

Abstract
Many organophosphorous esters synthesized for applications in industry, agriculture, or warfare irreversibly inhibit acetylcholinesterase, and acute poisoning with these compounds causes life-threatening cholinergic overstimulation. Following classical emergency treatment with atropine, an oxime, and a benzodiazepine, surviving victims often suffer brain neurodegeneration. Currently, there is no pharmacological treatment to prevent this brain injury. Here we show that a cyclic diterpenoid, (1S,2E,4R,6R,7E,11E)-cembra-2,7,11-triene-4,6-diol (4R) ameliorates the damage caused by diisopropylfluorophosphate (DFP) in the hippocampal area CA1.
DFP has been frequently used as a surrogate for the warfare nerve agent sarin. In rats, DFP is lethal at the dose used to cause brain damage. Therefore, to observe brain damage in survivors, the death rate was reduced by pre-administration of the peripherally acting antidotes pyridostigmine and methyl atropine or its analogue ipratropium. Pyridostigmine bromide, methyl atropine nitrate, and ipratropium bromide were dissolved in saline and injected intramuscularly at 0.1 mg/kg, 20 mg/kg, and 23 mg/kg, respectively. DFP (9 mg/kg) dissolved in cold water was injected intraperitoneally. 4R (6 mg/kg) dissolved in DMSO was injected subcutaneously, either 1 hour before or 5 or 24 hours after DFP. Neurodegeneration was assessed with Fluoro-Jade B and amino cupric silver staining; neuroinflammation was measured by the expression of nestin, a marker of activated astrocytes.
Forty-eight hours after DFP administration, 4R decreased the number of dead neurons by half when injected before or after DFP. 4R also significantly decreased the number of activated astrocytes. These data suggest that 4R is a promising new drug that could change the therapeutic paradigm for acute poisoning with organophosphorous compounds by the implementation of a second-stage intervention after the classical countermeasure treatment.
Keywords: diisopropylfluorophosphate; neurodegeneration; neuroprotection; cembranoid; (1S, 2E, 4R, 6R, 7E, 11E)-cembra-2, 7, 11-triene-4, 6-diol
1. Introduction
Organophosphorous compounds (OPs) are a diverse family of natural and man-made compounds used in industry, in agriculture as insecticides, and most notoriously, in warfare and terrorism as chemical warfare nerve agents (CWNA). OPs that inhibit acetylcholinesterase (AChE) (Pope et al., 2005) or the neuropathy target esterase (Damodaran et al., 2011) pose a threat to human life and health. Chronic exposure to low quantities of these compounds produces pathologies that appear to be less dependent on AChE inhibition than acute poisoning (Ray and Richards, 2001, Terry et al., 2011, Terry, 2012). In the case of acute OP poisoning, most, but not all, neurological damage caused by these compounds can be accounted for by inhibition of AChE, accumulation of acetylcholine, and the ensuing cholinergic crisis. For the last 50 years, medical countermeasures included administration of atropine to antagonize muscarinic effects, pralidoxime to reactivate AChE, and benzodiazepine to reduce seizures. These classical antidotes are effective in decreasing mortality but are ineffective in protecting against the delayed neurotoxicity that often follows. For example, the victims of the Tokyo 1995 sarin incident still suffer the consequences of OP poisoning. Many survivors showed severe neurological complications 7 years after the incident (Miyaki et al., 2005). To attenuate this delayed OP-mediated neurotoxicity, one should, therefore, target later steps along the pathway leading to brain damage, including inflammation and apoptosis (Chen, 2012).
The need for a neuroprotective compound capable of ameliorating neurodegenerative processes is evident. Our group discovered a diterpenoid called 4R-cembranoid (4R) that fits in this category (Fig. 1) (Ferchmin et al., 2009). Several natural and semisynthetic analogues of 4R are also promising neuroprotective compounds (Eterovic et al., 2013).
Figure 1. Structural formula of 4R-cembranoid.
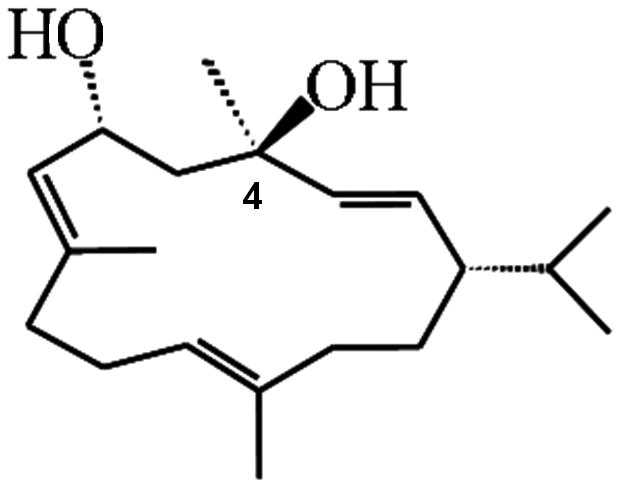
The chiral carbon 4, relevant to neuroprotection, is indicated.
The objective of this study was to determine whether 4R could decrease the neurodegeneration caused by diisopropylfluorophosphate (DFP) under conditions that simulate terrorist attacks or industrial accidents. DFP is an extremely toxic OP that has been used as a surrogate CWNA (Crawford et al., 2004, Deshpande et al., 2010). In vitro studies revealed that 4R is an antagonist of the α7 nicotinic acetylcholine receptor. Interestingly, by inhibition of this receptor, 4R triggers a nicotinic neuroprotective cell-signaling cascade (Ferchmin et al., 2005, Ferchmin et al., 2013). In vitro studies have shown that 4R rescues the electrophysiological activity of acute hippocampal slices measured as population spikes when applied 30 min after exposure to the OP paraoxon, an irreversible AChE inhibitor (Eterovic et al., 2011). Here we show that, as predicted by the above-mentioned in vitro studies, 4R reduces brain damage caused by DFP.
