


Abstract
Transcription elongation is a highly dynamic and discontinuous process, which includes frequent pausing of RNA polymerase II (RNAPII). RNAPII complexes that stall persistently on a gene during transcription elongation block transcription and thus have to be removed. It has been proposed that the cellular pathway for removal of these DNA damage-independently stalled RNAPII complexes is similar or identical to the removal of RNAPII complexes stalled due to DNA damage. Here, we show that—consistent with previous data—DNA damage-independent stalling causes polyubiquitylation and proteasome-mediated degradation of Rpb1, the largest subunit of RNAPII, using Saccharomyces cerevisiae as model system. Moreover, recruitment of the proteasome to RNAPII and transcribed genes is increased when transcription elongation is impaired indicating that Rpb1 degradation takes place at the gene. Importantly, in contrast to the DNA damage-dependent pathway Rpb1 degradation of DNA damage-independently stalled RNAPII is independent of the E3 ligase Elc1. In addition, deubiquitylation of RNAPII is also independent of the Elc1-antagonizing deubiquitylase Ubp3. Thus, the pathway for degradation of DNA damage-independently stalled RNAPII is overlapping yet distinct from the previously described pathway for degradation of RNAPII stalled due to DNA damage. Taken together, we provide the first evidence that the cell discriminates between DNA damage-dependently and -independently stalled RNAPII.
INTRODUCTION
Transcription elongation is a highly dynamic and discontinuous process that includes frequent pausing of RNA polymerase II (RNAPII), backtracking and arrest in vitro (1,2). In vivo transcription elongation is also discontinuous with frequent and prolonged arrests of RNAPII (3). Consequently, a multitude of transcription elongation factors are needed for efficient transcription elongation (4). When transcription elongation factors fail to ‘restart’ RNAPII, the persistently stalled RNAPII complex prevents transcription of the respective gene and thus has to be removed by the cell to free the gene for subsequent polymerases.
The major pathway for intracellular protein degradation is the ubiquitin-proteasome system (UPS) (5,6). For a protein to be degraded a polyubiquitin chain is covalently attached to it by the action of a ubiquitin-activating enzyme (also called E1), a ubiquitin-conjugating enzyme (E2) and a ubiquitin ligase (E3). The polyubiquitylated protein is then recognized and degraded by the proteasome, which recycles the ubiquitin moieties and cleaves the substrate protein into small peptides. The 26S proteasome consists of a core particle (CP or 20S complex), which contains the catalytic activity, and a regulatory particle (RP or 19S complex), which recognizes and prepares substrates for degradation by the CP.
Rpb1, the largest subunit of RNAPII, is polyubiquitylated and degraded in response to DNA damage. DNA damage in transcribed regions is efficiently repaired by transcription-coupled repair (TCR). However, if this fails RNAPII is thought to be degraded by the UPS as a ‘last resort’ mechanism (7–12). The switch from repair to degradation is mediated by the TCR protein Rad26 and the ubiquitylation promoting protein Def1 (13). Rpb1 is polyubiquitylated by the ubiquitin-conjugating enzymes (E2s) Ubc4 and Ubc5 and the ubiquitin ligases (E3s) Rsp5 and Elc1-Cul3 ((14–19) and references therein, summarized in Supplementary Figure S1, left panel). Polyubiquitylated Rpb1 is degraded by the 26S proteasome, which is facilitated by the AAA ATPase Cdc48 and its adaptor proteins Ufd1, Npl4, Ubx4 and Ubx5 (20). By degradation of the stalled RNAPII complex the damage becomes accessible for repair. However, when the DNA damage is repaired before Rpb1 is degraded, polyubiquitylated Rpb1 is deubiquitylated by the deubiquitylases Ubp2 and Ubp3 and spared from degradation ((18,21); summarized in Supplementary Figure S1, left panel). Although studied mainly in Saccharomyces cerevisiae, the pathway for degradation of DNA damage-dependently stalled RNAPII complexes is well conserved.
In contrast to RNAPII degradation caused by DNA damage the cellular pathway for removal of DNA damage-independently stalled RNAPII complexes is less well understood. However, removal of persistently stalled RNAPII complexes is important as it frees the gene for subsequent polymerases and thus ensures continued transcription. In vivo transcription elongation is inherently discontinuous (3). Adverse growth conditions such as lack of nutrients leading to low NTP levels most likely further impair transcription elongation as mimicked by treatment with the drug 6-azauracil (6AU). RNAPII complexes stalled during transcription elongation for a prolonged time might stall irreversibly. Thus, under natural growth conditions a pathway removing persistently stalled RNAPII from transcribed genes is likely to be of advantage. Since Ubc4, Ubc5, Def1 and Rsp5 are needed for polyubiquitylation of Rpb1 not only for DNA damage-dependent stalling of RNAPII but also in response to DNA damage-independent stalling (16,22), it was speculated that any stalled RNAPII complex—independent of the cause—is degraded by the same pathway (9,16). Here, we show that in S. cerevisiae the pathway for degradation of DNA damage-independently stalled RNAPII is largely overlapping yet distinct from the DNA damage-dependent pathway providing the first evidence that the cell distinguishes between RNAPII complexes stalled for different reasons. Specifically, we show that the E3 ligase Elc1, which adds K48-linked ubiquitin chains to Rpb1 that lead to its degradation in response to DNA damage, is not involved in the DNA damage-independent pathway. Instead, the E3 Rsp5 adds K63-linked polyubiquitin chains to Rpb1 of DNA damage-independently stalled RNAPII. Moreover, the catalytic 20S proteasome is recruited to RNAPII and transcribed genes when transcription elongation is impaired indicating that RNAPII complexes stalled are targeted for degradation at the gene. In addition, we identify Ubp2 and the proteasome associated deubiquitylase Ubp6 to deubiquitylate Rpb1, which might provide a rescue mechanism for Rpb1 when transcription elongation factors ‘restart’ the transcription elongation complex. Taken together, we provide the first evidence that the pathways for the degradation of RNAPII that stalled due to different reasons might be different (summarized in Supplementary Figure S1).
MATERIALS AND METHODS
Yeast strains and plasmids
Yeast strains and plasmids are listed in Supplemental Tables S1 and S2, respectively. TAP-tagged strains were generated by integration of the TAP-tag C-terminal of the respective gene by homologous recombination (23). Deletion strains were constructed by replacement of each ORF with a HIS3 or kanMX4/6 gene. Disrupted strains were complemented with plasmid pRS316 encoding a wild-type (wt) copy of the respective gene (shuffle strains). Double shuffle strains were created by mating the single shuffle strains and tetrad analysis.
Whole cell extracts
Denaturing protein extraction from yeast cells was carried out essentially as described (24). Briefly, 5 OD600 of cells were resuspended in 500 μl of H2O, 150 μl of pre-treatment solution (7.5% (v/v) β-mercaptoethanol, 1.85 M NaOH) were added and the mixture was incubated for 20 min on ice. One hundred fifty microliter of 55% (v/v) trichloroacetic acid was added, incubated for 20 min on ice and centrifuged for 20 min at 16 000 g at 4°C. The supernatant was removed and pellets were resuspended in 100 μl of 1×SB and 20 μl of 1 M Tris base. The samples were then incubated for 2 min at 95°C. For treatment with 6-azauracil (6AU), cells carrying pRS316 and grown in SDC(-ura) were incubated with 250 μg/ml 6AU for 2 h prior to harvesting. For the cycloheximide (CHX) chase experiment to determine the half-life of Rpb1 in wt and Δdst1 cells, protein synthesis was inhibited with 100 μg/ml CHX and samples were taken at the indicated time points.
