

Abstract
Hepatocellular carcinoma (HCC) has a poor prognosis due to widespread intrahepatic and extrahepatic metastases. There is an urgent need to understand signaling cascades that promote disease progression. Aspartyl-(Asparaginyl)-β-hydroxylase (ASPH) is a cell surface enzyme that generates enhanced cell motility, migration, invasion and metastatic spread in HCC. We hypothesize that inhibition of its enzymatic activity could have antitumor effects. Small molecule inhibitors (SMIs) were developed based on the crystal structure of the ASPH catalytic site followed by computer assisted drug design. Candidate compounds were tested for inhibition of β-hydroxylase activity and selected for their capability to modulate cell proliferation, migration, invasion and colony formation in vitro and to inhibit HCC tumor growth in vivo using orthotopic and subcutaneous murine models. The biologic effects of SMIs on the Notch signaling cascade were evaluated. The SMI inhibitor MO-I-1100 was selected since it reduced ASPH enzymatic activity by 80% and suppressed HCC cell migration, invasion and anchorage independent growth. Furthermore, substantial inhibition of HCC tumor growth and progression was observed in both animal models. The mechanism(s) for this antitumor effect was associated with reduced activation of Notch signaling both in vitro and in vivo.
Conclusions
These studies suggest that the enzymatic activity of ASPH was important for hepatic oncogenesis. Reduced β-hydroxylase activity generated by the SMI MO-I-1100 led to antitumor effects through inhibiting Notch signaling cascade in HCC. ASPH promotes the generation of an HCC malignant phenotype and represents an attractive molecular target for therapy of this fatal disease.
Keywords: ASPH, HCC, small molecule inhibitor, Notch, signaling pathway
INTRODUCTION
Hepatocellular carcinoma (HCC) is the fifth most prevalent and third most fatal type of cancer worldwide. HCC is currently being diagnosed in over 500,000 people each year globally and the incidence is increasing (1). Despite improvements in detection and management, the 5-year survival rate of HCC is 15% (localized 28%, regional 7% and distant 2%), and 80–90% of patients present with advanced unresectable disease at diagnosis due to widespread intra- and extrahepatic metastases. It is urgent to explore the molecular mechanisms of HCC development, progression and metastasis in an attempt to identify new molecular targets for therapy. Indeed, there is an intense search for identification and development of small molecules with anti-tumor effects against HCC; nevertheless, there is only one drug, Sorafenib, which is an inhibitor of multiple kinases such as Raf, VEGFR, PDGFRβ, c-Kit and RET, that has been approved for clinical use in this disease (2, 3).
Aspartate β-hydroxylase (ASPH) is an ~86KD Type II transmembrane protein and a member of the α-ketoglutarate-dependent dioxygenase family (4). It catalyzes the hydroxylation of aspartyl and asparaginyl residues in epidermal growth factor (EGF)-like domains of various proteins such as Notch and Jagged (JAG) which play critical roles in cell growth, differentiation, adhesion and migration (5). The DNA and protein sequences are highly conserved between human and murine orthologs, so that many of the same monoclonal antibodies (mAbs) and RNA/DNA primers can be used to identify and characterize ASPH expression in both species (6). ASPH was originally described to be overexpressed in HCC and cholangiocarcinoma patients (7). However, since the liver is derived from an early progenitor cell type (8) and ASPH is expressed in the embryo, we hypothesize that ASPH may reemerge during tumorigenesis and play a role in the pathogenesis of HCC. ASPH is rarely expressed in normal adult tissue, except for proliferating trophoblastic cells of the placenta, an invasive tissue (7). Clinical investigation has revealed that ASPH overexpression portends a poor prognosis with early disease reoccurrence of colon cancer (9). In HCC, there has been a significant association of ASPH expression with worse clinical outcome of patients who received surgery and ASPH has been considered to be an independent risk factor for a reduced survival (10). ASPH functions to enhance the HCC malignant phenotype by increasing cell migration, invasion and metastasis and therefore, could be considered as a potent therapeutic target for treatment of this disease.
ASPH expression is “shut off” in the adult only to re-emerge during oncogenesis where its function may be required for a generation of malignant phenotype (7, 10, 11). This study attempts to address several questions critical to the function of ASPH in HCC. Is ASPH a major activator of Notch signaling (which is known to promote cell migration, invasion and metastasis)? Can ASPH be directly linked to upstream growth factor signaling pathways important in pathogenesis? Furthermore, is the β-hydroxylase enzymatic activity of ASPH essential for its biologic/pathologic effects? We hypothesized that discovery of small molecule inhibitors (SMIs) against β-hydroxylase activity of ASPH may produce antitumor effects and their mechanism(s) of action involves inhibition of Notch signaling in HCC.
METHODS
Synthesis and characterization of SMIs of β-hydroxylase activity
We developed, characterized and evaluated a novel SMI for ASPH β-hydroxylase activity. Formulation of these compounds was based on the crystal structure of the ASPH catalytic site, followed by computer generated drug design that led to the synthesis of a series of parent compounds and derivatives likely to fit into the pocket of the catalytic site and inhibit enzymatic activity. The first step was a three component reaction including an aromatic aldehyde, glyoxal bisulfate addition product, and potassium cyanide to yield an arylhydroxytetronimide. In the second step arylhydroxytetronimide was sulfonylated with phenylmethansulfonyl chloride in dry tetrahydrofuran to yield compounds presented in S Fig. 1. Compounds were characterized by 1H and 13C nuclear magnetic resonance, high resolution mass spectroscopy, high performance liquid chromatography, infra-red spectroscopy, melting point, elemental analysis and binding to ASPH by isothermal titration calorimetry.
Cell lines
The FOCUS HCC cell line was derived from an HBV related poorly differentiated human HCC (12) and the subclone BNLT3 was derived from a murine hepatoma parenteral cell line following serial passage through immunodeficient mice to generate a clone with a highly aggressive malignant phenotype (13). The other human HCC cell lines included Huh7, Hep3B, and HepG2; the murine embryo fibroblast cell line NIH3T3 was used as a negative control for ASPH expression. The above cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA). Cell lines were maintained in DMEM supplement with 10% fetal bovine serum and 2mM L-glutamine; and cultivated in a humidified incubator at 37°C with 5% CO2. Cells were harvested with 0.25% Trypsin-0.03% EDTA for further investigations.
