoptosis. We demonstrate that specifically endogenous MYC is necessary for the induction of cell death after cellular stress caused by the inhibition of the proteasome. We show that the genes encoding for the BH3-only proteins NOXA and BIM are direct targets of MYC and we demonstrate that MYC and the transcription factor early growth response 1 (EGR1) act in a complex at both genes.
MATERIALS AND METHODS
Cell culture, reagents, RNAi, transfection, lentiviral transduction, viability assay, FACS and apoptosis assays
Bortezomib was purchased from LC Laboratories and 4-hydroxytamoxifen was obtained from Sigma-Aldrich. Culturing of DanG, MiaPaCa2, BxPc3 and p53-deficient and -proficient HCT116 cells was described (8–10). Mouse embryonic fibroblasts (MEFs) deficient for NOXA were provided by Dr A. Strasser and immortalized as described (10). HEK293FT cells were purchased from Invitrogen. Murine 3T9-MYClox/lox-CreER fibroblasts were provided by Dr B. Amati/Dr A. Trumpp (11). The murine p53-deficient primary dispersed pancreatic tumor (PPT) cell line PPT-W22 (genotype: Ptf1aCre/+; LSL-KrasG12D/+; p53lox/lox; LSL-R26Tva-lacZ/+) was described (8,12). The murine primary dispersed Cdkn2a-deficient pancreatic cancer cell line PPT-AA728 (genotype: Ptf1aCre/+; LSL-KrasG12D/+; Cdkn2alox/lox; LSL-R26Tva-lacZ/+) was isolated from a murine pancreatic cancer as described (13). The following genetically defined mouse strains were used to generate pancreatic cancers: LSL-KrasG12D/+ (14), Ptf1aCre/+ (15), LSL-R26Tva-lacZ (16), p53lox (17) and Cdkn2alox (18). Identity of the murine pancreatic cancer cell lines was verified using genotyping PCR and loss of ARF expression in PPT-AA728 cells was documented by qPCR (data not shown). All animal studies were conducted in compliance with European guidelines for the care and use of laboratory animals and were approved by the local authorities. Lentiviral transduction of cell lines was performed as described (10). siRNAs were purchased from Eurofins. Target sequences of shRNAs and siRNAs are shown in Supplementary Table S1. The following plasmids were purchased from Addgene (see also Supplementary Table S1): pcDNA3-Egr1 (#11729) (19), mEgr1/LZRS (#27783) (thanks to D. Wiest), MSCV-Myc-IRES-RFP (#35395) (20), pD40-His/V5-c-Myc (#45597) (21). pLKO.1, pLKO.1-shp53#1 and pLKO.1-shp53#2 were kindly provided by Dr von Burstin. PPT-AA728 cells transduced with pLKO.1, pLKO.1-shp53#1 and pLKO.1-shp53#2 were selected with puromycin (1 μg/ml) (Santa Cruz Biotechnology, sc-108071B). Further vectors are described in Supplementary Table S1. Plasmids and siRNAs were transfected as described (22). Viability of the cells was measured using MTT-assays (9). To determine Annexin V, cells were stained with propidium iodide (PI) and FITC-labeled anti-Annexin V using the Apoptosis Detection Kit I (BD Biosciences) and analyzed by FACS (23). Samples were analyzed using an FACScalibur flow cytometer (BD Biosciences) (23). A MoFlo (Beckman Coulter) cell sorter was used for sorting of red and/or green fluorescent cells. Caspase 3/7 activity was determined using Promega's Caspase-Glo 3/7 assay (23).
Total cell lysates, nuclear extracts, Western blot, immunoprecipitation and avidin-biotin-complex DNA assay
Whole cell lysates and nuclear extracts were prepared and western blots were carried out as described (9,24). A formaldehyde-based cross-linking protocol was used for immunoprecipitations as described by Klockenbusch and Kast (25). In brief, protein complexes were cross-linked with 1% formalin (Sigma-Aldrich) for 10 min at room temperature. The reaction was quenched with the addition of glycin (125 mM final concentration) for 5 min. Cells were washed in PBS at 4°C, scraped and lysed for 60 min on ice in RIPA buffer (50 mM Tris HCl (pH 8.0), 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, protease inhibitors (Complete mini, EDTA-free, Roche Diagnostics)). For immunoprecipitations, antibodies were coupled to Protein G Sepharose beads (GE Healthcare, 17-6002-35). Forty microliters of 100 mg/ml Protein G Sepharose beads, equilibrated in 50 mM Tris-HCl (pH7.5), 150 mM NaCl, 0.05% Triton-X 100 were coupled to 2 μg of the respective antibody and nutated at 4°C overnight. This antibody coupled beads were added to the protein lysate overnight, washed three times in Q1 (50 mM Tris-HCl, 150 mM NaCl, pH7.4) and eluted in E1 (0.1 M glycine HCl, pH 3). Supernatant was transferred into E2 (0.5 M Tris-HCl (pH 7.4) with 1.5 M NaCl) and protein loading buffer (60 mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol, 5% β-mercaptoethanol, 0.01% bromophenol blue) was added. For V5-tag immunoprecipitations, anti-V5 coupled beads were purchased from Sigma-Aldrich (Anti-V5 beads, Sigma-Aldrich A7345). Antibodies are shown in Supplementary Table S1. Avidin-Biotin-Complex DNA-(ABCD) assay was done as described (24). Sequences of the 5′ biotinylated oligonucleotides are listed in Supplementary Table S1.
Quantitative reverse-transcriptase PCR
Total RNA was isolated from pancreatic carcinoma cell lines using the RNeasy kit (Qiagen) (23). Quantitative mRNA analysis by qPCR was performed as previously described (StepOnePlus, PE Applied Biosystems) (23,26). Primer sequences are listed in Supplementary Table S1.
Quantitative chromatin immunoprecipitation and qChIP-Re-ChIP
Chromatin immunoprecipitations were performed using SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology, #9003) as described (23). Used antibodies are listed in Supplementary Table S1. For qChIP-Re-ChIP assays, the conventional ChIP assay was modified according to Metivier et al. (27). In brief, after the first precipitation the protein/DNA complexes were eluted in 10 mM DTT for 0.5 h at 37°C. After magnetic separation, supernatants were diluted with RC buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl, [pH 8.1]) and subsequently subjected to the second precipitation. Binding of the precipitated target proteins was determined by quantitative PCR and the fold difference between the experimental sample and the negative control (IgG) was determined as described (23). Histone marks were normalized to native histone H3. Primers with similar amplification efficiencies (E), calculated by E = 10−1/slope, were used and sequences are shown in Supplementary Table S1.
Gene expression profiling
Gene expression profiling was performed as described (26). In brief, DanG cells were treated for 2 h with 50 nM bortezomib or were treated with an equivalent volume of DMSO as a vehicle-treated control. Afterward, total RNA was prepared and labeled cRNA was produced and hybridized onto the GeneChip Human Genome U133 Plus 2.0 according to Affymetrix standard protocols. Expression profiles of murine pancreatic cancer cells were generated on an Affymetrix GeneChip Mouse Genome 430 2.0 platform and are in part described (26).
Virtual promoter analysis, gene set enrichment analysis (GSEA) and STRING analysis
For detection of CpG pattern rich sequence regions, the standard settings of the cpgplot program were used (28). Predicted CpG islands in the NOXA and BIM promoters are shown in Figure 4A and C. Using GSEA software (www.broadinstitute.org/gsea), microarray data were analyzed with the gene set matrix composed file c2.all.v4.0.symbols.gmt (29). Significant gene sets enriched by bortezomib treatment in DanG cells were identified using an FDR q-value < 0.05 and a nominal P-value < 0.05. For prediction of protein–protein interactions, STRING 9.1 was used (30) and a combined score of < 0.3 is considered to have a low confidence, 0.3–0.7 medium confidence, > 0.7 high confidence (31).
Figure 4.
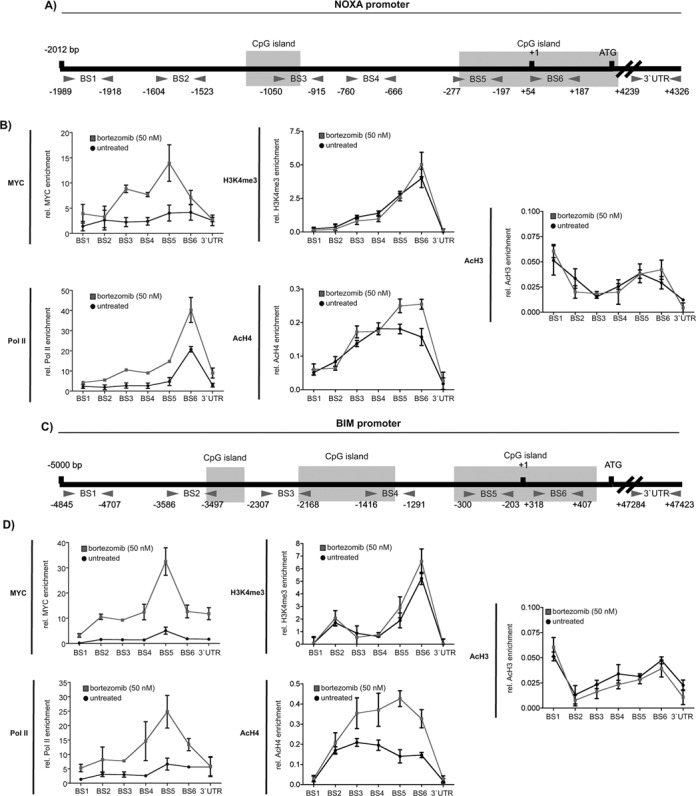
NOXA and BIM are direct MYC target genes. Schematic illustration of (A) the NOXA and (C) the BIM promoter. Location of the used primer pairs and CpG islands are depicted. (B) NOXA promoter qChIP and (D) BIM promoter qChIP of MYC, tri-methylated lysine 4 of histone H3, acetylated histone H4 and histone H3 and RNA Polymerase II (Pol II) binding after 6 h of bortezomib treatment. The enrichment of MYC and Pol II was normalized to input and IgG. Modified histones were normalized to pan histone H3 and IgG. Each point in the qChIP represents the mean and SEM from three independent immunoprecipitations.
