

Abstract
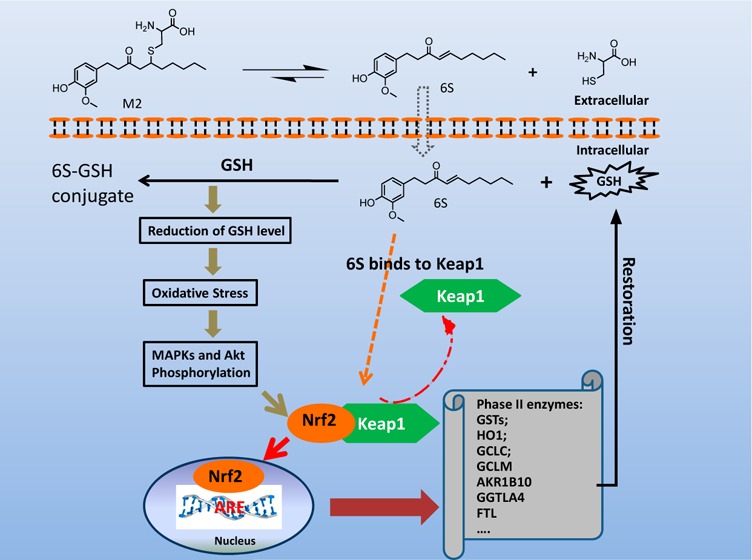
In this study, we identified Nrf2 as a molecular target of [6]-shogaol (6S), a bioactive compound isolated from ginger, in colon epithelial cells in vitro and in vivo. Following 6S treatment of HCT-116 cells, the intracellular GSH/GSSG ratio was initially diminished but was then elevated above the basal level. Intracellular reactive oxygen species (ROS) correlated inversely with the GSH/GSSG ratio. Further analysis using gene microarray showed that 6S upregulated the expression of Nrf2 target genes (AKR1B10, FTL, GGTLA4, and HMOX1) in HCT-116 cells. Western blotting confirmed upregulation, phosphorylation, and nuclear translocation of Nrf2 protein followed by Keap1 decrease and upregulation of Nrf2 target genes (AKR1B10, FTL, GGTLA4, HMOX1, and MT1) and glutathione synthesis genes (GCLC and GCLM). Pretreatment of cells with a specific inhibitor of p38 (SB202190), PI3K (LY294002), or MEK1 (PD098059) attenuated these effects of 6S. Using ultra-high-performance liquid chromatography–tandem mass spectrometry, we found that 6S modified multiple cysteine residues of Keap1 protein. In vivo 6S treatment induced Nrf2 nuclear translocation and significantly upregulated the expression of MT1, HMOX1, and GCLC in the colon of wild-type mice but not Nrf2–/– mice. Similar to 6S, a cysteine-conjugated metabolite of 6S (M2), which was previously found to be a carrier of 6S in vitro and in vivo, also activated Nrf2. Our data demonstrated that 6S and its cysteine-conjugated metabolite M2 activate Nrf2 in colon epithelial cells in vitro and in vivo through Keap1-dependent and -independent mechanisms.
Introduction
Ginger (Zingiber officinale Roscoe) has been used worldwide as a spice, dietary supplement, and traditional medicine for centuries.1 The major pharmacologically active compounds of ginger are gingerols and shogaols.2−6 [6]-Shogaol (6S), a major component of dried ginger, has received much attention because of its superior biological activity and enhanced stability compared to its counterpart in fresh ginger extract, [6]-gingerol.7−11 6S, with an electrophilic α,β-unsaturated carbonyl moiety, has been extensively reported for its various pharmacological effects including anti-inflammatory, analgesic, antipyretic, antioxidant, and anticancer properties.12−15 In particular, 6S induces autophagy by inhibiting the AKT/mTOR pathway in human nonsmall cell lung cancer A-549 cells.16 Additionally, Tan et al. showed that 6S inhibits breast and colon cancer cell proliferation through activation of peroxisomal proliferator activated receptor γ.17 Park et al. showed 6S inhibits the TRIF-dependent signaling pathway of TLRs by targeting TBK1.18 Furthermore, Ling et al. reported that 6S inhibits breast cancer cell invasion by reducing matrix metalloproteinase-9 expression via blockade of NFκB activation.19 A recent study showed 6S protects dopaminergic neurons in Parkinson’s disease models via inhibition of neuroinflammation.20 We have also demonstrated that 6S exhibits much higher antiproliferative potency than that of [6]-gingerol against human lung and colon cancer cells.21
The mercapturic acid pathway is one of the major routes for 6S metabolism in humans, mice, and cells.22,23 6S is initially conjugated with glutathione (GSH) by glutathione-S-transferases, and the GSH conjugate undergoes further enzymatic modifications to form cysteinylglycine conjugate, cysteine conjugate, and N-acetylcysteine conjugate. Among the thiol-conjugated metabolites, cysteine-conjugated 6S (M2) (Figure 1A) is one of the major metabolites of 6S and displays the highest potency against the growth of human colon and lung cancer cells, which is comparable to that of 6S. More importantly, M2 showed much lower toxicity toward human normal colon fibroblast cells and human normal lung cells than its parent compound. Both 6S and M2 trigger apoptosis in cancer cells.24 Further studies on the mechanisms of action indicated that it acts as a carrier of 6S in both cancer cells and in mice.25 Nevertheless, whether M2 acts on cells through mechanisms similar to those of 6S is yet to be determined.
Figure 1.

6S and its effects on glutathione metabolism, intracellular ROS, and gene expression in HCT-116 cells. (A) Structures of 6S and its cysteine conjugated metabolite, M2. (B) Effect of 6S on cellular GSH/GSSG ratio. HCT-116 cells were treated with 20 μM 6S for 0, 2, 4, 8, or 24 h, and cellular GSH/GSSG was measured using a commercial kit. Each bar represents the mean ± SD of six experiments. *p < 0.0001. (C) Effect of 6S on intracellular ROS. HCT-116 cells were treated with 20 μM 6S for 0, 2, 4, 8, or 24 h, ROS was determined using carboxy-DCFDA, and the starting ROS level was set as 1. *p < 0.0001. (D) A heatmap of differentially expressed genes due to 6S treatment with the brightest green, black, and brightest red of the color scale used for expression values of −3, 0, and +3, respectively. HCT-116 cells were treated with 20 μM 6S for 24 h, and mRNA was isolated for microarray analysis using Agilent two-channel human 8 × 60k microarrays. The heatmap was generated using Cluster 3.0 with data of 11 upregulated and 36 downregulated genes that were identified by SAM. Known Nrf2 target genes are marked with arrows.
Multiple mechanisms of action of 6S have been proposed in the literature; however, its molecular targets have not yet been elucidated. We previously found that treatment of human lung and colon cancer cells with 6S or M2 caused transient GSH depletion but then enhanced the intracellular GSH content.22,26 It is known that the enhanced GSH content may result from the induction of the phase II antioxidant enzymes, which are regulated by the antioxidant-response element (ARE),27 through the Keap1/Nrf2 signaling pathway.28,29 Under the normal quiescent cellular environment, Nrf2 is strictly regulated by its cytosolic inhibitor, Keap1. Alkylation of one or more of the cysteine residues of Keap1 by chemical compounds appears to be an important signaling mechanism for Nrf2 activation.30 Upon activation, Nrf2 is released from Keap1 and translocates to the nucleus, where it binds to ARE after heterodimerizing to other leucine zipper proteins to transcriptionally activate downstream genes.31,32 In addition, Nrf2 activity is also regulated by various transcriptional and post-translational modifications, such as phosphorylation by cellular kinases, which in turn results in its stabilization.33,34 Stabilization of Nrf2 is considered to be important to maintain the cellular defense system.35,36
Although 6S-rich ginger extract, 6S, and its analogues have been reported to induce ARE reporter activity and Nrf2 expression in vitro,37,38 the precise mechanisms by which 6S activates Nrf2 are poorly understood, especially in vivo. In the present study, we aimed to identify the molecular targets of 6S and to elucidate how 6S and its metabolite M2 modulate the cellular redox status.
Materials and Methods
Chemicals
6S was purified from ginger extract in our laboratory.21 M2 was synthesized in our laboratory as previously reported.24 PD98059 (MEK1 inhibitor), LY294002 (PI3K inhibitor), and SB202190 (p38 inhibitor) were acquired from Selleckchem (Houston, TX), Cayman Chemical (Ann Arbor, MI), and Thermo Fisher Scientific (Waltham, MA), respectively.