14CO2-α-Ketoglutarate-dependent capture assay for measurement of β-hydroxylase activity
ASPH β-hydroxylase activity was measured in protein lysates derived from FOCUS HCC cells treated with 5 μM of SMIs using an EGF-like domain containing peptide (DGDQCETSPCQNQGKCKDGLGYETCTCLEGFEGKNC) as the substrate. In this assay 14C-labeled α-ketoglutarate releases 14CO2, as described previously (4, 14, 15). Incubations were carried out at 37°C for 1 hour in a final volume of 40 μl containing 48 μg of clarified cell extract derived from FOCUS cells providing the source of ASPH β-hydroxylase protein and the 60 μM EGF containing peptide substrate. Radioactivity captured was quantified by a phosphoimager. We modified the assay to make it specific for the β-hydroxylase activity of ASPH since the reaction was run in a 96 well plate previously coated with mAb FB-50 prepared against the N-terminus of ASPH to first allow direct immunologic capture of the enzyme followed by the subsequent enzymatic assay measurement.
MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide) assay
All cell lines were cultured until they reached the logarithmic phase of growth and then were placed into a 96-well microtiter-plate at a density of 1×104 cells/well. To evaluate the effects of SMIs on cell viability, cell lines overexpressing ASPH or vector control were treated with SMIs at concentrations ranging from 0.3 to 5 μM for 24 hours, after which 10 μl of 5 mg/ml MTT (Sigma-Aldrich, MO, USA) was added to each well. After incubation for 3 hours and removing the supernatant, 50 μl of acidic isopropanol was added and the color reaction was quantified with a microplate reader at a test wavelength of 570 nm and a reference wavelength of 630 nm. Subsequently, the solutions were discarded and 50 μl of 10 μg/ml Hoechst 33342 solution (Molecular Probe, OR USA) was added into each well. After 5 minutes incubation at room temperature, the color reaction was quantified with a microplate reader at a test wavelength of 360 nm and a reference wavelength of 460 nm. Each experiment was carried out in triplicate.
Colony formation assay (Supplemental Methods)
Motility and invasion assays (Supplemental Methods)
Lentiviral infected stable cell lines with overexpression or knockdown of ASPH (Supplemental Methods)
Western blot analysis (Supplemental Methods)
In vivo studies
A subcutaneous (s.c.) tumor model was used to analyze the in vivo effects of SMIs of β hydroxylase activity, as described previously. (16) Six weeks old female nu/nu nude mice (Charles River Laboratories) were kept under pathogen-free conditions, fed standard chow, and given free access to sterilized water. Mice were anesthetized by isoflurane, and then s.c. xenografts were established by inoculating 1×107 FOCUS cells into the right dorsal flank. Palpable tumors were confirmed on day 3 following inoculation, and mice were randomized into treatment or control groups to receive a SMI (MO-I-1100) or saline, respectively. MO-I-1100 (50 mg/kg) was prepared in saline and administered by intraperitoneal (i.p.) injection. Tumor size was measured using calipers every other day and tumor volumes were calculated as A×B2×0.5 (A, length; B, width).
An orthotopic xenograft model was also created by direct intrahepatic inoculation of FOCUS cells, as described previously (16). On day 3 after inoculation, MO-I-1100 (50 mg/kg) or saline was administered to mice by i.p. injection on 5 consecutive days per week for 2 weeks. At 4 weeks after initiation of treatment, mice were sacrificed to assess the antitumor effects of MO-I-1100. All in vivo procedures were approved by the Institutional Animal Care and Use Committee of Rhode Island Hospital.
Immunohistochemistry (Supplemental Methods)
RESULTS
ASPH expression in human HCC
Levels of ASPH expression were evaluated by IHS using the FB-50 mAb that reacts to the N-terminus of ASPH (13), in human HCC tumors (n=27) of diverse etiologies [HBV (n=8), HCV (n=12), and alcohol cirrhosis (n=7) related], dysplastic nodules (n=9), and adjacent non-neoplastic normal liver (n=36) as shown in Fig. 1A and B. ASPH was expressed in 22 of 27 (81.48%) human HCC tumors derived from 86 TMA cores. No immunoreactivity was observed in normal liver parenchyma. Dysplastic regenerating nodules in cirrhotic liver were also negative for ASPH expression indicating that ASPH upregulation may be closely associated with malignant transformation. Most HCCs had a staining intensity of ASPH expression (bottom panels of Fig. 1A) with a mean semi-quantitative value of 1.37 (Fig. 1C). The high frequency of ASPH overexpression in human HCC suggests that it may be an important factor in HCC tumor development and progression.
Fig. 1. Expression of ASPH in HCC.

ASPH expression in human HCC tumors, dysplastic nodules and adjacent uninvolved normal liver. (A) represents IHS of normal liver (upper left), dysplastic nodules (upper right) and human HCCs (bottom two). (B) percent of ASPH positive expression in human HCC tumors (86 TMA cores) at 400×. ASPH was highly expressed in tumor tissues compared with normal liver. Negative staining was observed for all components of adjacent uninvolved liver and dysplastic (regenerating) nodules. (C) semiquantatative analysis of staining intensity distribution of ASPH levels in HCC (0, negative; 1+, moderate; 2+, strong; and 3+, very strong immunoreactivity).
Development and characterizations of β-hydroxylase SMIs
Fig. 2C depicted representatives of parent compounds synthesized and examined for inhibition of β-hydroxylase activity using a high throughput screening approach. In brief, Fig. 2A described the enzymatic reaction catalyzed by ASPH with the liberation of 14CO2 as the readout. Fig. 2B represented a “strong hit” with MO-I-1100 which inhibits the β-hydroxylase activity of ASPH by approximately 80%. Fig. 2C described the structure of representative parent compounds synthesized and examined for inhibition of β-hydroxylase activity. When inhibitors of ASPH enzymatic activity were identified, we performed a functional MTT assay to test their effects on cell viability over a range of drug concentration. Examples of positive and negative results among several candidate compounds are depicted in Fig. 2C (bottom). Fig. 2D suggested that the expression level of ASPH determined cellular response to the SMI since MO-I-1100 had no effect on viability of NIH3T3 cells which lacked ASPH on the cell surface (11), whereas FOCUS cells, which have high level ASPH expression (Fig. 3A), were inhibited by MO-I-1100 at 1 μM (Fig. 2D). MO-I-1100 had no effect on the proliferation rate of FOCUS or NIH3T3 cells over 24 hours (Fig. 2E) which was important to interpret its independent effects on cell migration and invasion presented in Fig. 4. In addition, MO-I-1100 was not cytotoxic to FOCUS cells. However, this SMI of ASPH β-hydroxylase activated pro-apoptosis mechanisms through Notch signaling (S Fig 2).
Fig. 2. Analysis of β-hydroxylase activity.
