

Abstract
Fourteen new resorcylic acid lactones (1–14) were isolated from an organic extract of a culture of a freshwater aquatic fungus Halenospora sp. originating from a stream in North Carolina. The structures were elucidated using a set of spectroscopic and spectrometric techniques. The absolute configuration of one representative member of the compounds (7) was assigned using X-ray crystallography of an analogue that incorporated a heavy atom, whereas for compounds 8–11, a modified Mosher’s ester method was utilized. The relative configurations of compounds 12–14 were determined on the basis of NOE data. Compounds 12–14 were proposed as artifacts produced by intramolecular cycloetherification of the ε-hydroxy-α,β-unsaturated ketone moieties of the parent compounds during the purification processes. The isolated compounds, except for 8 and 12, were tested against the MDA-MB-435 (melanoma) and HT-29 (colon) cancer cell lines. Compound 5 was the most potent, with IC50 values of 2.9 and 7.5 μM, respectively. The compounds were evaluated as TAK1–TAB1 inhibitors but were found to be inactive.
Fungi are one of the more diverse kingdoms of life, although they are not well investigated.1 For example, of the 1.5 to 5.1 million estimated species of fungi,2−4 less than 100 000 have been described in the literature.5,6 Interestingly, fungi from freshwater habitats, specifically ascomycetes that inhabit and decompose submerged woody and herbaceous organic matter in lotic and lentic habitats, represent an even less well studied area of mycology, resulting in slightly more than 3000 described species to date.7 Freshwater fungi also represent an understudied source of bioactive secondary metabolites, as approximately 125 compounds have been described, or less than 1% of the over 14 000 compounds that have been characterized from fungi.8,9 Hence, studies on freshwater fungi have been initiated10−12 in pursuit of new chemical diversity.13
An aquatic fungus, accessioned as G87, was sampled from a submerged wood substrate in a stream on the campus of the University of North Carolina at Greensboro and was identified putatively as a Halenospora sp., Helotiales, Leotiomycetes, Ascomycota. Promising bioactivity of the organic extract led to the isolation and characterization of 14 new, but structurally related, resorcylic acid lactones (RALs) [greensporone A (1), greensporone B (2), 8,9-dihydrogreensporone A (3), dechlorogreensporone A (4), greensporone C (5), O-desmethylgreensporone C (6), 8,9-dihydrogreensporone C (7), greensporone D (8), greensporone E (9), dechlorogreensporone D (10), 8,9-dihydrogreensporone D (11), greensporone F (12), dechlorogreensporone F (13), and greensporone G (14)]. There has been growing interest in macrocycles in drug discovery because of their interesting biological activities and unique properties, including cell permeability and oral bioavailability.14 Hence, these compounds, except for 8 and 12, were tested in the MDA-MB-435 (melanoma) and HT-29 (colon) cancer cell lines. Moreover, as similar polyketide-derived metabolites were reported to be potent inhibitors of ATPases and kinases, including TAK1,15,16 they were examined as TAK1–TAB1 inhibitors but were found to be inactive.
Results and Discussion
An organic extract [CHCl3–MeOH (1:1)] of the G87 culture that was grown on rice was partitioned with 1:1 CH3CN–MeOH and hexane. The brine shrimp test was utilized17−19 as an initial screen because fungi in aquatic environments may produce defensive secondary metabolites; a promising LC50 value of 20 μg/mL was observed. The organic extract was then fractionated using flash chromatography to afford five fractions. Of these, fractions 3 and 4 showed interesting HPLC profiles with PDA data that indicated a series of structurally related compounds (Figure S1, Supporting Information). Thus, these fractions were subjected to further purifications using preparative and semipreparative HPLC to yield 14 new compounds (1–14). The purity of the isolated compounds was measured by UPLC (Figure S2).
Compound 1 (13.9 mg) was obtained as a colorless solid. The molecular formula was determined as C19H21ClO6 by HRESIMS. The NMR (Tables 1 and 2), HRMS, and specific rotation data were similar to those for a recently reported RAL analogue, cryptosporiopsin A, described by Talontsi et al.20 in 2012 from a culture of Cryptosporiopsis sp., an endophytic fungus from leaves and branches of Zanthoxylum leprieurii (Rutaceae). Although the planar structure between 1 and cryptosporiopsin A was identical, a new trivial name for 1 has been proposed for two reasons. First, and as explained in more detail below, we have strong evidence that the absolute configuration for 1 at position 2 is S. In contrast, cryptosporiopsin A was drawn as 2R; however, those authors did not discuss their reasoning for this conclusion, although it could be due to similarities to ponchonin D.20,21 Moreover, the trivial name proposed by those authors20 was utilized in 1969 for a structurally unrelated compound.22,23 As such, compound 1 was ascribed the new trivial name, greensporone A.
Table 1. 1H NMR Data (500 MHz) for 1–4 in CDCl3a.
| position | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 1 | 1.33, d (6.3) | 1.30, d (6.3) | 1.33, d (6.3) | 1.36, d (6.3) |
| 2 | 5.16, m | 5.12, m | 5.23, m | 5.18, m |
| 3 | 1.73, m | 1.7, m | 1.61, m | 1.76, m |
| 1.97, m | 1.89, m | 2.14, m | 2.01, m | |
| 4 | 2.42, dt (19.5, 6.3) | 2.32, m | 2.30, m | 2.43, m |
| 2.57, m | 2.56, m | 2.62, m | 2.67, m | |
| 6 | 2.47, m | 2.36, m | 2.25, m | 2.47, m |
| 2.54, m | 2.62, m | 2.50, m | 2.53, m | |
| 7 | 2.48, m | 2.51, m | 1.71, m | 2.47, m |
| 2.54, m | 3.85, m | 2.56, m | ||
| 8 | 6.88, ddd | 6.06, ddd | 1.51, m | 6.79, m |
| (16.0, 6.3, 2.9) | (16.6, 11.5, 5.2) | 1.76, m | ||
| 9 | 6.06, d (16.0) | 6.29, dd | 2.41, ddd | 6.04, d (16.0) |
| (11.5, 1.7) | (16.0, 8.6, 2.9) | |||
| 2.61, m | ||||
| 11 | 3.84, d (16.0) | 4.01, d (18.3) | 3.90, d (18.3) | 3.33, d (14.3) |
| 4.19, d (16.0) | 4.11, d (18.3) | 4.02, d (18.3) | 4.31, d (14.3) | |
| 13 | 6.44, d (2.3) | |||
| 15 | 6.58, s | 6.58, s | 6.56, s | 6.31, d (2.3) |
| 19 | 3.78, s | 3.77, s | 3.77, s | 3.74, s |
| 14-OH | 6.02, br s | 5.77, s | 5.73, s | 6.33, br s |
δ in ppm, mult (J in Hz).
Table 2. 13C NMR Data for 12 (175 MHz) and for 1–11 and 13–14 (125 MHz) in CDCl3a.
| position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10b | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 20.3 | 19.8 | 20.3 | 20.5 | 20.6 | 20.3 | 20.5 | 20.1 | 19.9 | 20.1 | 20.3 | 21.5 | 20.9 | 18.3 |
| 2 | 71.8 | 71.6 | 71.4 | 71.2 | 71.3 | 73.9 | 70.9 | 71.3 | 71.0 | 69.3 | 70.8 | 73.4 | 72.7 | 72.3 |
| 3 | 28.5 | 28.5 | 28.7 | 28.5 | 34.9 | 34.6 | 34.9 | 31.0 | 31.0 | 30.2 | 30.7 | 33.8 | 33.0 | 30.1 |
| 4 | 39.8 | 34.1 | 35.6 | 39.4 | 23.3 | 23.8 | 26.3c | 29.4 | 29.1 | 29.2 | 29.8 | 34.7 | 31.3 | 30.0 |
| 5 | 210.0 | 210.4 | 211.3 | 209.9 | 25.6 | 28.3 | 22.4 | 69.1 | 69.0 | 66.1 | 68.7 | 79.3 | 79.5 | 77.2 |
| 6 | 40.8 | 43.0 | 42.8 | 40.5 | 25.8 | 25.8 | 25.5c | 35.0 | 36.7 | 34.6 | 36.1 | 31.6 | 33.5 | 33.5 |
| 7 | 29.0 | 24.8 | 22.9 | 28.6 | 30.9 | 33.1 | 25.4 | 28.8 | 24.3 | 28.2 | 23.2 | 29.8 | 30.5 | 32.2 |
| 8 | 146.4 | 147.2 | 23.2 | 147.1 | 150.5 | 150.3 | 23.3 | 147.7 | 148.3 | 148.2 | 23.7 | 76.3 | 76.1 | 76.5 |
| 9 | 129.9 | 127.1 | 41.3 | 130.7 | 129.9 | 129.7 | 41.8 | 128.1 | 127.1 | 128.4 | 41.7 | 46.6 | 47.9 | 47.2 |
| 10 | 194.7 | 196.8 | 206.3 | 198.0 | 199.6 | 198.1 | 211.1 | 195.1 | 196.7 | 195.9 | 205.9 | 203.9 | 207.7 | 210.1 |
| 11 | 42.7 | 45.9 | 44.7 | 43.7 | 44.1 | 47.2 | 46.3 | 44.2 | 45.7 | 44.6 | 44.3 | 48.6 | 49.0 | 48.6 |
| 12 | 132.1 | 131.6 | 131.8 | 135.0 | 135.0 | 139.0 | 133.8 | 132.6 | 131.7 | 135.6 | 131.8 | 132.1 | 134.2 | 134.3 |
| 13 | 113.6 | 113.0 | 112.8 | 109.6 | 109.5 | 113.3 | 110.3 | 113.4 | 112.9 | 109.7 | 113.4 | 113.1 | 109.2 | 108.1 |
| 14 | 153.7 | 153.3 | 153.2 | 158.4 | 159.1 | 160.8 | 158.6 | 153.7 | 153.2 | 159.8 | 153.4 | 153.3 | 157.7 | 158.0 |
| 15 | 99.1 | 99.1 | 98.9 | 98.5 | 98.7 | 103.1 | 98.6 | 99.2 | 100.0 | 98.5 | 99.0 | 99.1 | 98.3 | 98.3 |
| 16 | 157.0 | 156.6 | 156.7 | 159.5 | 159.6 | 165.6 | 159.0 | 157.6 | 156.6 | 159.2 | 157.0 | 157.0 | 159.0 | 158.8 |
| 17 | 117.9 | 118.4 | 118.8 | 116.1 | 115.8 | 106.1 | 116.1 | 118.2 | 118.5 | 114.1 | 118.2 | 119.2 | 117.3 | 118.0 |
| 18 | 167.2 | 167.0 | 167.4 | 168.1 | 168.5 | 170.8 | 168.7 | 167.1 | 167.6 | 167.3 | 167.7 | 166.9 | 167.7 | 167.8 |
| 19 | 56.2 | 56.2 | 56.2 | 56.0 | 56.0 | 55.7 | 56.3 | 56.1 | 55.9 | 56.1 | 56.4 | 56.0 | 56.0 |
δ in ppm, mult (J in Hz).