Cell Culture
HCT-116 cells derived from colon were obtained from ATCC (Manassas, VA). McCoy’s 5A medium was purchased from Thermo Fisher Scientific. Supplements of FBS and penicillin/streptomycin were purchased from Gemini Bio-Products (West Sacramento, CA).
Measurement of the Intracellular GSH, GSSG, and GSH/GSSG Ratio
HCT-116 cells were plated in 145 × 20 mm culture plates and were allowed to attach overnight at 37 °C with 5% CO2. Cells were treated with 20 μM 6S and incubated for 0, 2, 4, 8, or 24 h. Cells were harvested by gentle scraping with cold PBS, and proteins were precipitated with 5% (w/v) metaphosphoric acid. The GSH content was measured using a HT glutathione assay kit (Trevigen, Galthersburg, MD) and a Biotek plate reader (Winooski, VT). In brief, the sulfhydryl group of GSH reacts with 5,5′-dithiobis-2-nitrobenzoic acid (Ellman’s reagent) to produce yellow-colored 5-thio-2-nitrobenzoic acid (TNB) that absorbs at 405 nm. Mixed disulfide, which is concomitantly produced, is reduced by glutathione reductase to recycle glutathione and produce more TNB. The rate of TNB production is directly proportional to the concentration of glutathione in the sample. Glutathione is further normalized by protein concentration, which was determined using a Pierce BCA kit (Thermo Fisher Scientific).
For measurement of oxidized glutathione, samples and GSSG standards were treated with 2 M 4-vinylpyridine (1 μL/50 μL sample) at room temperature for 1 h. 4-Vinylpyridine (Sigma-Aldrich, St. Louis, MO) blocks free thiols present in the reaction, eradicating the contribution to the recycling reaction caused by GSH. A 2 M solution was freshly prepared by diluting 4-vinylpyridine in ethanol in a ratio of approximately 1:3.6. After incubation, samples were analyzed using the HT glutathione assay kit as above-described. Then, reduced glutathione was calculated by subtracting values derived from oxidized samples from those of total glutathione. The ratio of reduced to oxidized glutathione (GSH/GSSG) is calculated as an indicator of cellular redox status.
Measurement of Intracellular Reactive Oxygen Species (ROS)
This assay employed a cell-permeable fluorogenic probe, 2′,7′-dichlorodihydrofluorescin diacetate (Sigma-Aldrich), to measure the relative changes in O–2 and H2O2 levels in HCT-116 cells after treatment. HCT-116 cells were seeded in 96-well black-sided, clear-bottomed culture plates, with 5000 cells/well and were allowed to adhere for 24 h in a 37 °C incubator with 5% CO2. 6S (20 μM) or DMSO diluted in media was added to designated wells, which were run in triplicate. After 0, 2, 4, 8, or 24 h of incubation, media was aspirated. Cells were washed three times with 200 μL of PBS before addition of 100 μL of 1 mM probe. After incubation for 1 h at 37 °C, plates were immediately placed in a Biotek microplate reader to measure fluorescence at 485 excitation and 528 emission. Raw values were normalized to DMSO control for each time point and are presented as fold induction versus 0 h time point (n = 3).
Gene Microarray and Data Analysis
HCT-116 cells were plated in 100 × 20 mm culture plates and were allowed to attach overnight at 37 °C. Cells were treated with 20 μM 6S or DMSO control and incubated for 24 h. Cells were harvested, and total RNA was isolated using the RNeasy mini kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. The quantity of RNA was measured using a spectrophotometer (NanoDrop 2000c; Thermo Scientific). RNA quality was determined by gel electrophoresis and Bioanalyzer (Agilent Technologies, Santa Clara, CA).
Microarray experiments were performed at the Genomics Core Facility of Lineberger Comprehensive Cancer Center, UNC Chapel Hill, with Agilent two-channel human 8 × 60k microarrays. Red channel (Cy5) was used for samples, and green channel (Cy3), for human universal reference RNA. Hybridization was performed according to the standard protocol for two-color microarray-based gene expression analysis for Agilent Gene Expression Oligo microarrays, version 5.0.1. Briefly, a 2× target mix was generated containing 125 ng of cyanine 3-labeled cRNA, 125 ng of cyanine 5-labeled cRNA, appropriate amounts of labeled synthetic target, and 25 μL of Agilent’s 10× control solution in a final volume of 125 μL. The sample was then fragmented by the addition of 5 μL 25× fragmentation buffer followed by incubation at 60 °C for 30 min. Samples were moved to ice, and fragmentation was stopped by addition of 125 μL of Agilent’s 2× in situ hybridization buffer. Microarrays were hybridized in Agilent microarray hybridization chambers for 17 h at 60 °C with mixing on an Agilent rotator in a Robbin’s Scientific hybridization oven. After hybridization, the arrays were scanned by an Axon GenePix 4000B scanner (Axon Instruments; Foster City, CA). The images were analyzed using Gene Pix Pro 5.0 software (Axon Instruments). Gene expression values were quantified by log2 ratio of red channel intensity (mean) and green channel intensity (mean), followed by Lowess normalization to remove the intensity-dependent dye bias.
Data preprocessing was carried out via the UNC Microarray Database for quality filtering and data normalization. Agilent array data was extracted on the probe level. For probes spotted multiple times, the mean expression value was computed and retained. All probe sequences were searched via BLAST against the NCBI database and were annotated with their Entrez ID. When multiple probes were targeted on the same gene (with the same Entrez ID), these data were collapsed onto the Entrez ID, and mean values were computed as the gene expression value. To perform hierarchical clustering analysis, a data matrix with all the genes was extracted, row median-centered, and column-standardized. Clustering analysis was performed with Cluster 3.0. The microarray data have been submitted to the GEO database (GSE57006).
Significance analysis of microarrays (SAM) was used for identification of differentially expressed genes. Preprocessed data were used to construct a series of data matrix files for further analysis. For a given data matrix, the rows were excluded if more than 40% of missing values were observed. The rest of missing data was imputed with a K-nearest neighbor (k = 9) approach. Differentially expressed genes were obtained from two-class SAM in Excel with a median number of false positives of 1.45. To perform hierarchical clustering analysis, a data matrix with differentially expressed genes only was extracted, row median-centered, and column-standardized.
Gene set analysis (GSA) was carried out as an add-in in Excel to identify differentially expressed gene sets. One thousand permutations were applied to generate a null distribution for statistical testing, and significantly enriched gene sets were obtained at a false discovery rate cutoff of 0.5. Curated gene sets in three major categories, canonical pathway (880 gene sets), transcription factor targets (615 gene sets), and Gene Ontology (C5, 1454 gene sets), were downloaded from the GSEA web portal and used in this study (http://www.broadinstitute.org/gsea/index.jsp).
Determination of Alkylation of Cysteine Residues of Keap1 by UPLC–MS/MS
Human recombinant Keap1 (no. NM_012289, OriGene, Rockville, MD) was incubated with 6S [molar ratios of 1:10 (Keap1/6S)] or DMSO in 120 μL of 25 mM Tris-HCl buffer (pH 8) for 2 h at room temperature. The reaction was quenched by adding 1 mM DTT, and the mixture was incubated for an additional 15 min. Trypsin was added to the sample at a trypsin/Keap1 ratio of 1:50 (w/w) and incubated at 37 °C for 1.5 h. The samples were centrifuged, and the supernatant was pipetted into autosampler vials. The tryptic peptides were analyzed using ultra-high-performance liquid chromatography–tandem mass spectrometry (UPLC–MS/MS) to determine sites of modification by 6S.
The processed samples were analyzed on a Thermo Scientific LTQ Orbitrap XL mass spectrometry system coupled to a Waters nano-ACQUITY UPLC system. Peptides were separated on a Waters nano-ACQUITY UPLC Column (1.7 μm BEH 130 C18, 75 μm × 250 μm) using a linear gradient from 2 to 40% B by 60 min, to 90% B by 65 min, to 2% B by 67 min, then kept at 2% B for 23 min, where A is 99.9:0.1 water/formic acid and B is 99.9:0.1 acetonitrile/formic acid. Mass spectra were acquired using data dependent scans on the LTQ Orbitrap XL system over 90 min. The resulting data files were searched against the SwissProt (human) database in MASCOT. The following variable modifications were chosen for the search including: carbamidomethyl, deamidated, oxidation, and 6S. Peptide tolerances were 20 ppm for MS and 0.5 Da for MS/MS.