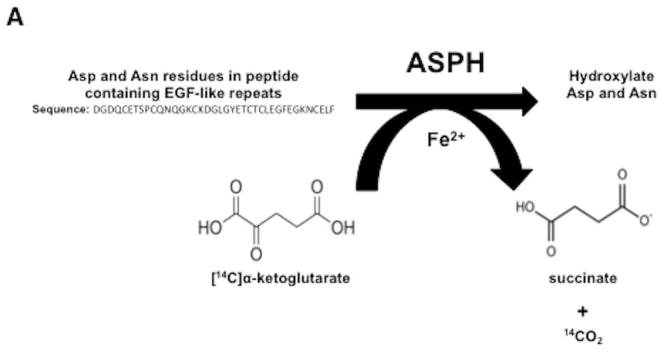

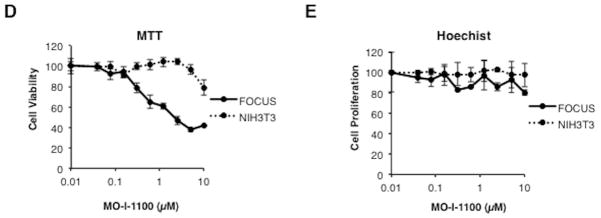
(A) The biochemical reaction catalyzed by ASPH. (B) Inhibition by 80% of β-hydroxylase activity by a SMI MO-I-1100. (C) Representative structures of compounds screening as potential inhibitors of β-hydroxylase activity (top). The MTT assay was used as a secondary screen for effects on FOCUS cell viability over a concentration range of 0.3–5 μM (bottom). Note that MO-I-1100 reduced both enzymatic activity and cell viability and was therefore, selected for further functional studies. (D) Dose-response curve of MO-I-1100 levels on FOCUS cells (high level ASPH expression) compared to NIH3T3 cells (no expression) with respect to cell viability using the MTT assay. (E) There was no effect on cell proliferation by MO-I-1100 over a 24 hour period at the concentrations indicated.
Fig. 3. Effects of MO-I-1100 on cell viability, growth and anchorage independent colony formation in HCC cell lines.
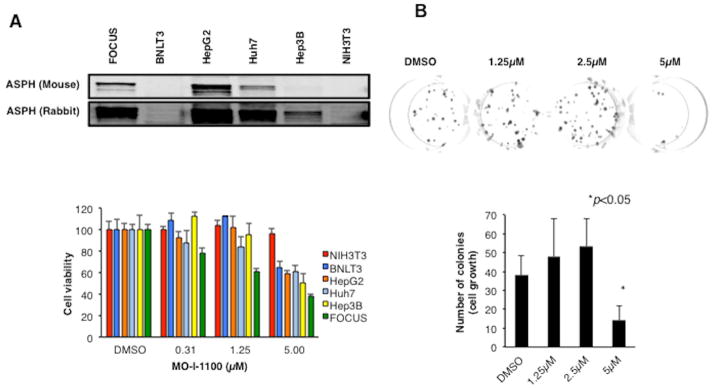

(A) (Top) HCC cell lines have variable levels of ASPH expression. Note that all HCC cell lines (including the murine BNLT3 hepatoma cells) studied expressed ASPH; NIH3T3 cells lack expression. (Bottom) Effects of different concentrations of MO-I-1100 on cell viability. There was a general correlation of ASPH expression with sensitivity to the β-hydroxylase inhibitor. (B) A three week exposure to MO-I-1100 (5 μM) reduced cell growth. (C) The β-hydroxylase inhibitor reduced colony formation in a dose dependent manner. (D) and (E) Graphic presentation of number and size of colony-forming units from three independent experiments. Vertical bar represents standard deviation (S.D.); *P < 0.05.
Fig. 4. β-hydroxylase inhibitor reduced cell motility and invasiveness in human and murine HCC cell lines.

Measurement of motility and invasiveness of HCC cell lines after treatment with MO-I-1100. (A) Representative examples of the microscopic image of the migrated FOCUS cells and graphic presentation of the results with MO-I-1100 treatment. (B) Representative examples of the microscopic image of migrated cells in BNLT3 murine HCC cells and their reduction following MO-I-1100 exposure. (C) Representative examples of FOCUS cell invasion; note reduction following MO-I-1100 treatment. (D) Effects of MO-I-1100 on invasive properties of murine BNLT3 cells. Magnification is 100×. All graphs depict the mean ± S.D. of results obtained from multiple independent culture experiments (*p < 0.05).
Additional human HCC cell lines including a murine BNLT3 hepatoma cell subclone were used to evaluate the inhibitory effects of SMIs on cell viability over 24 hours. ASPH expression levels were measured by Western blot analysis using both FB-50 mAb and polyclonal rabbit antibodies prepared against recombinant ASPH. As depicted in Fig. 3A (bottom), over a range of MO-I-1100 concentrations (0.3 to 5 μM), there was no effect on cell viability of NIH3T3 cells that lacked ASPH expression. In contrast, the murine BNLT3 hepatoma cells with very low levels of ASPH by Western blot but easily detectable levels using a more sensitive Fluorescence Activated Cell Sorting (FACS) analysis (13) or IHS with polyclonal rabbit anti-ASPH displayed in S Fig. 3 revealed reduced viability only at 5 μM concentration. Likewise Hep3B, with low ASPH levels only detectable with polyclonal anti-ASPH, exhibited reduced viability at 5 μM; whereas FOCUS, HepG2 and Huh7 with higher ASPH levels demonstrated reduced viability at a concentration as low as 1 or 1.25 μM of MO-I-1100. Thus, the level of ASPH expression correlated with response to MO-I-1100 with respect to cell viability which depended, in part, on the concentration of SMIs employed. However, when FOCUS cells were incubated with MO-I-1100 (5 μM) for 3 weeks, there was a substantial reduction of cell growth (Fig. 3B).
The ability to form colonies in soft agar is considered to be a reliable test for tumorigenic potential. In order to evaluate effects of MO-I-1100 on anchorage independent colony formation, cultivation of FOCUS cells in soft agar with the β-hydroxylase inhibitor was performed. After 3 weeks of incubation, MO-I-1100 reduced colony formation of FOCUS cells (Fig. 3C). As shown in Fig. 3D and E, treatment with MO-I-1100 revealed a dose-dependent reduction of colony number and size.
Overexpression of ASPH in Huh7 cells promotes a malignant phenotype
Huh7 cells have low level ASPH expression which was increased with lentiviral transfection of an ASPH expression plasmid and promoted cell migration and invasion as shown in S Fig. 4. Previous studies have demonstrated siRNA knockdown reduced this phenotype (17). More important, knockdown of ASPH expression in FOCUS cells by specific shRNAs reduced the malignant phenotype as characterized by decreased colony formation in soft agar as an index of reduced cellular transformation (S Fig. 5).