In DMSO-d6
Signals may be interchanged
Chart 1. New Compounds 1–14.

In addition to 1, a series of structurally related RALs (2–14) that differ in the geometry and substitution pattern of the 14-memebred macrocyclic lactone ring was isolated and characterized by analyses of HRMS and NMR data. Compound 2 (1.0 mg) was isolated as a colorless solid. The molecular formula was determined as C19H21ClO6 via HRESIMS along with 1H, 13C, and edited-HSQC NMR data (Tables 1 and 2 and Figure S4), establishing an index of hydrogen deficiency of 9. Analyses of the HRMS and NMR data suggested 2 as a RAL with structural similarity with that of 1. For example, compound 2 showed two ketone carbonyls, δC 210.4 and 196.8 ppm for C-5 and C-10, respectively. The upfield shift of C-10 relative to C-5 in conjunction with HMBC correlations from H-8 and H-9 to C-10 indicated conjugation with a double bond resulting from an α,β-unsaturated ketone. Additional similarities included NMR signals characteristic of six aromatic carbons and one singlet aromatic proton, suggesting a pentasubstituted benzene ring, with two downfield-shifted carbons (δC 153.3 and 156.6 for C-14 and C-16, respectively) indicating oxygenation, as also observed in 1. Other substituents included a methoxy group, a methyl doublet (JH3-1/H-2 = 6.3 Hz), deshielded methylene protons (JH-11a/H-11b = 18.3 Hz), and an ester group. COSY data identified two spin systems (H3-1/H-2/H2-3/H2-4 and H2-6/H2-7/H-8/H-9) that were connected by the ketone carbonyl C-5, as supported by HMBC correlations (Figure 1). A key difference between compounds 1 and 2 was the geometry of the C-8/C-9 double bond, being Z in 2 versus E in 1, as evidenced by the coupling constants of the olefinic protons (JH-8/H-9 = 11.5 Hz in 2 versus 16.0 Hz in 1). All of the benzene ring and the macrocyclic lactone ring substituents were confirmed by HMBC correlations (Figure 1). These data established the structure of 2, being the cis analogue of 1, and the trivial name greensporone B was ascribed.
Figure 1.

Key HMBC and COSY correlations of 2–14.
Compound 3 (1.2 mg), which was obtained as a colorless solid, had a molecular formula of C19H23ClO6, as evidenced by HRESIMS and analysis of 1H NMR, 13C NMR, and edited-HSQC NMR data (Tables 1 and 2 and Figure S5). The HRMS and NMR data indicated 3 to be a dihydro analogue of 1, as evidenced by both a 2 amu difference in the HRMS data and replacement of the H-8 and H-9 olefinic protons in 1 by four aliphatic protons (δH 1.51/1.76 and 2.41/2.61 for H2-8 and H2-9, respectively). The lack of conjugation of the C-8/C-9 double bond with the ketone carbonyl in 3 resulted in a diagnostic downfield shift of C-10 in 3 (δC 206.3) relative to that in 1 (δC 194.4). Further analyses of NMR data, including COSY and HMBC experiments (Figure 1), yielded the structure of 3, which was ascribed the trivial name 8,9-dihydrogreensporone A.
Compound 4 (2.1 mg) was obtained as a colorless solid with a molecular formula of C19H22O6, as evidenced by HRESIMS and analysis of 1H NMR, 13C NMR, and edited-HSQC NMR data (Tables 1 and 2 and Figure S6). The HRMS and NMR data indicated 4 as a dechlorinated analogue of 1, supported by both the absence of the characteristic isotopic pattern of the chlorine in the HRMS data of 4 and the appearance of an extra aromatic proton (δH 6.44), which, based on the coupling constant (JH-13/H-15 = 2.3 Hz), was meta coupled to H-15 (δH 6.31). The 13C and edited-HSQC NMR data were supportive of this conclusion, particularly the resonance at δC 109.6 ppm (C-13). Throughout this series of RALs, except for the desmethyl analogue (compound 6), a diagnostic resonance at either ∼δC 113 ppm or δC 109 could serve to differentiate the presence (former) or absence (latter) of the chlorine moiety on the aromatic ring. Analyses of NMR spectra, including COSY and HMBC data (Figure 1), yielded the structure of 4, which was ascribed the trivial name dechlorogreensporone A.
Compound 5 (22.7 mg) was also obtained as a colorless solid. The molecular formula was determined as C19H24O5 via HRESIMS, establishing an index of hydrogen deficiency of 8. The NMR data suggested structural similarity with that of 4. However, a key difference was replacement of the C-5 ketone carbonyl in 4 (δC 209.9) by a methylene moiety (δC/δH/δH 25.6/1.31/1.39), which resulted in an extended COSY spin system in 5 that spanned from H3-1 to H-9 (Figure 1). Further analyses of the NMR spectra, including HMBC data (Tables 2 and 3 and Figures 1 and S7), suggested the structure of 5, which was given the trivial name greensporone C.
Table 3. 1H NMR Data (500 MHz) for 5–8 in CDCl3a.
| position | 5 | 6 | 7 | 8 |
|---|---|---|---|---|
| 1 | 1.32, d (6.3) | 1.28, d (6.3) | 1.30, d (6.3) | 1.34, d (6.3) |
| 2 | 5.15, m | 5.13, m | 5.23, m | 5.08, m |
| 3 | 1.58, m | 1.50, m | 1.60, m | 1.74, m |
| 1.68, m | 1.66, m | 1.82, m | ||
| 4 | 1.24, m | 1.38, m | 1.33b, m | 1.25, m |
| 1.51, m | 1.50, m | 1.75, m | ||
| 5 | 1.31, m | 1.45, m | 1.21, m | 3.55, m |
| 1.39, m | 1.36, m | |||
| 6 | 1.56, m | 1.68, m | 1.34b, m | 1.61, m |
| 1.83, m | ||||
| 7 | 2.23, m | 2.25, m | 1.25, m | 2.25, m |
| 2.35, m | ||||
| 8 | 6.85, dt | 7.06, dt | 1.54, m | 6.78, ddd |
| (16.0, 7.5) | (16.0, 7.5) | 1.64, m | (15.5, 9.2, 6.9) | |
| 9 | 6.10, d (16.0) | 6.17, d (16.0) | 2.32, m | 6.08, d (15.5) |
| 2.56, m | ||||
| 11 | 3.44, d (14.9) | 3.92, d (17.2) | 3.48, d (17.8) | 3.85, d (17.2) |
| 4.34, d (14.9) | 4.39, d (17.2) | 4.29, d (17.8) | 4.21, d (17.2) | |
| 13 | 6.47, d (2.3) | 6.15, d (2.3) | 6.17, d (2.3) | |
| 15 | 6.31, d (2.3) | 6.32, d (2.3) | 6.19, d (2.3) | 6.59, s |
| 19 | 3.74, s | 3.71, s | 3.80, s | |
| 14–OH | 7.89, br s | 6.16, s | 7.25, br s | 5.86, s |
| 16–OH | 11.76, s |
δ in ppm, mult (J in Hz).
Signals may be interchanged.