Western Blotting
HCT-116 cells were plated in 145 × 20 mm flat-bottomed tissue culture dishes and grown to 70–80% confluence in a 37 °C incubator with 5% CO2. Cells were treated with 6S or M2 for various time points and doses. At the end of incubation period, cell lysates were prepared in ice-cold RIPA lysis buffer with a protease inhibitor cocktail (Thermo Fisher Scientific). Cell lysates (30 μg protein/lane) were resolve by SDS-PAGE. Proteins were then electro-transferred onto PVDF membranes, and blots were blocked for 1 h at room temperature in 1× TBS with 1% Casein (Bio-Rad Laboratories, Berkeley, CA). Blots were then incubated overnight at 4 °C with the desired primary antibody diluted in TBS with 0.5% Tween-20. Primary antibodies against AKR1B10, GGTLA4, MT1, GCLC, GCLM, Nrf2, and Keap1 (Santa Cruz Biotechnology, Santa Cruz, CA) or against FTL and p-Nrf2 (Abcam, Cambridge, MA) were used at 1:1000 dilutions. HMOX1 (Cell Signaling Technology, Beverly, MA) was used at a 1:1500 dilution. Blots were then washed with TBS–Tween-20 and probed for 1 h with the appropriate secondary antibody (1:1000). Protein bands were visualized with chemiluminescence using West Femto maximum detection substrate (Thermo Fisher Scientific).
Cytoplasmic proteins and nuclear proteins were fractionated with a nuclear extract kit (Active Motif, Carlsbad, CA), and colon tissue samples were homogenized with OMNI Bead Ruptor (OMNI International) for western blotting. To confirm equal protein loading in each lane, immunoblots were stripped and reprobed with an anti-β-actin (Cell Signaling Technology) or an anti-lamin B (Cell Signaling Technology) antibody. β-actin and lamin B served as loading controls of cytoplasmic and nuclear fractions, respectively.
Immunofluorescence Staining (IF)
Cells were grown on fibronectin-coated coverslips in the presence or absence of 6S. After treatment with test samples for the indicated times, cells were fixed with cold 2% (w/v) paraformaldehyde for 20 min, permeabilized in 0.1% (w/v) Triton X-100 in 1× PBS, washed, and blocked in 1% BSA at room temperature for 1 h. Tissue sample sections were deparaffinized and rehydrated. After being washed with PBS for 5 min three times, the cells were incubated with a rabbit anti-Nrf2 antibody (1:200; Santa Cruz Biotechnology) and tissue sections were incubated with a rabbit anti-Nrf2 antibody (1:200)39 overnight at 4 °C, followed by FITC-conjugated secondary antibody (1:200) for 1 h at room temperature. Samples were counterstained with DAPI (1 mg/mL) for visualization of the nuclei. Stained samples were mounted and visualized under a fluorescent microscope (Thermo Fisher Scientific).
Animal Study
Experiments with mice were carried out according to a protocol approved by the Institutional Animal Care and Use Committee, North Carolina Central University (protocol no. XC-12-03-2008). Wild-type (WT) C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and allowed to acclimate for at least 1 week prior to the start of the experiment. Nrf2–/– mice have been described in detail previously.40 Mice were housed 5 per cage and maintained in air-conditioned quarters with a room temperature of 20 ± 2 °C, relative humidity of 50 ± 10%, and an alternating 12 h light/dark cycle. Mice were fed Purina Rodent Chow no. 5001 (Research Diets) and water and were allowed to eat and drink ad libitum. Mice were divided into four groups: (a) control WT (n = 5); (b) control Nrf2–/– (n = 5); (c) WT + 6S (n = 5); and (d) Nrf2–/– + 6S (n = 5). The mice were then administered 6S at a dose of 100 mg/kg (dissolved in DMSO) for four consecutive days by oral gavage. The control group animals were administered vehicle only. Mice were sacrificed 2 h after 6S or vehicle treatment. Tissues (colon, liver, esophagus, stomach, and small intestine) were harvested with a part snap frozen in liquid nitrogen and another part fixed with 10% PBS buffered formalin, processed, and embedded in paraffin.
Immunohistochemical Staining (IHC)
Deparaffinized sections were submerged in methanol containing 0.3% hydrogen peroxide for 15 min at RT to inhibit endogenous peroxidase activity. Antigen retrieval was done prior to incubation with a rabbit anti-Nrf2 antibody (1:200),39 a mouse monoclonal anti-MT1 antibody (1:100; Accurate Chemical & Scientific, Westbury, NY), or a rabbit polyclonal anti-HMOX1 antibody (1:25; Abcam, Cambridge, MA) overnight at 4 °C. Tissue sections were then washed again in PBS and incubated with peroxidase-conjugated secondary antibody for 30 min at 37 °C. Detection of the antibody complex was done using the streptavidin–peroxidase reaction kit with DAB as a chromogen (ABC kit; Vector Laboratories, Burlingame, CA). To ensure the specificity of the primary antibody, control tissue sections were incubated in the absence of primary antibodies.
Reverse Transcription and Real-Time PCR
cDNA was prepared from DNase-treated total RNA using the Advantage RT-for-PCR Kit (Clontech; Mountain View, CA). TaqMan gene expression assays (FAM dye-labeled) with predesigned primers for each target gene were obtained from Applied Biosystems (Foster City, CA). The three target genes were Hmox1 (assay ID: Mm00516005_m1), Mt1 (assay ID: Mm00496660_g1), and Gclc (assay ID: Mm00802655_m1). 18S (18S rRNA; hypothetical LOC790964, assay ID: Mm03928990_g1) was used as the endogenous control. Relative quantitative real-time PCR was performed using an ABI 7900HT fast real-time PCR system (Applied Biosystems) with SDS v2.3 software. The real-time data exported from RQ Manager 1.2 were further analyzed by DataAssist 3.0 (Applied Biosystems) to generate the RQ Plot.
Statistical Analysis
For simple comparisons between two groups, a two-tailed Student’s t-test was used. A p value of less than 0.05 was considered to be statistically significant in all tests.
Results
Effects of 6S on Cellular Redox Status
The GSH/GSSG ratio was quantitated as an indicator of cellular redox status. In HCT-116 cells, the ratio of GSH/GSSG was initially reduced by 20 μM 6S to about 60% of the basal level at 4 h. It returned to the basal level within 8 h and then rose 2.5-fold higher than the basal level at 24 h (Figure 1B). In parallel with the GSH/GSSG ratio, intracellular ROS started to accumulate at 2 h. At 24 h, the ROS level was much lower than that of the basal level (Figure 1C).
Identification of Nrf2 as a Molecular Target of 6S
To identify the direct molecular targets of 6S, gene microarrays were used to profile gene expression in HCT-116 cells treated with 6S (20 μM for 24 h). Hierarchical clustering of gene microarray data for all genes separated the samples into two groups, control (1, 2, 3, 4) and 6S-treated (5, 6, 7, 8), indicating a good quality of the microarray data (data not shown). SAM analysis showed that 11 genes were upregulated and 36 genes were downregulated by 6S. Among the upregulated genes, there were four known Nrf2 target genes, AKR1B10, FTL, GGTLA4, and HMOX1 (Figure 1D). GSA analysis showed enrichment of two Nrf2-associated gene sets, V$NRF2_Q4 (transcription factor targets) and KEGG_GLUTATHIONE_METABOLISM (canonical pathway), in 6S-treated samples. In addition, GDF15/NAG1 was upregulated by 6S in HCT116 cells, as reported in MCF-7 cells.17 Nonsteroidal anti-inflammatory drugs like Sulindac inhibited intestinal tumorigenesis through induction of this gene.41,42 Transgenic overexpression of GDF15 suppressed intestinal neoplasia.43
We then decided to focus on Nrf2 and validated it as a molecular target of 6S. With western blotting, we confirmed that 6S upregulated five Nrf2 target genes (AKR1B10, FTL, GGTLA4, HMOX1, and MT1), and four of them were identified by gene microarray (Figure 2A). MT1, as an Nrf2-regulated gene, was analyzed to understand the transcriptional activity of Nrf2. MT1 gene is known to harbor at least one ARE in its promoter.44 Many studies have shown MT1 as a downstream gene of Nrf2 in multiple tissues. In the intestine, for example, Keap1 knockout would upregulate MT1 expression in human colon cancer HT29 cells.45 A classical Nrf2 activator, butylated hydroxyanisole, induced expression of MT1 in the small intestine of WT mice but not Nrf2–/– mice.46 In addition, we also examined expression of two Nrf2 target genes, GCLC and GCLM, with western blotting (Figure 2A). Upregulation of these two GSH synthesis genes explains why 6S treatment increased the GSH/GSSG ratio and reduced intracellular level of ROS at the later time points (Figure 1B,C).