Studying neurodegeneration and neuroprotection induced by administration of DFP to rats in doses near the LD50 is complicated by high mortality, caused by peripheral toxicity, and variable neurodegeneration in the survivors (Sparenborg et al., 1993, Crawford et al., 2004, Deshpande et al., 2010). Several peripherally acting drugs that do not cross the blood–brain barrier have been used to reduce mortality and at the same time allow for brain damage. We have chosen an experimental model in which pyridostigmine competes with DFP for the active site of AChE, protecting it from irreversible inhibition, while methyl atropine inhibits the overstimulation of muscarinic receptors (Kim et al., 1999, Li et al., 2011). At the beginning of these studies, methyl atropine was unavailable; therefore, we used the closest structural and functional analogue, N-isopropyl atropine, or ipratropium, in three of the four experiments presented here.
2. Materials and Methods
2.1 Drugs and reagents
Common laboratory chemicals and pyridostigmine bromide, ipratropium bromide monohydrate (N-isopropylatropine), 5,5-dithiobis(2-nitrobenzoic acid) (DTNB), acetylthiocholine iodide, sodium cacodylate, paraformaldehyde, xylene, potassium permanganate, dimethylsulfoxide, and diisopropylfluorophosphate were obtained from Sigma-Aldrich (St. Louis, MO). Methyl atropine nitrate was obtained from Spectrum Chemicals (New Brunswick, NJ), and Fluoro-Jade B was obtained from Millipore, (Billerica, MA, cat. no. AG310). The cembranoid (1S,2E,4R,6R,7E,11E)-cembra-2,7,11-triene-4,6-diol (4R) was prepared by Dr. K. El Sayed (School of Pharmacy, University of Louisiana, Monroe, LA) as previously described (El Sayed et al., 2008). The analytical criteria for characterization and purity were: 1) 1H NMR: integration of the H-6 proton at d 4.81 versus that of the 4S epimer (d 4.46). 2) 13C NMR: C-6 in the 4R (dC 67.5) versus C-6 in the 4S (dC 66.0), and 3) thin-layer chromatography (TLC): Rf value 0.42 (Si gel, n-hexane-ethyl acetate 1:1). The purity of the batch used for the present work was more than 98%.
2.2 Animals and treatments
Male Sprague-Dawley rats (240–315 g) from our colony derived from Taconic Farms were used. The animals were bred in the animal facility of Universidad Central del Caribe, Medical School. The animals were housed individually in positive-ventilation cages with filtered air (Allentown), controlled temperature and humidity, and a 12-hour light–dark cycle. Food and water were provided ad libitum. All protocols were approved by the Institutional Animal Care and Use Committee of Universidad Central del Caribe.
The design of the experiments is illustrated in Fig. 2. The peripherally protective drugs, pyridostigmine bromide (0.1 mg/kg), ipratropium bromide (23 mg/kg), and methyl atropine nitrate (20 mg/kg) were dissolved in saline and injected via the i.m. route. DFP (9 mg/kg) was dissolved in cold water and injected i.p., while 4R (6 mg/kg) was dissolved in DMSO and injected s.c. in the interscapular area. The volume of injections was 1 ml/kg in all cases.
Figure 2. Experimental design.
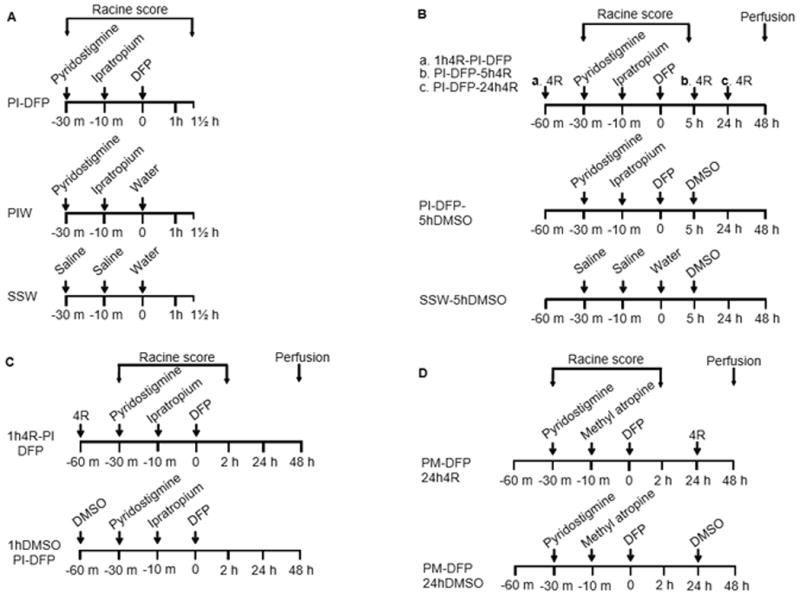
The figure presents the treatment groups and the protocols used for experiments 1–4. The treatment names are abbreviated as the first letter of each compound, except in the case of DFP and DMSO, where the standard abbreviations are used; the compounds are named in the same sequence as they were injected. The protocols indicate the times of injection of the various drugs as well as the period during which the Racine scores were obtained. In experiments 2–4, the animals were perfused 48 hours after DFP. (A) Experiment 1. Pyridostigmine (P) and ipratropium (I) were injected before DFP in the experimental group PI-DFP; this group tested for the effects of DFP when preceded by pyridostigmine and ipratropium. The PIW group received pyridostigmine, ipratropium, and water (W). Water was the vehicle of DFP; this group tested for the effects of pyridostigmine and ipratropium in the absence of DFP. The SSW group was the all-vehicles control, which received saline (S), saline, and water. (B) Experiment 2 tested for the neuroprotective effect of 4R against DFP. 4R was administered within the PI-DFP paradigm 1 hour before DFP (a. 1h4R-PI-DFP), 5 hours after DFP (b. PI-DFP-5h4R), and 24 hours after DFP (c. PI-DFP-24h4R). The PI-DFP-5hDMSO group tested for the effect of PI-DFP in the presence of DMSO, the vehicle of 4R. The SSW-5hDMSO group was the all-vehicles control. (C) Experiment 3 replicated the study of 4R when injected 1 hour before DFP (1h4R-PI-DFP). The 1 hDMSO-PI-DFP group was the 4R vehicle control. (D) Experiment 4 replicated the study of 4R injected 24 hours after DFP using methyl atropine (M) instead of ipratropium. The experimental group was injected with pyridostigmine and methyl atropine before DFP, followed 24 hours later by 4R (PM-DFP-24h4R). The control group that did not receive 4R (PM-DFP-24hDMSO) was injected 24 hours after DFP with DMSO.