Compound 6 (1.8 mg), which was also obtained as a colorless solid, had a molecular formula of C18H22O5, as determined by HRESIMS. Despite a 14 amu difference in the HRMS data, the NMR data suggested structural similarity with 5 (Tables 2 and 3 and Figure S8). However, a key difference was replacement of the CH3-19 (δC/δH 56.0/3.74) in 5 by a chelated phenolic proton (δH 11.76) in 6. Analyses of the NMR spectra, including COSY and HMBC data (Figure 1), established the structure of 6, which was given the trivial name O-desmethylgreensporone C.
Compound 7 (21.8 mg), which was isolated as a colorless solid, was identified as a dihydro derivative of 5, as suggested by a 2 amu difference in the HRMS data. NMR data indicated replacement of the H-8 and H-9 olefinic protons in 5 by four aliphatic protons (δH 1.54/1.64 and 2.32/2.56 for H2-8 and H2-9, respectively) (Tables 2 and 3 and Figure S9). A downfield shift of C-10 in 7 (δC 211.1), relative to that of 5 (δC 199.5), was indicative of the lack of an α,β-unsaturated ketone carbonyl in 7. These data, along with other NMR spectra, including COSY and HMBC data (Figure 1), identified the structure of 7, which was ascribed the trivial name 8,9-dihydrogreensporone C.
Compound 8 (2.4 mg) was obtained as a colorless solid with a molecular formula of C19H23ClO6, as determined via HRESIMS and analysis of 1H NMR, 13C NMR, and edited-HSQC NMR data (Tables 2 and 3 and Figure S10). The NMR data suggested structural similarity with 1. However, a key difference was replacement of the C-5 ketone carbonyl (δC 210.0) in 1 by a carbinol carbon in 8, as evidenced by the NMR chemical shift values (δH/δC 69.1/3.55) and a 2 amu difference in the HRMS data. Further analyses of the NMR spectra, including the extended COSY spin system from H3-1 to H-9 and HMBC data (Figure 1), established the structure of 8, to which the trivial name greensporone D was ascribed.
Compound 9 (1.2 mg) was also isolated as a colorless solid. The molecular formula was determined as C19H23ClO6 via HRESIMS, which was identical to that of greensporone D (8). Analyses of the NMR data suggested 9 to be a geometric isomer of 8, in reference to the C-8/C-9 double bond having a Z-configuration in 9 versus an E-configuration in 8, which was supported by the coupling constants of the olefinic protons (JH-8/H-9 = 11.5 Hz in 9 vs 15.5 Hz in 8) (Tables 2 and 4 and Figure S11). All other substituents, including the benzene ring and the macrocyclic lactone ring substituents, were confirmed by analyses of the COSY and HMBC data (Figure 1), establishing the structure of 9, to which the trivial name greensporone E was ascribed.
Table 4. 1H NMR Data (500 MHz) for 9–11a.
| position | 9b | 10c | 11b |
|---|---|---|---|
| 1 | 1.30, d (6.3) | 1.21, d (5.7) | 1.33, d (6.3) |
| 2 | 5.13, m | 4.91, m | 5.24, m |
| 3 | 1.75, m | 1.51, m | 1.69, m |
| 1.76, m | |||
| 4 | 1.11, m | 1.06, m | 1.24, m |
| 1.66, m | 1.52, m | 1.61, m | |
| 5 | 3.48, m | 3.35, m | 3.70, m |
| 6 | 1.61, m | 1.41, m | 1.47, m |
| 1.77, m | 1.68, m | 1.56, m | |
| 7 | 2.12, m | 2.15, m | 1.37, m |
| 3.48, m | |||
| 8 | 6.08, ddd | 6.64, ddd | 1.53, m |
| (16.0, 11.5, 4.6) | (16.0, 8.0, 7.5) | 1.75, m | |
| 9 | 6.30, dd | 5.95, d (16.0) | 2.29, ddd |
| (11.5, 1.7) | (13.8, 9.7, 3.4) | ||
| 2.66, ddd | |||
| (13.8, 8.6, 2.9) | |||
| 11 | 4.03, d (18.3) | 3.36, d (16.0) | 3.88, d (18.3) |
| 4.16, d (18.3) | 4.03, d (16.0) | 4.25, d (18.3) | |
| 13 | 6.25, d (2.3) | ||
| 15 | 6.58, s | 6.35, d (2.3) | 6.56, s |
| 19 | 3.77, s | 3.68, s | 3.77, s |
| 5-OH | 1.2 br s | 4.48, br s | 3.76 |
| 14-OH | 5.80, s | 9.99, br s | 5.87, s |
δ in ppm, mult (J in Hz).
In CDCl3.
In DMSO-d6.
Compound 10 (13.2 mg) was obtained as a colorless solid. The HRESIMS and 1H NMR, 13C NMR, and edited-HSQC NMR data suggested a molecular formula of C19H24O6 (Tables 2 and 4 and Figure S12). The HRMS and NMR data designated 10 as a dechlorinated analogue of 8, as evidenced by a 34 amu difference in the HRMS data and absence of the characteristic isotopic pattern of the chlorine. 1H NMR data of 10 relative to 8 indicated an extra aromatic proton (δH 6.25, J = 2.3 Hz), which was meta coupled to H-15 (δH 6.35, J = 2.3 Hz). Further analyses of the NMR spectra, including COSY and HMBC data (Figure 1), suggested the structure of 10, which was ascribed the trivial name dechlorogreensporone D.
Similar to the analysis of compounds 7 vs 5, compound 11 (4.0 mg), which was isolated as a colorless solid, was identified as a dihydro derivative of 8, as evidenced by a 2 amu difference in the HRMS data. NMR data indicated replacement of the H-8 and H-9 olefinic protons in 8 by four aliphatic protons (δH 1.53/1.75 and 2.29/2.66 for H2-8 and H2-9, respectively). A downfield shift of C-10 in 11 (δC 205.9) relative to that of 8 (δC 195.1) was indicative of the lack of conjugation of the C-8/C-9 double bond with the ketone carbonyl. These data, along with those from COSY and HMBC experiments (Figure 1), identified the structure of 11, which was ascribed the trivial name 8,9-dihydrogreensporone D.
In solution (CDCl3), compound 8 converted to 12. The molecular formula of 12 was determined as C19H23ClO6 via HRESIMS along with 1H, 13C, and edited-HSQC NMR data (Tables 2 and 5 and Figure S14), establishing an index of hydrogen deficiency of 9. Analyses of the HRMS and NMR data of 12 suggested structural similarity with 8, sharing the same molecular formula. Key differences were the lack of the α,β-conjugated double bond in 12, as evidenced by a downfield shift of the ketone carbonyl (δC 203.9) relative to that of 8 (δC 195.1) and the replacement of the C-8 and C-9 olefinic protons in 8 by three aliphatic protons in 12 (δC/δH 76.3/4.32 and 46.6/2.53/2.81 for CH-8 and CH2-9, respectively), suggesting oxygenation of C-8 in 12. Another key difference was the downfield shift of C-5/H-5 in 12 (δC/δH 79.3/3.79) relative to 8 (δC/δH 69.1/3.55). Key HMBC correlations included H2-6 to C-4 and C-8; H2-7 to C-5; H2-9 to C-7 and C-10; and H-8 to C-5 and C-10 (Figure 1). COSY identified one extended spin system, H3-1/H-2/H2-3/H2-4/H-5/H2-6/H2-7/H-8/H2-9 (Figure 1). These data suggested a C-5 to C-8 tetrahydrofuran ring in 12 relative to 8, and the formation of a ring was consistent with the index of hydrogen deficiency. The observed NOESY correlation from H-5 to H-8 indicated a cis-THF ring, thereby establishing the relative configuration at C-5 and C-8 (Figure 2). These data established the structure of 12, to which the trivial name greensporone F was ascribed. We hypothesize that 12 was produced by intramolecular cycloetherification of the ε-hydroxy-α,β-unsaturated ketone moiety in 8 (Figure S19); this is the first report of an RAL undergoing an intramolecular cycloaddition.
Table 5. 1H NMR Data (500 MHz) for 12–14a.
| position | 12 | 13 | 14 |
|---|---|---|---|
| 1 | 1.32, d (6.2) | 1.32, d (6.3) | 1.32, d (6.9) |
| 2 | 5.21, m | 5.26, m | 5.41, m |
| 3 | 1.72, m | 1.83, m | 1.62, m |
| 1.90, m | 2.33, m | ||
| 4 | 1.38, m | 1.51, m | 1.48, m |
| 1.79, m | 1.96, m | 1.92, m | |
| 5 | 3.79, m | 3.81, m | 3.99, tdd |
| (10.3, 4.6, 2.3) | |||
| 6 | 1.55, m | 1.50, m | 1.30, m |
| 2.02, m | 1.89, m | ||
| 7 | 1.75, m | 1.65, m | 1.57, m |
| 1.91, m | 1.94, m | 2.10, m | |
| 8 | 4.33, m | 4.14, m | 4.27, qd (8.6, 2.9) |
| 9 | 2.53, dd | 2.55, dd | 2.27, m |
| (13.5, 6.9) | (13.2, 8.0) | ||
| 2.81, dd | 2.62, dd | 2.49, dd | |
| (13.5, 4.6) | (13.2, 3.4) | (12.0, 8.6) | |
| 11 | 3.99, d (18.5) | 3.90, d (17.2) | 3.55, d (16.0) |
| 4.13, d (18.5) | 3.99, d (17.2) | 4.21, d (16.0) | |
| 13 | 6.25, d (2.3) | 6.10, d (2.3) | |
| 15 | 6.59, s | 6.34, d (2.3) | 6.34, d (2.3) |
| 19 | 3.77, s | 3.77, s | 3.78, s |
| 14-OH | 5.70, s | 5.62, br s | 6.07, br s |
δ in ppm, mult (J in Hz).