Figure 2.

Effects of 6S on expression of Nrf2 and Nrf2 target genes in HCT-116 cells. (A) Effect of 6S on expression of AKR1B10, GGTLA4, FTL, HMOX1, GCLC, GCLM, and MT1 in HCT-116 cells. (B) Effect of 6S on the expression of Keap1, Nrf2, and phosphorylated Nrf2 (p-Nrf2). The protein levels of AKR1B10, GGTLA4, FTL, HMOX1, GCLC, GCLM, MT1, Keap1, Nrf2, and p-Nrf2 were determined by western blotting at the indicated time points after treatment of HCT-116 cells with 6S (20 μM). β-Actin was used as an internal standard. (C) Time-dependent effect of 6S on Nrf2 nuclear translocation. HCT-116 cells were treated with 20 μM 6S for 0, 2, 4, 6, 12, and 24 h. (D) Dose-dependent effect of 6S on Nrf2 nuclear translocation. HCT-116 cells were treated with 0, 5, 10, 20, and 40 μM 6S for 6 h. Lamin B and β-actin were used as internal controls for nuclear and cytoplasmic fractions, respectively. (E) IF staining of Nrf2. HCT-116 cells were treated with 20 μM 6S for 12 or 24 h and then fixed and labeled with anti-Nrf2 and appropriate FITC-conjugated secondary antibodies. Cells were counterstained with DAPI for visualization of the nuclei. Slides were viewed using fluorescent microscopy (DAPI, blue; Nrf2, red).
Under normal conditions, Nrf2 is sequestered in the cytoplasm via binding to its repressor molecule, Keap1. Oxidative, electrophilic, and ER stress as well as Nrf2 phosphorylation by protein kinases cause dissociation of the Nrf2–Keap1 complex, which culminates in ubiquitination of Keap1 and nuclear translocation of Nrf2. We next decided to examine whether the expression levels of Nrf2 and Keap1 changed. With western blotting, we confirmed that 6S significantly increased expression of both Nrf2 and phosphorylated Nrf2 in HCT-116 whole-cell lysates (Figure 2B). Oppositely, the expression of the Nrf2 repressor Keap1 was decreased (Figure 2B).
We further investigated subcellular localization of Nrf2 with western blotting and IF. Cells were treated with 20 μM 6S for 0, 2, 4, 6, 12, or 24 h. Nuclear Nrf2 increased in a time-dependent manner in parallel with a time-dependent decrease of cytoplasmic Nrf2 (Figure 2C). In addition, 6S dose-dependently activated nuclear translocation of Nrf2 (Figure 2D). Furthermore, IF clearly showed that Nrf2 translocated from cytoplasm to nucleus in a time-dependent manner (Figure 2E). These data clearly demonstrate that 6S activated nuclear translocation of Nrf2 and upregulated Nrf2 target genes in colon epithelial cells.
6S Activates Nrf2 via Phosphorylation Induced by the Kinase Cascade
It is known that phosphorylation of serine/threonine residues of Nrf2 by protein kinases facilitates nuclear translocation of Nrf2 and its subsequent transcriptional activities.30,47,48 As shown in Figure 2B, increased phosphorylated Nrf2 (p-Nrf2) was observed in 6S-treated HCT-116 cells in a time-dependent manner. Therefore, HCT-116 cells were pretreated with pharmacological inhibitors of PI3K (LY294002), MEK1 (PD098059), or p38 (SB202190) for 30 min before being exposed to 6S at 20 μM. After 24 h, the cells were lysed and analyzed for nuclear and cytoplasmic Nrf2 or p-Nrf2 by western blotting. Nrf2 translocation and phosphorylation was partially blocked by all three inhibitors, with SB202190 being the most potent inhibitor (Figure 3A). In addition, transcriptional activity of Nrf2, as measured by HMOX1 expression, was also significantly inhibited by these inhibitors (Figure 3B). These data show that 6S activated Nrf2 nuclear translocation through phosphorylation induced by protein kinases.
Figure 3.

Effects of kinase inhibitors on 6S-induced Nrf2 translocation, phosphorylation, and HMOX1 expression in HCT-116 cells. (A) Effect of kinase inhibitors on 6S induced Nrf2 translocation and phosphorylation. PD098059 (50 μM, a MEK1 inhibitor), LY294002 (50 μM, a PI3K inhibitor), or SB202190 (50 μM, a p38 inhibitor) was used to pretreat the cells for 30 min before they were exposed to 20 μM 6S. After another 24 h of incubation, cytosolic and nuclear Nrf2 as well as p-Nrf2 was determined using western blotting with the appropriate specific antibodies. Lamin B and β-actin were used as internal controls for nuclear and cytosolic extracts, respectively. *p < 0.05. (B) Effect of kinase inhibitors on 6S-induced HMOX1 expression. Inhibitor was used to pretreat the cells for 30 min before they were exposed to 20 μM 6S. After another 24 h of incubation, whole-cell lysates were prepared and assessed for HMOX1 expression by western blotting. *p < 0.05.
6S Activates Nrf2 via Alkylation of Cysteine Residues of Keap1 Protein
Alkylation of one or more of the sulfhydryl groups of the 27 cysteine residues of human Keap1 has been proposed to be one of the activating mechanisms for Nrf2 nuclear translocation.47 To test whether 6S can modify the cysteine residues of Keap1 protein, human recombinant Keap1 (10 μM) was treated with 100 μM 6S for 2 h at room temperature. Three separate experiments were performed, and the adducts were mapped by UPLC–MS/MS. Among the 17 cysteines modified by 6S, modification of 4 cysteines (Cys23, Cys38, Cys395, and Cys406) were detected in all three experiments. Six cysteines (Cys77, Cys171, Cys196, Cys368, Cys583, and Cys613) were detected in two of the three experiments, and 7 cysteines (Cys226, Cys297, Cys319, Cys434, Cys489, Cys622, and Cys624), in one of the three experiments. These cysteine residues are scattered in the N-terminal, BTB, central linker, Kelch, and C-terminal domains of Keap1 protein (Table 1).
Table 1. Cysteine Residues of Keap1 Modified by 6S as Determined by UPLC–MS/MS Analysisa.
| domain | cysteine | exp. 1 | exp. 2 | exp. 3 |
|---|---|---|---|---|
| N-terminal | C13 | |||
| C14 | ||||
| C23 | × | × | × | |
| C38 | × | × | × | |
| BTB | C77 | × | × | |
| C151 | ||||
| C171 | × | × | ||
| Central linker | C196 | × | × | |
| C226 | × | |||
| C241 | ||||
| C249 | ||||
| C257 | ||||
| C273 | ||||
| C288 | ||||
| C297 | × | |||
| C319 | × | |||
| Kelch | C368 | × | × | |
| C395 | × | × | × | |
| C406 | × | × | × | |
| C434 | × | |||
| C489 | × | |||
| C513 | ||||
| C518 | ||||
| C583 | × | × | ||
| C-terminal | C613 | × | × | |
| C622 | × | |||
| C624 | × |
These data represent the results of UPLC–MS/MS analyses of three independent experiments.