Purification of RNAPII
To assess the ubiquitylation levels of Rpb1, RPB3-TAP cells were used to purify RNAPII. Limiting amounts of beads were used for all experiments in order to purify equal amounts of Rpb1, and purifications were performed in the presence of proteasome and deubiquitylase inhibitors. Briefly, 100 OD600 of logarithmically growing cells were resuspended in 1× lysis buffer (0.1 M NaCl, 0.05 M Tris-Cl, pH 7.5, 15 mM MgCl2, 0.15% NP40, 2 mM N-ethylmaleimide (Sigma) and 20 μM lactacystine (Boston Biochemicals)) and lysed with an equal volume of glass beads by vortexing at full speed for 5 × 3 min with 3 min breaks on ice. After centrifugation, the clear lysate was incubated with 50 μl of IgG slurry for 2 h at 4ºC. Samples were washed four times with 1× lysis buffer and bound proteins eluted with 2x sodium dodecyl sulphate (SDS) sample buffer and incubation for 3 min at 65ºC. For the 6AU experiment (Figure 2B) RNAPII was purified by tandem affinity purification (TAP) until the TEV eluate according to (25) after treatment with 250 μg/mg 6AU for 2.5 h. For purification of proteins carrying K63-linked polyubiquitin chains the TEV eluate of an Rpb3-TAP purification was incubated for 1 h at 16°C with 50 μl of RAP80 UIM-agarose (BostonBiochemTM), washed three times with 1× lysis buffer and eluted with 2× SDS samples buffer. To detect polyubiquitylation of Rpb1 upon UV irradiation, cells with an OD600 of 0.7 were pretreated with 50 μg/ml CHX for 45 min, UV irradiated with 400 J/m2 and grown for 30 min prior to harvesting. To rescue the lethality of the Δrsp5 mutation, the medium was supplemented with 0.2% NP40 and 2 mM oleic acid (free acid; Sigma) for all strains of this experiment.
Figure 2.
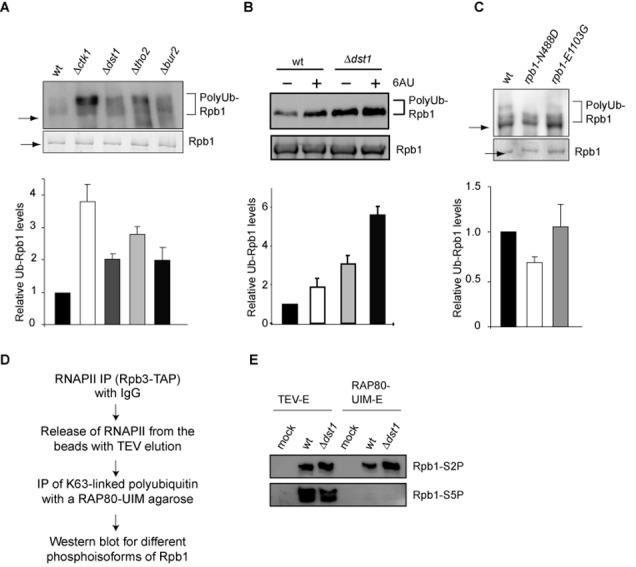
Rpb1 is polyubiquitylated upon impairment of transcription elongation. (A) Polyubiquitylation of Rpb1 is increased in Δctk1, Δdst1, Δtho2 and Δbur2 cells. Approximately equal amounts of RNAPII were purified using Rpb3-TAP and limiting amounts of beads in the presence of DUB and proteasome inhibitors. Polyubiquitylation of Rpb1 was assessed by western blotting using a ubiquitin specific antibody (upper panel). The approximate positions of Rpb1 (arrow) and polyubiquitylated Rpb1 (brackets) are indicated. (B) Polyubiquitylation of Rpb1 is increased after treatment with 6AU. RNAPII was purified using Rpb3-TAP until the TEV eluate from wt and Δdst1 cells in the presence of DUB and proteasome inhibitors. (C) rpb1-N488D, a slow polymerization allele of RPB1, has lower Rpb1 polyubiquitylation levels compared to wt. In contrast, rpb1-E1103G, a fast polymerization allele of RPB1, had polyubiquitylation levels of Rpb1 comparable to wt. Experiment as in A. Levels of polyubiquitylated Rpb1 were normalized to the amount of purified Rpb1. Columns and error bars represent the mean ± standard deviation from three to four independent experiments (bottom panels). P-values are listed in Supplementary Table S3. (D) Scheme for the purification of K63-linked proteins from an RNAPII enriched fraction. Proteins carrying K63-linked ubiquitin chains were purified from the TEV eluate of an RPB3-TAP strain and a non-tagged strain that served as a negative control (mock) from a wt or Δdst1 background using a RAP80-UIM agarose matrix in the presence of DUB and proteasome inhibitors. The phosphorylation status of the CTD was assessed by western blotting using antibodies specific for phosphorylated S2 (H5) or phosphorylated S5 (H14) as shown in (E).
Quantitative western blot analysis
To determine total cellular levels and the level of polyubiquitylation of Rpb1 quantitative western blot analysis was performed. Whole cell extracts or purified RNAPII were subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis and western blotting and probed with anti-Rpb1 (8WG16, Covance), anti-Pgk1 (Invitrogen), anti-ubiquitin (Enzo Life Sciences), linkage-specific anti-ubiquitin (Apu2.07 and Apu3.A8, Genentech) or anti-20S (‘core’ subunits, Enzo Life Sciences) antibodies, respectively. Western blot signals were in the linear range of the antibody reaction, acquired using a Fujifilm Mini-LAS300 System (Fujifilm Life Sciences) and quantified with MultiGauge ScienceLab2005Ver3 (Fujifilm Life Sciences). Levels of Rpb1 were calculated relative to the loading control Pgk1 and ubiquitin signals relative to purified Rpb1 from at least three biologically independent experiments.
Chromatin Immunoprecipitation
To analyze the occupancy of RNAPII and the proteasome at the genes ADH1, PMA1 and ACT1, chromatin Immunoprecipitation (ChIP) experiments using Rpb3-TAP and Pre1-TAP strains were performed essentially as described (25). Briefly, crosslinked cells were lysed with an equal volume of glass beads by vortexing at full speed for 6 × 3 min with 3 min breaks on ice. The lysate was sonified in a BioruptorTM UCD-200 (Diagenode) using 25 × 30 s cycles with 30 s breaks at an output of 200 W to produce chromatin fragments of an average size of ∼250 bp. Chromatin lysate corresponding to 3 A280 (Rpb3) or 18 A280 (Pre1) was used for immunoprecipitation with 15 μl of IgG-coupled Dynabeads for 3.5 h at 20°C or O/N at 4°C, respectively. After washing and elution from the beads according to (26), the IP eluates as well as 0.08 A280 for input samples were treated with protease type XIV (Sigma) and crosslinks were reversed by overnight incubation at 65°C. DNA was purified over spin columns (Macherey-Nagel). Quantitative PCR with input and IP samples was performed on an ABI Prism 7000 cycler (Applied Biosystems). As a control, primers for a nontranscribed region (NTR) of chromosome V were used (sequences available on request). PCR efficiencies (E) were determined with standard curves. Enrichment of Rpb3 and Pre1 over the NTR was calculated according to [E∧(CT Input – CT IP)]gene / [E∧(CT Input – CT IP)]NTR. The enrichment of Pre1 relative to Rpb3 in wt cells was set to 1. Means were calculated from three to four biologically independent experiments.
RESULTS
Impairment of transcription elongation causes degradation of Rpb1, the largest subunit of RNAPII
RNAPII complexes stalling persistently on a gene during transcription elongation impair transcription and thus have to be removed to free the gene to be transcribed by subsequent polymerases. It has been previously shown that polyubiquitylation of Rpb1, the largest subunit of RNAPII, is increased when transcription elongation is impaired, e.g. in mutants lacking the transcription factor Dst1 or after addition of the transcription elongation inhibitor 6-azauracil (6AU), which impairs transcription elongation by decreasing cellular GTP/UTP levels (16). Thus, it is likely that Rpb1 is degraded and consequently RNAPII removed from the gene, when transcription complexes stall persistently during transcription elongation. To study the mechanism of removal of persistently stalled RNAPII, we used a set of four diverse transcription elongation mutants: Δctk1, Δdst1, Δtho2 and Δbur2. Ctk1 is the kinase subunit of the CTDK-I complex that phosphorylates the C-terminal domain (CTD) of Rpb1, Dst1 is the cleavage factor TFIIS, Tho2 is a subunit of the THO complex, and Bur2 is the cyclin of the Bur1-Bur2 kinase complex. First, we assessed the stability of Rpb1. Deletion of any one of these four transcription elongation factors reduced total cellular Rpb1 levels to ∼50–70% (Figure 1A). A similar reduction was caused by treatment of the cells with 6AU (Figure 1A). The decrease of total Rpb1 levels is not caused by reduced levels of RPB1 mRNA as these remained unchanged (Supplementary Figure S2A). Furthermore, since deletion of CTK1, THO2 and BUR2 causes a growth defect, we assessed Rpb1 levels after depletion of the respective protein. The same reduction of total Rpb1 levels was observed at a time point when growth of the cells was not yet impaired indicating that the reduction of Rpb1 levels is a direct effect (Supplementary Figure S2B–D).
Figure 1.