Figure 2.
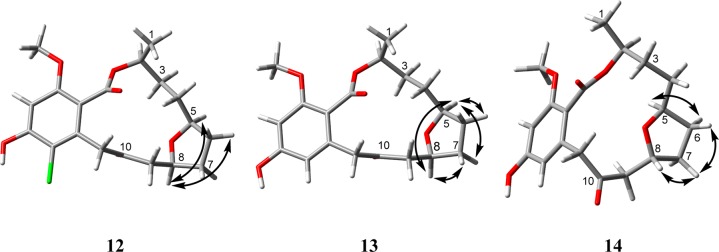
Key NOESY correlations of 12–14.
Compound 13 (1.4 mg) was obtained as a colorless solid. HRESIMS and analysis of 1H, 13C, and edited-HSQC NMR data suggested a molecular formula of C19H24O6 (Tables 2 and 5 and Figure S15). The HRMS and NMR data suggested 13 to be a dechlorinated analogue of 12, as evidenced by a 34 amu difference in the HRMS data of 13 and absence of the characteristic isotopic pattern of the chlorine atom. 1H NMR data of 13 relative to 12 indicated an extra aromatic proton (δH 6.25) that was meta coupled to H-15 (δH 6.34) (JH-13/H-15 = 2.3 Hz). Further analyses of the NMR data, including COSY and HMBC data (Figure 1), suggested the structure of 13, which was ascribed the trivial name dechlorogreensporone F. Similar to 12, NOESY correlations between H-5 and H-8 supported a cis-THF ring (Figure 2). As for 12, compound 13 was likely an artifact produced by intramolecular cycloetherification of the ε-hydroxy-α,β-unsaturated ketone moiety in 10 during the extraction and purification processes. To test this hypothesis, an aliquot of 10 was suspended (not soluble) in CDCl3, and a proton NMR spectrum was collected; a poor signal was observed due to insolubility (Figure S16). The suspension was left to stand at room temperature for 3 days, and then another proton NMR spectrum was collected. The latter NMR spectrum showed signals of good intensity that matched those of 13 (Figure S16).
Compound 14 (1.8 mg) was obtained as a colorless solid. HRESIMS and analysis of 1H NMR, 13C NMR, and edited-HSQC NMR data suggested a molecular formula of C19H24O6 (Tables 4 and 5 and Figure S17). The HRMS and NMR data suggested structural similarity to 13, both sharing the same molecular formula and the same planar structure, as evidenced from analyses of the 2D NMR data of 14, including COSY and HMBC spectra (Figure 1). However, a key difference was the absence of the H-5 to H-8 NOESY correlations in 14 relative to 13. This observation was supported by NOESY correlations observed from H-8 to H-7b, H-7b to H-6a, and H-5 to H-6b, indicating that H-8, H-7b, and H-6a were on the same face of the tetrahydrofuran ring (Figure 2). Hence, 14 and 13 differed in the spatial arrangement of the tetrahydrofuran ring, being cis in 13 versus trans in 14. As for 12 and 13, compound 14 was likely an artifact produced by intramolecular cycloetherification of the ε-hydroxy-α,β-unsaturated ketone moiety in 10 but by attacking the α,β-unsaturated ketone moiety in an opposite manner from the way that 13 was formed. The trivial name greensporone G was ascribed to compound 14.
Determination of the Absolute Configuration
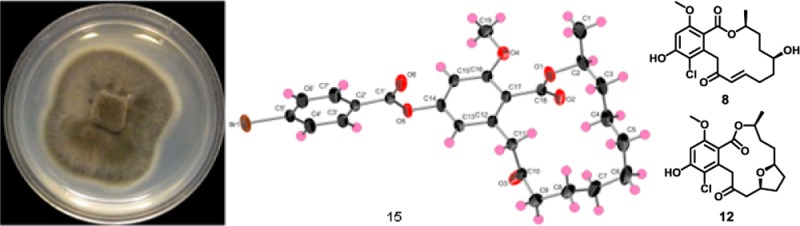
The absolute configuration of the stereogenic center in 7 was established as 2S by single-crystal X-ray diffraction analysis of the bromobenzoyl derivative (compound 15, Figures 3 and S18). The configuration of the C-2 asymmetric carbon in compounds 1–6 and 8–14 was proposed to be analogous to that of 7; as described below, this supposition was supported by Mosher’s esters in compounds 8–11. Cryptosporiopsin A, a RAL with the same planar structure as that of 1, was reported to have a 2R configuration20 by analogy to pochonin D;21 however, no direct measurements were made to confirm this. The RAL analogues identified in this study were structurally related to the well-known RAL monorden (synonym, radicicol)24,25 and its analogues pochonins A–F21 and K–P.26 For monorden, single-crystal X-ray diffraction via analysis of anomalous scattering established the configuration of the C-2 asymmetric carbon as R.25 Then, to assign the relative configuration of pochonins,261H–1H coupling constants and analysis of NOESY data were used in conjunction with comparisons to the specific rotation data of monorden.21 However, there are challenges in the use of specific rotation data for assignment of absolute configuration, except in the case of enantiomers.27 The use of X-ray crystallography and Mosher’s ester in this study provided a more comprehensive assignment of the absolute configuration of this series of RALs.
Figure 3.
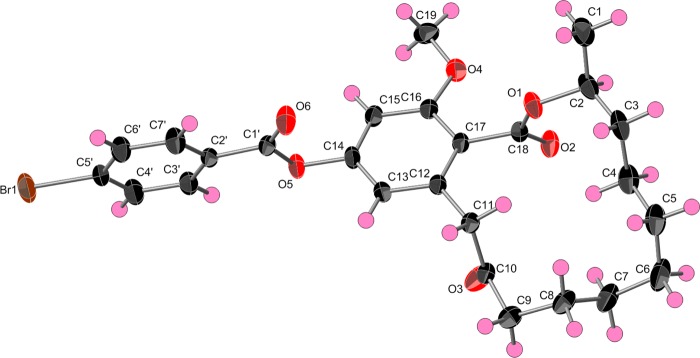
X-ray crystallographic structure of 14-(4-bromobenzoyl)-8,9-dihydrogreensporone C (15).
The absolute configurations of 8–11 were assigned via a modified Mosher’s ester method,28 establishing the configurations as 2S and 5S (Figure 4). However, a challenge arose in this analysis due to the instability of compound 8 in solution, i.e., intramolecular cycloetherification of the ε-hydroxy-α,β-unsaturated ketone moiety (Figure S19). The 1H NMR peaks that corresponded to the (S)- and (R)-MTPA esters of 8 were of low intensity; thus, the chemical shifts of H-2 and H-9 were the only ones that could be identified readily. Regardless, biogenetic considerations support the consistency of the configurations at the C-2 and C-5 asymmetric carbons throughout the series.
Figure 4.

ΔδH values [Δδ (in ppm) = δS – δR] obtained for (S)- and (R)-MTPA esters (A) 8a and 8b, respectively, of greensporone D (8), (B) 9a and 9b, respectively, of greensporone E (9), (C) 10a and 10b, respectively, of dechlorogreensporone D (10), and (D) 11a and 11b, respectively, of 8,9-dihydrogreensporone D (11) in pyridine-d5.
Biological Evaluation
The cytotoxicities of the isolated compounds, except for 8 and 12, were tested against MDA-MB-435 (melanoma) and HT-29 (colon) cancer cell lines; compound 8 was not tested due to the aforementioned instability. Compound 5 was the most potent, with IC50 values of 2.9 and 7.5 μM, respectively (Table 6). The cytotoxicity data of the 12 analogues permitted preliminary conclusions regarding structure–activity relationships. Compounds 3, 7, and 11–14, all with a reduced enone, and compounds 2 and 9 with a (Z)-enone were inactive, demonstrating the importance of the (E)-enone. Replacing the C-16 methoxy moiety in 5 by a chelated phenolic functionality in 6 reduced the activity against the MDA-MB-435 and HT-29 cell lines by factors of 5 and 2, respectively. Similarly, hydroxylation of position C-5, as noted in compounds 5 vs 10, and oxidation of the C-5 to a ketone, as in 5 vs 1 and 4, diminished cytotoxicity in MDA-MB-435 cells by a factor of ∼4 and ∼5, respectively.
Table 6. Activities of Compounds 1–7, 9–11, 13, and 14 against Two Human Tumor Cell Lines.
| IC50 values (μM)b |
||
|---|---|---|
| compounda | MDA-MB-435c | HT-29c |
| 1 | 14.1 | >20 |
| 4 | 14.1 | >20 |
| 5 | 2.9 | 7.5 |
| 6 | 14.5 | 13.8 |
| 10 | 11.2 | 25.4 |
Compounds 2, 3, 7, 9, 11, 13, 14 were inactive (IC50 values > 20 μM).