In vitro effects of β-hydroxylase on motility and invasiveness of HCC cell lines
We determined whether the β-hydroxylase activity was required for ASPH to mediate its effects on cell motility. Directional motility and invasiveness were measured using Boyden chamber-type culture inserts equipped with a porous membrane. FOCUS cells were pretreated with 5 μM of MO-I-1100 for 24 hours, and then placed into the upper chamber. Migration and invasion were evaluated after 24 hours. As shown in Fig. 4A and C, migration and invasion were reduced following MO-I-1100 treatment of FOCUS cells. SMIs exerted a direct inhibitory effect on this malignant phenotype, which was not secondary to cell proliferation as shown in Fig. 2E. Migration and invasion assays were also inhibited in BNLT3 hepatoma cells expressing a low level of ASPH (a highly conserved murine ortholog) (2) (Fig. 4B and D). The catalytic site is identical in protein sequence between human and murine orthologs of ASPH. These observations suggest that the β-hydroxylase of both species is susceptible to the inhibitory effect of MO-I-1100.
Effects of the β-hydroxylase inhibitor MO-I-1100 on Notch signaling
We determined the expression of Notch receptors and ligands as well as the activated Notch intracellular domains (NICDs) which, when cleaved from the full length receptors and transported to the nucleus, serve as transcription factors to upregulate Notch responsive genes in several HCC cell lines. Notch receptors 1, 2, 3 and 4 were variably expressed in these cell lines as shown in Fig. 5A. In contrast, JAG1 was expressed in all cell lines and was particularly prominent in HepG2, Huh7 and Hep3B whereas JAG 2, DLL 1 and 4 had little expression. There was generation of NICDs in many of the cell lines and activated Notch 2 intracellular domain was particularly prominent in human and murine HCC cell lines tested suggesting that Notch signaling were present in HCC cell lines and could contribute to generation of a highly migrated, invasive, proliferative and metastatic phenotype.
Fig. 5. Alterations in Notch signaling produced by MO-I-1100.
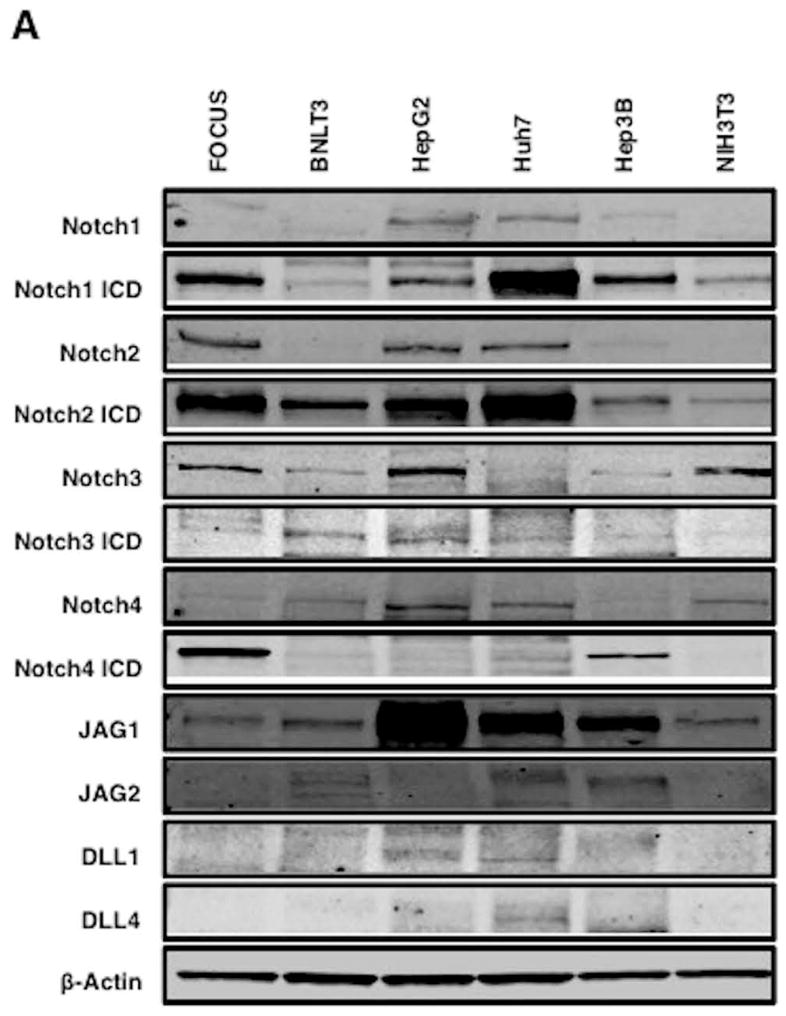
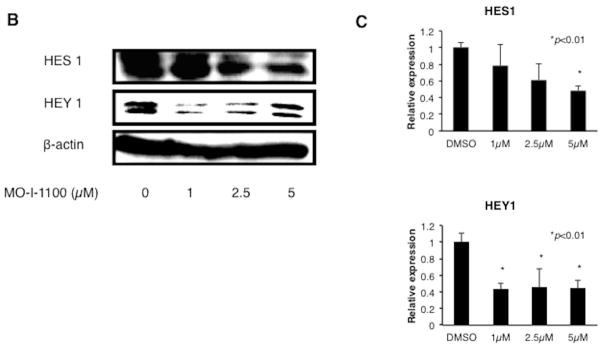
(A) Notch protein expression in HCC cell lines. Note that there is expression of Notch receptors, activated NICDs and ligands (JAG1) in most HCC cell lines. (B) and (C) Treatment of FOCUS cells with a range of MO-I-1100 concentrations reduces the expression of downstream Notch regulated HES1 and HEY1 genes compared to DMSO control (*p < 0.01).
To test this hypothesis, we explored the effects of MO-I-1100 on Notch signaling in FOCUS cells. Expression levels of HES1 and HEY1 known to be downstream genes regulated by Notch signaling were evaluated by Western blot analysis. Cell lysates were prepared from FOCUS cells treated with MO-I-1100 for 24 hours at various concentrations. The results showed a dose inhibitory effect of MO-I-1100 on HES1 and HEY1 protein expression (Fig. 5B and C). These results suggest that the β-hydroxylase activity may be important in modulating Notch signaling since inhibition of enzymatic activity by 80% (MO-I-1100) reduced expression of Notch responsive genes.