Statistical methods
A two-sided Student's t-test with a correction for multiple testing according to Bonferroni or a one-way ANOVA test followed by a Tukey′s multiple comparison test was used to investigate statistical significance, as indicated. P-values were calculated with GraphPad Prism5 and * in the figures denotes a P-value of at least < 0.05. Spearman r was calculated with GraphPad Prism5 using collapsed microarray data sets for MYC and pro-death BCL2 family probes. All results are presented as mean and SEM.
RESULTS
MYC mediates cell death upon proteasome inhibition
Proteasome inhibitors like bortezomib are potent inducers of apoptosis (32). When we analyzed gene expression profiles of bortezomib-treated pancreatic cancer cells (DanG, Figure 1A and Supplementary Figure S1), we found a significant enrichment of various apoptosis gene signatures. In addition, MYC signatures were significantly enriched in bortezomib-treated cells (Figure 1A and Supplementary Figure S1)
Figure 1.
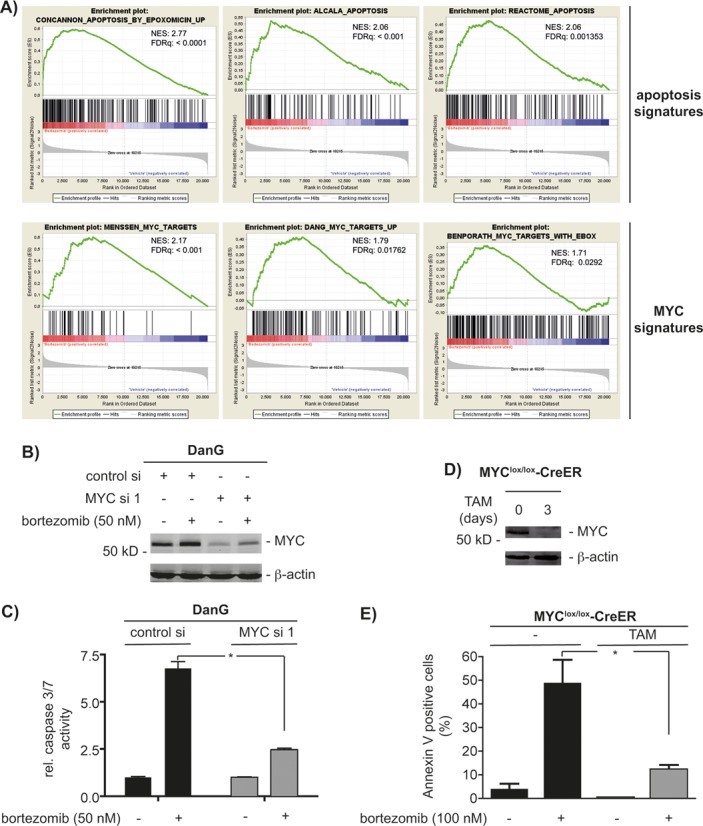
Stabilized MYC induces apoptosis. (A) Gene set enrichment analysis (GSEA) of transcriptome profiles of DanG cells. Cells were treated with 50 nM bortezomib (left) or vehicle (right) for 2 h. NES: normalized enrichment score; q: false discovery rate. All p values were <0.05. (B) DanG cells were siRNA transfected for 66 h as indicated. Afterward, cells were treated with 50 nM bortezomib for additional 6 h or were left as a vehicle-treated control. Western blot analysis of MYC expression (β-actin: loading control). (C) DanG cells were transfected as indicated. After 48 h, cells were treated with bortezomib for additional 24 h or were left as a vehicle-treated control. Caspase activity was measured using caspase 3/7 activity assays. Student′s t-test *P < 0.05. (D) 3T9 MYClox/lox-CreER mouse embryonic fibroblasts cells were treated for 3 days with 4-hydroxytamoxifen (TAM; 250 nM). Western blot analysis of MYC expression (β-actin: loading control). (E) 3T9 MYClox/lox-CreER MEFs were treated for 3 days with TAM, followed by bortezomib for additional 24 h. Cells were stained with propidium iodide (PI) and FITC-labeled anti-Annexin V and subsequently analyzed by FACS. Depicted is the Annexin V positive fraction (early apoptosis = Annexin V+/PI− and late apoptosis = Annexin V+/PI+). Student′s t-test *P < 0.05, n = 3.
In line, increased MYC protein expression (Figure 1B) and binding of MYC to an E-box oligonucleotide (Supplementary Figure S2C) upon proteasome inhibition was observed in DanG cells. Stabilization of MYC was also found in another pancreatic cancer cell line (MiaPaCa2, Supplementary Figure S2A) and in immortalized MEFs (Supplementary Figure S2B). Therefore, we conclude that increased MYC levels are generally seen upon proteasomal inhibition.
The elimination of MYC by siRNA (Figure 1B) significantly reduced caspase 3/7 activity in DanG cells upon bortezomib treatment (Figure 1C), demonstrating a pro-apoptotic contribution of endogenous MYC. To further prove the relevance of MYC for stress-induced apoptosis caused by proteasome inhibition, we used 3T9-MYClox/lox-CreER MEFs. This conditional model enables inactivation of the floxed MYC gene due to tamoxifen-induced CreER activation. In line, MYC expression was completely blocked in these cells after treatment with 4-hydroxytamoxifen (TAM) (Figure 1D). Remarkably, the loss of MYC distinctly reduced apoptosis (Figure 1E) induced by proteasome inhibition. The observation that endogenous MYC is necessary for the apoptotic effects of bortezomib in different systems and species shows that this death modality is a robust model to investigate MYC's pro-death functions.
NOXA and BIM contribute to MYC's pro-death function
The BH3-only protein NOXA is controlled by MYC in response to proteasome inhibition (33). In agreement, increased NOXA expression in DanG cells treated with bortezomib correlated well with MYC expression levels (Supplementary Figure S3A). Bortezomib induced NOXA protein and mRNA expression in a dose-dependent fashion (Supplementary Figure S3B and S3C). Thus, proteasomal blockade induces both, the accumulation of NOXA as well as the expression of the NOXA gene.
NOXA siRNAs efficiently reduced bortezomib-induced NOXA levels (Supplementary Figure S3D) and diminished apoptosis (Supplementary Figure S3E). Consistently, immortalized NOXA knock-out MEFs (Supplementary Figure S3F) were partially protected from bortezomib-induced loss of viability (Supplementary Figure S3G). However, compared to the MYC siRNA experiments (Figure 1C), siRNA mediated knockdown of NOXA (Supplementary Figure S3E) reduced bortezomib-induced caspase activation in DanG cells less efficiently. Such findings suggest a contribution of additional MYC-regulated factors.
In NOXA-deficient MEFs, we detected a time-dependent induction of the BIM protein (Supplementary Figure S4A). Knockdown of BIM (Supplementary Figure S4B) in NOXA-proficient and -deficient MEFs decreased bortezomib-induced caspase activation (Supplementary Figure S4C), arguing for the contribution of BIM to apoptosis due to proteasome inhibition. Induction of BIM occurred transcriptonally in NOXA-proficient and deficient MEFs (Supplementary Figure S4D) as well as in DanG cells (Supplementary Figure S4E). Microarray data of bortezomib-treated DanG cells revealed the upregulation of several pro-death BCL2 family members (Supplementary Figure S1). Consistently, using microarray expression data of 53 murine pancreatic cancer cell lines, we detected a significant positive correlation of basal MYC mRNA expression with basal mRNA expression of BAX, NOXA, BIM and BID (Supplementary Table S2). However, the potential MYC target gene BAX was not regulated at the protein level in DanG cells upon the blockade of the proteasome (Supplementary Figure S4F), suggesting that bortezomib induces regulatory circuits beyond the mRNA level. Since NOXA and BIM are regulated at the protein and mRNA level upon bortezomib treatment and significantly contribute to bortezomib-induced apoptosis, we focused on these BH3-only genes.
To test whether MYC contributes to the bortezomib-induced expression of BIM and NOXA, we used RNAi. Expression of BIM and NOXA protein (Figure 2A) and mRNA (Figure 2B) was blocked by two different MYC siRNAs in DanG and MiaPaCa2 cells (Supplementary Figure S5) after proteasome inhibition. In addition, bortezomib-induced BIM expression was decreased in 4-hydroxytamoxifen (TAM) treated 3T9-MYClox/lox-CreER MEFs (Figure 2C). The induction of MYC mRNA levels in the investigated human pancreatic cancer cell lines suggests a positive feed-forward loop occurring upon proteasome inhibition (Figure 2B and Supplementary Figure S5A).