6S Activates Nrf2 in Colon Epithelial Cells in Vivo
To determine whether 6S activates Nrf2 and its target genes in colon epithelial cells in vivo, we treated Nrf2–/– and WT mice with 6S and then examined expression of Nrf2 and its target genes in mouse colon. 6S treatment for four consecutive days (100 mg/kg p.o., once per day) increased nuclear Nrf2 in colon epithelial cells as detected by IF (Figure 4A). IHC also showed increased expression of Nrf2 target genes, MT1 and HMOX1, in colon epithelial cells of WT mice but not Nrf2–/– mice (Figure 4B). Western blotting and semiquantitation further confirmed increased expression of HMOX1 and GCLC due to 6S treatment in WT mice but not in Nrf2–/– mice (Figure 4C). Real-time PCR also showed that 6S treatment significantly upregulated mRNA expression of Gclc and Mt1 in colon epithelial cells of WT mice but not Nrf2–/– mice. Hmox1 mRNA was also increased by 6S treatment, yet without statistical significance (Figure 4D), in colon epithelial cells of WT mice. In addition, we also observed an increase of nuclear Nrf2, MT1, and HMOX1 in other gastrointestinal tissues, particularly in the liver (data not shown). Taken together, our data clearly indicate that 6S activated Nrf2 in vivo and further induced expression of Nrf2 target genes in an Nrf2-dependent manner.
Figure 4.

6S induced Nrf2 expression in mouse colon. (A) 6S treatment increases nuclear Nrf2 levels in the colon of wild-type (WT) mice. Nrf2 expression is shown by IF and IHC staining. (B) IHC staining of HMOX1 and MT1 in the colon of WT and Nrf2–/– mice after 6S treatment. Scale bar = 50 μm (C) Effects of 6S on HMOX1 and GCLC expression in the colon of WT and Nrf2–/– mice. Colon lysates from five animals of each genotype were analyzed; graph represents the average; *p < 0.05, **p < 0.01. (D) Effect of 6S on the expression of Gclc, Hmox1, and Mt1 mRNA in the colon of WT and Nrf2–/– mice as determined by real-time PCR. *p < 0.05, **p < 0.01.
Similar to 6S, M2 Activates Nrf2 in Colon Epithelial Cells
As a cysteine-conjugated metabolite of 6S, M2 is known to be metabolized as a carrier of 6S both in vitro and in vivo.25 To determine whether M2 shares biological activities of 6S on Nrf2, we treated HCT-116 cells with M2 using an experimental design similar to that for the above experiments with 6S. As expected, M2 time-dependently upregulated the expression of Nrf2 target genes, AKR1B10, GGTLA4, FTL, HMOX1, GCLC, GCLM, and MT1 (Figure 5A), upregulated the expression of Nrf2 and p-Nrf2 while downregulating Keapl (Figure 5B), induced nuclear translocation of Nrf2 in a time- and dose-dependent manner (Figure 5C,D), and activated Nrf2 via phosphorylation by the kinase cascade (Figure 5E). HMOX1 expression was significantly inhibited by three kinase inhibitors as well (Figure 5F). These data suggested that M2 activated Nrf2 through similar mechanisms as those of 6S.
Figure 5.

Effects of M2 on expression of Nrf2 and Nrf2 target genes in HCT-116 cells. (A) Effect of M2 on the expression of AKR1B10, GGTLA4, FTL, HMOX1, GCLC, GCLM, and MT1. (B) Effect of M2 on the expression of Keap1, Nrf2, and p-Nrf2. The protein levels of AKR1B10, GGTLA4, FTL, HMOX1, GCLC, GCLM, MT1, Keap1, Nrf2, and p-Nrf2 were determined by western blotting at the indicated time points after the treatment of HCT-116 cells with M2 (20 μM). β-Actin was used as internal standard. (C) Time-dependent effect of M2 on Nrf2 nuclear translocation. HCT-116 cells were treated with 20 μM M2 for 0, 2, 4, 6, 12, and 24 h. (D) Dose-dependent effect of M2 on Nrf2 nuclear translocation. HCT-116 cells were treated with 0, 5, 10, 20, and 40 μM M2 for 6 h. Cytosolic and nucleic Nrf2 were determined using western blotting with the appropriate specific antibodies. Lamin B and β-actin were used as internal controls for nuclear and cytoplasmic fractions, respectively. (E, F) Effects of kinase inhibitors on M2-induced Nrf2 translocation and phosphorylation (E) and HMOX1 (F) expression. PD098059 (50 μM, a MEK1 inhibitor), LY294002 (50 μM, a PI3K inhibitor), or SB202190 (50 μM, a p38 inhibitor) was used to pretreat the cells for 30 min before they were exposed to 20 μM M2. After another 24 h of incubation, whole-cell lysates were prepared and assessed for Nrf2, p-Nrf2, and HMOX1 expression by western blotting. *p < 0.05.
Discussion
In the present study, we identified Nrf2 as a molecular target of 6S in colon epithelial cells both in vitro and in vivo. Our data obtained with gene expression profiling, western blotting, and immunostaining clearly demonstrate that 6S induced nuclear translocation of Nrf2 and activated Nrf2 target genes in an Nrf2-dependent manner. Although 6S induced transient glutathione depletion and oxidative stress in colon epithelial cells at early time points, upregulation of two glutathione synthesis genes as well as Nrf2 target genes, GCLC and GCLM, increased intracellular the GSH/GSSG ratio and reduced the ROS level at latter time points. Modification of multiple cysteine residues of Keap1 protein and the expression of Keap1 by 6S suggested a Keap1-dependent mechanism for Nrf2 activation. Inhibition of Nrf2 activation and Nrf2 phosphorylation by kinase inhibitors suggested that 6S also activated Nrf2 through a Keap1-independent mechanism. Moreover, our data demonstrate that M2 activated Nrf2 in a way similar to that of 6S.
Nrf2 is known to be regulated by Keap1-independent and -dependent mechanisms.48 Phosphorylation of serine/threonine residues of Nrf2 by protein kinases facilitates nuclear translocation of Nrf2 and its subsequent transcriptional activities.30,49,50 MAPKs have been associated with the Nrf2 pathway in an inducer- and cell type-dependent manner.30 PI3K has also been extensively investigated for its regulation of Nrf2.30,49,50 For example, tBHQ has been shown to activate PI3K, inhibition of which caused a significant decrease in ARE-dependent gene expression.51 To identify which signaling cascade controls Nrf2 activation by 6S, we examined the effects of inhibitors of MAPKs or PI3K on Nrf2 nuclear translocation and HMOX1 upregulation. The inhibitors of MEK1 and p38 attenuated the 6S-induced phosphorylation and nuclear translocation of Nrf2 as well as HMOX1 overexpression. In addition, inhibition of PI3K/AKT signaling by LY294002 led to a decrease of 6S-induced Nrf2 phosphorylation and translocation as well as HMOX1 expression. These findings suggest that MAPKs and PI3K/AKT pathways participated in 6S-induced Nrf2 activation in HCT-116 cells.