Anti-tumor effects of MO-I-1100 on HCC growth and progression in immunodeficient mice
The FOCUS cell line generates a highly aggressive tumor when injected s.c. into nude mice. Nude mice bearing FOCUS s.c. established tumors were treated with MO-I-1100 (20 mg/kg per day) on 5 consecutive days for 2 weeks and then MO-I-1100 was administered every other day. As depicted in Fig. 6A, treatment of tumor bearing mice significantly reduced HCC growth. Tumor growth and progression were substantially reduced by day 21 of treatment; mean tumor volume in treated mice was 31.7% of tumors observed in control mice (Fig. 6B). However, the size of HCC tumors at the initiation of treatment was 50 mm3 and we cannot exclude that MO-I-1100 may have other off target effects on angiogenesis, for example. None of the MO-I-1100 treated mice showed signs of wasting or other toxicity relative to control animals.
Fig. 6. In vivo effects of MO-I-1100 on HCC growth and progression.
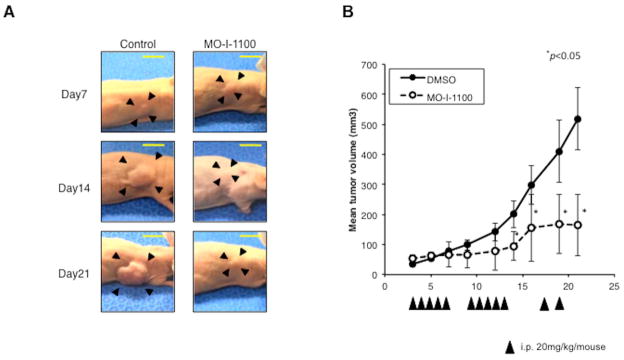
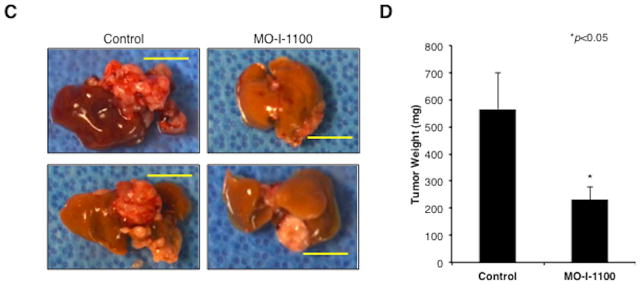
Animals with established s.c. tumor xenografts generated by FOCUS cells were treated with i.p. injection of MO-I-1100 at 20 mg/kg or saline (control) on 5 consecutive days per week for 2 weeks, and then kept on an every other day regimen until the end of study. (A) Examples of subcutaneous grown FOCUS tumors in mice on days 7, 14, and 21 following treatment with MO-I-1100 (right) compared to the saline injected controls (left). (B) Tumor volumes were measured and plotted every other day in MO-I-1100-treated or control mice (n = 10). Arrows indicate timing of administration; vertical bars, standard error (*p < 0.05). (C) Appearance of intrahepatic grown tumors 4 weeks after administration of MO-I-1100 (right) compared to the controls (left). Horizontal yellow bars indicate 1 cm length. (D) Liver tumor weight was analyzed at 4 weeks after administration of MO-I-1100 compared to control (n = 12). HCC tumor size and weight was significantly reduced by MO-I-1100 treatment (*p < 0.05).
β-hydroxylase inhibitor MO-I-100 reduces tumor growth in an intrahepatic orthotopic xenograft model of HCC
In order to further establish the efficacy of MO-I-1100 on HCC growth and progression, an orthotopic xenograft model of HCC was developed. MO-I-1100 at 50 mg/kg was administered to mice bearing FOCUS generated tumors in the liver for 5 consecutive days per week for 2 weeks and then administered every other day. Due to large tumor size in controls, all mice were sacrificed at 3 weeks and HCC intrahepatic growth was significantly inhibited by MO-I-1100 (Fig. 6C). Mean liver tumor weight in the treated group was 42.6% of the control animals (Fig. 6D).
Effects of MO-I-1100 on Notch signaling in HCC tumors
Nude mice with established s.c. tumor (approximately 200 mm3) xenografts of FOCUS cells were treated i.p. with MO-I-1100 (50 mg/kg) or saline for 5 consecutive days and sacrificed on day 5. Expression levels of Notch related proteins were analyzed by Western blot analysis. The results demonstrated that the activated Notch 1 (cleaved fragment) as well as downstream HES1 and HEY1 gene expression were reduced by MO-I-1100 administration (Fig. 7A). Densitometric analysis revealed significantly reduced levels of activated Notch 1, HES1 and HEY1 by 69.8%, 42% and 39.8%, respectively; suggesting that inhibition of β-hydroxylase activity reduces Notch signaling activation in HCC tumors (Fig. 7B). Furthermore, immunohistochemical analysis of murine xenograft tumor tissue revealed that Notch 1 ICD was observed prominently in about 50% of the nuclei of tumor cells in control mice and there was a significant reduction in nuclear localization of NICD following treatment of established HCC tumors for 5 days with MO-I-1100 (Fig. 7C). Indeed, percent of NICD positive nuclei was substantially decreased following MO-I-1100 administration by 60.6% (Fig. 7C). The enzymatic activity of ASPH appeared important in regulation of Notch proteins by MO-I-1100 as shown in Fig. 3D.
Fig. 7. Inhibition of Notch signaling by a SMI in HCC tumors.

We determined the length of time that may be necessary to generate inhibitor effects on Notch signaling by MO-I-1100 treatment. (A) Western blot analysis of activated NICD, HES1 and HEY1 expression from xenograft tumors generated by FOCUS cells after treatment with MO-I-1100 at 50 mg/kg/day for 5 days compared to the control. (B) Densitometric analyses of protein expression. (C) Graft depicts IHS of NICD in xenograft tumor nuclei following treatment with MO-I-1100 compared to control. Note that MO-I-1100 therapy of established tumors strikingly reduced the generation and nuclear translocation of NICD and was associated with downregulation of HES1 and HEY1 gene expression. (D) The enzymatic activity of ASPH is involved in the regulation of Notch proteins. Increasing concentrations of MO-I-1100 enhanced Notch 1 but reduced Jagged1 and downstream HES1 gene expression after a 24 hour exposure of FOCUS HCC cells in vitro (*p < 0.05; **p<0.001).
DISCUSSION
ASPH is highly expressed in HCC tumors as suggested by several previous studies (7, 10, 18) and increased ASPH expression predicts a worse prognosis due to disease reoccurrence following surgical resection (10). Interestingly, ASPH was not expressed in dysplastic regenerating nodules suggesting an association with a fully transformed hepatic phenotype as shown in Fig. 1. The biologic function of ASPH is associated with increased cell motility, migration, invasion and anchorage independent cell growth which are characteristics of malignant cells (11, 19). Thus, its role in hepatic oncogenesis is proposed to promote tumor growth, progression and metastases. Blocking its biologic activities in this context may have therapeutic potential.