Figure 2.
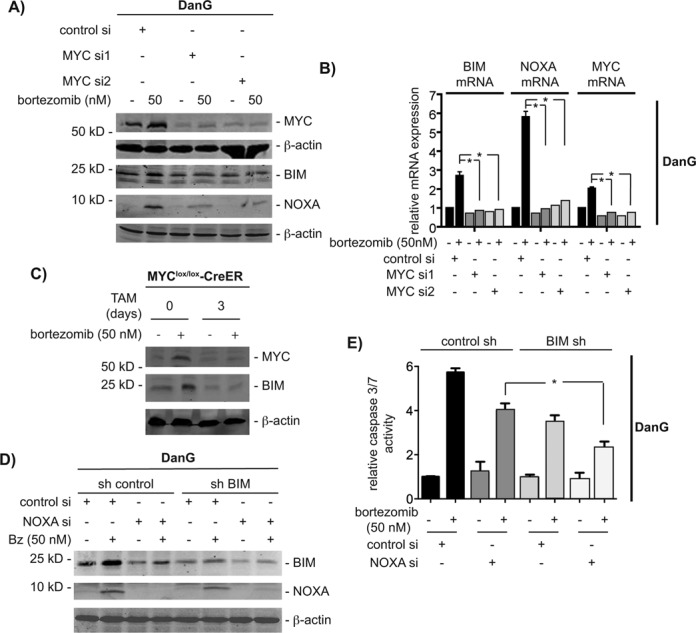
Both NOXA and BIM contribute to MYC-dependent apoptosis. (A) DanG cells were siRNA transfected as indicated. Sixty hours after the transfection, cells were treated with bortezomib for additional 12 h. Western blot of MYC, BIM and NOXA. Same extracts were transferred to two membranes and both were controlled by β-actin for equal loading. (B) DanG cells were treated as in (A). NOXA, BIM and MYC mRNA expression was determined by qPCR using PPIA mRNA as reference. Student's t-test *P < 0.05, n = 3. (C) 3T9 MYClox/lox-CreER mouse embryonic fibroblasts cells were treated for 3 days with 4-hydroxytamoxifen (TAM; 250 nM) and subsequently treated with bortezomib for additional 12 h or left as vehicle-treated controls. Western blot analysis of MYC and BIM expression (β-actin: loading control). (D) Stable transfected sh control and shBIM DanG cells were transiently transfected with a control siRNA or a NOXA siRNA. After 60 h, cells were treated with bortezomib for additional 12 h. Western blot of BIM, NOXA and β-actin (loading control). (E) Relative caspase 3/7 activity of stable transduced sh control and shBIM DanG cells, which were transiently transfected with a control siRNA or a NOXA siRNA for 48 h. Caspase 3/7 activity was measured 24 h after bortezomib treatment. One-way ANOVA *P < 0.05, n = 3.
To elucidate the impact of both MYC controlled BH3-only proteins toward bortezomib-induced apoptosis, we used stable BIM shRNA expressing cells and transfected these cells transiently with a NOXA-specific siRNA (Figure 2D). As shown in Figure 2E, apoptosis induction of DanG cells in response to proteasome inhibition was decreased upon attenuation of BIM. Interestingly, combined depletion of BIM and NOXA reduced caspase activation in DanG cells to the same extent as depletion of MYC (compare Figures 1E and 2C), arguing that NOXA and BIM are important MYC-dependent contributors to cell death induced by proteasome inhibition.
Bortezomib-induced expression of NOXA and BIM is not restricted in p53- and Cdkn2a-deficient models
MYC-dependent apoptosis involves p53-dependent and -independent mechanisms (6,7). Furthermore, the tumor suppressor Arf, encoded by the Cdkn2a locus, can contribute to both arms of MYC's pro-death response by either increasing p53 protein levels or by directly binding and modulating MYC (34–37). To investigate whether bortezomib-induced expression of NOXA and BIM depends on p53 and/or ARF, we used well-characterized models and generated novel cell systems (Supplementary Table S3). Since an unbiased siRNA screen demonstrated the contribution of MYC to bortezomib-induced apoptosis in HCT116 cells (38) and due to the availability of an isogenic p53-deficient HCT116 cell line (39), we first used this colon cancer cell line (Figure 3A). Protein levels of p53 become stabilized in p53-proficient HCT116 cells upon bortezomib treatment (Figure 3A). Interestingly, independent of the p53 status, the BIM and NOXA mRNAs are induced upon bortezomib treatment (Figure 3B). As in the human pancreatic cancer cell lines, MYC mRNA is increased in bortezomib treated p53-proficient and -deficient HCT116 cells, arguing afresh for a positive feed forward loop activated by the proteasome inhibitor (Figure 3B). HCT116 cells express ARF mRNA, which is reduced upon bortezomib treatment (Figure 3B). Together, these data argue that bortezomib treatment activates the expression of NOXA and BIM independent of p53. To further explore the contribution of p53 and ARF toward bortezomib-induced apoptosis, we used cell lines isolated from genetically engineered pancreatic cancer mouse models. Bortezomib treatment induced BIM expression in the murine p53-deficient pancreatic cancer cell line PPT-W22, isolated from a Ptf1aCre/+; LSL-KrasG12D/+; p53lox/lox; R26LSL-TVA-LacZ/+ mouse (Figure 3C) (9,12). To investigate the contribution of ARF, we generated a murine pancreatic cancer cell line from a Ptf1aCre/+; LSL-KrasG12D/+; Cdkn2alox/lox;R26LSL-TVA-LacZ/+ mouse, PPT-AA728. BIM protein expression was robustly induced by the proteasome inhibitor in this cell line (Figure 3C). To analyze the contribution of p53 in this Arf-deficient cell line, we used RNAi. Treatment with the proteasome inhibitor stabilized p53 in control cells (Figure 3D); this effect was diminished in p53 shRNA transduced cells. The appearance of cleaved caspase 3 in response to the treatment with bortezomib was increased upon the p53 knockdown in PPT-AA728 cells (Figure 3D). Absolut basal as well as induced NOXA and BIM mRNA expression levels were lower in p53 shRNA expressing PPT-AA728 cells, compared to controls (data not shown). However, NOXA and BIM mRNA is comparably induced upon the treatment with bortezomib in control PPT-AA728 cells as well as in cells expressing two different p53 shRNAs (Figure 3E). Together, these data support the conclusion that in the investigated models, bortezomib can activate NOXA and BIM expression independent of the p53 or ARF status.
Figure 3.
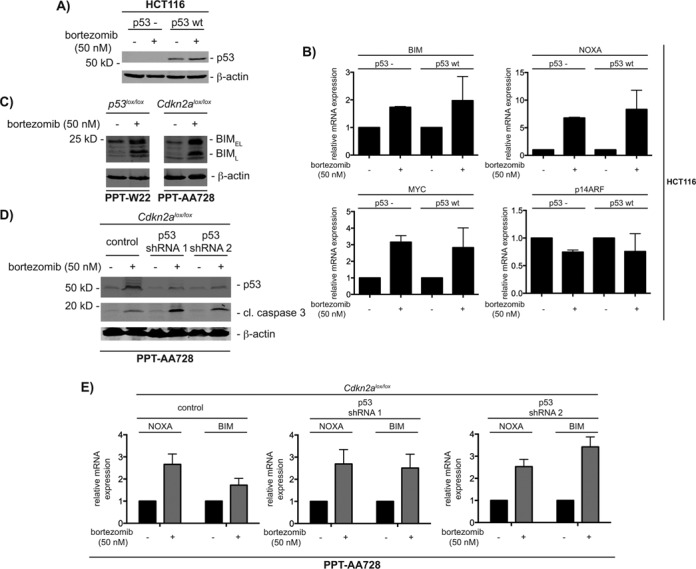
Proteasome inhibition induces BIM and NOXA independent of ARF and p53. (A) p53-deficient and -proficient HCT116 cells were treated with vehicle control and 50 nM bortezomib for 6 h. Western blot of p53 and β-actin (loading control) expression. (B) p53-proficient and -deficient HCT116 cells were treated with vehicle control and 50 nM bortezomib for 12 h. Relative BIM, NOXA, MYC and p14ARF mRNA expression was determined by qPCR using PPIA mRNA as reference, n = 3. (C) The murine pancreatic cancer cells PPT-W22 (p53-deficient) and PPT-AA728 (Cdkn2a-deficient) were treated with vehicle control and 50 nM bortezomib for 12 h. Western blot of BIM and β-actin (loading control) expression. (D) PPT-AA728 (deficient for Cdkn2a) were lentiviral transduced with pLKO.1, pLKO.1-shp53#1 and pLKO.1-shp53#2 and selected with puromycin (1 μg/ml). Cells were treated with 50 nM bortezomib for 12 h. Western blot of p53, cleaved caspase 3 and β-actin (loading control) expression. (E) Transduced PPT-AA728 cells described in (D), were treated with 50 nM bortezomib for 12 h. Relative BIM and NOXA mRNA expression was determined by qPCR using Ppia mRNA as reference, n = 3.