Activation of Nrf2 through modification of Keap1 cysteine residues is another important mechanism of cellular defense against oxidative stress. Under basal conditions, interaction between cytosolic Keap1 with Nrf2 results in a low level of expression of Nrf2 target genes. Keap1 serves as a substrate linker protein for interaction of Cul3-based E2-ubiquitin ligase complex with Nrf2, leading to continuous ubiquitination of Nrf2 and its proteasomal degradation.52 Targeted disruption of the Keap1 gene in mice clearly demonstrated the crucial role of Keap1 in the regulation of Nrf2.53 Keap1 is a cysteine-rich protein possessing 27 cysteine residues in the human protein. Alkylation of one or more of the cysteine residues of Keap1 by ROS and xenobiotic electrophiles appears to be an important signaling mechanism for the regulation of ARE activity through Nrf2.47 In this study, we found that 6S modified Keap1 at 17 cysteine residues and that these cysteine residues were located in all five domains of Keap1 protein (Table 1). Although only some cysteine residues (e.g., Cys151, Cys273, and Cys288) have been validated by mutagenesis experiments as being critical modification sites of Keap1,54 many cysteine residues of Keap1 are known to be modified by chemicals as a potential mechanism of Nrf2 activation. Some 6S-modified cysteine residues identified in this study have been shown to be modified by other chemicals, for example, Cys23, Cys226, and Cys368 by tert-butylbenzoquinone,55 Cys77 and Cys368 by andrographolide,55 Cys297 by BM31,56 and Cys434 by nitric oxide and ROS.57 Even GSSG can modify cysteines of Keap1 to form type I disulfides (Cys77, Cys297, Cys319, Cys368, and Cys434) and type II disulfides (Cys23–Cys38, Cys257–Cys297) and subsequently activate Nrf2.58
Although this study, for the first time, demonstrated that 6S has the capability to modify Keap1 and activate Nrf2, exactly which cysteines are the critical sensors of 6S is still unknown. In the literature, [10]-shogaol, an analogue of 6S with difference of side chain length, has been reported to modify a different set of cysteines.59 This is probably due to different experimental conditions (e.g., sample preparation, ratio of chemical to protein) and different lengths of the side chain of these two compounds.60 In fact, 6S can easily react with cysteine as a Michael reaction acceptor.24 Cysteine residues of tubulin61 and TRPA1,62 and even serine residues of eIF2α63 and Akt1,64 are modified by 6S. Further study is warranted to pinpoint the sensor cysteines in Keap1 modified by 6S and the structure–activity relationship of shogaols in Keap1 modification. It would also be very interesting to elucidate concentration-dependent effects of 6S on cysteine-rich protein targets other than Keap1, such as PPARγ, NFκB, and c-Jun, to better understand its molecular targets, mechanisms of action, and potential applications.65,66
Our data, showing Nrf2 activation in colon epithelial cells by 6S in vitro and in vivo, suggest that 6S and ginger may be potentially used for prevention and treatment of colonic diseases such as colitis, colorectal cancer, and radiation injury. Nrf2 deficiency enhanced dextran sulfate-induced colitis, aberrant crypt foci, and colitis-associated colorectal cancer in mice.67,68 Treatment with Nrf2 activators mitigated dextran sulfate-induced acute colitis and inhibited azoxymethane-induced colorectal cancer.69,70 Furthermore, activation of Nrf2 protected colonic epithelial cells from ionizing radiation in vitro and in vivo.71 It should be noted, though, that Nrf2 has dual roles in both cancer prevention and carcinogenesis.72 Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs and radiation therapy,73 promotes colon tumor angiogenesis through hypoxia-induced activation of HIF1α,74 and promotes cancer cell proliferation.75 These observations suggest that benefits and risks need to be adequately weighted when 6S or ginger are given to human subjects for colonic diseases.
In summary, our data clearly demonstrated that Nrf2 is a molecular target of 6S in colon epithelial cells in vitro and in vivo through Keap1-dependent and -independent mechanisms. In addition, the similar property of its metabolite M2 suggested that 6S may activate Nrf2 for an extended period even after being quickly metabolized.
Glossary
Abbreviations
- 6S
[6]-shogaol
- AKR1B10
aldo-keto reductase family member 1B10
- ARE
antioxidant response elements
- FTL
ferritin
- GCLC
glutamate cysteine ligase catalytic
- GCLM
glutamate cysteine ligase modulatory
- GGTLA4
gamma-glutamyltransferase light chain
- GSA
gene set analysis
- GSH
glutathione
- GSSG
oxidized glutathione
- HMOX1
heme oxygenase 1
- Keap1
Kelch-like ECH associated protein 1
- M2
5-cysteinyl-[6]-shogaol
- MS
mass spectrometry
- MT1
metallothionein-1
- Nrf2
NF-E2-related factor 2
- p-Nrf2
phosphorylated Nrf2
- ROS
reactive oxygen species
- SAM
significance analysis of microarrays
- TNB
5-thio-2-nitrobenzoic acid
- UPLC
ultra-high-performance liquid chromatography
Author Contributions
⊥ H.C. and J.F. contributed equally to this work.
This investigation was partially supported by grant nos. CA138277 (S. Sang) and CA138277S1 (S. Sang) from the National Cancer Institute and the Office of Dietary Supplement and by the Diagnostic and Molecular Histopathology Facility of a U54 grant (CA156735) from the National Cancer Institute.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Leung A. Y., and Foster S. (1996) Encyclopedia of Common Natural Ingredients Used in Food, Drugs, and Cosmetics, 2nd ed., pp 271–274, Wiley, New York. [Google Scholar]
- Jiang H.; Solyom A. M.; Timmermann B. N.; Gang D. R. (2005) Characterization of gingerol-related compounds in ginger rhizome (Zingiber officinale Rosc.) by high-performance liquid chromatography/electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 19, 2957–2964. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Timmermann B. N.; Gang D. R. (2007) Characterization and identification of diarylheptanoids in ginger (Zingiber officinale Rosc.) using high-performance liquid chromatography/electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 21, 509–518. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Xie Z.; Koo H. J.; McLaughlin S. P.; Timmermann B. N.; Gang D. R. (2006) Metabolic profiling and phylogenetic analysis of medicinal Zingiber species: Tools for authentication of ginger (Zingiber officinale Rosc). Phytochemistry 67, 1673–1685. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Huang T.; Yang B.; Liu X.; Duan G. (2007) Development of gas chromatography-mass spectrometry with microwave distillation and simultaneous solid-phase microextraction for rapid determination of volatile constituents in ginger. J. Pharm. Biomed. Anal. 43, 24–31. [DOI] [PubMed] [Google Scholar]
- Masada Y.; Inoue T.; Hashimoto K.; Fujioka M.; Uchino C. (1974) [Studies on the constituents of ginger (Zingiber officinale Roscoe) by GC-MS (author’s transl)]. Yakugaku Zasshi 94, 735–738. [DOI] [PubMed] [Google Scholar]
- Zick S. M.; Djuric Z.; Ruffin M. T.; Litzinger A. J.; Normolle D. P.; Alrawi S.; Feng M. R.; Brenner D. E. (2008) Pharmacokinetics of 6-gingerol, 8-gingerol, 10-gingerol, and 6-shogaol and conjugate metabolites in healthy human subjects. Cancer Epidemiol., Biomarkers Prev. 17, 1930–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Li C. Y.; Wen X. D.; Li P.; Qi L. W. (2009) Simultaneous determination of 6-gingerol, 8-gingerol, 10-gingerol and 6-shogaol in rat plasma by liquid chromatography-mass spectrometry: application to pharmacokinetics. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 877, 671–679. [DOI] [PubMed] [Google Scholar]
- Shukla Y.; Singh M. (2007) Cancer preventive properties of ginger: a brief review. Food Chem. Toxicol. 45, 683–690. [DOI] [PubMed] [Google Scholar]
- Kawai T.; Kinoshita K.; Koyama K.; Takahashi K. (1994) Anti-emetic principles of Magnolia obovata bark and Zingiber officinale rhizome. Planta Med. 60, 17–20. [DOI] [PubMed] [Google Scholar]