A series of experimental treatments comprising various combinations of drugs and times of 4R injection were tested. To describe each treatment in a succinct and unequivocal manner, we created the following system of nomenclature: 1) Each drug is abbreviated as follows: pyridostigmine (P), ipratropium (I), methyl atropine (M), dimethylsulfoxide (DMSO), diisopropylfluorophosphate (DFP), and 4R-cembranoid (4R). 2) The order of these abbreviations within the treatment name indicates the order in which the drugs were administered; e.g., PI-DFP indicates that the animal received first P, then I, and finally DFP. 3) The time of 4R injection relative to DFP injection is indicated, e.g., 1h4R-PI-DFP indicates that 4R was administered 1 hour before DFP; 4R was then followed by the PI-DFP protocol as explained above.
2.3 Determination of acetylcholinesterase activity
AChE activity was measured as previously described (Ellman et al., 1961). The brains were dissected into the neocortex, hippocampus, striatum, remaining subcortex, cerebellum, and pons and medulla (Craigie et al., 1963). After dissection, the brain areas were weighed, frozen on dry ice, and stored at −80°C. The tissue was homogenized in sodium phosphate buffer (0.1 M, pH 8.0 + 1% Triton X-100) at a concentration of 100 mg of wet weight per ml of buffer. The homogenates were centrifuged at 12,000 × g for 1 min, the supernatant collected, and tetraisopropyl pyrophosphoramide (100 μM) added to inhibit butyrylcholinesterase. AChE activity was measured in triplicate wells, and the color changes were read in a spectrophotometer at 405 nM with 16 kinetic cycles using a minimal kinetic interval. Enzyme activity was normalized to protein concentration, which was determined using Bradford reagent (Bradford, 1976). Data were expressed as μmoles of substrate transformed/min/mg of protein using the following formula:
2.4 Recording of behavioral convulsions
Behavioral convulsions were quantified using a modified Racine scale (Racine, 1972) (Table 1) in which the definition of behavioral categories was revised by a clinical neurologist on the basis of video recordings of our experimental subjects undergoing DFP-caused convulsions. The scores were recorded independently by two observers. Discrepancies between observers were minor and were resolved by inspection of video recordings. The observation time was divided into 10-min periods; each rat was observed every minute, and the highest score obtained during the 10-min period was taken as the score for this period. To compare the intensity of convulsions across the experiments the “total convulsions” were calculated by multiplying the score for each period by 10 and adding up the scores of 9 periods comprising 90 minutes of observation.
Table 1.
The table defines the behavior to which each score value was assigned.
| Behavioral Stages | Score |
|---|---|
| No behavioral response | 0 |
| Agitation/Compulsive grooming | 1 |
| Behavioral arrest | 2 |
| Orofacial movements | 3 |
| Chewing | |
| Head nodding | |
| Paralysis/Weakness | |
| Unilateral/bilateral forelimb clonus without rearing | 4 |
| Straub tail | |
| Extended body posture | |
| Bilateral forelimb clonus plus rearing | 5 |
| Rearing and falling | 6 |
| Full tonic seizures | 7 |
| Death | 8 |
2.5 Histological methods
2.5.1 Experiment 2
The animals were deeply anesthetized with isoflurane, perfused transcardially with 4% formaldehyde in phosphate-buffered saline pressurized to match rat blood pressure. Brains were cryoprotected by treatment with cold 10% sucrose in phosphate-buffered saline for 24 h, followed by 30% sucrose until they sank. Coronal slices (18-μm) were cut in a cryostat (Micron) at −20°C and mounted on 12.5% gelatin-coated glass slides. All slides were simultaneously stained with cresyl violet on a staining rack, which was passed through ethanol (95%, 70%, and 50%), distilled water, and 0.5% cresyl violet in glacial acetic acid, washed with ethanol solutions, and finally treated with Histoclear. Slides were mounted with Permount, and sections were viewed in a Zeiss Axioscope 2 microscope at 5× magnification and photographed with a Hamamatsu Orca camera under brightfield illumination. Cell density was calculated using the densitometric module of MCID elite image-analysis software (Imaging Research, Inc.). Alternate slices were stained with Fluoro-Jade B as described below.
2.5.2 Experiments 3 and 4
For Experiments 3 and 4, perfusion and other histological procedures were performed following the instructions provided by NeuroScience Associates (NSA). Briefly, subjects were deeply anesthetized with isoflurane and perfused transcardially with isotonic saline solution, 0.4% glucose, 0.8% sucrose, 0.023% CaCl2, and 0.034% Na cacodylate. The perfusion was done with a solution pressurized to match rat blood pressure to avoid the rupture of capillaries and extravasation. This was followed by addition of the fixing solution containing 4% formaldehyde, 4% sucrose, and 1.4% sodium cacodylate. The skulls with brains in situ were stored in fixing solution for 12–16 hours at 4–10 °C, after which the brains were removed and stored in 1.4% sodium cacodylate (pH 7.3) until shipped for processing by NSA. Using NSA procedures, 16 rat brains were embedded in a block of proprietary gel and freeze-sectioned at 40-μm intervals in the coronal plane throughout the entire brain. All sections were collected in antigen-preservation solution. Every eighth section was stained using the de Olmos' amino cupric silver staining method (de Olmos et al, 1994), and the anatomical structures were made visible by neutral red counterstaining; adjacent sections were stained using an antibody for nestin, as described in http://neuroscienceassociates.com/. The adjacent sections were then stained with Fluoro-Jade B in our laboratory, as described below. An Olympus 1X71 inverted microscope with Olympus UPlan F1 10X/0.30 ph1 objective was used with MetaMorph® (Meta Imaging Series 7.5 and 7.1) software for imaging analysis (Molecular Devices®). The results were presented as the number of labeled cells per square millimeter.