MYC is recruited to NOXA and BIM gene promoters
Since the regulation of pro-death genes downstream of MYC is understood incompletely, we investigated whether MYC is recruited to both genes using promoter-scanning quantitative chromatin immunoprecipitations (qChIP). Figure 4A and C show location of the primer pairs used and CpG islands predicted by cpgplot (28) for the NOXA and BIM promoters. As shown in Figure 4B, MYC was recruited 6 h after proteasome inhibition to the NOXA promoter in DanG cells with a peak at the binding site (BS) 5. Binding of MYC correlated with the recruitment of RNA polymerase II (Pol II), demonstrating transcriptional activation (Figure 4B). In addition, MYC was recruited to the proximal BIM promoter (BS 5) 6 h after proteasome inhibition in DanG cells (Figure 4D). Again, MYC binding correlated with Pol II binding to the BIM promoter (Figure 4D). These data demonstrate that both BH3-only genes are directly bound by MYC. In agreement with these results, genome-wide MYC ChIP assays of the ENCODE project revealed MYC binding to the proximal promoters of the NOXA and BIM gene (40). Also in accordance with previous data, all MYC peaks are located in CpG islands, which are thought to have high affinity for MYC (41) (Figure 4A and C). Furthermore, MYC binds efficiently to promoters within euchromatic marks, like tri-methylated lysine 4 of histone H3 (H3K4me3) (42,43). Consistently, we detected binding of H3K4me3 to the proximal regions of both promoters (Figure 4B and D). Fitting to transcriptional activation, we observed preferential acetylation of histone H4 after MYC binding to both promoters, whereas acetylation of histone H3 was not significantly changed upon treatment with bortezomib (Figure 4B and D).
EGR1 contributes to bortezomib-induced apoptosis
The BS5 amplicon of the NOXA promoter contains no E-box sequences. Since the Ets motif was recently shown to be significantly bound by MYC (44), we investigated the BS5 amplicon of the human NOXA promoter for Ets binding sites. In addition, EGR1 binding sites are overrepresented in MYC target gene promoters (45) and as some EGR1 motifs are significantly enriched in MYC immunoprecipitations (44). Therefore, we also scanned for EGR1 motifs. As shown in Figure 5A, the BS5 amplicon of the NOXA promoter contains Ets and EGR1 binding motifs. Although a noncanonical E-box is located 3′ of the BS5 amplicon in the BIM promoter, the core BS5 amplicon of the BIM promoter also contains potential Ets and EGR1 binding sequences (Figure 5A). To test the contribution of Ets factors and of EGR1 in MYC-dependent transcription of both genes, we used RNAi. Among Ets factors, we tested Ets1 due to a previously described role in apoptosis (46). However, we detected no significant influence of Ets1 on bortezomib-induced expression of NOXA and BIM (Supplementary Figure S6A and S6B). In contrast, RNAi targeting EGR1 prevented NOXA and BIM protein (Figure 5B) and mRNA (Figure 5C) induction upon proteasome inhibition in DanG cells, respectively. The influence of the EGR1 knockdown toward bortezomib-induced expression of NOXA and BIM was also observed in MiaPaCa2 cells (Supplementary Figure S7A). Consequently, bortezomib-induced activation of caspase 3/7 is prevented in EGR1 siRNA-transfected DanG (Figure 5D) and MiaPaCa2 cells (Supplementary Figure S7B).
Figure 5.
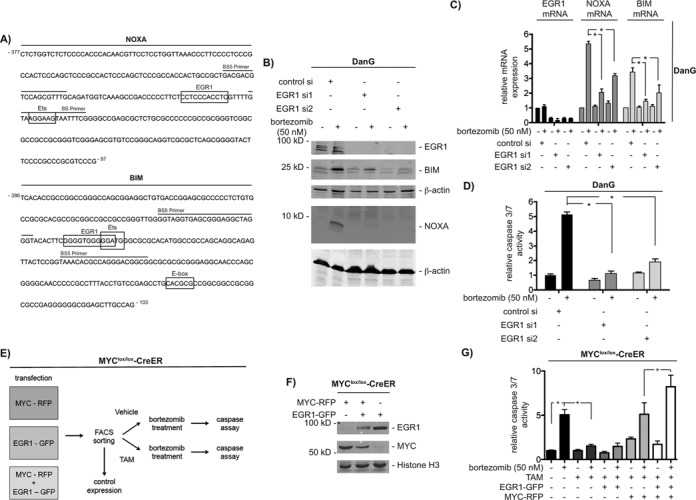
EGR1 contributes to bortezomib-induced apoptosis. (A) DNA sequences of the BS5 amplicon of human NOXA and BIM promoters. Primers are indicated by lines. Boxes show potential binding motifs for EGR1, Ets and MYC transcription factors. (B) DanG cells were transfected with control siRNA or two specific EGR1 siRNAs. After 66 h, cells were treated for 12 h as indicated. Western blot detected expression of EGR1, BIM and NOXA. Same extracts were transferred to two membranes and both were controlled by β-actin for equal loading. (C) DanG cells were treated as described in (B). Relative BIM, NOXA and EGR1 mRNA expression was determined by qPCR using PPIA mRNA as reference. Student's t-test *P < 0.05, n = 3. (D) DanG cells were transfected as indicated. Forty eight hours after the transfection, cells were treated with bortezomib for additional 24 h. Caspase activity was measured using caspase 3/7 activity assays. Student's t-test *P < 0.05, n = 3. (E) Working scheme: 3T9-MYClox/lox-CreER cells were transfected each with MYC-IRES-RFP, EGR1-IRES-GFP and combined. After 72 h, cells were FACS sorted. Cells were passaged and analyzed for expression of MYC and EGR1. Afterward, cells were treated with vehicle (EtOH) or 4-Hydroxytamoxifen (250 nM, TAM) for 72 h and subsequently treated with vehicle (DMSO) or bortezomib for additional 24 h and assayed for caspase 3/7 activity. (F) 3T9-MYClox/lox-CreER cells were transfected each with MYC-IRES-RFP, EGR1-IRES-GFP as outlined in (E). Western blot detected expression of EGR1, MYC and histone H3 (loading control). (G) 3T9-MYClox/lox-CreER cells were treated as described in (E) and relative caspase 3/7 activity was measured 24 h after bortezomib treatment (100 nM). One-way ANOVA *P < 0.05, n = 3.
To further corroborate the functional interaction of EGR1 and MYC to execute bortezomib-induced apoptosis, we performed an experiment outlined in Figure 5E. In short, 3T9-MYClox/lox-CreER MEFs were transiently transfected with bicistronic IRES vectors driving MYC and red fluorescent protein (RFP) expression, EGR1 and green fluorescent protein (GFP) expression or both vectors. Transfected cells were FACS sorted and propagated. Before the treatment with 4-hydroxytamoxifen (TAM), cells were probed for expression of MYC and EGR1 (Figure 5F). Afterward, the cells were treated with 4-hydroxytamoxifen (TAM) for 3 days to delete the floxed MYC alleles. Finally, both cell populations were treated with bortezomib for 24 h and caspase 3/7 activity was measured. As expected, genetic deletion of MYC in this setting distinctly reduced bortezomib-induced caspase activity (Figure 5G). Although, EGR1 alone was insufficient to substitute MYC in bortezomib-induced apoptosis, reconstitution of MYC alone completely restored proteasome inhibitor-induced caspase activation (Figure 5G). The observation that co-transfection of EGR1 and MYC further increased bortezomib-induced caspase activity (Figure 5G), argues for the synergistic function of both transcription factors in the investigated apoptosis model.
MYC binds to the EGR1 binding site of the NOXA and BIM gene promoters
Due to the functional cooperation of MYC and EGR1 in bortezomib-induced apoptosis, we tested for their DNA binding and interaction in a complex. To investigate the binding of MYC to the EGR1 binding sites of the NOXA and BIM promoters, we used DNA-pull down (ABCD-) assays. The sequences of the biotinylated oligonucleotides corresponding to the wild-type and mutated EGR1 binding sites of the NOXA and BIM promoters are shown in Figure 6A. Low basal binding of MYC to the EGR1 sites of both promoters was increased after treatment with bortezomib in DanG cells (Figure 6B). Oligonucleotides with mutations in the EGR1 consensus were not bound by MYC, demonstrating specificity (Figure 6B). To investigate the direct binding of EGR1 to both promoters, we performed qChIP in control and EGR1 siRNA transfected DanG cells. A significant recruitment of EGR1 to the BS5 of the NOXA and the BIM promoter was observed (Figure 6C). Bortezomib-induced binding of EGR1 to both promoters was reduced by the EGR1 siRNA. Furthermore, we detected a clear drop in the loading of RNA polymerase II, arguing for a role of EGR1 in transcriptional activation (Figure 6C). In addition, recruitment of MYC to both promoters is considerably impaired in EGR1 siRNA transfected DanG cells (Figure 6C). These data suggest the simultaneous binding of EGR1 and MYC to the BS5 region of the NOXA and BIM promoter. Since bortezomib-induced MYC stabilization is only slightly reduced in DanG cells transfected with the EGR1 siRNA used in the qChIP assay (Supplementary Figure S8A), the presence of EGR1 seems to be specifically required for the proper MYC recruitment and activation of both genes.
Figure 6.