- Surh Y. J. (2002) Anti-tumor promoting potential of selected spice ingredients with antioxidative and anti-inflammatory activities: a short review. Food Chem. Toxicol. 40, 1091–1097. [DOI] [PubMed] [Google Scholar]
- Kubra I. R.; Rao L. J. (2012) An impression on current developments in the technology, chemistry, and biological activities of ginger (Zingiber officinale Roscoe). Crit. Rev. Food Sci. Nutr. 52, 651–688. [DOI] [PubMed] [Google Scholar]
- Mashhadi N. S.; Ghiasvand R.; Askari G.; Hariri M.; Darvishi L.; Mofid M. R. (2013) Anti-oxidative and anti-inflammatory effects of ginger in health and physical activity: review of current evidence. Int. J. Prev. Med. 4, S36–42. [PMC free article] [PubMed] [Google Scholar]
- Onogi T.; Minami M.; Kuraishi Y.; Satoh M. (1992) Capsaicin-like effect of (6)-shogaol on substance P-containing primary afferents of rats: a possible mechanism of its analgesic action. Neuropharmacology 31, 1165–1169. [DOI] [PubMed] [Google Scholar]
- Li F.; Nitteranon V.; Tang X.; Liang J.; Zhang G.; Parkin K. L.; Hu Q. (2012) In vitro antioxidant and anti-inflammatory activities of 1-dehydro-[6]-gingerdione, 6-shogaol, 6-dehydroshogaol and hexahydrocurcumin. Food Chem. 135, 332–337. [DOI] [PubMed] [Google Scholar]
- Hung J. Y.; Hsu Y. L.; Li C. T.; Ko Y. C.; Ni W. C.; Huang M. S.; Kuo P. L. (2009) 6-Shogaol, an active constituent of dietary ginger, induces autophagy by inhibiting the AKT/mTOR pathway in human non-small cell lung cancer A549 cells. J. Agric. Food Chem. 57, 9809–9816. [DOI] [PubMed] [Google Scholar]
- Tan B. S.; Kang O.; Mai C. W.; Tiong K. H.; Khoo A. S.; Pichika M. R.; Bradshaw T. D.; Leong C. O. (2013) 6-Shogaol inhibits breast and colon cancer cell proliferation through activation of peroxisomal proliferator activated receptor gamma (PPARgamma). Cancer Lett. 336, 127–139. [DOI] [PubMed] [Google Scholar]
- Park S. J.; Lee M. Y.; Son B. S.; Youn H. S. (2009) TBK1-targeted suppression of TRIF-dependent signaling pathway of Toll-like receptors by 6-shogaol, an active component of ginger. Biosci., Biotechnol., Biochem. 73, 1474–1478. [DOI] [PubMed] [Google Scholar]
- Ling H.; Yang H.; Tan S. H.; Chui W. K.; Chew E. H. (2010) 6-Shogaol, an active constituent of ginger, inhibits breast cancer cell invasion by reducing matrix metalloproteinase-9 expression via blockade of nuclear factor-kappaB activation. Br. J. Pharmacol. 161, 1763–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park G.; Kim H. G.; Ju M. S.; Ha S. K.; Park Y.; Kim S. Y.; Oh M. S. (2013) 6-Shogaol, an active compound of ginger, protects dopaminergic neurons in Parkinson’s disease models via anti-neuroinflammation. Acta Pharmacol. Sin. 34, 1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang S.; Hong J.; Wu H.; Liu J.; Yang C. S.; Pan M. H.; Badmaev V.; Ho C. T. (2009) Increased growth inhibitory effects on human cancer cells and anti-inflammatory potency of shogaols from Zingiber officinale relative to gingerols. J. Agric. Food Chem. 57, 10645–10650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Soroka D. N.; Hu Y.; Chen X.; Sang S. (2013) Characterization of thiol-conjugated metabolites of ginger components shogaols in mouse and human urine and modulation of the glutathione levels in cancer cells by [6]-shogaol. Mol. Nutr. Food Res. 57, 447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Lv L.; Soroka D.; Warin R. F.; Parks T. A.; Hu Y.; Zhu Y.; Chen X.; Sang S. (2012) Metabolism of [6]-shogaol in mice and in cancer cells. Drug Metab. Dispos. 40, 742–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Warin R. F.; Soroka D. N.; Chen H.; Sang S. (2013) Metabolites of ginger component [6]-shogaol remain bioactive in cancer cells and have low toxicity in normal cells: chemical synthesis and biological evaluation. PLoS One 8, e54677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Soroka D. N.; Zhu Y.; Hu Y.; Chen X.; Sang S. (2013) Cysteine-conjugated metabolite of ginger component [6]-shogaol serves as a carrier of [6]-shogaol in cancer cells and in mice. Chem. Res. Toxicol. 26, 976–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warin R. F.; Chen H.; Soroka D. N.; Zhu Y.; Sang S. (2014) Induction of lung cancer cell apoptosis through a p53 pathway by [6]-shogaol and its cysteine-conjugated metabolite M2. J. Agric. Food Chem. 62, 1352–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes J. D.; McMahon M. (2001) Molecular basis for the contribution of the antioxidant responsive element to cancer chemoprevention. Cancer Lett. 174, 103–113. [DOI] [PubMed] [Google Scholar]
- Chen C.; Kong A. N. (2005) Dietary cancer-chemopreventive compounds: from signaling and gene expression to pharmacological effects. Trends Pharmacol. Sci. 26, 318–326. [DOI] [PubMed] [Google Scholar]
- Wasserman W. W.; Fahl W. E. (1997) Functional antioxidant responsive elements. Proc. Natl. Acad. Sci. U.S.A. 94, 5361–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird L.; Dinkova-Kostova A. T. (2011) The cytoprotective role of the Keap1–Nrf2 pathway. Arch. Toxicol. 85, 241–272. [DOI] [PubMed] [Google Scholar]
- Itoh K.; Chiba T.; Takahashi S.; Ishii T.; Igarashi K.; Katoh Y.; Oyake T.; Hayashi N.; Satoh K.; Hatayama I.; Yamamoto M.; Nabeshima Y. (1997) An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 236, 313–322. [DOI] [PubMed] [Google Scholar]
- Nguyen T.; Yang C. S.; Pickett C. B. (2004) The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress. Free Radical Biol. Med. 37, 433–441. [DOI] [PubMed] [Google Scholar]
- Yu R.; Lei W.; Mandlekar S.; Weber M. J.; Der C. J.; Wu J.; Kong A. N. (1999) Role of a mitogen-activated protein kinase pathway in the induction of phase II detoxifying enzymes by chemicals. J. Biol. Chem. 274, 27545–27552. [DOI] [PubMed] [Google Scholar]
- Huang H. C.; Nguyen T.; Pickett C. B. (2002) Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 277, 42769–42774. [DOI] [PubMed] [Google Scholar]
- Hayes J. D.; McMahon M.; Chowdhry S.; Dinkova-Kostova A. T. (2010) Cancer chemoprevention mechanisms mediated through the Keap1–Nrf2 pathway. Antioxid. Redox Signaling 13, 1713–1748. [DOI] [PubMed] [Google Scholar]
- Itoh K.; Wakabayashi N.; Katoh Y.; Ishii T.; O’Connor T.; Yamamoto M. (2003) Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 8, 379–391. [DOI] [PubMed] [Google Scholar]
- Bak M. J.; Ok S.; Jun M.; Jeong W. S. (2012) 6-Shogaol-rich extract from ginger up-regulates the antioxidant defense systems in cells and mice. Molecules 17, 8037–8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan F. F.; Ling H.; Ang X.; Reddy S. A.; Lee S. S.; Yang H.; Tan S. H.; Hayes J. D.; Chui W. K.; Chew E. H. (2013) A novel shogaol analog suppresses cancer cell invasion and inflammation, and displays cytoprotective effects through modulation of NF-kappaB and Nrf2–Keap1 signaling pathways. Toxicol. Appl. Pharmacol. 272, 852–862. [DOI] [PubMed] [Google Scholar]
- Suvorova E. S.; Lucas O.; Weisend C. M.; Rollins M. F.; Merrill G. F.; Capecchi M. R.; Schmidt E. E. (2009) Cytoprotective Nrf2 pathway is induced in chronically txnrd 1-deficient hepatocytes. PLoS One 4, e6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Li J.; Li H.; Hu Y.; Tevebaugh W.; Yamamoto M.; Que J.; Chen X. (2012) Transcript profiling identifies dynamic gene expression patterns and an important role for Nrf2/Keap1 pathway in the developing mouse esophagus. PLoS One 7, e36504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmers T. A.; Gutierrez J. C.; Koniaris L. G. (2010) Loss of GDF-15 abolishes sulindac chemoprevention in the ApcMin/+ mouse model of intestinal cancer. J. Cancer Res. Clin. Oncol. 136, 571–576. [DOI] [PubMed] [Google Scholar]
- Wang X.; Kingsley P. J.; Marnett L. J.; Eling T. E. (2011) The role of NAG-1/GDF15 in the inhibition of intestinal polyps in APC/Min mice by sulindac. Cancer Prev. Res. (Phila.) 4, 150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek S. J.; Okazaki R.; Lee S. H.; Martinez J.; Kim J. S.; Yamaguchi K.; Mishina Y.; Martin D. W.; Shoieb A.; McEntee M. F.; Eling T. E. (2006) Nonsteroidal anti-inflammatory drug-activated gene-1 over expression in transgenic mice suppresses intestinal neoplasia. Gastroenterology 131, 1553–1560. [DOI] [PubMed] [Google Scholar]