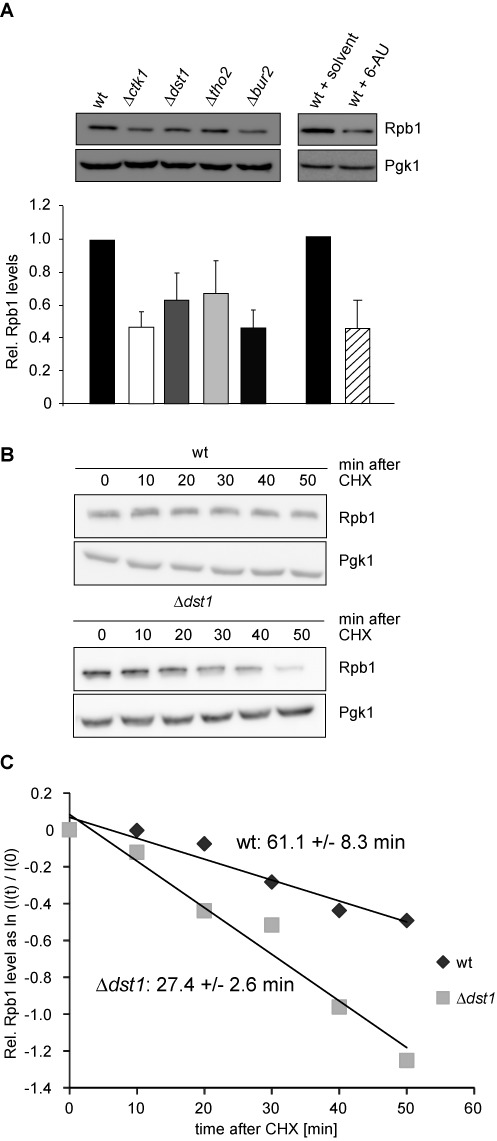
Rpb1, the largest subunit of RNAPII, is degraded when transcription elongation is impaired. (A) Total Rpb1 levels are reduced in strains lacking the transcription elongation factors Ctk1, Dst1, Tho2 or Bur2 or treated with 6AU as assessed by western blotting against Rpb1 (upper panels). Columns and error bars represent the mean ± standard deviation from four independent experiments (bottom panels). P-values are listed in Supplementary Table S3. (BandC) The half-life of Rpb1 decreases to about half in Δdst1 cells compared to wt cells. The half-life of Rpb1 in wt and Δdst1 cells was assessed by quantification of Rpb1 levels in the respective strain after treatment with CHX at the indicated time points (n = 8). (B) Western blots of Rpb1 and Pgk1, which served as a loading control. (C) Rpb1 half-lifes in wt and Δdst1 were calculated by linear regression on the logarithmized relative protein levels determined by western blot (normalized signal intensities at each time point (I(t)) relative to signal intensity at time point zero (I(0)) before CHX addition). The standard error of the linear regression is indicated.
In order to assess whether these lower steady state levels of Rpb1 in the transcription elongation mutants and after treatment with 6AU are caused by enhanced degradation of Rpb1, we assessed the half-life of Rpb1 in wt and Δdst1 cells after CHX treatment. Indeed, the half-life of Rpb1 decreased from ∼61 min in wt cells to ∼27 min in Δdst1 cells (Figure 1C). Taken together, levels of Rpb1 are reduced by enhanced degradation when transcription elongation is impaired.
Rpb1 is polyubiquitylated upon DNA damage-independent stalling
Targeted cellular proteolysis is primarily mediated by the UPS (5,6). Interestingly, several connections between the proteasome and RNAPII transcription have been reported. The proteasome has been shown to function in transcription initiation, in the removal of RNAPII complexes stalled due to DNA damage and—in a non-proteolytic manner—in transcription elongation ((7–11,27–29) and references therein). Previously, the UPS has also been suggested to function in degradation of DNA damage-independently stalled RNAPII ((8–10,30,31) and references therein). Thus, the lower Rpb1 levels observed upon impairment of transcription elongation are most likely due to UPS-mediated degradation. To test this, we first assessed the levels of Rpb1 ubiquitylation in wt cells and the four transcription elongation mutants or after treatment with 6AU. In order to facilitate quantification of Rpb1 ubiquitylation levels, we purified approximately equal levels of RNAPII and thus Rpb1 from the respective strain backgrounds by using limiting amounts of beads. In addition, to inhibit deubiquitylating enzymes (DUBs) and the proteasome all purifications were performed in the presence of DUB and proteasome inhibitors. Increased Rpb1 ubiquitylation was observed previously in Δdst1 cells or after treatment with 6AU (16). In addition, it has been shown recently that Rpb1 of RNAPII that is unable to resume transcription after backtracking due to a mutation in Dst1 also has increased Rpb1 polyubiquitylation levels (32). Consistently, wt cells displayed a low level of polyubiquitylated Rpb1, which increased ∼2-fold when transcription elongation was impaired by deletion of one of the four transcription elongation factors (Figure 2A) or treatment with the transcription elongation inhibitor 6AU (Figure 2B). In order to exclude that a protein associated with RNAPII, i.e. other than Rpb1, is responsible for the observed ubiquitylation signal, we purified RNAPII using Rpb3-TAP from wt and Δckt1 cells under high salt conditions (1 M NaCl). Under these conditions, TFIIF, the transcription factor with the highest affinity for RNAPII, was no longer associated with RNAPII (Supplementary Figure S3). However, ubiquitylation of Rpb1 was still increased in the transcription elongation mutant (Supplementary Figure S3) indicating that ubiquitylation of Rpb1 itself is increased when transcription elongation is impaired. Interestingly, the level of Rpb1 ubiquitylation differs in the four transcription elongation mutants. This might reflect the differing effects these mutants have on transcription elongation (33).
In order to assess whether the polyubiquitylation of Rpb1 observed in wt cells is due to DNA damage-independent stalling we used rpb1-N488D, an allele of RPB1 that causes a slower polymerization rate in vitro, is not synthetic lethal with Δdst1 and thus might be less prone to stalling (34). In this mutant background Rpb1 ubiquitylation should therefore be reduced compared to wt cells. Indeed, rpb1-N488D cells had lower Rpb1 polyubiquitylation levels compared to the wt enzyme (Figure 2C), indicating that Rpb1 polyubiquitylation is caused by DNA damage-independent stalling occurring in wt cells. Alternatively, the reduced polyubiquitylation of Rpb1 in the rpb1-N488D mutant might be due to a lower processivity of RNAPII (34). RNAPII that dissociates from the template during elongation might thus escape ubiquitylation.
According to our model, Rpb1 of elongating RNAPII should be the substrate for polyubiquitylation and its CTD thus phosphorylated on S2. Indeed, in a reconstituted in vitro RNAPII ubiquitylation system, an arrested RNAPII elongation complex is the preferred substrate for ubiquitylation, and serine 5 phosphorylation of the CTD, a hallmark of transcription initiation, inhibits polyubiquitylation (16). In order to assess the phosphorylation status of the polyubiquitylated Rpb1, proteins carrying K63-linked ubiquitin chains were purified from an RNAPII enriched fraction (TEV-E of an Rpb3-TAP purification) (for scheme of the purification see Figure 2D). K63-linked proteins were purified since polyubiquitin chains on Rpb1 are mainly K63-linked in DNA damage-independent stalling (Figure 5). Expectedly, the TEV eluate of an Rpb3-TAP purification contained Rpb1 with S2- and S5-phosphorylated CTDs (Figure 2E). In contrast, the CTD of polyubiquitylated Rpb1 was exclusively phosphorylated on S2 (Figure 2E). In addition, ubiquitylation of Rpb1 phosphorylated on S2 increased in the Δdst1 mutant (Figure 2E). Thus, consistent with previous results DNA damage-independent stalling leads to increased Rpb1 polyubiquitylation of elongating RNAPII.
Figure 5.