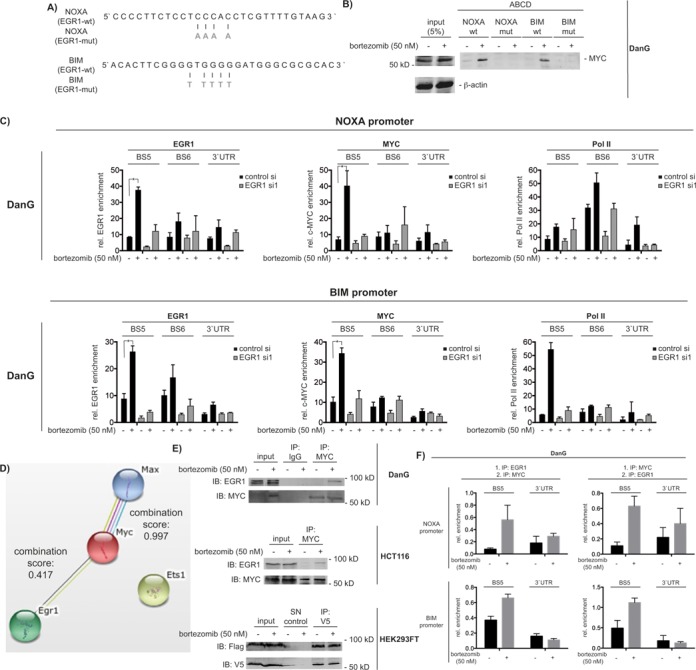
MYC interacts with EGR1 and binds to the EGR1 binding sites in the NOXA and BIM promoter. (A) DNA sequence of the EGR1 binding site of human NOXA and BIM gene promoters. Mutations of the EGR1 binding sites are depicted in gray. (B) ABCD assay of MYC binding to the wild-type and mutated EGR1 binding site of the NOXA and BIM promoters in nuclear extracts of DanG cells after 0 and 1 h of 50 nM bortezomib treatment. (C) DanG cells were transfected with the indicated siRNAs. After 48 h, cells were treated with bortezomib for 6 h or were left as a vehicle-treated control. NOXA and BIM promoter qChIP of EGR1, MYC and RNA Polymerase II (Pol II). One way ANOVA *P < 0.05, n = 3. (D) STRING analysis of murine Myc, Max, Egr1 and Ets1 proteins. The combination score for murine Myc-Max and Egr1-Myc is depicted. (E) Co-immunoprecipitations of endogenous MYC and EGR1 in DanG, HCT116 cells and Flag-tagged EGR1/V5-tagged MYC in HEK293FT cells after 6-h vehicle control or 50 nM bortezomib treatment. HEK293FT were transfected as indicated and after 48 h, the cells were treated with 50 nM bortezomib for additional 6 h. SN: supernatant. (F) NOXA and BIM promoter qChIP-Re-ChIP of EGR1 and MYC in DanG after 6-h bortezomib (50 nM) treatment. Graph represents the mean and SEM from three independent qChIP-Re-ChIP assays.
To further corroborate a functional linkage of MYC with EGR1, we performed a STRING database analysis, including MYC, MAX, Ets1 and EGR1 (Figure 6D) (30). Here, a combination score of 0.417 was calculated for murine MYC and EGR1, suggesting a protein–protein association with medium confidence in mice. As a comparison, the combination score was 0.997 for murine MAX with MYC and no functional link was suggested by the STRING analysis for murine Ets1. To test the direct interaction of MYC with EGR1, we used MYC immunoprecipitations. Using conventional immunoprecipitation protocols, we failed to detect a physical interaction of MYC with EGR1. However, using a formaldehyde-based cross-linking protocol (25), we observed that bortezomib-induced a complex containing MYC and EGR1 in extracts of DanG cells and HCT116 cells (Figure 6E). Accordingly, we detected both proteins in a complex in whole cell extracts of HEK293FT cells, which were transiently transfected with V5-tagged MYC and Flag-tagged EGR1 (Figure 6E). In this overexpression model, the MYC-EGR1 interaction was independent of the treatment of HEK293FT cells with bortezomib (Figure 6E).
To further determine the co-localization of EGR1 and MYC in a complex at the same chromatinized DNA sites in cis, we used sequential qChIP assays (qChIP-Re-ChIP) with antibodies specifically recognizing EGR1 and MYC. The precipitation of EGR1 followed by an MYC precipitation as well as the precipitation of MYC followed by an EGR1 precipitation enriched the BS5 of the NOXA and the BIM promoter in bortezomib-treated DanG cells (Figure 6F). A similar result was obtained in p53-deficient HCT116 cells (Supplementary Figure S8B), arguing that EGR1 and MYC co-localize in a complex in cis upon bortezomib treatment in vivo.
DISCUSSION
Aberrant expression of MYC induces apoptosis in both, cultured cells (47) and animals (48,49). On the other hand, the loss of endogenous MYC dramatically reduces apoptosis induction upon treatment with topoisomerase II inhibitors in cultured cells (50) and upon ionizing irradiation and cisplatin treatment in vivo (51). Such findings disclose a particular relevance of endogenous MYC in apoptotic pathways. Many genes of the pro-death BCL2 family, like BIM (37,52,53), BID (54), NOXA (33,55) or BAX (56,57), were described to act downstream of MYC to execute its pro-death function. However, how these genes are regulated by MYC is in many cases unclear. In an appropriate model of apoptosis signaling, we show here that two pro-death BCL2 family genes, NOXA and BIM, are direct MYC targets. Furthermore, we demonstrate an interaction and functional cooperation of MYC with the transcription factor EGR1 in apoptosis signaling.
To induce apoptosis, MYC expression must exceed a threshold, which is defined by BCL2 family proteins in a cell type and milieu-specific fashion (49). For investigation of MYC's pro-death function, we increased MYC levels with the proteasome inhibitor bortezomib. Our data, which were collected in human and murine pancreatic cancer cells, colon cancer cells and, genetically defined murine fibroblasts reveal an important contribution of endogenous MYC toward bortezomib-induced apoptosis. A genome-wide siRNA screen supports our conclusions. It defines MYC as an essential pro-death mediator upon bortezomib treatment in HCT116 cells (58). Likewise, malignant pleural mesothelioma cell lines with secondary resistance toward bortezomib revealed lower MYC expression accompanied with a loss of MYC binding to the NOXA promoter (55), further corroborating the notion that endogenous MYC is necessary in the apoptosis model investigated and that this model is relevant to study aspects of endogenous MYC's pro-death (patho)-biology.
The development of sophisticated techniques in the last decade demonstrated that several thousand sites in the human genome were occupied by MYC. This makes it extraordinarily complex to define biologically important MYC target genes. Moreover, the existence of definite and specific MYC target genes was challenged by recent ChIP-Seq experiments demonstrating that MYC can bind to each actively transcribed gene in a specific cellular context, causing the sole amplification of a pre-existing genetic program (59,60). However, existing data equally suggest that some MYC target genes have an outstanding role to execute MYC biology, since their ectopic expression is able to substitute MYC in a certain biological process or their loss definitively impairs a specific function of MYC. For instance, MYC-deficient Rat1 fibroblasts exhibit a distinct extension of the G1-phase of the cell cycle and reveal an insufficient induction of the MYC target gene CDK4 upon mitogenic stimulation (61). The observation that restoring CDK4 expression in these cells partially corrects the cell cycle defect (61) supports the notion that CDK4 is important for MYC-dependent proliferation.
Considering MYC-induced tumorigenesis, Nilsson and colleagues discovered a critical role of the ornithine decarboxylase (Odc) gene, an MYC-target gene involved in the polyamine biosynthesis pathway (62). A marked delay in lymphomagenesis in the Eμ-Myc model after the genetic or pharmacological interference with normal expression/function of Odc was detected (62). In addition, an MYC-repressed gene, Tristetraprolin, was demonstrated to be of noteworthy importance in the Eμ-Myc model. Tristetraprolin is an AU-rich element binding protein, leading to the destabilization of mRNAs important for tumorigenesis. The rescue of the MYC-dependent repression of this gene in the Eμ-Myc model distinctly impaired lymphomagenesis in vivo (63). Together these studies bring forward the argument that Odc and Tristetraprolin belong to a core set of target genes contributing to MYC-driven lymphomagenesis. Consistent with a concept of core MYC target genes, which drive certain biological processes, we defined NOXA and BIM as critical mediators out of numerous regulated genes with the potential to induce cell death. The observations that (i) NOXA and BIM are upregulated upon proteasome inhibition in human and murine models, (ii) MYC directly binds to the promoters of NOXA and BIM and (iii) a similar rescue of bortezomib-induced apoptosis by the MYC knockdown and the combined NOXA/BIM knockdown occurs, argues that NOXA and BIM are core genes mediating MYC's pro-death function in the investigated model. The concept of the regulation of a specific subset of genes by MYC, important to change the state of a cell, is also supported by recent ChIP-Seq data (64).
Recruitment of MYC to a +87 (relative to the transcriptional start) binding site in the human NOXA promoter upon proteasome inhibition in melanoma cells was described (33). In DanG cells, we detected an MYC peak in the −277 to −197 region of the proximal NOXA promoter after proteasome inhibition. These observations indicate an important contribution of this promoter region for MYC-dependent regulation. MYC binds to canonical as well as to noncanonical E-box sequences (44,65–67). Interestingly, the −277 to −197 region of the human NOXA promoter contains none of the six E-box sequences, arguing for an E-box-independent transcriptional activation. Consistently, genome wide analysis of MYC target genes suggested that up to 40% of MYC bound promoters lack E-box sequences (68). In silico analysis of the MYC bound NOXA promoter regions revealed the existence of conserved binding sites for EGR1 and Ets transcription factors. In agreement, recent ChIP-sequencing experiments showed a significant enrichment of the Ets motif and some EGR1 motifs in MYC precipitations (44). In addition, EGR1 binding sites are overrepresented on MYC target gene promoters (45). Whereas we observed no influence of ETS1 for MYC-driven NOXA transcription and apoptosis, we detected a significant contribution of EGR1. As predicted by the STRING database for murine MYC and EGR1, we observed low-affinity interaction of MYC with EGR1 upon proteasome inhibition. Furthermore, qChIP-Re-ChIP data provide evidence that MYC and EGR1 act in the same complex in cis and that EGR1 expression is needed for binding of MYC to the NOXA promoter. This is necessary for a functional cooperation of both transcription factors.