IC50 values were determined as the concentration required to inhibit growth to 50% of control with a 72 h incubation.
Positive control was vinblastine tested at concentration of 1 nM in MDA-MB-435 cells and 10 nM in HT-29 cells, which had 21% and 44% viable cells, respectively.
Transforming growth factor-β activated kinase-1 (TAK1), a member of the mitogen-activated protein kinase kinase kinase (MAP3K) family, along with its activator TAK1-binding protein 1 (TAB1), regulates several cellular signaling pathways, including activation of nuclear factor kappa B (NF-κB) and JNK/p38 MAPKs.29,30 Hence, TAK1 is considered to be a potential target for combating several diseases, including cancer and inflammation. An RAL, (5Z)-7-oxozeaenol, has been reported to be a highly potent and selective inhibitor of TAK1.31 Hence, the isolated compounds, except for 8 and 12, were evaluated as TAK1–TAB1 inhibitors using (5Z)-7-oxozeaenol32 as a positive control. Although all RALs in this series were inactive as TAK1–TAB1 inhibitors, the data extended our understanding of the structure–activity relationship of the RAL family of compounds.
Fungal Strain
In 1963, Halenospora sp. was described originally as the marine fungus Zalerion varia.33 Later in 2009, it was assigned to a new genus, Halenospora, due to phylogenetic affinities to the Leotiaceae, Ascomycota based on ITS rDNA sequence data.34,35 Alternatively, the type species, Zalerion maritima, groups with a recently established marine fungal order Lulworthiales, Ascomycota.36 Ecologically, aquatic fungi are defined as those occurring in freshwater, brackish, and marine habitats.7 Marine fungi can further be classified into obligate (fungi that grow and sporulate exclusively in marine or estuarine habitats) or facultative marine fungi (fungi that occur both in freshwater and marine habitats).37 The fungal strain used in this study was collected in a freshwater stream more than 100 linear miles from the Atlantic Ocean. Thus, H. varia can be classified as a facultative marine fungus, as has been reported from marine33 and freshwater habitats. To better understand if physiological conditions could affect secondary metabolite production, fungal strain G87 was grown on rice using seed culture grown in saltwater and found to produce a similar chemical profile to that of the freshwater isolate of Halenospora sp. (data not shown).
In conclusion, 14 new RALs (1–14) were isolated and characterized from a Halenospora sp. The absolute configuration of compound 7 was assigned using X-ray crystallography of an analogue that incorporated a heavy atom. NOE data were used to assign the relative configuration of compounds (12–14), whereas for compounds 8–11, a modified Mosher’s ester method was utilized. The cytotoxicity data of this series of RALs established the importance of the (E)-enone for activity. Finally, this study demonstrated the value of examining fungi from understudied habitats, regardless of their geographical origin. While traveling the world in search of unique niches has merits, the present study originated from a sample in a stream that is less than 300 m from our chemistry laboratories and is traversed daily by hundreds of students; understudied biodiversity is everywhere.
Experimental Section
General Experimental Procedures
Optical rotations, UV, and IR data were obtained using a Rudolph Research Autopol III polarimeter (Rudolph Research Analytical), a Varian Cary 100 Bio UV–vis spectrophotometer (Varian Inc.), and a PerkinElmer Spectrum One with Universal ATR attachment (PerkinElmer). NMR data were collected using either a JEOL ECA-500 NMR spectrometer operating at 500 MHz for 1H and 125 MHz for 13C (JEOL Ltd.) or an Agilent 700 MHz NMR spectrometer (Agilent Technologies) equipped with a cryoprope, operating at 700 MHz for 1H and 175 MHz for 13C. Residual solvent signals were utilized for referencing. HRMS utilized a Thermo LTQ Orbitrap XL mass spectrometer equipped with an electrospray ionization source (Thermo Fisher Scientific). A Waters Acquity UPLC system (Waters Corp.) utilizing a Phenomenex Kinetex C18 column (1.3 μm; 50 × 2.1 mm) was used to evaluate the purity of the isolated compounds with data collected and analyzed using Empower 3 software. Phenomenex Gemini-NX C18 analytical (5 μm; 250 × 4.6 mm), preparative (5 μm; 250 × 21.2 mm), and semipreparative (5 μm; 250 × 10.0 mm) columns (all from Phenomenex) were used on a Varian Prostar HPLC system equipped with ProStar 210 pumps and a Prostar 335 photodiode array detector (PDA), with data collected and analyzed using Galaxie Chromatography Workstation software (version 1.9.3.2, Varian Inc.). Flash chromatography was performed on a Teledyne ISCO CombiFlash Rf 200 using Silica Gold columns (both from Teledyne Isco) and monitored by UV and evaporative light-scattering detectors. Single-crystal X-ray diffraction data were collected using a Bruker APEX CCD diffractometer (Mo Kα radiation, graphite monochromator). The crystallographic images were generated using CrystalMaker (CrystalMaker Software). All other reagents and solvents were obtained from Fisher Scientific and were used without further purification.
Fungal Strain Isolation and Identification
The fungal strain, G87, was isolated from a sample of submerged wood collected in July of 2011 from a freshwater stream on the campus of the University of North Carolina at Greensboro (36° 4′ 16″, 79° 48′ 28″W). A culture of strain G87 is preserved in the Department of Chemistry and Biochemistry culture collection at the same university. Previously outlined collection methods and culturing conditions were followed.38,39 Molecular identification of strain G87 was carried out by sequencing the internal transcribed spacer regions 1 and 2 and 5.8S nrDNA (ITS)40 along with the D1 and D2 regions of the 28S nuclear ribosomal large subunit rRNA gene (LSU).41,42 DNA extraction, PCR amplification, and sequencing were performed according to previously published procedures.10,11,43−45 A BLAST search in GenBank using the complete ITS rDNA sequence showed 98% sequence similarity of the strain G87 with several members of the Leotiomycetes, Ascomycota. In particular, strain G87 displayed homology with a number of sequences belonging to Halenospora varia (Anastasiou) (E.B.G. Jones34) GenBank KF156329; identities = 474/483 (98%); gaps = 0/483 (0%), Halenospora varia; GenBank AJ608987; identities = 524/534 (98%); gaps = 1/534 (0%), Zalerion varium (Halenospora varia). Zalerion varium was placed originally in the genus Zalerion (Lulworthiales, Ascomycota). However, based on ITS data, Bills and colleagues35 found that Z. varium had phylogenetic affinities to the Leotiaceae, while the type species Z. maritima belonged to the Lulworthiales. Jones and colleagues34 subsequently transferred Z. varium to a new genus, Halenospora varia, Leotiomycetes, Ascomycota. Fungal strain G87 also displayed 97–98% sequence similarity in ITS data to several other genera of aquatic fungi, such as Spirosphaera, Tricladium, Lambertella, and Mycofalcella. To better understand the phylogenetic affinities of G87 to these taxa in the Leotiomycetes, a phylogenetic analysis using maximum likelihood was employed using combined ITS–LSU data for strain G87, along with sequences downloaded from the top hits in the BLAST search using RAxML v. 7.0.446 run on the CIPRES Portal v. 2.047 with the default rapid hill-climbing algorithm and GTR model employing 1000 fast bootstrap searches. Clades with bootstrap values ≥70% were considered to be significant and strongly supported.48 Results of both BLAST and phylogenetic analysis suggested strain G87 to show affinities with H. varia (Figure S20), which resides in Clade 7 sensu Baschien and co-workers.49 On the basis of these data, G87 was identified putatively as Halenospora aff. varia, Helotiales, Leotiomycetes, Ascomycota. The combined ITS and LSU sequence was deposited in GenBank (accession no. KJ803850).
Fermentation, Extraction, and Isolation
A fresh culture of strain G87 was prepared in a slant and was inoculated into a 50 mL culture tube containing a seed liquid culture consisting of 2% MEA, potato dextrose agar (PDA, Difco), and YESD media. The culture tube was shaken at room temperature using a rotary shaker at 125 rpm until the strain showed good growth, typically within 14–21 days. Afterward, the seed culture was used to inoculate a 250 mL Erlenmeyer flask containing 30 mL of autoclaved rice medium, prepared using 10 g of rice and twice the volume of rice with H2O. Fermentation was carried out by incubating the flask at rt until showing good growth (approximately 14 days). Large-scale cultures were prepared by the parallel processing of four such cultures.