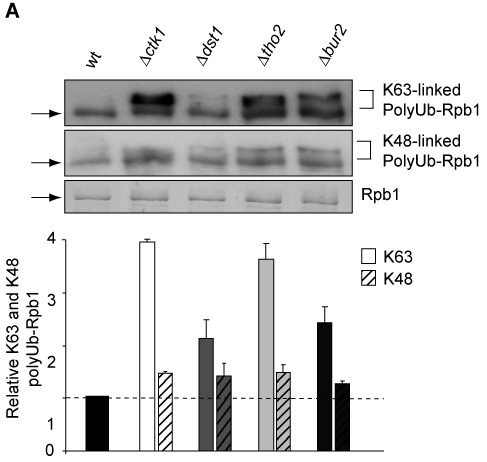
The levels of K48- and K63-linked polyubiquitin chains on Rpb1 increases upon DNA damage-independent stalling. Polyubiquitin chains formed on Rpb1 in Δctk1, Δdst1, Δtho2 and Δbur2 cells are mainly K63-linked. Experiment as in Figure 2A. The nature of polyubiquitin chains on Rpb1 was assessed by western blotting using K48- and K63-ubiquitin specific antibodies (Apu2.07 and Apu3.A8 (Genentech), respectively; upper panels). Columns representing the level of K63-chains are plain and the ones for K48-chains hatched. Levels of polyubiquitylated Rpb1 were normalized to the amount of purified Rpb1. P-values are listed in Supplementary Table S3.
The proteasome associates with RNAPII at the transcribed gene when transcription elongation is impaired
Polyubiquitylated Rpb1 of persistently stalled transcription elongation complexes is most likely the substrate of the proteasome. Thus, we examined a potential association of the proteasome with RNAPII. The 20S proteolytic core of the proteasome copurified with RNAPII in the presence of proteasome inhibitors from wt cells (Figure 3A and (31)). Importantly, the association of the proteasome with RNAPII increased when transcription elongation was impaired (Figure 3A).
Figure 3.
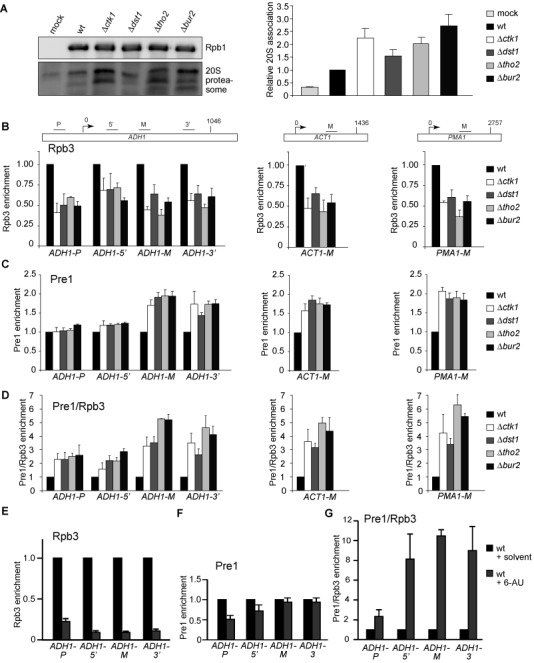
The proteasome associates with RNAPII and transcribed genes. (A) RNAPII was purified in the presence of DUB and proteasome inhibitors using an RPB3-TAP strain and levels of Rpb1 and co-purifying proteasome detected with 8WG16 and an anti-20S core antibodies, respectively (upper panel). Columns and error bars represent the mean ± standard deviation from four independent experiments (bottom panel). (B–D) To assess recruitment of the proteasome to actively transcribed genes, ChIP experiments were performed with a RPB3-TAP and a PRE1-TAP strain. Enrichment of Rpb3 (B), Pre1 (C) and Pre1 relative to Rpb3 (RNAPII) (D) at the genes ADH1, ACT1 and PMA1 was quantified by real-time PCR with the indicated primers (upper panel). (E–G) Experiment as in B–D after inhibition of transcription by treatment with 250 μg/ml 6AU for 2.5 h. Columns and error bars represent the mean ± standard deviation from three independent experiments (bottom panels). P-values are listed in Supplementary Table S3.
The proteasome is present at the coding regions of genes (29,35,36). In addition, it has been shown that ubiquitylated Rpb1 and the proteasome accumulate on chromatin after UV irradiation suggesting that the degradation of DNA damage-dependently stalled RNAPII takes place on chromatin (20). If Rpb1 of RNAPII complexes stalled in a DNA damage-independent manner is also degraded by the proteasome at the transcribed gene, one would expect that the association of the proteasome with coding regions increases, when transcription elongation is impaired. In order to test this, we performed ChIP experiments with four subunits of the 20S core of the proteasome (Pre1, Pre2, Pre4 and Pup1) since previously the 19S regulatory particle has been implicated in transcription elongation independently of degradation (29). In the transcription elongation mutants, RNAPII occupancy decreases ∼2-fold compared to wt (Figure 3B). This decrease most likely reflects the decrease in Rpb1 levels (Figure 1A). Importantly, recruitment of the 20S proteasome relative to RNAPII is increased, especially at the middle (M) and 3’ region of the ADH1 gene and the middle (M) regions of ACT1 and PMA1 (Figure 3C and D and Supplementary Figure S4A–C). Inhibition of transcription elongation by treatment with 6AU also increased recruitment of the 20S proteasome to the gene relative to RNAPII (Figure 3E–G). However, since recent data suggest that the canonical 26S proteasome complex is recruited during transcription (36) most likely recruitment of the whole 26S particle is increased when transcription elongation is impaired. These data are consistent with the finding that elongating RNAPII complexes are the preferred substrate for degradation ((16,22,37,38) and Figure 2E). Taken together, Rpb1 of transcription complexes stalled in a DNA damage-independent manner is polyubiquitylated and degraded by the proteasome at the site of transcription.
Molecular mechanism of Rpb1 ubiquitylation
We next wanted to further elucidate the ubiquitylation machinery of this DNA damage-independent pathway. In general, polyubiquitylation is mediated by a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2) and a ubiquitin ligase (E3). As there is only one E1 enzyme (Uba1) in yeast, we aimed to assess the E2 and E3 enzymes. In the case of DNA damage within the coding region of an actively transcribed gene the DNA lesion is efficiently repaired by TCR. If this fails RNAPII is thought to be disassembled by specific degradation of Rpb1 triggered by complex formation with the TCR protein Rad26 and the ubiquitylation promoting protein Def1 (13,14,39,40). Ubiquitylation of Rpb1 in this DNA damage-dependent pathway is mediated by Def1, the RNAPII subunit Rpb9, the E2s Ubc4 and Ubc5 and the E3s Rsp5 and Elc1 ((15–19,41) and references therein, depicted in Supplementary Figure S1, left panel). The E2s Ubc4 and Ubc5 function redundantly, whereas the E3s Rsp5 and Elc1 function sequentially, with Rsp5 mono-ubiquitylating Rpb1 and Elc1 adding a K48-linked polyubiquitin chain that leads to degradation. In addition, Rsp5 produces non-functional K63-linked polyubiquitin chains (18). As Ubc4, Ubc5, Def1 and Rsp5 are also required for polyubiquitylation of Rpb1 in response to DNA damage-independent stalling (16,22), it was speculated that DNA damage-independently stalled RNAPII complexes are degraded by the same pathway (9,16,22).
We thus tested these proteins for a function in the DNA damage-independent pathway using one of the transcription elongation mutants, Δdst1, since this mutant does not have a growth defect. We again purified approximately equal levels of Rpb1 from the corresponding strain backgrounds by using limiting amounts of beads in order to facilitate quantification of the Rpb1 ubiquitylation levels—in the presence of DUB and proteasome inhibitors. Deletion of DEF1, UBC4 or UBC5 in the Δdst1 background reduces polyubiquitylation of Rpb1 showing that Def1 and the E2s Ubc4 and Ubc5 are needed for polyubiquitylation of Rpb1 (Figure 4A and B). Ubc4 and Ubc5 have overlapping, partially redundant functions, which is reflected by the partial reduction of Rpb1 ubiquitylation observed by deletion of only one of these two E2s (Figure 4B). Deletion of RAD26 increases Rpb1 degradation in response to DNA damage (13), probably due to a defect in TCR. However, loss of Rad26 function does not affect polyubiquitylation of Rpb1 in the Δdst1 background indicating that Rad26 is not necessary in the DNA damage-independent pathway (data not shown). As in the DNA damage-dependent pathway the E3 Rsp5 polyubiquitylates Rpb1 in response to DNA damage-independent stalling (Figure 4C). Importantly, however, the E3 Elc1 is not required for polyubiquitylation of Rpb1 in response to DNA damage-independent stalling in the Δdst1 strain (Figure 4D, compare Δdst1−UV with Δdst1Δelc1−UV). This is the first evidence that the mechanism for polyubiquitylation of Rpb1 is different depending on the cause of RNAPII stalling. In order to corroborate in one experiment and with identical strains that the DNA damage-independent degradation of Rpb1 is indeed distinct from the well-studied DNA damage-dependent pathway, we assessed Rpb1 polyubiquitylation after induction of DNA damage by UV in wt, Δelc1 and Δdst1Δelc1 strains. As published previously, Rpb1 ubiquitylation increased by UV treatment (Figure 4D, compare wt−UV and wt+UV), and this increase is dependent on Elc1 (Figure 4D, compare wt+UV with Δelc1+UV and Δdst1Δelc1−UV with Δdst1Δelc1+UV). Rpb1 ubiquitylation increased 2-fold and the kind of ubiquitin signal was similar both in wt cells treated with UV and in Δdst1 cells (Figure 4D, compare wt+UV and Δdst1). Consistent with the finding that Rpb1 polyubiquitylation of RNAPII stalled in a DNA damage-independent manner is independent of Elc1, Cul3, a ubiquitin-protein ligase that forms a complex with Elc1 and is required for Rpb1 ubiquitylation of Rpb1 after DNA damage (17), is also not required for Rpb1 polyubiquitylation of DNA damage-independently stalled RNAPII complexes (Figure 4E). Thus, the pathways of polyubiquitylation and thus likely degradation of Rpb1 in response to DNA damage versus DNA damage-independent stalling diverge in their requirement of the E3 ligase Elc1. Taken together, polyubiquitylation of Rpb1 of DNA damage-independently stalled RNAPII complexes is mediated by an overlapping but different set of enzymes than polyubiquitylation of Rpb1 in the DNA damage-dependent pathway (Supplementary Figure S1).