Consistent with published observations (69), we describe here that neither NOXA knock-out MEFs nor NOXA siRNA transfected cancer cells are completely protected from bortezomib-induced apoptosis, which argues that further pro-death programs are executed. We demonstrate here that BIM is an important contributor. Wild-type MYC can induce BIM expression in MEFs, murine pro-B-cell lymphoid FL5.12 cells, hematopoietic stem cells and transgenic B cells of Eμ-Myc mice (37,52,53). However, in most instances the mode of regulation of BIM -direct or indirect- is still unclear. We demonstrate now that BIM is a direct target gene of MYC in pancreatic cancer and in colon cancer cells upon bortezomib treatment. A proteasome inhibitor-induced MYC peak was observed at the −300 to −203 amplicon of the BIM promoter and analog to the NOXA promoter, in silico analysis shows the presence of potential EGR1 as well as potential ETS binding sites. Again, we observed dependency of MYC recruitment to the BIM promoter onto EGR1 expression. We provide evidence that MYC and EGR1 act in the same complex in cis and that the cooperation of MYC and EGR1 is needed to activate the BIM gene. In line, activation of the BIM gene by EGR1 was demonstrated in a model of deprivation-induced neuronal apoptosis (70). Our data show a striking similarity in the promoter structure, in the epigenetic shape and in functional cooperativeness of MYC with EGR1 to activate the NOXA and the BIM gene, arguing for a common mode of MYC-dependent transcriptional activation during bortezomib-induced apoptotic signaling. Consistent with our observations, MYC recruitment to the BIM promoter was detected in unstimulated BT474 breast cancer cells (71). This is also in agreement with our observation that the expression of MYC and BIM mRNAs is positively correlated in murine pancreatic cancer cell lines. Consistently, recent ChIP-Seq data show that the NOXA and BIM genes are bound by MYC in murine pancreatic cancer cells (72).
MYC is known to interact with several transcription factors (2). Here we demonstrate a functionally relevant low-affinity interaction of MYC with EGR1 in response to stress inducing apoptosis. In agreement with these data, EGR1 knockout MEFs are protected from MYC-induced apoptosis (36,37) and bortezomib-induced death depends on EGR1 in the myeloma cell lines U266 and OCI-My5 (38). These data suggest a common involvement of the MYC-EGR1 pathway in MYC's pro-death functions. Nevertheless, we are aware that proteasome inhibition increases MYC protein expression to unphysiological levels and induces specific intracellular stress signaling pathways, which in sum might limit the generality of the described pathway. However, projects such as the The Cancer Genome Atlas compellingly demonstrate that MYC belongs to the top three genes with a high frequency of copy number variations across human cancers (73). MYC amplification is correlated with high protein expression and work in a large cell line resource demonstrated that MYC amplified cell lines respond significantly different from not amplified cell lines toward certain cancer therapeutics (74) (see also: http://www.cancerrxgene.org/translation/Gene/1250). These observations argue that cellular responses induced by high MYC levels are important and engaged by specific drugs. Furthermore, in such a scenario it is likely that E-boxes are saturated (60,75) and MYC uses low affinity interaction partners, like EGR1, to drive a tumor cell response. Therefore, it is important to decipher and understand molecular vulnerabilities induced by high MYC expression.
The MYC-EGR1 pathway was reported to operate in a p53-independent manner (36). In agreement, we observed induction of the MYC target genes NOXA and BIM in human and murine p53-deficient as well as in p53-mutated cell lines upon bortezomib treatment. The zinc finger transcription factor EGR1 can induce apoptosis upon various cellular stresses (76). Of note, Boone and colleagues demonstrated that MYC induces the EGR1 gene p53 independently, but dependent on the tumor suppressor ARF (36). ARF is known to interact with MYC (34,35) and to increase binding of MYC to the EGR1 promoter (36). We did not detect a significantly increased expression of EGR1 upon MYC activation in the human pancreatic cancer cell lines DanG and MiaPaCa2, which might be explained by the lack of ARF expression in these cellular models. Congruently, murine Arf-deficient pancreatic cancer cells activate the expression of pro-apoptotic MYC target genes in response to bortezomib. In sum, our data support the notion that endogenous MYC can induce context-specific apoptosis by acting in a complex with EGR1 in a p53- or ARF-independent manner. Continuation of this concept awaits the investigation of genetically defined p53-/Arf-double knock-out models.
The carboxy-terminal basic helix-loop-helix leucine zipper (bHLH/LZ) domain of MYC is responsible for its hetrodimerization with MAX. In addition to DNA binding, the MYC-MAX dimeric bHLH/LZ provides a docking platform for the interaction with other proteins, like the zinc finger transcription factor MIZ-1 (77,78). Interestingly, a chimeric MYC protein containing the bHLH/LZ of the MYC-antagonistic MAX dimerization partner MAD1, fails to induce apoptosis, arguing that MYCs bHLH/LZ confer a specific pro-apoptotic characteristic (79). Indeed, enforced MYC expression under serum deprivation induces apoptosis in human fibroblasts, a feature not conducted by the MYC V394D mutant, defective in binding to MIZ-1 (80). This shows that proteins binding to the MYC-MAX docking platform can modulate a specific cellular response. Therefore it is attractive to speculate that in the bortezomib-induced apoptosis models, the zinc finger protein EGR1 uses the MYC-MAX dimeric bHLH/LZ docking platform to allow recruitment of the complex to the investigated BH3-only gene promoters. However, this speculation needs further experimentation including the mapping of potential interaction domains in EGR1 and MYC as well as the demonstration of a direct contribution of MAX to bortezomib-induced apoptosis, since some MYC directed cell death pathways depend on its dimerization with MAX (81).
Taken together our results show for the first time a functional cooperation of MYC with EGR1, leading to the activation of BH3-only gene promoters in a model of bortezomib-driven apoptosis. Increasing the knowledge about such MYC-dependent pathways might face the avenue to novel therapeutic strategies.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
Acknowledgments
We thank Dr B. Amati, Dr A. Berns, Dr R. DePinho, Dr T. Jacks, Dr A. Strasser, Dr A. Trumpp, Dr D. Tuveson and Dr B. Vogelstein for providing mice and cell lines. We thank all colleagues providing plasmids via the Addgene platform (Supplementary Table S1) and Dr J. von Burstin for providing p53 shRNA vectors. Furthermore, we thank Dr M. Schiemann and Lynette Henkel for help with FACS sorting.
FUNDING
Wilhelm-Sander Foundation [2012.084.1 to G.S., 2010.078.1 to O.H.K]; Deutsche Forschungsgemeinschaft (DFG) [SCHN 959/1-2, SCHN 959/2-1 to G.S. and SFB824 to G.S.; D.S.]; Deutsche Krebshilfe [110908 to G.S. and 110909 to O.H.K.]. Funding for open access charge: DFG.
Conflict of interest statement. None declared.