To each of the four solid fermentation cultures that constitute a large-scale culture of G87, 60 mL of 1:1 MeOH–CHCl3 were added. The cultures were broken down into small pieces with a spatula and shaken using a rotary shaker overnight (∼16 h) at ∼125 rpm at rt. The samples were filtered with vacuum, and the remaining residues were washed with small volumes of 1:1 MeOH–CHCl3. To the combined filtrates, 360 mL of CHCl3 and 360 mL of H2O were added; the mixture was stirred for 30 min and then transferred into a separatory funnel. The organic layer was evaporated to dryness under reduced pressure. This organic extract was partitioned between 100 mL of 1:1 MeOH–CH3CN and 100 mL of hexanes. The MeOH–CH3CN layer was evaporated to dryness in vacuum. The defatted extract (∼439 mg) was dissolved in a mixture of CHCl3–MeOH, adsorbed onto Celite 545 and fractionated via flash chromatography using a gradient solvent system of hexane–CHCl3–MeOH at a 30 mL/min flow rate and 61 column volumes over 34.1 min to afford five fractions. Fraction 3 (153 mg) was subjected to preparative HPLC using a gradient system of 40:60 to 60:40 of CH3CN–H2O (acidified with 0.1% formic acid) over 30 min at a flow rate of 21.24 mL/min to yield 15 subfractions. Subfractions 4, 5, 7, 10, 12, and 13 yielded compounds 1 (19.7 mg), 2 (2.0 mg), 3 (1.3 mg), 5 (27.7 mg), 6 (3.0 mg), and 7 (30.2 mg), which eluted at ∼9.8, 11.5, 13.5, 20.5, 22.7, and 23.5 min, respectively. Fraction 4 (99.7 mg) was subjected to preparative HPLC using a gradient system of 20:80 to 40:60 of CH3CN–H2O (acidified with 0.1% formic acid) over 40 min then to 60:40 over 10 min at the same flow rate to yield 19 subfractions. Subfractions 3, 9, 11, 12, and 16 yielded compounds 10 (18.7 mg), 8 (5.2 mg), 11 (6.4 mg), 13 (2.6 mg) and 14 (1.8 mg), which eluted at 19.0, 25.0, 57.5, 28.5, and 37.3 min, respectively. Subfraction 10 (6.7 mg), which eluted at 26.0 min, was subjected to further preparative HPLC purification using a gradient system of 50:50 to 60:40 of MeOH–H2O (0.1% formic acid) over 15 min at the same flow rate to yield compounds 4 (2.1 mg) and 9 (1.2 mg), which eluted at ∼8.3 and 9.7 min, respectively.
The isolated compounds, except for compounds 4 and 9, which were already of appropriate purity, were further purified by preparative and semipreparative HPLC to generate material of high purity for biological evaluation using a mobile phase of MeOH–H2O (0.1% formic acid) at different gradient systems to yield compounds 1 (13.9 mg), 2 (1.0 mg), 3 (1.2 mg), 5 (22.7 mg), 6 (1.8 mg), 7 (21.8 mg), 8 (2.4 mg), 10 (13.2 mg), 11 (4.0 mg), 13 (1.4 mg), and 14 (2.8 mg).
Greensporone A (1):
colorless solid; [α]D20 = +5 (c 0.20, MeOH); −24 (c 0.26, CHCl3); UV (MeOH) λmax (log ε) 293 (3.32), 233 (3.70) nm; IR (diamond) vmax 2971, 1725, 1682, 1628, 1583, 1439, 1329, 1246, 1213 cm–1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 1 and 2; HRESIMS m/z 381.1090 [M + H]+ (calcd for C19H2235ClO6, 381.1099) and 383.1061 [M + H]+ (calcd for C19H2237ClO6, 383.1070).
Greensporone B (2):
colorless solid; [α]D20 = −37 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 292 (3.12), 225 (3.64) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 1 and 2; HRESIMS m/z 381.1110 [M + H]+ (calcd for C19H2235ClO6, 381.1099) and 383.1059 [M + H]+ (calcd for C19H2237ClO6, 383.1070).
8,9-Dihydrogreensporone A (3):
colorless solid; [α]D20 = +30 (c 0.12, MeOH); UV (MeOH) λmax (log ε) 292 (3.25), 221 (3.62) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 1 and 2; HRESIMS m/z 383.1267 [M + H]+ (calcd for C19H2435ClO6, 383.1256) and 385.12170 [M + H]+ (calcd for C19H2437ClO6, 385.1226).
Dechlorogreensporone A (4):
colorless solid; [α]D20 = +56 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 287 (3.28), 234 (3.64) nm; IR (diamond) vmax 3354, 2930, 2855, 1678, 1589, 1432, 1258, 1161, 1077, 985, 832, 725 cm–1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 1 and 2; HRESIMS m/z 347.1477 [M + H]+ (calcd for C19H23O6, 347.1489).
Greensporone C (5):
colorless solid; [α]D20 = +112 (c 0.33, MeOH); UV (MeOH) λmax (log ε) 288 (3.32), 238 (3.65) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 2 and 3; HRESIMS m/z 333.1685 [M + H]+ (calcd for C19H25O5, 333.1697).
O-Desmethylgreensporone C (6):
colorless solid; [α]D20 = +30 (c 0.09, MeOH); UV (MeOH) λmax (log ε) 302 (3.52), 263 (3.63), 237 (3.63) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 2 and 3; HRESIMS m/z 319.1534 [M + H]+ (calcd for C18H23O5, 319.1540).
8,9-Dihydrogreensporone C (7):
colorless solid; [α]D20 = +79 (c 0.29, MeOH); UV (MeOH) λmax (log ε) 286 (3.34), 252 (3.48), 224 (3.60) nm; IR (diamond) vmax 3413, 2929, 2857, 1682, 1606, 1461, 1432, 1281, 1258, 1159, 1079, 948, 843 cm–1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 2 and 3; HRESIMS m/z 335.1844 [M + H]+ (calcd for C19H27O5, 335.1853).
Greensporone D (8):
colorless solid; [α]D20 = −18 (c 0.12, MeOH); UV (MeOH) λmax (log ε) 292 (3.34), 227 (3.61) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 2 and 3; HRESIMS m/z 383.1246 [M + H]+ (calcd for C19H2435ClO6 383.1256) and 385.1220 [M + H]+ (calcd for C19H2437ClO6, 385.1226).
Greensporone E (9):
colorless solid; [α]D20 = −32 (c 0.12, MeOH); UV (MeOH) λmax (log ε) 292 (3.24), 230 (3.67) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 2 and 4; HRESIMS m/z 383.1245 [M + H]+ (calcd for C19H2435ClO6 383.1256) and 385.1220 [M + H]+ (calcd for C19H2437ClO6, 385.1226).
Dechlorogreensporone D (10):
colorless solid; [α]D20 = +116 (c 0.27, MeOH); UV (MeOH) λmax (log ε) 288 (3.33), 236 (3.64) nm; 1H NMR (DMSO, 500 MHz) and 13C NMR (DMSO, 125 MHz), see Tables 2 and 4; HRESIMS m/z 349.1639 [M + H]+ (calcd for C19H25O6, 349.1646).
8,9-Dihydrogreensporone D (11):
colorless solid; [α]D20 = +26 (c 0.20, MeOH); UV (MeOH) λmax (log ε) 292 (3.43), 226 (3.67) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 2 and 4; HRESIMS m/z 385.1402 [M + H]+ (calcd for C19H2635ClO6, 385.1412) and 387.1373 [M + H]+ (calcd for C19H2637ClO6, 387.1383).
Greensporone F (12):
colorless solid; [α]D20 = −38 (c 0.01, MeOH); UV (MeOH) λmax (log ε) 292 (3.01), 247 (3.11), 227 (3.27) nm; 1H NMR (CDCl3, 700 MHz) and 13C NMR (CDCl3, 175 MHz), see Tables 2 and 5; HRESIMS m/z 383.1242 [M + H]+ (calcd for C19H2435ClO6, 383.1256) and 385.1212 [M + H]+ (calcd for C19H2437ClO6, 385.1226).
Dechlorogreensporone F (13):
colorless solid; [α]D20 = −31 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 286 (3.20), 248 (3.35), 224 (3.54) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 2 and 5; HRESIMS m/z 349.1637 [M + H]+ (calcd for C19H25O6, 349.1646).
Greensporone G (14):
colorless solid; [α]D20 = +150 (c 0.07, MeOH); UV (MeOH) λmax (log ε) 285 (3.17), 223 (3.55) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Tables 2 and 5; HRESIMS m/z 349.1639 [M + H]+ (calcd for C19H25O6, 349.1646).
14-(4-Bromobenzoyl)-8,9-dihydrogreensporone C (15)
To a sample of compound 7 (7.7 mg, 1 equiv) dissolved in 1 mL of anhydrous THF, 4-bromobenzoyl chloride (6.1 mg, 1.2 equiv) and 4-dimethylaminopyridine (2.8 mg, 1 equiv) were added, and the mixture was stirred at rt under nitrogen. To enhance the solubility of the reagents, 0.3 mL of anhydrous CH2Cl2 was added. The mixture was left stirring overnight and monitored for completion using TLC. The reaction mixture (16.8 mg) was purified via a reversed-phase preparative HPLC using a Phenomenex Gemini-NX C18 (5 μm; 250 × 21.2 mm) column and a gradient system of 70:30 to 90:10 of CH3CN–H2O (acidified with 0.1% formic acid) over 15 min at a flow rate of 21.24 mL/min to yield compound 15 (8.8 mg), which eluted at 17.3 min. 1H NMR (CDCl3, 500 MHz) δ 8.02 (2H, d, J = 8.6), 7.65 (2H, d, J = 8.6), 6.74 (1H, d, J = 2.3), 6.67 (1H, d, J = 2.3), 5.27 (1H, m), 4.18 (1H, d, J = 17.8), 3.81 (3H, s), 3.61 (1H, d, J = 17.8), 2.55 (1H, ddd, J = 13.2, 8.6, 4.6), 2.33 (1H, ddd, J = 13.2, 8.6, 4.6), 1.71–1.51 (5H, m), 1.43–1.19 (7H, m), 1.34 (3H, d, J = 6.3) (Figure S18). HRESIMS m/z 517.1205 [M + H]+ (calcd for C26H3079BrO6, 517.1220) and 519.1185 [M + H]+ (calcd for C26H3081BrO6, 519.1200).