2.5.3 Fluoro-Jade B staining
Fluoro-Jade B labeling of degenerating neurons (Schmued and Hopkins, 2000) was performed following the manufacturer instructions with modifications reported by (Li et al., 2011) and their personal instructions. Briefly, the slices were dried for 30 min at 50 °C, postfixed with 4% formaldehyde for 5–10 min, and washed twice with saline. Next, the slices adhering to the glass slides were incubated with 0.06% potassium permanganate and washed with distilled water before incubation with 0.4‰ Fluoro-Jade B. Finally; the slices were rinsed, dried, and cleared with xylene before cover-slipping with DPX mounting medium.
2.5.4 Overlap of nestin and glial fibrillary acidic protein
The confocal photomicrographs were taken from coronal cortical sections from rats treated with DFP. Immunostaining for glial fibrillary acidic protein (GFAP) was performed using mouse antibody conjugated with Cy3 (Sigma) at a dilution of 1:750. Immunostaining for nestin was done with antibody (Abcam, 1:1000) raised in rabbit using the secondary antibody anti-rabbit Alexa Fluor 488. Pictures were taken with an Olympus Fluoview Laser Confocal Microscope.
2.6 Statistical analysis
The effect of 4R was determined by using a series of generalized linear models in which the primary study endpoints were the number of dead neurons or of activated astrocytes. The differences between experimental groups were determined by a series of generalized linear models in which the primary outcome was the number of dead neurons or activated astrocytes, the main independent variable was the study group, and covariate factors included slice and hemisphere.
Therefore, the total number of “cell level data” used for the analysis was the number of rats × 2 hemispheres × number of slices. The number of slices analyzed in experiment 2 was two and in experiments 3 and 4 it was five. For the graphical examination of the effect of 4R, we generated bar charts with the mean and standard error of the mean (SEM). The mean value for each rat was obtained by averaging all the measurements for that rat; that is, the measurements obtained from each slice on each hemisphere. The overall mean for the group was obtained by averaging the mean values of each rat. The SEM of the individual means was calculated as usual (standard deviation divided by the square root of the number of rats in that group).
Total convulsion values generated form the Racine scores were analyzed by the Kruskal-Wallis nonparametric test followed by Dunn's multiple comparison method or the Mann-Whitney rank sum test. All statistical tests were performed using SAS v.9.3.
3. Results
3.1 Animal model
As mentioned above, pyridostigmine and methyl atropine must be administered before acute administration of DFP at a dose high enough to cause observable brain damage in survivors. However, at the time we started this work, methyl atropine was unavailable, and we replaced it with ipratropium in experiments 1, 2, and 3, generating the PI-DFP protocol. Methyl atropine became available for experiment 4, in which the experimental protocol used was pyridostigmine, methyl atropine, and DFP (PM-DFP).
3.2 Experiment 1. The pyridostigmine, ipratropium, and DFP protocol
The goal of experiment 1 was to determine the effect of DFP on the inhibition of AChE and Racine scores in rats treated with the PI-DFP protocol. The animals were distributed at random among three treatment groups: PI-DFP, PIW, and SSW (Fig.2A). One set of rats was used for the determination of the inhibition of AChE and another set for the determination of Racine scores.
3.2.1 Brain AChE activity
The inhibition of AChE by DFP and the rate of recovery of enzyme activity in six brain areas are shown in Fig. 3. Rats that received the PI-DFP treatment were euthanized from 15 min to 21 days after DFP vehicle injection. There were two control groups without DFP, the DFP vehicle control (PIW), and the all-vehicles control (SSW). The AChE activity of both control groups was the same as expected from the fact that pyridostigmine does not readily reach the brain; therefore, both control groups were pooled together. The enzyme activity in controls varied within brain regions from 16–219 μmoles substrate/min/mg protein, with the highest activity recorded in striatum and the lowest in cerebellum. Fifteen minutes after DFP injection, enzyme activity was reduced to less than 10% of control in all brain areas. The recovery of enzyme activity was slow: in cortex and cerebellum, it had recovered completely by 21 days after DFP administration, while the other regions did not reach complete recovery during this time.
Figure 3. Experiment 1: Effect of DFP on brain AChE activity.
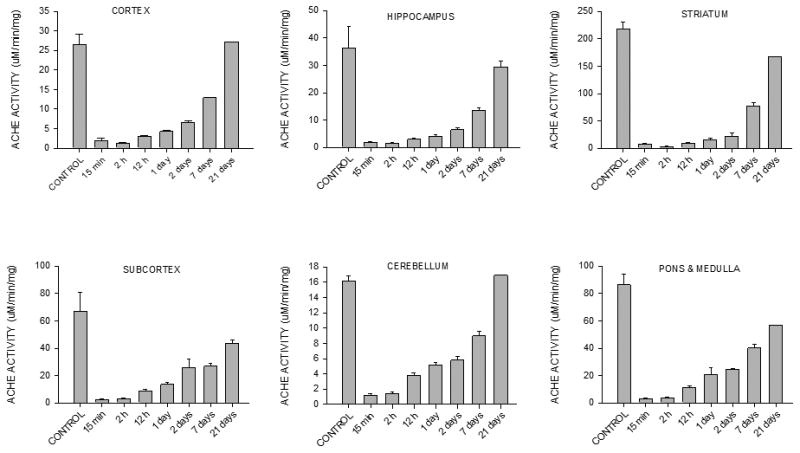
AChE activity was measured in six brain areas of rats treated with the pyridostigmine-ipratropium-DFP protocol (PI-DFP); pyridostigmine-ipratropium-water (PIW), or saline-saline-water (SSW) (see Fig. 2A). Enzyme activity was the same in the PIW and SSW groups, which were pooled together in the “CONTROL” group. The bars represent the mean ± SEM of 3-6 rats per time point, except at 21 days when there were only two rats, and therefore SEM was not calculated.