REFERENCES
- 1.Strasser A., Cory S., Adams J.M. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30:3667–3683. doi: 10.1038/emboj.2011.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eilers M., Eisenman R.N. Myc's broad reach. Genes Dev. 2008;22:2755–2766. doi: 10.1101/gad.1712408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pelengaris S., Khan M., Evan G. c-MYC: more than just a matter of life and death. Nat. Rev. Cancer. 2002;2:764–776. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- 4.Nilsson J.A., Cleveland J.L. Myc pathways provoking cell suicide and cancer. Oncogene. 2003;22:9007–9021. doi: 10.1038/sj.onc.1207261. [DOI] [PubMed] [Google Scholar]
- 5.Larsson L.G., Henriksson M.A. The Yin and Yang functions of the Myc oncoprotein in cancer development and as targets for therapy. Exp. Cell Res. 2010;316:1429–1437. doi: 10.1016/j.yexcr.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 6.Meyer N., Penn L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer. 2008;8:976–990. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- 7.Zindy F., Eischen C.M., Randle D.H., Kamijo T., Cleveland J.L., Sherr C.J., Roussel M.F. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schneider G., Henrich A., Greiner G., Wolf V., Lovas A., Wieczorek M., Wagner T., Reichardt S., von Werder A., Schmid R.M., et al. Cross talk between stimulated NF-kappaB and the tumor suppressor p53. Oncogene. 2010;29:2795–2806. doi: 10.1038/onc.2010.46. [DOI] [PubMed] [Google Scholar]
- 9.Conradt L., Godl K., Schaab C., Tebbe A., Eser S., Diersch S., Michalski C.W., Kleeff J., Schnieke A., Schmid R.M., et al. Disclosure of erlotinib as a multikinase inhibitor in pancreatic ductal adenocarcinoma. Neoplasia. 2011;13:1026–1034. doi: 10.1593/neo.111016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weber A., Kirejczyk Z., Potthoff S., Ploner C., Hacker G. Endogenous noxa determines the strong proapoptotic synergism of the BH3-mimetic ABT-737 with chemotherapeutic agents in human melanoma cells. Transl. Oncol. 2009;2:73–83. doi: 10.1593/tlo.08223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trumpp A., Refaeli Y., Oskarsson T., Gasser S., Murphy M., Martin G.R., Bishop J.M. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature. 2001;414:768–773. doi: 10.1038/414768a. [DOI] [PubMed] [Google Scholar]
- 12.Conradt L., Henrich A., Wirth M., Reichert M., Lesina M., Algul H., Schmid R.M., Kramer O.H., Saur D., Schneider G. Mdm2 inhibitors synergize with topoisomerase II inhibitors to induce p53-independent pancreatic cancer cell death. Int. J. Cancer. 2013;132:2248–2257. doi: 10.1002/ijc.27916. [DOI] [PubMed] [Google Scholar]
- 13.von Burstin J., Eser S., Paul M.C., Seidler B., Brandl M., Messer M., von Werder A., Schmidt A., Mages J., Pagel P. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology. 2009;137:361–371. doi: 10.1053/j.gastro.2009.04.004. 371, e1–e5. [DOI] [PubMed] [Google Scholar]
- 14.Hingorani S.R., Petricoin E.F., Maitra A., Rajapakse V., King C., Jacobetz M.A., Ross S., Conrads T.P., Veenstra T.D., Hitt B.A., et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 15.Nakhai H., Sel S., Favor J., Mendoza-Torres L., Paulsen F., Duncker G.I., Schmid R.M. Ptf1a is essential for the differentiation of GABAergic and glycinergic amacrine cells and horizontal cells in the mouse retina. Development. 2007;134:1151–1160. doi: 10.1242/dev.02781. [DOI] [PubMed] [Google Scholar]
- 16.Seidler B., Schmidt A., Mayr U., Nakhai H., Schmid R.M., Schneider G., Saur D. A Cre-loxP-based mouse model for conditional somatic gene expression and knockdown in vivo by using avian retroviral vectors. Proc. Natl Acad. Sci. USA. 2008;105:10137–10142. doi: 10.1073/pnas.0800487105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jonkers J., Meuwissen R., van der Gulden H., Peterse H., van der Valk M., Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 2001;29:418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 18.Aguirre A.J., Bardeesy N., Sinha M., Lopez L., Tuveson D.A., Horner J., Redston M.S., DePinho R.A. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu J., de Belle I., Liang H., Adamson E.D. Coactivating factors p300 and CBP are transcriptionally crossregulated by Egr1 in prostate cells, leading to divergent responses. Mol. Cell. 2004;15:83–94. doi: 10.1016/j.molcel.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 20.Kawauchi D., Robinson G., Uziel T., Gibson P., Rehg J., Gao C., Finkelstein D., Qu C., Pounds S., Ellison D.W., et al. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell. 2012;21:168–180. doi: 10.1016/j.ccr.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeh E., Cunningham M., Arnold H., Chasse D., Monteith T., Ivaldi G., Hahn W.C., Stukenberg P.T., Shenolikar S., Uchida T., et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 2004;6:308–318. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 22.Wirth M., Fritsche P., Stojanovic N., Brandl M., Jaeckel S., Schmid R.M., Saur D., Schneider G. A simple and cost-effective method to transfect small interfering RNAs into pancreatic cancer cell lines using polyethylenimine. Pancreas. 2011;40:144–150. doi: 10.1097/MPA.0b013e3181f7e41c. [DOI] [PubMed] [Google Scholar]
- 23.Labisso W.L., Wirth M., Stojanovic N., Stauber R.H., Schnieke A., Schmid R.M., Kramer O.H., Saur D., Schneider G. MYC directs transcription of MCL1 and eIF4E genes to control sensitivity of gastric cancer cells toward HDAC inhibitors. Cell Cycle. 2012;11:1593–1602. doi: 10.4161/cc.20008. [DOI] [PubMed] [Google Scholar]
- 24.Brandl M., Seidler B., Haller F., Adamski J., Schmid R.M., Saur D., Schneider G. IKK(alpha) controls canonical TGF(ss)-SMAD signaling to regulate genes expressing SNAIL and SLUG during EMT in panc1 cells. J. Cell Sci. 2010;123:4231–4239. doi: 10.1242/jcs.071100. [DOI] [PubMed] [Google Scholar]
- 25.Klockenbusch C., Kast J. Optimization of formaldehyde cross-linking for protein interaction analysis of non-tagged integrin beta1. J. Biomed. Biotechnol. 2010;2010:927585. doi: 10.1155/2010/927585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diersch S., Wenzel P., Szameitat M., Eser P., Paul M.C., Seidler B., Eser S., Messer M., Reichert M., Pagel P., et al. Efemp1 and p27Kip1 modulate responsiveness of pancreatic cancer cells towards a dual PI3K/mTOR inhibitor in preclinical models. Oncotarget. 2013;4:277–288. doi: 10.18632/oncotarget.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Metivier R., Penot G., Hubner M.R., Reid G., Brand H., Kos M., Gannon F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 28.Rice P., Longden I., Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 29.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., Lin J., Minguez P., Bork P., von Mering C., et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jensen L.J., Kuhn M., Stark M., Chaffron S., Creevey C., Muller J., Doerks T., Julien P., Roth A., Simonovic M., et al. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fennell D.A., Chacko A., Mutti L. BCL-2 family regulation by the 20S proteasome inhibitor bortezomib. Oncogene. 2008;27:1189–1197. doi: 10.1038/sj.onc.1210744. [DOI] [PubMed] [Google Scholar]
- 33.Nikiforov M.A., Riblett M., Tang W.H., Gratchouck V., Zhuang D., Fernandez Y., Verhaegen M., Varambally S., Chinnaiyan A.M., Jakubowiak A.J., et al. Tumor cell-selective regulation of NOXA by c-MYC in response to proteasome inhibition. Proc. Natl. Acad. Sci. USA. 2007;104:19488–19493. doi: 10.1073/pnas.0708380104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qi Y., Gregory M.A., Li Z., Brousal J.P., West K., Hann S.R. p19ARF directly and differentially controls the functions of c-Myc independently of p53. Nature. 2004;431:712–717. doi: 10.1038/nature02958. [DOI] [PubMed] [Google Scholar]
- 35.Datta A., Nag A., Pan W., Hay N., Gartel A.L., Colamonici O., Mori Y., Raychaudhuri P. Myc-ARF (alternate reading frame) interaction inhibits the functions of Myc. J. Biol. Chem. 2004;279:36698–36707. doi: 10.1074/jbc.M312305200. [DOI] [PubMed] [Google Scholar]
- 36.Boone D.N., Qi Y., Li Z., Hann S.R. Egr1 mediates p53-independent c-Myc-induced apoptosis via a noncanonical ARF-dependent transcriptional mechanism. Proc. Natl. Acad. Sci. USA. 2011;108:632–637. doi: 10.1073/pnas.1008848108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Q., Spears E., Boone D.N., Li Z., Gregory M.A., Hann S.R. Domain-specific c-Myc ubiquitylation controls c-Myc transcriptional and apoptotic activity. Proc. Natl. Acad. Sci. USA. 2013;110:978–983. doi: 10.1073/pnas.1208334110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen L., Wang S., Zhou Y., Wu X., Entin I., Epstein J., Yaccoby S., Xiong W., Barlogie B., Shaughnessy J.D., Jr, et al. Identification of early growth response protein 1 (EGR-1) as a novel target for JUN-induced apoptosis in multiple myeloma. Blood. 2010;115:61–70. doi: 10.1182/blood-2009-03-210526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bunz F., Dutriaux A., Lengauer C., Waldman T., Zhou S., Brown J.P., Sedivy J.M., Kinzler K.W., Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 40.Consortium E.P., Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernandez P.C., Frank S.R., Wang L., Schroeder M., Liu S., Greene J., Cocito A., Amati B. Genomic targets of the human c-Myc protein. Genes Dev. 2003;17:1115–1129. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guccione E., Martinato F., Finocchiaro G., Luzi L., Tizzoni L., Olio V., Zardo G., Nervi C., Bernard L., Amati B. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat. Cell Biol. 2006;8:764–770. doi: 10.1038/ncb1434. [DOI] [PubMed] [Google Scholar]