The Notch signaling cascades consist of 4 large transmembrane receptors (Notch 1, 2, 3 and 4) coupled with ligands such as JAG 1 and 2 as well as DLL 1, 3, and 4; and requires cell-cell interaction for activation. A key molecular event is the cleavage of full-length receptors and the release of the intracellular domain (NICD) where it translocates to the nucleus to form a transcriptional complex that upregulates Notch responsive genes, such as HES and HEY, which are important not only in cell fate determination principally during embryogenesis but also in maintaining organ and tissue homeostasis (20–22). Notch signaling may play a critical role in HCC development as outlined in a concise review (22). Constitutive activation of Notch by conditional overexpression at the NICD produced HCC in 88–100% of the animals (23, 24). Notch activation has been described in 30–90% of human HCC tumors (18, 22, 24) although its exact role and mechanisms of activation that contribute to HCC development and progression are controversial (22–26). However, it is a major regulator of cell proliferation, migration, invasion and plays a prominent role in the regulation of apoptosis.
Previous studies have established that ASPH overexpression leads to a direct interaction with Notch 1 and JAG followed by activation of downstream Notch responsive genes (18). We have recently demonstrated that reduction in the enzymatic activity of ASPH by the H675Q mutation does not prevent this interaction but blocks upregulation of Notch activated genes (unpublished observations). Furthermore, transient inhibition of ASPH expression by siRNA reduces cell motility, migration and invasion (17). It has not been previously clarified if the enzymatic activity was necessary for ASPH biologic function. The development of a SMI MO-I-1100 addressed this issue since an 80% reduction in β-hydroxylase activity had an impact on HCC viability and there was a general correlation between the expression level and sensitivity to this drug as shown in Fig. 2 and 3. This finding may have clinical relevance for selecting high ASPH expressing tumors for targeted therapy with specific β-hydroxylase inhibitors. ASPH could serve as a biomarker to predict likelihood of anti-tumor responses since there was variable expression of ASPH protein levels in HCC tumors as shown in Fig. 1. The specificity of MO-I-1100 for ASPH β-hydroxylase but no other hydroxylases was established through a capture step employing a mAb (FB-50) directed against the N-terminus of the protein prior to measurement of β-hydroxylase activity. We have found that ASPH overexpression is associated with Notch activation both in vitro and in vivo and the enzymatic activity of the β-hydroxylase is important since treatment with a SMI MO-I-1100 (Fig. 2) downregulates this signaling cascade as shown in Fig. 6 and 7.
Notch receptors, ligands and cleaved NICDs are expressed in HCC cell lines suggesting that constitutive activation of this signaling cascade may be important in promoting a highly invasive and metastatic phenotype (27). Treatment with MO-I-1100 was particularly revealing since it reduced generation of NICD and subsequent nuclear localization as well as inhibited Notch responsive HES1 and HEY1 gene expression both in vitro and in established HCC tumors as shown in Fig. 5 and 7. More important, MO-I-1100 treatment of established FOCUS cell generated tumors with aggressive characteristics was substantial for anti-tumor effects using two animal models of s.c. and intrahepatic growth, which suggests that the enzymatic activity of ASPH was important for tumor progression.
Several key signaling pathways are commonly activated in HCC tumors irrespective of viral or other etiologies, including insulin (IN)/insulin-like growth factor 1 (IGF1)/insulin-receptor substrate type 1 (IRS1)/MAPK/ERK, IN/IGF1/IRS1/PI3K/AKT/GSK3β and WNT/β-catenin (18, 20, 21, 28, 29) among many others (30). Indeed, activation of these pathways in a double transgenic murine model was necessary and sufficient to transform the normal liver to HCC without the participation of known genetic mutations (31). ASPH is a downstream target of these pathways and is upregulated at both mRNA and protein levels (31). Since upregulation of ASPH promotes cell proliferation, migration, invasion, malignant transformation and growth of tumors in immunodeficient mice, it is essential to establish what the downstream effector signaling pathway that produces such an ASPH associated malignant phenotype. Previous studies suggest that Notch activation may play a role in this regard (18).
Transcriptional regulation of ASPH is provided by tripartite signaling pathways: IN/IGF1/IRS1/MAPK/ERK, IN/IGF1/IRS1/PI3K/AKT and WNT/β-catenin (18, 32). Post-transcriptional regulation of ASPH is mediated by phosphorylation of GSK3β-related motifs located in the N-terminal region of the molecule (17). IN/IGF1/IRS1/MAPK/ERK, PI3K/AKT and WNT/β-catenin signaling-upregulated ASPH expression has been implicated to increase motility and invasion of HCC cells (17, 18, 32, 33). Accordingly, siRNA inhibition of ASPH expression reduces cell motility and growth (19, 33–35). Collectively, ASPH has been hypothesized to exert its effector function by activating Notch signaling and its downstream target genes to promote cell migration and invasion (17, 18, 32).
Our working hypothesis on the role of ASPH in HCC pathogenesis as well as formulate a potential therapeutic strategy is based on this signaling pathway as shown in Fig. 8. In this scheme, overexpression of ASPH through activation of upstream growth factor signaling cascade (18, 20, 21, 28–31) facilitates an interaction with Notch receptors and ligands (27, 36) followed by two cleavage events to generate NICDs (37) which serve as transcription factors following translocation into the nucleus (Fig. 8A). The level of β-hydroxylase activity is essential for its biologic function since it may link upstream growth factor signaling pathways to reveal how Notch signaling cascade may be subsequently activated in HCC through ASPH overexpression. Inhibition of enzymatic activity with SMIs like MO-I-1100 prevents activation of Notch signaling (Fig. 8B) and downstream gene activation involved in cell migration, invasion and metastasis to produce anti-tumor effects on HCC growth, progression and distant spread.
Fig. 8.