3.2.2 Racine scores
Another group of animals was used to record Racine scores. The rats were distributed at random among the three treatment groups, and Racine scores were recorded for two hours starting after pyridostigmine injection (Fig. 2A). All animals injected with DFP (PI-DFP group) developed prolonged behavioral convulsions (scores 3–8), which ended in death in three cases (3/11). The convulsions lasted for the 90-min observation period, but only a few subjects had sporadic convulsions 24 hours after PI-DFP treatment. Injection of pyridostigmine followed by ipratropium and water (PIW) produced only weak and transient behavioral manifestations (scores 0–3). The vehicles control (SSW) displayed no reaction to the injections or only a transient agitation (scores 0–1, Fig. 4). Total convulsions for the 90-min period were significantly higher for the PI-DFP group than for the PIW and SSW groups, which did not differ among themselves (mean ± SEM: PI-DFP, 536 ± 57; PIW, 61 ± 19; SSW, 13 ± 3; p<0.05, n=11 rats per group).
Figure 4. Experiment 1: Effect of pyridostigmine, ipratropium, and DFP on the Racine scores.
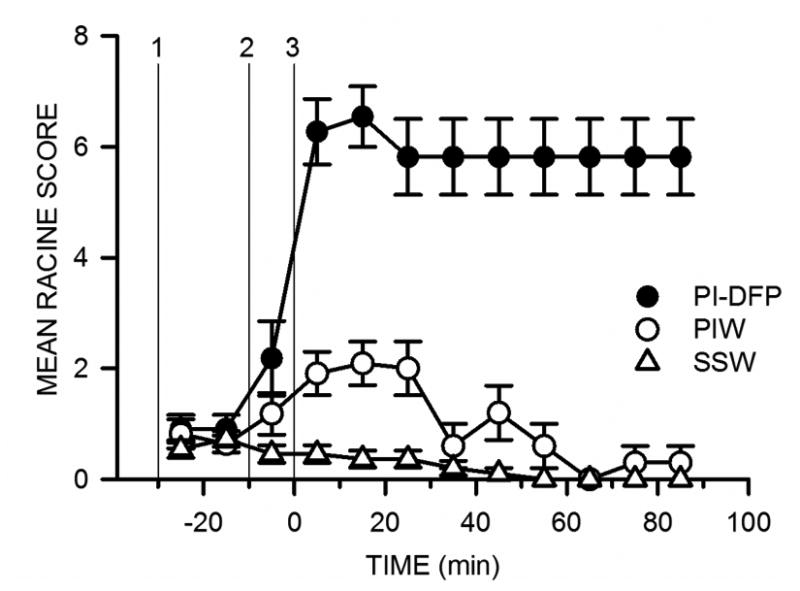
Racine scores were determined as described in Methods (experimental protocol in Fig. 2A). The symbols are the mean ± SEM of the scores for 10-min observation periods. The vertical lines labeled 1, 2, and 3 indicate the time of the three injections for each group. PI-DFP: (1) pyridostigmine, (2) ipratropium, and (3) DFP. PIW: (1) pyridostigmine, (2) ipratropium, and (3) water. SSW: (1) saline, (2) saline, and (3) water.
3.3 Experiment 2. 4R ameliorates neurodegeneration induced by DFP
The purpose of this pilot experiment was to test whether 4R decreases DFP-induced neuronal death and determine the window of therapeutic opportunity for 4R. The subjects were distributed at random among five treatment groups (Fig. 2B). Three groups received the PI-DFP treatment plus 4R, which was administered 1 hour before DFP (1h4R-PI-DFP), 5 hours after DFP (PI-DFP-5h4R) or 24 hours after DFP (PI-DFP-24h4R). The 4R vehicle control received the PI-DFP treatment plus DMSO (PI-DFP-5hDMSO). The all-vehicles control received saline, saline, and water plus DMSO (SSW-5hDMSO). In this experiment, animals appearing to be in immediate danger of death were injected with additional doses of ipratropium. Forty-eight hours after DFP injection, the animals were euthanized, and alternate brain slices stained with either Fluoro-Jade B or cresyl violet.
3.3.1 Racine scores
The behavioral observations started immediately after the injection of pyridostigmine and continued for the next 5 hours (Fig.2B). During this time, two of the groups injected with 4R (PI-DFP-5h4R and PI-DFP-24h4R) had not yet received the 4R injection, and so their treatment did not differ from the vehicle control (PI-DFP-DMSO). For this reason, the Racine scores for these three groups were consolidated into the PI-DFP group.
Minutes after DFP injection, all animals, presented convulsions, which lasted for the whole observation period (Fig. 5). Total convulsions for the first 90 min were not significantly different between rats that received 4R or DMSO, (mean ± SEM): 1h4R-PI-DFP, 323 ± 26 (n=8); PI-DFP, 388 ± 26 (n=27). Therefore, 4R administered 1hour before DFP did not affect convulsions.
Figure 5. Experiment 2: Pre-application of 4R does not affect Racine scores.
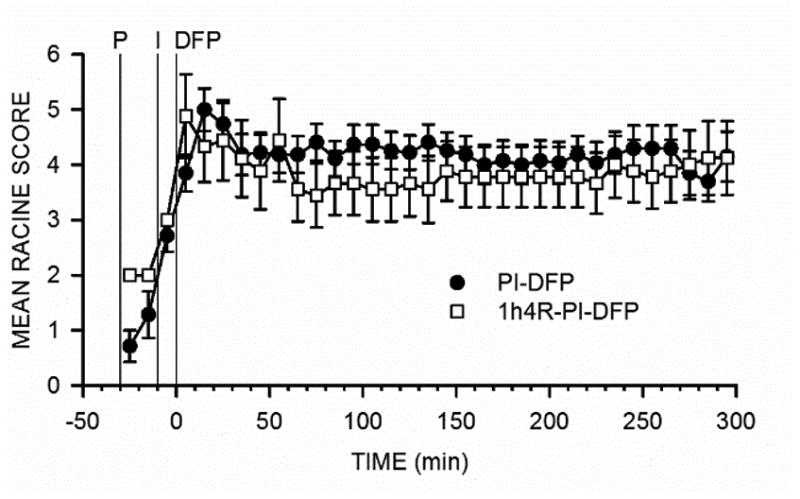
Racine scores were determined as described in Methods; for the experimental protocol, see Fig. 2B. The symbols are the mean ± SEM values of the scores for 10-min observation periods. PI-DFP stands for the combined PI-DFP-DMSO, PI-DFP-5h4R, and PI-DFP-24h4R groups and 1h4R-PI-DFP corresponds to the subjects injected with 4R 1 hour before DFP.