Figure 4.
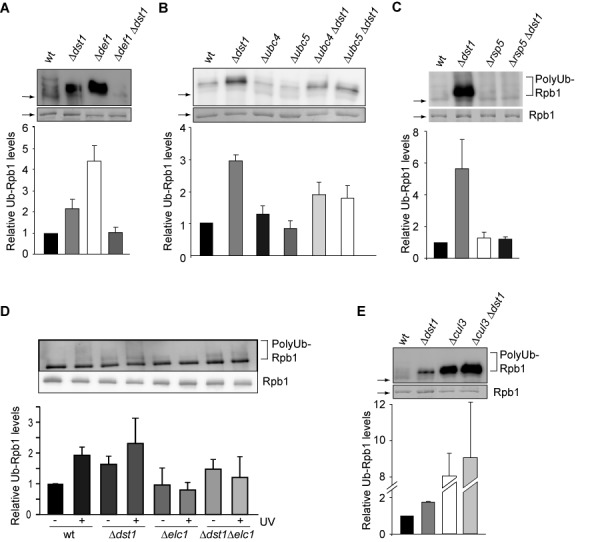
Mechanism of polyubiquitylation of Rpb1 of DNA damage-independently stalled RNAPII. (A–E) Polyubiquitylation of Rpb1 in Δdst1 and Δubc4, Δubc5, Δdef1, Δrsp5, Δelc1, Δcul3 and the respective double mutant strains was assessed after purification of approximately equal levels of RNAPII using Rpb3-TAP and limiting amounts of beads in the presence of DUB and proteasome inhibitors. Rpb1 levels were assessed by Coomassie staining and polyubiquitylation by western blotting using a ubiquitin specific antibody (upper panel). In (D) cells were also treated with UV (+UV). Note that Rpb1 ubiquitylation is unusually high in the Δrsp5 strain, which is probably due to the oleic acid and NP40 containing media needed for viability of the Δrsp5 strain. An explanation for the increased Rpb1 ubiquitylation in the Δcul3 and Δcul3Δdst1 strains could be that impairment of the E3 ligase Cul3 most likely leads to decreased ubiquitylation of Cul3-dependent substrates, which might lead to higher levels of free ubiquitin causing in turn unspecific ubiquitylation of Rpb1. However, there is no satisfactory explanation for the observed increase in Rpb1 ubiquitylation in the Δdef1 strain, which contradicts the current model. Columns and error bars represent the mean ± standard deviation from four independent experiments (bottom panels). P-values are listed in Supplementary Table S3.
Polyubiquitin chains on Rpb1 of DNA damage-independently stalled RNAPII are K48- and K63-linked
To investigate the nature of the polyubiquitin chains attached to Rpb1 of DNA damage-independently stalled RNAPII, we purified RNAPII from wt cells and the four transcription elongation mutants as above in the presence of DUB and proteasome inhibitors and assessed the nature of the polyubiquitin chains using antibodies specific for K48- or K63-linked ubiquitin chains (Supplementary Figure S5). Interestingly, the polyubiquitin chains attached to Rpb1 when transcription elongation is impairment are mainly K63-linked (Figure 5). This is consistent with our finding that polyubiquitylation of Rpb1 is mediated by Rsp5 (Figure 4C) since Rsp5 produces K63-linked polyubiquitin chains (42).
This finding underlines the notion that the pathways for degradation of stalled RNAPII are different depending on the cause of stalling since polyubiquitin chains targeting Rpb1 for degradation in the DNA damage-dependent pathway are mainly K48-linked ((18) and references therein). Thus, K63-linked polyubiquitin chains could be a prerequisite for Rpb1 degradation caused by DNA damage-independent stalling. In addition to K48-linked polyubiquitin chains, which serve as the main signal for degradation, K63-linked polyubiquitin chains can target a protein for degradation by the proteasome in vitro and in vivo (42–44). Moreover, Rsp5-mediated K63-polyubiquitylated Rpb1 is readily degraded by the 26S proteasome in vitro whereas nonphysiological polyubiquitylated substrates are not (42). However, the level of K48-linked polyubiquitin chains also increases. Thus, the K63-linked polyubiquitin chains could be remodeled by deubiquitylases (also see below) and a K48-linked chain added by a yet unknown E3 ligase for degradation of Rpb1, which would be consistent with the increase in K48-linked chains (Figure 5). Thus, a backup system for the function of Elc1/Cul3 in K48-linked polyubiquitylation of Rpb1 could exist that is more active during DNA damage-independent stalling of RNAPII than during DNA damage. Alternatively and not mutually exclusively, Rsp5 could be more active when Elc1 is missing. It will be interesting to assess in the future which role the K48- and K63-linked polyubiquitin chains play in the degradation of DNA damage-independently stalled RNAPII and by which enzymes these polyubiquitylations are catalyzed.
Deubiquitylation of Rpb1 as a rescue mechanism
Since the proteasome degrades polyubiquitylated proteins, deubiquitylation is an important rescue mechanism, especially for a subunit of an enzyme as essential as RNAPII. Furthermore, deubiquitylation of already ubiquitylated Rbp1 can rescue Rpb1 from unnecessary degradation when RNAPII resumes transcription after a prolonged pause (also see below). Two deubiquitylases have been described to function in the DNA damage-dependent pathway: Ubp2 deubiquitylates excess K63-linked polyubiquitin chains formed by Rsp5, whereas Ubp3 removes Rsp5-mediated monoubiquitin and Elc1-mediated K48-linked polyubiquitin chains from Rpb1 ((18,21), also see Supplementary Figure S1). In order to identify the deubiquitylases responsible for Rpb1 deubiquitylation in the DNA damage-independent pathway, we tested deubiquitylases, deletion of which causes sensitivity to 6AU (21) for an effect on Rpb1 ubiquitylation when transcription elongation is impaired: Ubp2, Ubp3, Ubp6, Ubp10 and Ubp12. To do this, we assessed the levels of Rpb1 polyubiquitylation in Δdst1 cells in combination with deletion of one of the abovementioned deubiquitylases after purification of approximately equal levels of Rpb1 by using limiting amounts of beads in the presence of DUB and proteasome inhibitors. Deletion of UBP10 or UBP12 does not affect Rpb1 polyubiquitylation levels (data not shown). Similarly, in contrast to the DNA damage-dependent pathway, Ubp3 is not required for deubiquitylation of Rpb1 (Figure 6, right panel). Unexpectedly, polyubiquitylation of Rpb1 also does not increase in the Δubp3 background when DST1 is deleted. This might be caused by a disturbed ubiquitin homeostasis when Ubp3 function is lacking. However, consistent with a function of Rsp5 in ubiquitylating Rpb1 in DNA damage-independently stalled RNAPII complexes, Ubp2 is required for deubiquitylation of Rpb1 (Figure 6, middle panel).
Figure 6.