Hypothesis of how ASPH may be involved in HCC growth and progression through Notch activation (38). The catalytic domain of ASPH (Yellow) contains the catalytic site sequence M670HPGTH675. Both Notch and JAG are substrates of ASPH since they have EGF-like domains: thus, ASPH acts as an “adaptor” for direct physical interaction with Notch1 with JAG2 on the surface of two neighbored cells. (A) In the presence of β-hydroxylase activity of ASPH, binding of the JAG2 ligand (pink) on one cell to the Notch receptor (blue) on another cell results in two sequential proteolytic cleavage of the receptor. The ADAM10 or TACE (TNF-α-converting enzyme; ADAM17) metalloprotease (black) catalyzes the S2 cleavage and generates a substrate for S3 cleavage by the γ-secretase complex (red). This proteolytic process mediates release of the Notch intracellular domain (NICD), which enters the nucleus and interacts with the DNA-binding CSL [CBF1, Su(H) and LAG-1] protein. The co-activator Mastermind (MAM) and other transcription factors are recruited to the CSL complex, whereas co-repressors (Co-R) are released in this context. The Notch signaling is “ON” and the downstream target genes are expressed: HES1 and HEY1 are involved in cell proliferation, migration, invasion, tumor growth and metastasis in HCC. (B) Small molecule inhibitors (SMIs) such as MO-I-1100 can block the catalytic site of ASPH, which leads to loss of β-hydroxylase activity and failure of Notch activation as manifested by reduced generation and nuclear translocation of NICDs and downregulation of responsive genes such as HES1 and HEY1. Therefore, Notch signaling is “OFF” and its oncogenic effects are reduced in HCC.
Supplementary Material
Acknowledgments
Financial Support: This work was supported by grants from Ruth L. Kirschstein National Research Service Award (NRSA) Institutional Research Training (IT-32 DK60415), NIH/NCI (R01CA123544), National Institute for General Medical Sciences (NIGMS), National Institutes of Health (NIH) RI-INBRE Faculty Development Research Project Grant (8P20GM103430-12), 2012 URI Division of Research & Economic Development and URI Council for Research Proposal Development Grants, 2011 AACR-FNAB Fellows Grant for Translational Pancreatic Cancer Research (11-30-14-DONG).
Tissue samples were provided by Dr. Murray Resnick from the Department of Pathology and Laboratory Medicine, Rhode Island and Miriam Hospitals, Alpert Medical School of Brown University. The authors also gratefully acknowledge the assistance of Drs. Tong and de le Monte in the construction of the β-hydroxylase assay.
Abbreviations
- ASPH
Aspartyl-(Asparaginyl)-β-hydroxylase
- DLL
delta-like
- EGF
epidermal growth factor
- FACS
Fluorescence Activated Cell Sorting
- JAG
Jagged
- HCC
hepatocellular carcinoma
- IHS
immunohistochemical staining
- i.p
intraperitoneal
- mAb
monoclonal antibody
- mRNA
messenger RNA
- MTT
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide
- NICD
Notch intracellular domain
- s.c
subcutaneous
- S.D
standard deviation
- S.E
standard error
- SMI
small molecule inhibitor
- TMA
tissue microarray
Footnotes
Conflict of interest: We have no conflict of interest.
Contributor Information
Arihiro Aihara, Email: Aaihara0@gmail.com.
Chiung-Kuei Huang, Email: Chiung-Kuei_Huang@brown.edu.
Mark J. Olsen, Email: molsen@midwestern.edu.
Qiushi Lin, Email: qiushilin@hotmail.com.
Waihong Chung, Email: Waihongchung@gmail.com.
Qi Tang, Email: qitang@my.uri.edu.
Xiaoqun Dong, Email: xiaoqun_dong@mail.uri.edu.
Jack R. Wands, Email: Jack_Wands_MD@brown.edu.
References
- 1.Fong Y, Sun RL, Jarnagin W, Blumgart LH. An analysis of 412 cases of hepatocellular carcinoma at a Western center. Ann Surg. 1999;229:790–799. doi: 10.1097/00000658-199906000-00005. discussion 799–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 3.Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, Luo R, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- 4.Jia S, VanDusen WJ, Diehl RE, Kohl NE, Dixon RA, Elliston KO, Stern AM, et al. cDNA cloning and expression of bovine aspartyl (asparaginyl) beta-hydroxylase. J Biol Chem. 1992;267:14322–14327. [PubMed] [Google Scholar]
- 5.Engel J. EGF-like domains in extracellular matrix proteins: localized signals for growth and differentiation? FEBS Lett. 1989;251:1–7. doi: 10.1016/0014-5793(89)81417-6. [DOI] [PubMed] [Google Scholar]
- 6.Shimoda M, Tomimaru Y, Charpentier KP, Safran H, Carlson RI, Wands J. Tumor progression-related transmembrane protein aspartate-beta-hydroxylase is a target for immunotherapy of hepatocellular carcinoma. Journal of hepatology. 2012 doi: 10.1016/j.jhep.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lavaissiere L, Jia S, Nishiyama M, de la Monte S, Stern AM, Wands JR, Friedman PA. Overexpression of human aspartyl(asparaginyl)beta-hydroxylase in hepatocellular carcinoma and cholangiocarcinoma. J Clin Invest. 1996;98:1313–1323. doi: 10.1172/JCI118918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaret KS, Grompe M. Generation and regeneration of cells of the liver and pancreas. Science. 2008;322:1490–1494. doi: 10.1126/science.1161431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J, de la Monte SM, Sabo E, Kethu S, Tavares R, Branda M, Simao L, et al. Prognostic value of humbug gene overexpression in stage II colon cancer. Hum Pathol. 2007;38:17–25. doi: 10.1016/j.humpath.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 10.Wang K, Liu J, Yan ZL, Li J, Shi LH, Cong WM, Xia Y, et al. Overexpression of aspartyl-(asparaginyl)-beta-hydroxylase in hepatocellular carcinoma is associated with worse surgical outcome. Hepatology. 2010;52:164–173. doi: 10.1002/hep.23650. [DOI] [PubMed] [Google Scholar]
- 11.Ince N, de la Monte SM, Wands JR. Overexpression of human aspartyl (asparaginyl) beta-hydroxylase is associated with malignant transformation. Cancer Res. 2000;60:1261–1266. [PubMed] [Google Scholar]
- 12.Nishiyama M, Wands JR. Cloning and increased expression of an insulin receptor substrate-1-like gene in human hepatocellular carcinoma. Biochem Biophys Res Commun. 1992;183:280–285. doi: 10.1016/0006-291x(92)91640-c. [DOI] [PubMed] [Google Scholar]