3.3.2 Brain histology: the effect of 4R on neuronal death
Neuronal degeneration in the CA1 hippocampal area (bregma, −2.4 to −4.2) was quantified using two different stains applied to adjacent brain slices: cresyl violet, which reveals live cells and Fluoro-Jade B, which stains dead cells.
The effect of 4R on DFP-induced neuronal death in brain slices stained with Fluoro-Jade B is shown in Fig. 6. Many dead cells were observed in stratum pyramidale of rats exposed to DFP without 4R (PI-DFP-5hDMSO, Fig. 6A), while all three groups injected with DFP and 4R as well as the all-vehicles control (SSW-5hDMSO) displayed few or no dead cells (Fig. 6B–E). The quantitative analysis of these data is shown in Fig. 6F. The group exposed to DFP without 4R had significantly more dead neurons than the vehicles control or the groups where 4R was applied 1 hour before or 24 hours after DFP; the animals injected with 4R 5 hours after DFP showed a 70% decrease in dead cells but barely missed the 0.05 significance level (p<0.07). The three groups that received 4R did not significantly differ among themselves.
Figure 6. Experiment 2: 4R decreased neuronal death produced by the PI-DFP treatment.
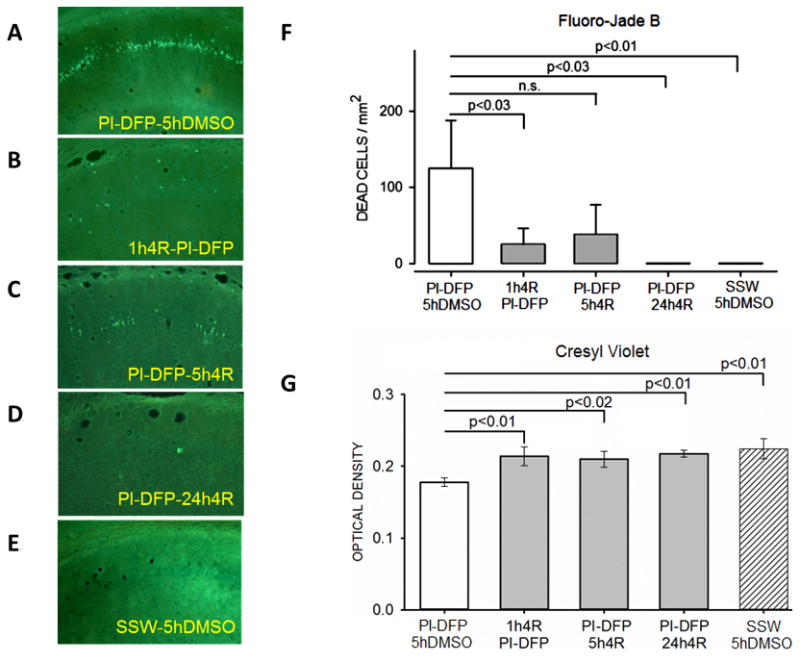
See Fig. 2B for experimental design. There were five experimental groups. The determinations were done in adjacent slices of the same brains. (A-E) Representative micrographs from the CA1 hippocampal area stained with Fluoro-Jade B. (F) The bar graph represents the mean ± SEM of dead cell density determined with Fluoro-Jade B. (G) The bars represent the mean ± SEM of the optical density of live cells stained with cresyl violet. The number of rats per group was PI-DFP-DMSO (10), 1h4R-PI-DFP (6), PI-DFP-5h4R (5), PI-DFP-24h4R (4), and SSW-5hDMSO (6). See statistical analysis in Methods.
Alternate slices were stained with cresyl violet, which shows live cells. Results from the stratum pyramidale of the CA1 area are shown Fig. 6G. The cell density of the group injected with DFP without 4R (PI-DFP-5hDMSO) was significantly reduced from the level found in the all-vehicles control (SSW-5hDMSO, p<0.01). By contrast, the cell density of all three groups that received DFP and 4R was increased to the level of the vehicles control group, and each was significantly higher than the PI-DFP-5hDMSO group.
Taken together, these results show that 4R significantly protected the cells from DFP at all three times tested.
3.4 Experiment 3. 4R administered 1 hour before DFP protected against brain neurodegeneration
The goal of experiment 3 was to confirm the neuroprotective effect of 4R injected 1 hour before DFP. An additional goal was to expand our understanding of DFP and 4R effects on brain degeneration by using three different histological methods on alternate slices of each brain: Fluoro-Jade B staining, amino cupric silver staining, and nestin immunocytochemistry. Fluoro-Jade B and amino cupric silver staining detect degenerating neurons, but the latter provides better resolution, showing axons, dendrites, and synaptic terminals, in addition to cell bodies.
Twenty rats were used, 10 preinjected with 4R then treated with DFP (1h4R-PI-DFP) and 10 rats preinjected with DMSO before DFP (1hDMSO-PI-DFP). Racine scores were determined for 2½ hours after pyridostigmine injection. Forty-eight hours after DFP, both groups, were perfused for histological analysis (Fig. 2C).
3.4.1 Racine scores
All animals displayed strong behavioral convulsions after DFP injection, which lasted for the whole observation period and were not affected by the preinjection of 4R (data not shown). Total convulsions for the first 90 min period were (mean ± SEM): 534 ± 32 for 1h4R-PI-DFP and 519 ± 25 for 1hDMSO-PI-DFP. There was no significant difference between the groups (n=10 for both groups). These results reproduced those from Experiment 2 shown in Fig. 5.