Preparation of the (R)- and (S)-MTPA Ester Derivatives of Greensporone D (8), Greensporone E (9), Dechlorogreensporone D (10), and 8,9-Dihydrogreensporone D (11)
To 0.31, 0.20, 0.31, and 0.45 mg of compounds 8–11, respectively, was added 400 μL of pyridine-d5, and the contents were transferred into NMR tubes. To initiate the reactions, 20 μL of S-(+)-α-methoxy-α-(trifluoromethyl)phenylacetyl (MTPA) chloride were added into each NMR tube with careful shaking and then monitored immediately by 1H NMR at 5, 10, and 15 min. The reactions were complete within 5 min, yielding the mono (R)-MTPA ester derivatives (8b) of 8, (9b) of 9, (10b) of 10, and (11b) of 11. 1H NMR data (500 MHz, pyridine-d5) of 8b: δH 6.13 (1H, m, H-2), 6.39 (1H, d, J = 17.8, H-9); of 9b: 1.24 (3H, d, J = 6.3, H3–1), 5.13 (1H, m, H-2), 5.27 (1H, m, H-5), 6.13 (1H, td, J = 12.0, 5.7, H-8), and 6.42 (1H, d, J = 12.0, H-9); of 10b: 1.30 (3H, d, J = 6.3, H3-1), 5.22 (1H, m, H-2), 5.30 (1H, m, H-5), 6.97 (1H, dd, J = 15.5, 7.5, H-8), and 6.33 (1H, d, J = 15.5, H-9); of 11b: 1.28 (3H, d, J = 6.3, H3-1), 5.36 (1H, m, H-2), 5.41 (1H, m, H-5), 2.37 (1H, m, H-9a), 2.79 (1H, m, H-9b), 4.19 (1H, d, J = 18.9, H-11a), and 4.39 (1H, J = 18.9, H-11b). In an analogous manner, 0.31, 0.20, 0.31, and 0.45 mg of compounds 8–11, respectively, dissolved in 400 μL of pyridine-d5 was reacted in NMR tubes with 20 μL of (R)-(−)-α-MTPA chloride for 15 min to afford the mono (S)-MTPA esters (8a, 9a, 10a, and 11a). 1H NMR data (500 MHz, pyridine-d5) of 8a: δH 6.17 (1H, m, H-2), 6.38 (1H, d, J = 17.8, H-9); of 9a: 1.32 (3H, d, J = 6.3, H3-1), 5.15 (1H, m, H-2), 5.41 (1H, m, H-5), 6.12 (1H, dd, J = 12.0, 4.0, H-8), and 6.41 (1H, d, J = 12.0, H-9); of 10a: 1.36 (3H, d, J = 6.3, H3-1), 5.25 (1H, m, H-2), 5.44 (1H, m, H-5), 6.96 (1H, dd, J = 15.5, 7.5, H-8), and 6.29 (1H, d, J = 15.5, H-9); of 11a: 1.33 (3H, d, J = 6.3, H3-1), 5.37 (1H, m, H-2), 5.52 (1H, m, H-5), 2.39 (1H, m, H-9a), 2.76 (1H, m, H-9b), 4.20 (1H, d, J = 18.9, H-11a), and 4.35 (1H, J = 18.9, H-11b).
X-ray Crystallography
Crystallographic data for compound 15 has been deposited with the Cambridge Crystallographic Data Centre, deposition number 1005143. Crystals of 15 were grown in a mixture of EtOAc and hexane at rt. A clear, colorless, rectangular-parallelepiped-like specimen of C26H29BrO6, approximate dimensions 0.12 mm × 0.19 mm × 0.52 mm, was used for the X-ray crystallographic analysis. The X-ray intensity data were measured on a Bruker APEX CCD system equipped with a graphite monochromator and a Mo Kα sealed X-ray tube (λ = 0.71073 Å). The total exposure time was 18.57 h. The frames were integrated with the Bruker SAINT software package using a narrow-frame algorithm. The integration of the data using a monoclinic unit cell yielded a total of 23 715 reflections to a maximum θ angle of 30.20 (0.71 Å resolution), of which 7231 were independent (average redundancy 3.270, completeness = 99.4%, Rint = 3.10%) and 6524 (90.22%) were greater than 2σ(F2). The final cell constants of a = 22.9481(11) Å, b = 5.2631(3) Å, c = 21.5301(11) Å, β = 108.666(1), volume = 2463.6(2) Å3 were based on the refinement of the XYZ centroids of 9887 reflections above 20σ(I) with 7.114 < 2θ < 61.72. Data were corrected for absorption effects using the multiscan method (SADABS). The ratio of minimum to maximum apparent transmission was 0.774. The structure was solved and refined using the Bruker SHELXTL Software Package, using the space group C2, with Z = 4 for the formula unit, C26H29BrO6. The final anisotropic full-matrix least-squares refinement on F2 with 300 variables converged at R1 = 3.69% for the observed data and wR2 = 9.56% for all data. The goodness-of-fit was 1.033. The largest peak in the final difference electron density synthesis was 1.225 e–/Å3, and the largest hole was −0.811 e–/Å3 with an RMS deviation of 0.057 e–/Å3 (the top three peaks were within 0.90 Å of a bromine atom). On the basis of the final model, the calculated density was 1.395 g/cm3 and F(000), 1072 e–. For absolute structure determination, Flack x, determined using 2675 quotients [(I+) – (I–)]/[(I+) + (I–)],50 refined to a final value of −0.011(3). Crystal data, data collection, structure solution, and refinement details are summarized in Table S1.
Cytotoxicity Assay
Human melanoma cancer cell line designated MDA-MB-435 and human colon cancer cell line designated HT-29 were purchased from the American Type Culture Collection. The cell lines were propagated at 37 °C in 5% CO2 in RPMI 1640 medium supplemented with fetal bovine serum (10%), penicillin (100 units/mL), and streptomycin (100 μg/mL). Cells in log-phase growth were harvested by trypsinization followed by washing (2×) to remove all traces of enzyme. A total of 5000 cells were seeded per well of a 96-well clear, flat-bottomed plate (Microtest 96, Falcon) and incubated overnight (37 °C in 5% CO2). Compounds dissolved in DMSO were then diluted and added to the appropriate wells (concentrations: 20, 4, 0.8, 0.16, and 0.032 μM; total volume: 100 μL; DMSO: 0.5%). The cells were incubated in the presence of test substance for 72 h at 37 °C and evaluated for viability with a commercial absorbance assay (CellTiter 96 AQueous One Solution Cell Proliferation Assay, Promega Corp) that measured viable cells. IC50 values are expressed in micromolar relative to the solvent (DMSO) control. Vinblastine was used as a positive control in both cell lines.
Tak1–TAB1 (Transforming Growth Factor-β Activated Kinase-1/TAK-1 Binding Protein 1) Inhibitor Assays
The assay was performed at BPS Bioscience Inc. (5Z)-7-Oxozeaenol32 was used as a positive control. Detailed experimental procedures are provided in the Supporting Information.
Acknowledgments
This research was supported in part via program project grant P01 CA125066 from the National Cancer Institute/National Institutes of Health, Bethesda, MD, USA. The high-resolution mass spectrometry data were acquired in the Triad Mass Spectrometry Laboratory at the University of North Carolina at Greensboro. Sequence data were generated at the Mycology laboratory of Dr. Andrew N. Miller, Illinois Natural History Survey, University of Illinois at Urbana–Champaign. The Wake Forest University X-ray Facility thanks the National Science Foundation (grant CHE-0234489) for funds to purchase the X-ray instrument and computers. We thank Dr. T. Prisinzano of the University of Kansas, Dr. A. Horswill of the University of Iowa, and Dr. C. Pearce of Mycosynthetix, Inc. for helpful discussions. We also thank L. Al-Fakhouri of the University of North Carolina at Greensboro for technical assistance with the bromobenzoylation reaction.