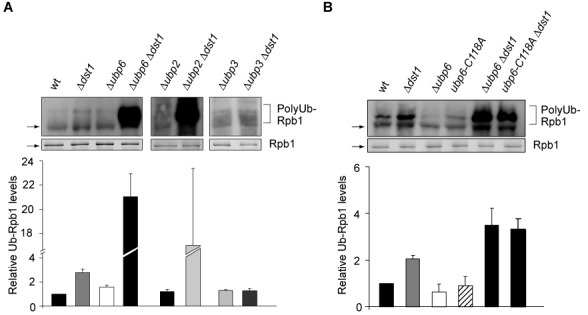
Deubiquitylation of Rpb1. (A) Deubiquitylation of Rpb1 is mediated by Ubp6 and Ubp2, but not Ubp3. (B) The catalytic activity of Ubp6 is needed for deubiquitylation of Rpb1. Experiment as in Figure 4. P-values are listed in Supplementary Table S3.
In addition to Ubp2, loss of Ubp6 function leads to an increase in the levels of polyubiquitylated Rpb1 (Figure 6A, left panel). Ubp6 is one of two proteasome associated deubiquitylases in S. cerevisiae. Interestingly, Ubp6 has a catalytic as well as a non-catalytic activity: It deubiquitylates ubiquitylated proteins before their degradation thereby recycling ubiquitin and delays degradation of polyubiquitylated proteins by the proteasome in a mainly non-catalytic manner (45). To determine whether the catalytic activity of Ubp6 is indeed needed for deubiquitylation of Rpb1 (Figure 6A) we assessed Rpb1 ubiquitylation in the catalytically inactive ubp6-C118A mutant that still inhibits the proteasome (45). Rpb1 ubiquitylation levels are increased in ubp6-C118A Δdst1 cells to a similar extent as in Δubp6 Δdst1 cells showing that the deubiquitylase activity of Ubp6 is necessary for Rpb1 deubiquitylation (Figure 6B). Thus, Ubp6 deubiquitylates Rpb1 of RNAPII complexes stalled during transcription elongation. Taken together, consistent with the lack of the E3 ligase Elc1 the Elc1-antagonizing deubiquitylase Ubp3 is not involved in the pathway for degradation of DNA damage-independently stalled RNAPII. Thus, not only the mechanism for ubiquitylation but also for deubiquitylation of Rpb1 caused by DNA damage-independent stalling differs from the DNA damage-dependent pathway.
DISCUSSION
In summary, we demonstrate here that the pathways for degradation of DNA damage-dependently and -independently stalled RNAPII complexes are overlapping yet distinct (Figure 7). Thus, we provide the first evidence that the cell distinguishes between RNAPII complexes persistently stalled by different causes. In contrast to the DNA damage-dependent pathway, the E3 ligase Elc1 does not ubiquitylate Rpb1 of DNA damage-independently stalled RNAPII. Consistently, Ubp3, the deubiquitylase that removes ubiquitin added by Elc1 in the DNA damage-dependent pathway, is not involved in the DNA damage-independent pathway. Instead, polyubiquitylation of Rpb1 occurs by the E3 Rsp5 and consistently the polyubiquitin chains are mainly K63-linked. Interestingly, elongating RNAPII complexes that collide due to convergent transcription are degraded in an Elc1-dependent manner (46). As these RNAPII complexes also stall in a DNA damage-independent manner, the mechanisms for degradation of RNAPII complexes persistently stalled during transcription elongation or due to collision with another RNAPII complex seem to be different.
Figure 7.
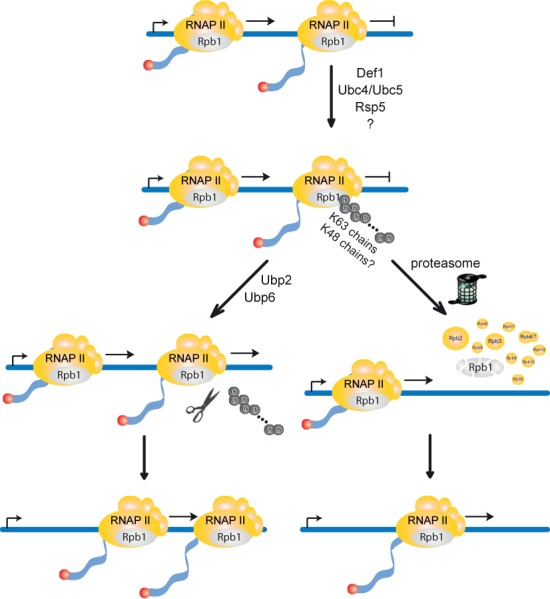
Model for the degradation of DNA damage-independently stalled RNA polymerase II (RNAPII). Persistently stalled RNAPII complexes are polyubiquitylated with K63-specific polyubiquitin chains by Def1, the E2s Ubc4 and Ubc5 and the E3 Rsp5. When transcription factors restart RNAPII, Rpb1 is deubiquitylated by Ubp2 or Ubp6 (left pathway) and RNAPII resumes transcription. Alternatively, as a failsafe mechanism when RNAPII remains persistently stalled, polyubiquitylated Rpb1 is recognized by the proteasome and degraded (right pathway). For Rpb1 degradation a remodeling of the K63-linked polyubiquitin chains and conjugation of K48-linked polyubiquitin chains by a yet to be identified E3 ligase might be necessary. This pathway for degradation of RNAPII complexes stalled persistently during transcription elongation frees the way for following RNAPII complexes. The two pathways for Rpb1 degradation, DNA damage-dependent and -independent, are compared in Supplementary Figure S1.
Different cellular pathways to eliminate stalled RNAPII dependent on the cause of stalling might be necessary. Whenever a DNA damage within a transcribed gene cannot be repaired the stalled polymerase should be degraded quickly. In contrast, since stalling of RNAPII occurs frequently during transcription elongation (3) it is crucial that not every paused polymerase is immediately degraded. Thus, once RNAPII is recognized as stalled ubiquitin moieties could be added slowly by Rsp5. Slow polyubiquitylation of Rbp1 could ensure that only prolonged stalling leads to degradation. Slow ubiquitylation would also open a time window for transcription elongation factors to ‘restart’ RNAPII. In this case, i.e. when RNAPII resumes transcription after stalling, the polyubiquitin chain can be removed by Ubp2 or Ubp6 and Rpb1 spared from degradation.
Along the same lines, it is surprising that the proteasome interacts stably with its substrate RNAPII. As degradation of Rpb1 is potentially very deleterious, the proteasome might have to stay associated with RNAPII for some time before it can degrade Rpb1. This would open another time window for transcription elongation factors to rescue the stalled RNAPII complex. This model also fits well with our finding of Ubp6 being a deubiquitylase of Rpb1 to counteract degradation. Ubp6 is one of two proteasome-associated deubiquitylases ((45,47) and references therein). In addition to its catalytic activity, Ubp6 delays proteasomal degradation directly by binding to the proteasome (45). One function of Ubp6 is to recycle ubiquitin from ubiquitylated substrates before their degradation (48). In addition, the proteasome inhibitory function of Ubp6 is thought to delay the decision to degrade a substrate (45). Since Ubp6 progressively deubiquitylates the substrate protein during its delay of proteasomal degradation, the length of inhibition is crucial. When the polyubiquitin chain is shortened beyond a critical length the substrate will be released from the proteasome and thus spared from degradation (45). As Ubp2 has been suggested to maintain short ubiquitin chains on Rsp5 substrates in order to spare them from degradation (42), both deubiquitylases, Ubp2 and Ubp6, uncovered here to deubiquitylate Rpb1 upon DNA damage-independent stalling, could act as a ‘fail-safe mechanism’ to spare Rpb1 from degradation; when RNAPII resumes transcription after arrest or when Rpb1 has been erroneously ubiquitylated.
How stalled RNAPII is recognized by the cell in either pathway remains an open question. However, independent of the cause of stalling—DNA damage-dependent or -independent—XPG/Rad2 and CSB/Rad26 bind to the stalled RNAPII forming a so-called ‘supracomplex’ (49). Rad26 in turn forms a stable complex with the ubiquitylation promoting protein Def1 (13). In vivo Rad26 is unlikely to be essential for recognition of DNA damage-independently stalled RNAPII as deletion of RAD26 does not affect polyubiquitylation of Rpb1 (data not shown). The first steps of Rpb1 polyubiquitylation are mediated by Def1, Ubc4 and Ubc5 and are thus identical in both pathways (Supplementary Figure S1). Thus, in contrast to Rad26, Def1 could be involved in recognition of stalled RNAPII because it promotes ubiquitylation of Rpb1 in both pathways (Figure 4A and (19)). Since Def1 has been proposed to recruit the cullin in the DNA damage-dependent pathway (50), it might also play a crucial role in distinguishing between the two degradation pathways. This is consistent with the finding that activation of Def1 is induced by DNA damage as well as impairment of transcription elongation by 6AU (50). According to such a model, the two pathways could diverge by the action of DNA repair factors and/or Rad26 that are only present in case of DNA damage-dependent stalling and could be required for Def1 to recruit Elc1/Cul3. Thus, Def1 might play a pivotal role in distinguishing between different pathways for degradation of persistently stalled RNAPII.