3.4.2 Brain histology: the effect of 4R on neuronal death and astrocyte activation
Staining with either Fluoro-Jade B or amino cupric silver showed that neurodegeneration was not present in all brains. Forty percent (4/10) of subjects displayed dead neurons in the CA1 hippocampal area irrespective of whether the animals received 4R or DMSO. However, the density of dead cells was significantly lower in the group preinjected with 4R (Fluoro-Jade B, Fig. 7A, p<0.02; amino cupric silver, Fig. 7B, p<0.006).
Figure 7. 4R injected 1 hour before DFP decreased neuronal death and astrocyte activation.
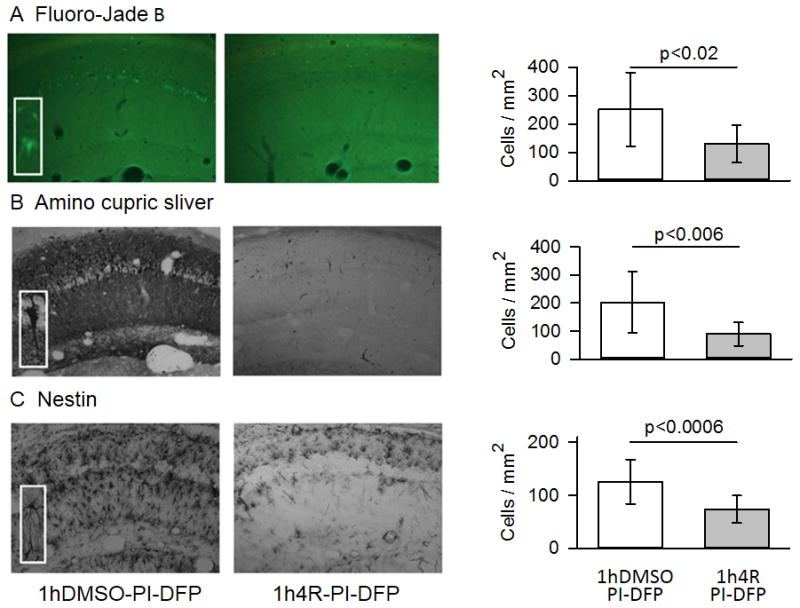
Micrographs (10X) are from a 4R vehicle control (1hDMSO-PI-DFP) and a rat preinjected with 4R (1h4R-PI-DFP). The magnification of the insets for Fluoro-Jade B was 30X and for amino cupric silver and nestin, 40X. (A) Fluoro-Jade B staining. The number of Fluoro-Jade B stained cells in the stratum pyramidale was significantly lower in the 4R group than in the 4R vehicle control (n =10 rats per group). (B) Amino cupric silver staining. The number of amino cupric silver-stained cells in the stratum pyramidale was significantly lower in the 4R group than in the 4R vehicle controls (n=10 per group). (C) Immunostaining for nestin. The number of activated astrocytes in the stratum radiatum was significantly lower in the 4R group than the 4R vehicle control which was injected with DMSO (n= 6 rats per group). The bars are means ± SEM. See statistics in Methods. See Fig. 2C for experimental design.
Nestin immunostaining revealed a large number of labeled cells in the stratum radiatum, but few in the stratum pyramidale (Fig. 7C). Judging from their localization, morphology, and cross-reactivity with GFAP, these cells appear to be activated astrocytes (see insert in Fig. 7C and Fig. 8). Activated astrocytes were seen in 83% (5/6) of subjects in both experimental groups. The density of activated astrocytes was significantly lower in the 4R group (1h4R-PI-DFP) than in the 4R vehicle control (1hDMSO-PI-DFP) group (Fig. 7C, p<0.0006).
Figure 8. Nestin and GFAP immunostaining overlap in DFP-activated astrocytes.
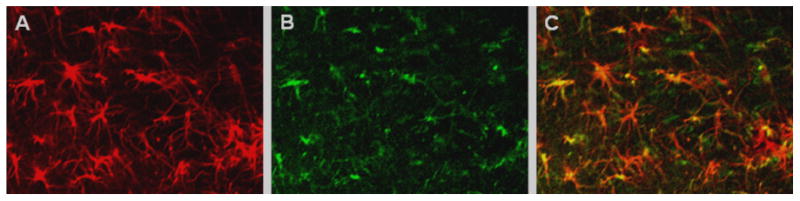
Confocal photomicrographs from coronal cortical sections from rats treated with DFP in the PI-DFP paradigm. (A) Immunostaining with antibody against GFAP. (B) Immunostaining with antibody against nestin. (C) Most GFAP-labeled cells overlap with nestin-positive cells. See Methods.
The hippocampal CA1 area of four rats injected with saline, saline and water were immunostained for nestin to demonstrate that DFP was needed for the expression of nestin. No activated astrocytes were observed. The scarce cells labeled with the antibody did not present the morphology of activated astrocytes. A representative micrograph is shown in supplemental material.
In summary, 40% of subjects showed extensive neuronal death and 83% displayed significant neuroinflammation. 4R injected 1 hour before DFP significantly decreased the density of dead neurons in stratum pyramidale and the density of activated astrocytes in stratum radiatum.
3.4.3 Nestin antibody detected activated astrocytes
Initially, nestin was considered to be a selective marker of stem cells; however, numerous studies have shown that in adult brains nestin is predominantly a marker of activated astrocytes (Clarke et al., 1994, Holmin et al., 1997). To settle this question under our conditions, we double-labeled a cortical brain slice with nestin and GFAP antibodies. Nearly all nestin-labeled cells were also labeled by GFAP antibody, and these cells displayed the morphology of activated astrocytes (Fig. 8). In addition, the labeled cells were located in the stratum radiatum and were rarely present in the stratum pyramidale, as expected from astrocytes (Fig.7 and 9).
Figure 9. 4R was neuroprotective when injected 24 hours after DFP.