- Ohtsuji M.; Katsuoka F.; Kobayashi A.; Aburatani H.; Hayes J. D.; Yamamoto M. (2008) Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J. Biol. Chem. 283, 33554–33562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung K. A.; Kwak M. K. (2013) Enhanced 4-hydroxynonenal resistance in KEAP1 silenced human colon cancer cells. Oxid. Med. Cell. Longevity 2013, 423965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S.; Xu C.; Shen G.; Hebbar V.; Gopalakrishnan A.; Hu R.; Jain M. R.; Lin W.; Keum Y. S.; Liew C.; Chan J. Y.; Kong A. N. (2006) Pharmacogenomics of phenolic antioxidant butylated hydroxyanisole (BHA) in the small intestine and liver of Nrf2 knockout and C57BL/6J mice. Pharm. Res. 23, 2621–2637. [DOI] [PubMed] [Google Scholar]
- Zhang D. D.; Hannink M. (2003) Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 23, 8137–8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan H. K.; Olayanju A.; Goldring C. E.; Park B. K. (2013) The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 85, 705–717. [DOI] [PubMed] [Google Scholar]
- Feng J.; Zhang P.; Chen X.; He G. (2011) PI3K and ERK/Nrf2 pathways are involved in oleanolic acid-induced heme oxygenase-1 expression in rat vascular smooth muscle cells. J. Cell. Biochem. 112, 1524–1531. [DOI] [PubMed] [Google Scholar]
- Na H. K.; Kim E. H.; Jung J. H.; Lee H. H.; Hyun J. W.; Surh Y. J. (2008) (−)-Epigallocatechin gallate induces Nrf2-mediated antioxidant enzyme expression via activation of PI3K and ERK in human mammary epithelial cells. Arch. Biochem. Biophys. 476, 171–177. [DOI] [PubMed] [Google Scholar]
- Lee J. M.; Hanson J. M.; Chu W. A.; Johnson J. A. (2001) Phosphatidylinositol 3-kinase, not extracellular signal-regulated kinase, regulates activation of the antioxidant-responsive element in IMR-32 human neuroblastoma cells. J. Biol. Chem. 276, 1–20016. [DOI] [PubMed] [Google Scholar]
- Kobayashi A.; Kang M. I.; Okawa H.; Ohtsuji M.; Zenke Y.; Chiba T.; Igarashi K.; Yamamoto M. (2004) Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 24, 7130–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi N.; Itoh K.; Wakabayashi J.; Motohashi H.; Noda S.; Takahashi S.; Imakado S.; Kotsuji T.; Otsuka F.; Roop D. R.; Harada T.; Engel J. D.; Yamamoto M. (2003) Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 35, 238–245. [DOI] [PubMed] [Google Scholar]
- Yamamoto T.; Suzuki T.; Kobayashi A.; Wakabayashi J.; Maher J.; Motohashi H.; Yamamoto M. (2008) Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell. Biol. 28, 2758–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abiko Y.; Miura T.; Phuc B. H.; Shinkai Y.; Kumagai Y. (2011) Participation of covalent modification of Keap1 in the activation of Nrf2 by tert-butylbenzoquinone, an electrophilic metabolite of butylated hydroxyanisole. Toxicol. Appl. Pharmacol. 255, 32–39. [DOI] [PubMed] [Google Scholar]
- Wu J. H.; Miao W.; Hu L. G.; Batist G. (2010) Identification and characterization of novel Nrf2 inducers designed to target the intervening region of Keap1. Chem. Biol. Drug Des. 75, 475–480. [DOI] [PubMed] [Google Scholar]
- Fujii S.; Akaike T. (2013) Redox signaling by 8-nitro-cyclic guanosine monophosphate: nitric oxide- and reactive oxygen species-derived electrophilic messenger. Antioxid. Redox Signaling 19, 1236–1246. [DOI] [PubMed] [Google Scholar]
- Holland R.; Hawkins A. E.; Eggler A. L.; Mesecar A. D.; Fabris D.; Fishbein J. C. (2008) Prospective type 1 and type 2 disulfides of Keap1 protein. Chem. Res. Toxicol. 21, 2051–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y.; Eggler A. L.; Liu D.; Liu G.; Mesecar A. D.; van Breemen R. B. (2007) Sites of alkylation of human Keap1 by natural chemoprevention agents. J. Am. Soc. Mass Spectrom. 18, 2226–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C.; Eggler A. L.; Mesecar A. D.; van Breemen R. B. (2011) Modification of keap1 cysteine residues by sulforaphane. Chem. Res. Toxicol. 24, 515–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro K.; Ando T.; Watanabe O.; Goto H. (2008) Specific reaction of alpha,beta-unsaturated carbonyl compounds such as 6-shogaol with sulfhydryl groups in tubulin leading to microtubule damage. FEBS Lett. 582, 3531–3536. [DOI] [PubMed] [Google Scholar]
- Riera C. E.; Menozzi-Smarrito C.; Affolter M.; Michlig S.; Munari C.; Robert F.; Vogel H.; Simon S. A.; le Coutre J. (2009) Compounds from Sichuan and Melegueta peppers activate, covalently and non-covalently, TRPA1 and TRPV1 channels. Br. J. Pharmacol. 157, 1398–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Peng Y. B.; Zhou P.; Qi L. W.; Zhang M.; Gao N.; Liu E. H.; Li P. (2013) 6-Shogaol induces apoptosis in human leukemia cells through a process involving caspase-mediated cleavage of eIF2alpha. Mol. Cancer 12, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. O.; Lee M. H.; Oi N.; Kim S. H.; Bae K. B.; Huang Z.; Kim D. J.; Reddy K.; Lee S. Y.; Park S. J.; Kim J. Y.; Xie H.; Kundu J. K.; Ryoo Z. Y.; Bode A. M.; Surh Y. J.; Dong Z. (2014) [6]-Shogaol inhibits growth and induces apoptosis of non-small cell lung cancer cells by directly regulating Akt1/2. Carcinogenesis 35, 683–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liby K. T.; Sporn M. B. (2012) Synthetic oleanane triterpenoids: multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol. Rev. 64, 972–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M.; Li L.; Iwamoto N.; Nakajima-Takagi Y.; Kaneko H.; Nakayama Y.; Eguchi M.; Wada Y.; Kumagai Y.; Yamamoto M. (2009) The antioxidant defense system Keap1–Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol. Cell. Biol. 29, 493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor T. O.; Huang M. T.; Prawan A.; Liu Y.; Hao X.; Yu S.; Cheung W. K.; Chan J. Y.; Reddy B. S.; Yang C. S.; Kong A. N. (2008) Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev. Res. 1, 187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osburn W. O.; Karim B.; Dolan P. M.; Liu G.; Yamamoto M.; Huso D. L.; Kensler T. W. (2007) Increased colonic inflammatory injury and formation of aberrant crypt foci in Nrf2-deficient mice upon dextran sulfate treatment. Int. J. Cancer 121, 1883–1891. [DOI] [PubMed] [Google Scholar]
- Wagner A. E.; Will O.; Sturm C.; Lipinski S.; Rosenstiel P.; Rimbach G. (2013) DSS-induced acute colitis in C57BL/6 mice is mitigated by sulforaphane pre-treatment. J. Nutr. Biochem. 24, 2085–2091. [DOI] [PubMed] [Google Scholar]
- Pandurangan A. K.; Ananda Sadagopan S. K.; Dharmalingam P.; Ganapasam S. (2014) Luteolin, a bioflavonoid inhibits Azoxymethane-induced colorectal cancer through activation of Nrf2 signaling. Toxicol. Mech. Methods 24, 13–20. [DOI] [PubMed] [Google Scholar]
- Kim S. B.; Pandita R. K.; Eskiocak U.; Ly P.; Kaisani A.; Kumar R.; Cornelius C.; Wright W. E.; Pandita T. K.; Shay J. W. (2012) Targeting of Nrf2 induces DNA damage signaling and protects colonic epithelial cells from ionizing radiation. Proc. Natl. Acad. Sci. U.S.A. 109, E2949–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo M. C.; Zhang D. D. (2013) The emerging role of the Nrf2–Keap1 signaling pathway in cancer. Genes Dev. 27, 2179–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. J.; Sun Z.; Villeneuve N. F.; Zhang S.; Zhao F.; Li Y.; Chen W.; Yi X.; Zheng W.; Wondrak G. T.; Wong P. K.; Zhang D. D. (2008) Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 29, 1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T. H.; Hur E. G.; Kang S. J.; Kim J. A.; Thapa D.; Lee Y. M.; Ku S. K.; Jung Y.; Kwak M. K. (2011) NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. Cancer Res. 71, 2260–2275. [DOI] [PubMed] [Google Scholar]
- Mitsuishi Y.; Taguchi K.; Kawatani Y.; Shibata T.; Nukiwa T.; Aburatani H.; Yamamoto M.; Motohashi H. (2012) Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79. [DOI] [PubMed] [Google Scholar]