Since RNAPII stalls frequently and for prolonged times during transcription elongation (3), degradation of DNA damage-independently stalled RNAPII most likely has an essential function in wt cells. Consistently, the proteasome stably associates with RNAPII and is recruited to transcribed genes in wt cells (Figure 3 and (31,35,51)). In addition, we observe a basic level of polyubiquitylated Rpb1 in wt cells that is reduced in cells carrying rpb1-N488D, an allele causing a slower polymerization rate of RNAPII that might be less prone to stalling (Figure 2C). Moreover, Rpb1 is degraded and polyubiquitylated upon treatment with 6AU, a drug that causes increased transcriptional stalling due to depletion of nucleotide pools, a situation that might mimic natural conditions such as starvation. Thus, this pathway could be essential for survival under suboptimal growth conditions. Finally, the pathway for degradation of DNA damage-independently stalled RNAPII is most likely evolutionarily conserved as RNAPII is ubiquitylated and degraded after transcriptional arrest by α-amanitin in higher eukaryotes and the polyubiquitin chains formed are also K63-linked (30,38,52).
Taken together, we provide the first evidence that RNAPII stalled by different causes—DNA damage-dependent versus -independent—is polyubiquitylated and degraded by overlapping but distinct pathways.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank S. Röther for the initial experiments that led to this project and P. Cramer and A. Buchberger for critical reading of the manuscript. We are grateful to V. M. Dixit (Genentech, Inc.) for the K48- and K63-specific antibodies, to Jeffrey N. Strathern (National Cancer Institute, National Institutes of Health) for the RPB1 mutants, to D. Finley (Harvard Medical School, Boston) for plasmids pJH80 (URA3, UBP6) and pJH81 (URA3, ubp6-C118A) and yeast strains SUB280 and SUB413 and to A. Aguilera (University of Sevilla) for plasmid pRS315-THO2.
Footnotes
Present address: Institute of Biochemistry, Justus Liebig University Giessen, Heinrich-Buff-Ring 58, 35392 Giessen, Germany.
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
FUNDING
The Boehringer Ingelheim Fonds (to E.K.); the German Research Foundation (SFB646); the Center for Integrated Protein Science, Munich (CIPSM) and the EMBO Young Investigator Programme (to K.S.). Funding for open access charge: SFB646.
Conflict of interest statement. None declared.
REFERENCES
- 1.Neuman K.C., Abbondanzieri E.A., Landick R., Gelles J., Block S.M. Ubiquitous transcriptional pausing is independent of RNA polymerase backtracking. Cell. 2003;115:437–447. doi: 10.1016/s0092-8674(03)00845-6. [DOI] [PubMed] [Google Scholar]
- 2.Landick R. The regulatory roles and mechanism of transcriptional pausing. Biochem. Soc. Trans. 2006;34:1062–1066. doi: 10.1042/BST0341062. [DOI] [PubMed] [Google Scholar]
- 3.Darzacq X., Shav-Tal Y., de Turris V., Brody Y., Shenoy S.M., Phair R.D., Singer R.H. In vivo dynamics of RNA polymerase II transcription. Nat. Struct. Mol. Biol. 2007;14:796–806. doi: 10.1038/nsmb1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saunders A., Core L.J., Lis J.T. Breaking barriers to transcription elongation. Nat. Rev. Mol. Cell Biol. 2006;7:557–567. doi: 10.1038/nrm1981. [DOI] [PubMed] [Google Scholar]
- 5.Varshavsky A. Regulated protein degradation. Trends Biochem. Sci. 2005;30:283–286. doi: 10.1016/j.tibs.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 6.Navon A., Ciechanover A. The 26 S proteasome: from basic mechanisms to drug targeting. J. Biol. Chem. 2009;284:33713–33718. doi: 10.1074/jbc.R109.018481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ratner J.N., Balasubramanian B., Corden J., Warren S.L., Bregman D.B. Ultraviolet radiation-induced ubiquitination and proteasomal degradation of the large subunit of RNA polymerase II. Implications for transcription-coupled DNA repair. J. Biol. Chem. 1998;273:5184–5189. doi: 10.1074/jbc.273.9.5184. [DOI] [PubMed] [Google Scholar]
- 8.Svejstrup J.Q. Contending with transcriptional arrest during RNAPII transcript elongation. Trends Biochem. Sci. 2007;32:165–171. doi: 10.1016/j.tibs.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 9.Daulny A., Tansey W.P. Damage control: DNA repair, transcription, and the ubiquitin-proteasome system. DNA Repair (Amst.) 2009;8:444–448. doi: 10.1016/j.dnarep.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 10.Wilson M.D., Harreman M., Svejstrup J.Q. Ubiquitylation and degradation of elongating RNA polymerase II: the last resort. Biochim. Biophys. Acta. 2012;1829:151–157. doi: 10.1016/j.bbagrm.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Geng F., Wenzel S., Tansey W.P. Ubiquitin and proteasomes in transcription. Annu. Rev. Biochem. 2012;81:177–201. doi: 10.1146/annurev-biochem-052110-120012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edenberg E.R., Downey M., Toczyski D. Polymerase stalling during replication, transcription and translation. Curr. Biol. 2014;24:R445–R452. doi: 10.1016/j.cub.2014.03.060. [DOI] [PubMed] [Google Scholar]
- 13.Woudstra E.C., Gilbert C., Fellows J., Jansen L., Brouwer J., Erdjument-Bromage H., Tempst P., Svejstrup J.Q. A Rad26-Def1 complex coordinates repair and RNA pol II proteolysis in response to DNA damage. Nature. 2002;415:929–933. doi: 10.1038/415929a. [DOI] [PubMed] [Google Scholar]
- 14.Malik S., Bagla S., Chaurasia P., Duan Z., Bhaumik S.R. Elongating RNA polymerase II is disassembled through specific degradation of its largest but not other subunits in response to DNA damage in vivo. J. Biol. Chem. 2008;283:6897–6905. doi: 10.1074/jbc.M707649200. [DOI] [PubMed] [Google Scholar]
- 15.Beaudenon S.L., Huacani M.R., Wang G., McDonnell D.P., Huibregtse J.M. Rsp5 ubiquitin-protein ligase mediates DNA damage-induced degradation of the large subunit of RNA polymerase II in Saccharomyces cerevisiae. Mol. Cell. Biol. 1999;19:6972–6979. doi: 10.1128/mcb.19.10.6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Somesh B.P., Reid J., Liu W.F., Sogaard T.M., Erdjument-Bromage H., Tempst P., Svejstrup J.Q. Multiple mechanisms confining RNA polymerase II ubiquitylation to polymerases undergoing transcriptional arrest. Cell. 2005;121:913–923. doi: 10.1016/j.cell.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 17.Ribar B., Prakash L., Prakash S. ELA1 and CUL3 are required along with ELC1 for RNA polymerase II polyubiquitylation and degradation in DNA-damaged yeast cells. Mol. Cell. Biol. 2007;27:3211–3216. doi: 10.1128/MCB.00091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harreman M., Taschner M., Sigurdsson S., Anindya R., Reid J., Somesh B., Kong S.E., Banks C.A., Conaway R.C., Conaway J.W., et al. Distinct ubiquitin ligases act sequentially for RNA polymerase II polyubiquitylation. Proc. Natl Acad. Sci. U.S.A. 2009;106:20705–20710. doi: 10.1073/pnas.0907052106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reid J., Svejstrup J.Q. DNA damage-induced Def1-RNA polymerase II interaction and Def1 requirement for polymerase ubiquitylation in vitro. J. Biol. Chem. 2004;279:29875–29878. doi: 10.1074/jbc.C400185200. [DOI] [PubMed] [Google Scholar]
- 20.Verma R., Oania R., Fang R., Smith G.T., Deshaies R.J. Cdc48/p97 mediates UV-dependent turnover of RNA Pol II. Mol. Cell. 2011;41:82–92. doi: 10.1016/j.molcel.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kvint K., Uhler J.P., Taschner M.J., Sigurdsson S., Erdjument-Bromage H., Tempst P., Svejstrup J.Q. Reversal of RNA polymerase II ubiquitylation by the ubiquitin protease Ubp3. Mol. Cell. 2008;30:498–506. doi: 10.1016/j.molcel.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 22.Somesh B.P., Sigurdsson S., Saeki H., Erdjument-Bromage H., Tempst P., Svejstrup J.Q. Communication between distant sites in RNA polymerase II through ubiquitylation factors and the polymerase CTD. Cell. 2007;129:57–68. doi: 10.1016/j.cell.2007.01.046. [DOI] [PubMed] [Google Scholar]