Supporting Information Available
UV–PDA spectra and UPLC chromatograms of compounds 1–14, 1H and 13C NMR spectra for compound 1–15, experimental procedures of the Tak1–TAB1 inhibitor assays, phylogram of the most likely tree, crystal data, data collection, and structure refinement details. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Primack R. B.Essentials of Conservation Biology, 5th ed.; Sinauer Associates, Inc.: Sunderland, MA, 2010; p 601. [Google Scholar]
- Hawksworth D. L. Mycol. Res. 1991, 95, 641–655. [Google Scholar]
- Hawksworth D. L.; Rossman A. Y. Phytopathology 1997, 87, 888–91. [DOI] [PubMed] [Google Scholar]
- Blackwell M. Am. J. Bot. 2011, 98, 426–438. [DOI] [PubMed] [Google Scholar]
- Kirk P. M.; Cannon P. F.; Minter D. W.; Staplers J. A.. Ainsworth & Bisby’s Dictionary of the Fungi, 10th ed.; CABI: Oxon, UK, 2008; p 784. [Google Scholar]
- Scheffers B. R.; Joppa L. N.; Pimm S. L.; Laurance W. F. Trends in Ecology & Evolution 2012, 27, 501–510. [DOI] [PubMed] [Google Scholar]
- Shearer C. A.; Descals E.; Kohlmeyer B.; Kohlmeyer J.; Marvanova L.; Padgett D.; Porter D.; Raja H. A.; Schmit J. P.; Thorton H. A.; Voglymayr H. Biodiversity Conserv. 2007, 16, 49–67. [Google Scholar]
- Hernández-Carlos B.; Gamboa-Angulo M. Phytochem. Rev. 2011, 10, 261–286. [Google Scholar]
- Dong J. Y.; Shen K. Z.; Sun R. Mycosystema 2011, 30, 206–217. [Google Scholar]
- Raja H. A.; Oberlies N. H.; El-Elimat T.; Miller A. N.; Zelski S. E.; Shearer C. A. Mycoscience 2013, 54, 353–361. [Google Scholar]
- Raja H. A.; Oberlies N. H.; Figueroa M.; Tanaka K.; Hirayama K.; Hashimoto A.; Miller A. N.; Zelski S. E.; Shearer C. A. Mycologia 2013, 105, 959–976. [DOI] [PubMed] [Google Scholar]
- El-Elimat T.; Raja H. A.; Figueroa M.; Falkinham J. O. III; Oberlies N. H. Phytochemistry 2014, 104, 114–120. [DOI] [PubMed] [Google Scholar]
- El-Elimat T.; Zhang X. L.; Jarjoura D.; Moy F. J.; Orjala J.; Kinghorn A. D.; Pearce C. J.; Oberlies N. H. ACS Med. Chem. Lett. 2012, 3, 645–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordanetto F.; Kihlberg J. J. Med. Chem. 2013, 57, 278–295. [DOI] [PubMed] [Google Scholar]
- Serba C.; Winssinger N. Eur. J. Org. Chem. 2013, 2013, 4195–4214. [Google Scholar]
- Wu J. Q.; Powell F.; Larsen N. A.; Lai Z. W.; Byth K. F.; Read J.; Gu R. F.; Roth M.; Toader D.; Saeh J. C.; Chen H. W. ACS Chem. Biol. 2013, 8, 643–650. [DOI] [PubMed] [Google Scholar]
- Alali F. Q.; El-Elimat T.; Li C.; Qandil A.; Alkofahi A.; Tawaha K.; Burgess J. P.; Nakanishi Y.; Kroll D. J.; Navarro H. A.; Falkinham J. O.; Wani M. C.; Oberlies N. H. J. Nat. Prod. 2005, 68, 173–178. [DOI] [PubMed] [Google Scholar]
- McLaughlin J. L.; Rogers L. L. Drug Inf. J. 1998, 32, 513–524. [Google Scholar]
- Meyer B. N.; Ferrigni N. R.; Putnam J. E.; Jacobsen L. B.; Nichols D. E.; McLaughlin J. L. Planta Med. 1982, 45, 31–34. [PubMed] [Google Scholar]
- Talontsi F. M.; Facey P.; Tatong M. D. K.; Islam M. T.; Frauendorf H.; Draeger S.; von Tiedemann A.; Laatsch H. Phytochemistry 2012, 83, 87–94. [DOI] [PubMed] [Google Scholar]
- Hellwig V.; Mayer-Bartschmid A.; Muller H.; Greif G.; Kleymann G.; Zitzmann W.; Tichy H. V.; Stadler M. J. Nat. Prod. 2003, 66, 829–837. [DOI] [PubMed] [Google Scholar]
- Strunz G. M.; Court A. S.; Komlossy J.; Stillwell M. A. Can. J. Chem. 1969, 47, 2087–2094. [Google Scholar]
- Strunz G. M.; Court A. S.; Komlossy J.; Stillwell M. A. Can. J. Chem. 1969, 47, 3700–3701. [Google Scholar]
- Mirrington R. N.; Ritchie E.; Shoppee C. W.; Taylor W. C.; Sternhell S. Tetrahedron Lett. 1964, 5, 365–370. [Google Scholar]
- Cutler H. G.; Arrendale R. F.; Springer J. P.; Cole P. D.; Roberts R. G.; Hanlin R. T. Agric. Biol. Chem. 1987, 51, 3331–3338. [Google Scholar]
- Shinonaga H.; Kawamura Y.; Ikeda A.; Aoki M.; Sakai N.; Fujimoto N.; Kawashima A. Tetrahedron 2009, 65, 3446–3453. [Google Scholar]
- Polavarapu P. L. Chirality 2002, 14, 768–781. [DOI] [PubMed] [Google Scholar]
- Hoye T. R.; Jeffrey C. S.; Shao F. Nat. Protoc. 2007, 2, 2451–2458. [DOI] [PubMed] [Google Scholar]
- Takaesu G.; Surabhi R. M.; Park K.-J.; Ninomiya-Tsuji J.; Matsumoto K.; Gaynor R. B. J. Mol. Biol. 2003, 326, 105–115. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K.; Shirakabe K.; Shibuya H.; Irie K.; Oishi I.; Ueno N.; Taniguchi T.; Nishida E.; Matsumoto K. Science 1995, 270, 2008–11. [DOI] [PubMed] [Google Scholar]
- Ninomiya-Tsuji J.; Kajino T.; Ono K.; Ohtomo T.; Matsumoto M.; Shiina M.; Mihara M.; Tsuchiya M.; Matsumoto K. J. Biol. Chem. 2003, 278, 18485–18490. [DOI] [PubMed] [Google Scholar]
- Ayers S.; Graf T. N.; Adcock A. F.; Kroll D. J.; Matthew S.; Carcache de Blanco E. J.; Shen Q.; Swanson S. M.; Wani M. C.; Pearce C. J.; Oberlies N. H. J. Nat. Prod. 2011, 74, 1126–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasiou C. J. Can. J. Bot. 1963, 41, 1135–1139. [Google Scholar]
- Jones E. B. G.; Sakayaroj J.; Suetrong S.; Somrithipol S.; Pang K. L. Fungal Diversity 2009, 35, 1–187. [Google Scholar]
- Bills G. F.; Platas G.; Pelaez F.; Masurekar P. Mycol. Res. 1999, 103, 179–192. [Google Scholar]
- Campbell J.; Volkmann-Kohlmeyer B.; Grafenhan T.; Spatafora J. W.; Kohlmeyer J. Mycol. Res. 2005, 109, 556–568. [DOI] [PubMed] [Google Scholar]
- Kohlmeyer J.; Kohlmeyer E.. Marine Mycology: The Higher Fungi; Academic Press: New York, 1979; p 690. [Google Scholar]
- Shearer C. A.; Langsam D. M.; Longcore J. E. In Measuring and Monitoring Biological Diversity: Standard Methods for Fungi; Mueller G. M., Bills G. F., Foster M. S., Eds.; Smithsonian Institution Press: Washington, DC, 2004; pp 513–531. [Google Scholar]
- Raja H.; Schmit J.; Shearer C. Biodiversity Conserv. 2009, 18, 419–455. [Google Scholar]
- Schoch C. L.; Seifert K. A.; Huhndorf S.; Robert V.; Spouge J. L.; Levesque C. A.; Chen W.; Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 6241–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porras-Alfaro A.; Liu K. L.; Kuske C. R.; Xie G. Appl. Environ. Microbiol. 2014, 80, 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K. L.; Porras-Alfaro A.; Kuske C. R.; Eichorst S. A.; Xie G. Appl. Environ. Microbiol. 2012, 78, 1523–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Elimat T.; Figueroa M.; Raja H. A.; Adcock A. F.; Kroll D. J.; Swanson S. M.; Wani M. C.; Pearce C. J.; Oberlies N. H. Tetrahedron Lett. 2013, 54, 4300–4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Elimat T.; Figueroa M.; Raja H. A.; Graf T. N.; Adcock A. F.; Kroll D. J.; Day C. S.; Wani M. C.; Pearce C. J.; Oberlies N. H. J. Nat. Prod. 2013, 76, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa M.; Raja H.; Falkinham J. O.; Adcock A. F.; Kroll D. J.; Wani M. C.; Pearce C. J.; Oberlies N. H. J. Nat. Prod. 2013, 76, 1007–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A.; Hoover P.; Rougemont J. Syst. Biol. 2008, 57, 758–771. [DOI] [PubMed] [Google Scholar]
- Miller M. A.; Pfeiffer W.; Schwartz T.. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees, Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, November 14, 2010; pp 1–8.
- Hillis D. M.; Bull J. J. Syst. Biol. 1993, 42, 182–192. [Google Scholar]
- Baschien C.; Tsui C. K. M.; Gulis V.; Szewzyk U.; Marvanova L. Fungal Biol. 2013, 117, 660–672. [DOI] [PubMed] [Google Scholar]
- Parsons S.; Flack H. Acta Crystallogr., Sect. A: Found. Crystallogr. 2004, 60, s61. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.