- 43.Kim J., Chu J., Shen X., Wang J., Orkin S.H. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perna D., Faga G., Verrecchia A., Gorski M.M., Barozzi I., Narang V., Khng J., Lim K.C., Sung W.K., Sanges R., et al. Genome-wide mapping of Myc binding and gene regulation in serum-stimulated fibroblasts. Oncogene. 2011;31:1695–709. doi: 10.1038/onc.2011.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elkon R., Zeller K.I., Linhart C., Dang C.V., Shamir R., Shiloh Y. In silico identification of transcriptional regulators associated with c-Myc. Nucleic Acids Res. 2004;32:4955–4961. doi: 10.1093/nar/gkh816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dittmer J. The biology of the Ets1 proto-oncogene. Mol. Cancer. 2003;2:2–29. doi: 10.1186/1476-4598-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Evan G.I., Wyllie A.H., Gilbert C.S., Littlewood T.D., Land H., Brooks M., Waters C.M., Penn L.Z., Hancock D.C. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 48.Pelengaris S., Khan M., Evan G.I. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell. 2002;109:321–334. doi: 10.1016/s0092-8674(02)00738-9. [DOI] [PubMed] [Google Scholar]
- 49.Murphy D.J., Junttila M.R., Pouyet L., Karnezis A., Shchors K., Bui D.A., Brown-Swigart L., Johnson L., Evan G.I. Distinct thresholds govern Myc's biological output in vivo. Cancer Cell. 2008;14:447–457. doi: 10.1016/j.ccr.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adachi S., Obaya A.J., Han Z., Ramos-Desimone N., Wyche J.H., Sedivy J.M. c-Myc is necessary for DNA damage-induced apoptosis in the G(2) phase of the cell cycle. Mol. Cell Biol. 2001;21:4929–4937. doi: 10.1128/MCB.21.15.4929-4937.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Phesse T.J., Myant K.B., Cole A.M., Ridgway R.A., Pearson H., Muncan V., van den Brink G.R., Vousden K.H., Sears R., Vassilev L.T., et al. Endogenous c-Myc is essential for p53-induced apoptosis in response to DNA damage in vivo. Cell Death Differ. 2014;21:956–966. doi: 10.1038/cdd.2014.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hemann M.T., Bric A., Teruya-Feldstein J., Herbst A., Nilsson J.A., Cordon-Cardo C., Cleveland J.L., Tansey W.P., Lowe S.W. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436:807–811. doi: 10.1038/nature03845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Egle A., Harris A.W., Bouillet P., Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc. Natl. Acad. Sci. USA. 2004;101:6164–6169. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iaccarino I., Hancock D., Evan G., Downward J. c-Myc induces cytochrome c release in Rat1 fibroblasts by increasing outer mitochondrial membrane permeability in a Bid-dependent manner. Cell Death Differ. 2003;10:599–608. doi: 10.1038/sj.cdd.4401211. [DOI] [PubMed] [Google Scholar]
- 55.Busacca S., Chacko A.D., Klabatsa A., Arthur K., Sheaff M., Gunasekharan V.K., Gorski J.J., El-Tanani M., Broaddus V.C., Gaudino G., et al. BAK and NOXA are critical determinants of mitochondrial apoptosis induced by bortezomib in mesothelioma. PLoS One. 2013;8:e65489. doi: 10.1371/journal.pone.0065489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dansen T.B., Whitfield J., Rostker F., Brown-Swigart L., Evan G.I. Specific requirement for Bax, not Bak, in Myc-induced apoptosis and tumor suppression in vivo. J. Biol. Chem. 2006;281:10890–10895. doi: 10.1074/jbc.M513655200. [DOI] [PubMed] [Google Scholar]
- 57.Juin P., Hunt A., Littlewood T., Griffiths B., Swigart L.B., Korsmeyer S., Evan G. c-Myc functionally cooperates with Bax to induce apoptosis. Mol. Cell Biol. 2002;22:6158–6169. doi: 10.1128/MCB.22.17.6158-6169.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen S., Blank J.L., Peters T., Liu X.J., Rappoli D.M., Pickard M.D., Menon S., Yu J., Driscoll D.L., Lingaraj T., et al. Genome-wide siRNA screen for modulators of cell death induced by proteasome inhibitor bortezomib. Cancer Res. 2010;70:4318–4326. doi: 10.1158/0008-5472.CAN-09-4428. [DOI] [PubMed] [Google Scholar]
- 59.Nie Z., Hu G., Wei G., Cui K., Yamane A., Resch W., Wang R., Green D.R., Tessarollo L., Casellas R., et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151:68–79. doi: 10.1016/j.cell.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin C.Y., Loven J., Rahl P.B., Paranal R.M., Burge C.B., Bradner J.E., Lee T.I., Young R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hermeking H., Rago C., Schuhmacher M., Li Q., Barrett J.F., Obaya A.J., O'Connell B.C., Mateyak M.K., Tam W., Kohlhuber F., et al. Identification of CDK4 as a target of c-MYC. Proc. Natl. Acad. Sci. USA. 2000;97:2229–2234. doi: 10.1073/pnas.050586197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nilsson J.A., Keller U.B., Baudino T.A., Yang C., Norton S., Old J.A., Nilsson L.M., Neale G., Kramer D.L., Porter C.W., et al. Targeting ornithine decarboxylase in Myc-induced lymphomagenesis prevents tumor formation. Cancer Cell. 2005;7:433–444. doi: 10.1016/j.ccr.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 63.Rounbehler R.J., Fallahi M., Yang C., Steeves M.A., Li W., Doherty J.R., Schaub F.X., Sanduja S., Dixon D.A., Blackshear P.J., et al. Tristetraprolin impairs myc-induced lymphoma and abolishes the malignant state. Cell. 2012;150:563–574. doi: 10.1016/j.cell.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sabo A., Kress T.R., Pelizzola M., de Pretis S., Gorski M.M., Tesi A., Morelli M.J., Bora P., Doni M., Verrecchia A., et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature. 2014;511:488–492. doi: 10.1038/nature13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blackwell T.K., Huang J., Ma A., Kretzner L., Alt F.W., Eisenman R.N., Weintraub H. Binding of myc proteins to canonical and noncanonical DNA sequences. Mol. Cell Biol. 1993;13:5216–5224. doi: 10.1128/mcb.13.9.5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zeller K.I., Jegga A.G., Aronow B.J., O'Donnell K.A., Dang C.V. An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol. 2003;4:R69. doi: 10.1186/gb-2003-4-10-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim J., Lee J.H., Iyer V.R. Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS One. 2008;3:e1798. doi: 10.1371/journal.pone.0001798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zeller K.I., Zhao X., Lee C.W., Chiu K.P., Yao F., Yustein J.T., Ooi H.S., Orlov Y.L., Shahab A., Yong H.C., et al. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc. Natl. Acad. Sci. USA. 2006;103:17834–17839. doi: 10.1073/pnas.0604129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fernandez Y., Verhaegen M., Miller T.P., Rush J.L., Steiner P., Opipari A.W., Jr, Lowe S.W., Soengas M.S. Differential regulation of noxa in normal melanocytes and melanoma cells by proteasome inhibition: therapeutic implications. Cancer Res. 2005;65:6294–6304. doi: 10.1158/0008-5472.CAN-05-0686. [DOI] [PubMed] [Google Scholar]
- 70.Xie B., Wang C., Zheng Z., Song B., Ma C., Thiel G., Li M. Egr-1 transactivates Bim gene expression to promote neuronal apoptosis. J. Neurosci. 2011;31:5032–5044. doi: 10.1523/JNEUROSCI.5504-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Campone M., Noel B., Couriaud C., Grau M., Guillemin Y., Gautier F., Gouraud W., Charbonnel C., Campion L., Jezequel P. c-Myc dependent expression of pro-apoptotic Bim renders HER2-overexpressing breast cancer cells dependent on anti-apoptotic Mcl-1. Mol. Cancer. 2011;10:1–14. doi: 10.1186/1476-4598-10-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Walz S., Lorenzin F., Morton J., Wiese K.E., von Eyss B., Herold S., Rycak L., Dumay-Odelot H., Karim S., Bartkuhn M., et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature. 2014;511:483–487. doi: 10.1038/nature13473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ciriello G., Miller M.L., Aksoy B.A., Senbabaoglu Y., Schultz N., Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013;45:1127–1133. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garnett M.J., Edelman E.J., Heidorn S.J., Greenman C.D., Dastur A., Lau K.W., Greninger P., Thompson I.R., Luo X., Soares J., et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sabo A., Amati B. Genome recognition by MYC. Cold Spring Harbor Perspect. Med. 2014;4:2–4. doi: 10.1101/cshperspect.a014191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thiel G., Cibelli G. Regulation of life and death by the zinc finger transcription factor Egr-1. J. Cell Physiol. 2002;193:287–292. doi: 10.1002/jcp.10178. [DOI] [PubMed] [Google Scholar]
- 77.Herold S., Wanzel M., Beuger V., Frohme C., Beul D., Hillukkala T., Syvaoja J., Saluz H.P., Haenel F., Eilers M. Negative regulation of the mammalian UV response by Myc through association with Miz-1. Mol. Cell. 2002;10:509–521. doi: 10.1016/s1097-2765(02)00633-0. [DOI] [PubMed] [Google Scholar]
- 78.Conacci-Sorrell M., McFerrin L., Eisenman R.N. An overview of MYC and its interactome. Cold Spring Harbor Persp. Med. 2014;4:a014357. doi: 10.1101/cshperspect.a014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.James L., Eisenman R.N. Myc and Mad bHLHZ domains possess identical DNA-binding specificities but only partially overlapping functions in vivo. Proc. Natl. Acad. Sci. USA. 2002;99:10429–10434. doi: 10.1073/pnas.162369299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Patel J.H., McMahon S.B. Targeting of Miz-1 is essential for Myc-mediated apoptosis. J. Biol. Chem. 2006;281:3283–3289. doi: 10.1074/jbc.M513038200. [DOI] [PubMed] [Google Scholar]
- 81.Amati B., Littlewood T.D., Evan G.I., Land H. The c-Myc protein induces cell cycle progression and apoptosis through dimerization with Max. EMBO J. 1993;12:5083–5087. doi: 10.1002/j.1460-2075.1993.tb06202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.