- 23.Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Seraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- 24.Knop M., Finger A., Braun T., Hellmuth K., Wolf D.H. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 1996;15:753–763. [PMC free article] [PubMed] [Google Scholar]
- 25.Strasser K., Masuda S., Mason P., Pfannstiel J., Oppizzi M., Rodriguez-Navarro S., Rondon A.G., Aguilera A., Struhl K., Reed R., et al. TREX is a conserved complex coupling transcription with messenger RNA export. Nature. 2002;417:304–308. doi: 10.1038/nature746. [DOI] [PubMed] [Google Scholar]
- 26.Kuras L., Struhl K. Binding of TBP to promoters in vivo is stimulated by activators and requires Pol II holoenzyme. Nature. 1999;399:609–613. doi: 10.1038/21239. [DOI] [PubMed] [Google Scholar]
- 27.Conaway R.C., Brower C.S., Conaway J.W. Emerging roles of ubiquitin in transcription regulation. Science. 2002;296:1254–1258. doi: 10.1126/science.1067466. [DOI] [PubMed] [Google Scholar]
- 28.Collins G.A., Tansey W.P. The proteasome: a utility tool for transcription. Curr. Opin. Genet. Dev. 2006;16:197–202. doi: 10.1016/j.gde.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 29.Kodadek T. No Splicing, no dicing: non-proteolytic roles of the ubiquitin-proteasome system in transcription. J. Biol. Chem. 2010;285:2221–2226. doi: 10.1074/jbc.R109.077883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee K.B., Wang D., Lippard S.J., Sharp P.A. Transcription-coupled and DNA damage-dependent ubiquitination of RNA polymerase II in vitro. Proc. Natl Acad. Sci. U.S.A. 2002;99:4239–4244. doi: 10.1073/pnas.072068399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gillette T.G., Gonzalez F., Delahodde A., Johnston S.A., Kodadek T. Physical and functional association of RNA polymerase II and the proteasome. Proc. Natl Acad. Sci. U.S.A. 2004;101:5904–5909. doi: 10.1073/pnas.0305411101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sigurdsson S., Dirac-Svejstrup A.B., Svejstrup J.Q. Evidence that transcript cleavage is essential for RNA polymerase II transcription and cell viability. Mol. Cell. 2010;38:202–210. doi: 10.1016/j.molcel.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mason P.B., Struhl K. Distinction and relationship between elongation rate and processivity of RNA polymerase II in vivo. Mol. Cell. 2005;17:831–840. doi: 10.1016/j.molcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 34.Malagon F., Kireeva M.L., Shafer B.K., Lubkowska L., Kashlev M., Strathern J.N. Mutations in the Saccharomyces cerevisiae RPB1 gene conferring hypersensitivity to 6-azauracil. Genetics. 2006;172:2201–2209. doi: 10.1534/genetics.105.052415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Auld K.L., Brown C.R., Casolari J.M., Komili S., Silver P.A. Genomic association of the proteasome demonstrates overlapping gene regulatory activity with transcription factor substrates. Mol. Cell. 2006;21:861–871. doi: 10.1016/j.molcel.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 36.Geng F., Tansey W.P. Similar temporal and spatial recruitment of native 19S and 20S proteasome subunits to transcriptionally active chromatin. Proc. Natl Acad. Sci. U.S.A. 2012;109:6060–6065. doi: 10.1073/pnas.1200854109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitsui A., Sharp P.A. Ubiquitination of RNA polymerase II large subunit signaled by phosphorylation of carboxyl-terminal domain. Proc. Natl Acad. Sci. U.S.A. 1999;96:6054–6059. doi: 10.1073/pnas.96.11.6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arima Y., Nitta M., Kuninaka S., Zhang D., Fujiwara T., Taya Y., Nakao M., Saya H. Transcriptional blockade induces p53-dependent apoptosis associated with translocation of p53 to mitochondria. J. Biol. Chem. 2005;280:19166–19176. doi: 10.1074/jbc.M410691200. [DOI] [PubMed] [Google Scholar]
- 39.Malik S., Chaurasia P., Lahudkar S., Durairaj G., Shukla A., Bhaumik S.R. Rad26p, a transcription-coupled repair factor, is recruited to the site of DNA lesion in an elongating RNA polymerase II-dependent manner in vivo. Nucleic Acids Res. 2010;38:1461–1477. doi: 10.1093/nar/gkp1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van den Boom V., Jaspers N.G., Vermeulen W. When machines get stuck–obstructed RNA polymerase II: displacement, degradation or suicide. Bioessays. 2002;24:780–784. doi: 10.1002/bies.10150. [DOI] [PubMed] [Google Scholar]
- 41.Chen X., Ruggiero C., Li S. Yeast Rpb9 plays an important role in ubiquitylation and degradation of Rpb1 in response to UV-induced DNA damage. Mol. Cell. Biol. 2007;27:4617–4625. doi: 10.1128/MCB.00404-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saeki Y., Kudo T., Sone T., Kikuchi Y., Yokosawa H., Toh-e A., Tanaka K. Lysine 63-linked polyubiquitin chain may serve as a targeting signal for the 26S proteasome. EMBO J. 2009;28:359–371. doi: 10.1038/emboj.2008.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim H.T., Kim K.P., Lledias F., Kisselev A.F., Scaglione K.M., Skowyra D., Gygi S.P., Goldberg A.L. Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J. Biol. Chem. 2007;282:17375–17386. doi: 10.1074/jbc.M609659200. [DOI] [PubMed] [Google Scholar]
- 44.Kirkpatrick D.S., Hathaway N.A., Hanna J., Elsasser S., Rush J., Finley D., King R.W., Gygi S.P. Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat. Cell Biol. 2006;8:700–710. doi: 10.1038/ncb1436. [DOI] [PubMed] [Google Scholar]
- 45.Hanna J., Hathaway N.A., Tone Y., Crosas B., Elsasser S., Kirkpatrick D.S., Leggett D.S., Gygi S.P., King R.W., Finley D. Deubiquitinating enzyme Ubp6 functions noncatalytically to delay proteasomal degradation. Cell. 2006;127:99–111. doi: 10.1016/j.cell.2006.07.038. [DOI] [PubMed] [Google Scholar]
- 46.Hobson D.J., Wei W., Steinmetz L.M., Svejstrup J.Q. RNA polymerase II collision interrupts convergent transcription. Mol. Cell. 2012;48:365–374. doi: 10.1016/j.molcel.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reyes-Turcu F.E., Ventii K.H., Wilkinson K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009;78:363–397. doi: 10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leggett D.S., Hanna J., Borodovsky A., Crosas B., Schmidt M., Baker R.T., Walz T., Ploegh H., Finley D. Multiple associated proteins regulate proteasome structure and function. Mol. Cell. 2002;10:495–507. doi: 10.1016/s1097-2765(02)00638-x. [DOI] [PubMed] [Google Scholar]
- 49.Sarker A.H., Tsutakawa S.E., Kostek S., Ng C., Shin D.S., Peris M., Campeau E., Tainer J.A., Nogales E., Cooper P.K. Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: insights for transcription-coupled repair and Cockayne Syndrome. Mol. Cell. 2005;20:187–198. doi: 10.1016/j.molcel.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 50.Wilson M.D., Harreman M., Taschner M., Reid J., Walker J., Erdjument-Bromage H., Tempst P., Svejstrup J.Q. Proteasome-mediated processing of Def1, a critical step in the cellular response to transcription stress. Cell. 2013;154:983–995. doi: 10.1016/j.cell.2013.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sikder D., Johnston S.A., Kodadek T. Widespread, but non-identical, association of proteasomal 19 and 20 S proteins with yeast chromatin. J. Biol. Chem. 2006;281:27346–27355. doi: 10.1074/jbc.M604706200. [DOI] [PubMed] [Google Scholar]
- 52.Lee K.B., Sharp P.A. Transcription-dependent polyubiquitination of RNA polymerase II requires lysine 63 of ubiquitin. Biochemistry. 2004;43:15223–15229. doi: 10.1021/bi048719x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.