- 13.Shimoda M, Tomimaru Y, Charpentier KP, Safran H, Carlson RI, Wands J. Tumor progression-related transmembrane protein aspartate-beta-hydroxylase is a target for immunotherapy of hepatocellular carcinoma. J Hepatol. 2012;56:1129–1135. doi: 10.1016/j.jhep.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang QP, VanDusen WJ, Petroski CJ, Garsky VM, Stern AM, Friedman PA. Bovine liver aspartyl beta-hydroxylase. Purification and characterization. J Biol Chem. 1991;266:14004–14010. [PubMed] [Google Scholar]
- 15.Gronke RS, Welsch DJ, VanDusen WJ, Garsky VM, Sardana MK, Stern AM, Friedman PA. Partial purification and characterization of bovine liver aspartyl beta-hydroxylase. J Biol Chem. 1990;265:8558–8565. [PubMed] [Google Scholar]
- 16.Aihara A, Tanaka S, Yasen M, Matsumura S, Mitsunori Y, Murakata A, Noguchi N, et al. The selective Aurora B kinase inhibitor AZD1152 as a novel treatment for hepatocellular carcinoma. J Hepatol. 52:63–71. doi: 10.1016/j.jhep.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 17.de la Monte SM, Tong M, Carlson RI, Carter JJ, Longato L, Silbermann E, Wands JR. Ethanol inhibition of aspartyl-asparaginyl-beta-hydroxylase in fetal alcohol spectrum disorder: potential link to the impairments in central nervous system neuronal migration. Alcohol. 2009;43:225–240. doi: 10.1016/j.alcohol.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cantarini MC, de la Monte SM, Pang M, Tong M, D’Errico A, Trevisani F, Wands JR. Aspartyl-asparagyl beta hydroxylase over-expression in human hepatoma is linked to activation of insulin-like growth factor and notch signaling mechanisms. Hepatology. 2006;44:446–457. doi: 10.1002/hep.21272. [DOI] [PubMed] [Google Scholar]
- 19.Sepe PS, Lahousse SA, Gemelli B, Chang H, Maeda T, Wands JR, de la Monte SM. Role of the aspartyl-asparaginyl-beta-hydroxylase gene in neuroblastoma cell motility. Lab Invest. 2002;82:881–891. doi: 10.1097/01.lab.0000020406.91689.7f. [DOI] [PubMed] [Google Scholar]
- 20.Branda M, Wands JR. Signal transduction cascades and hepatitis B and C related hepatocellular carcinoma. Hepatology. 2006;43:891–902. doi: 10.1002/hep.21196. [DOI] [PubMed] [Google Scholar]
- 21.Calvisi DF, Frau M, Tomasi ML, Feo F, Pascale RM. Deregulation of signalling pathways in prognostic subtypes of hepatocellular carcinoma: novel insights from interspecies comparison. Biochim Biophys Acta. 2012;1826:215–237. doi: 10.1016/j.bbcan.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 22.Strazzabosco M. Notch signaling in hepatocellular carcinoma: guilty in association! Gastroenterology. 2012;143:1430–1434. doi: 10.1053/j.gastro.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dill MT, Tornillo L, Fritzius T, Terracciano L, Semela D, Bettler B, Heim MH, et al. Constitutive Notch2 signaling induces hepatic tumors in mice. Hepatology. 2013;57:1607–1619. doi: 10.1002/hep.26165. [DOI] [PubMed] [Google Scholar]
- 24.Villanueva A, Alsinet C, Yanger K, Hoshida Y, Zong Y, Toffanin S, Rodriguez-Carunchio L, et al. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology. 2012;143:1660–1669. e1667. doi: 10.1053/j.gastro.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tschaharganeh DF, Chen X, Latzko P, Malz M, Gaida MM, Felix K, Ladu S, et al. Yes-associated protein up-regulates Jagged-1 and activates the Notch pathway in human hepatocellular carcinoma. Gastroenterology. 2013;144:1530–1542. e1512. doi: 10.1053/j.gastro.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou L, Zhang N, Song W, You N, Li Q, Sun W, Zhang Y, et al. The significance of Notch1 compared with Notch3 in high metastasis and poor overall survival in hepatocellular carcinoma. PLoS One. 2013;8:e57382. doi: 10.1371/journal.pone.0057382. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29:4989–5005. doi: 10.1038/onc.2010.236. [DOI] [PubMed] [Google Scholar]
- 29.Ito Y, Sasaki Y, Horimoto M, Wada S, Tanaka Y, Kasahara A, Ueki T, et al. Activation of mitogen-activated protein kinases/extracellular signal-regulated kinases in human hepatocellular carcinoma. Hepatology. 1998;27:951–958. doi: 10.1002/hep.510270409. [DOI] [PubMed] [Google Scholar]
- 30.Wands JR, Kim M. Hepatology. 2014. WNT/beta-catenin signaling and hepatocellular carcinoma. [DOI] [PubMed] [Google Scholar]
- 31.Longato L, de la Monte S, Kuzushita N, Horimoto M, Rogers AB, Slagle BL, Wands JR. Overexpression of insulin receptor substrate-1 and hepatitis Bx genes causes premalignant alterations in the liver. Hepatology. 2009;49:1935–1943. doi: 10.1002/hep.22856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomimaru Y, Koga H, Yano H, de la Monte SM, Wands JR, Kim M. Up-regulation of TCF-4 isoform-responsive target genes in hepatocellular carcinoma. Liver International. 2013 doi: 10.1111/liv.12188. (Revised version under review) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de la Monte SM, Tamaki S, Cantarini MC, Ince N, Wiedmann M, Carter JJ, Lahousse SA, et al. Aspartyl-(asparaginyl)-beta-hydroxylase regulates hepatocellular carcinoma invasiveness. J Hepatol. 2006;44:971–983. doi: 10.1016/j.jhep.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 34.Gundogan F, Bedoya A, Gilligan J, Lau E, Mark P, De Paepe ME, de la Monte SM. siRNA inhibition of aspartyl-asparaginyl beta-hydroxylase expression impairs cell motility, Notch signaling, and fetal growth. Pathol Res Pract. 2011;207:545–553. doi: 10.1016/j.prp.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maeda T, Sepe P, Lahousse S, Tamaki S, Enjoji M, Wands JR, de la Monte SM. Antisense oligodeoxynucleotides directed against aspartyl (asparaginyl) beta-hydroxylase suppress migration of cholangiocarcinoma cells. J Hepatol. 2003;38:615–622. doi: 10.1016/s0168-8278(03)00052-7. [DOI] [PubMed] [Google Scholar]
- 36.Zender S, Nickeleit I, Wuestefeld T, Sorensen I, Dauch D, Bozko P, El-Khatib M, et al. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell. 2013;23:784–795. doi: 10.1016/j.ccr.2013.04.019. [DOI] [PubMed] [Google Scholar]
- 37.Shih Ie M, Wang TL. Notch signaling, gamma-secretase inhibitors, and cancer therapy. Cancer Res. 2007;67:1879–1882. doi: 10.1158/0008-5472.CAN-06-3958. [DOI] [PubMed] [Google Scholar]
- 38.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–689. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.