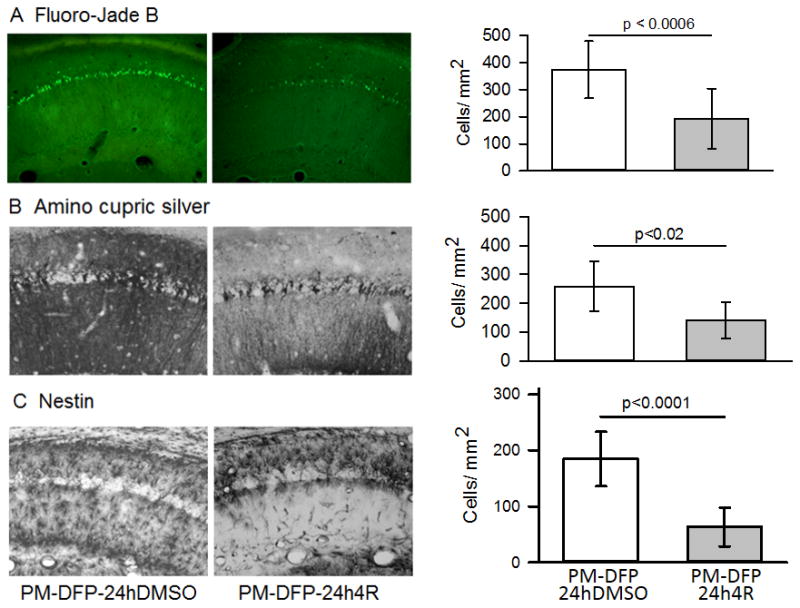
Experiment 4 (see Fig. 2D for experimental design and abbreviations). Fluoro-Jade B staining (A), amino cupric silver staining (B), and nestin immunostaining (C) were applied to adjacent slices of the same brains. The images show representative micrographs from two rats; a rat injected 24 hours after DFP with DMSO, the vehicle of 4R (PM-DFP-24hDMSO), and the experimental rat injected at the same time with 4R (PM-DFP-24h4R). The bar graphs show dead neurons in (A) and (B) and activated astrocytes in (C). The number of rats used for Fluoro-Jade B staining included 9 4R vehicle controls and 10 rats injected with 4R; for ACS and nestin there were 8 rats per group. The bars are means ± SEM. See statistics in Methods.
3.5 Experiment 4. 4R decreased neurodegeneration when administered 24 hours after DFP
The goal of experiment 4 was to expand the study of neuroprotection by 4R administered 24 hours after DFP. Such a long window of therapeutic opportunity is particularly relevant for potential therapeutic applications of 4R in situations of CWNA attack or industrial accidents.
Experimental and control animals were injected with pyridostigmine and methyl atropine followed by DFP. Methyl atropine instead of ipratropium was injected in this experiment at a dose equimolar to the dose of ipratropium used in experiments 1, 2, and 3. Twenty-four hours after DFP, the controls were injected with DMSO (PM-DFP-24hDMSO), while the experimental animals were injected with 4R (PM-DFP-24h4R). (See Fig. 2D for experimental design).
3.5.1 Racine scores
Racine scores were recorded to test whether pyridostigmine, methyl atropine, and DFP produced a comparable intensity of convulsions to the treatment with pyridostigmine, ipratropium, and DFP in experiment 3. Racine scores were recorded for 2 hours, starting 30 min before DFP. Since 4R or DMSO were injected 24 hours after DFP, at the time of Racine score recording both groups were identical. Total Racine scores of pyridostigmine-, methyl atropine-, and DFP-treated rats were practically identical to those of rats treated with pyridostigmine, ipratropium, and DFP (mean ± SEM): PM-DFP, 572 ± 22 (n=22); 1hDMSO-PI-DFP, 519 ± 25 (n=10); p=n.s.
3.5.2 Brain histology: the effect of 4R on neuronal death and astrocyte activation
Fluoro-Jade B staining showed neuronal death in 6/9 control rats (PM-DFP-24hDMSO) and 4/10 in 4R injected rats (PM-DFP-24h4R). 4R decreased the density of dead neurons in the stratum pyramidale by 48% (p < 0.0006, Fig. 9A).
Amino cupric silver staining showed that 5/8 rats from the control group and 2/8 4R rats presented extensive neurodegeneration in many areas of the forebrain but not in cerebellum or pons and medulla. The remaining rats did not show neuronal death in any brain area (data not shown). These observations coincided with those from experiment 3. In the CA1 hippocampal area, neuronal death was prominent in the stratum pyramidale of controls but was reduced in the 4R injected group (p < 0.02, Fig. 9B). Thus, the amino cupric silver and the Fluoro-Jade B stains applied to adjacent slices showed a similar degree of neuronal degeneration.
Immunostaining for nestin revealed a large number of activated astrocytes (Fig. 9C). Activated astrocytes were observed in 8/8 brains of 4R vehicle controls and in 7/8 brains from the 4R-treated group. The average number of activated astrocytes per square millimeter of stratum radiatum was decreased by 66% in the 4R group versus the 4R vehicle control group (p<0.0001).
In summary, application of 4R 24 hours after DFP significantly decreased the prevalence of neurodegeneration, the extent of neuronal death in the stratum pyramidale, and the density of activated astrocytes in the stratum radiatum.
3.6 Mortality and neurodegeneration under PI-DFP and PM-DFP conditions
The overall mortality of rats treated with 9 mg/kg of DFP was 13%. There was no difference in mortality between the PI-DFP and PM-DFP paradigms. The time of death ranged from 3 min to 27 hours after DFP. None of the control rats that did not receive DFP died.
Experiments 2 and 3 showed that the PI-DFP protocol produced extensive neuronal death in 40% of subjects, while the PM-DFP treatment produced neurodegeneration in 63% of subjects. Neuroinflammation, assessed as enhanced expression of nestin by active astrocytes, was detected in 90% of PI-DFP- and 100% of PM-DFP-treated subjects.
4. Discussion
The objective of this study was to determine whether 4R could ameliorate the neurodegeneration caused by acute DFP poisoning within a realistic time window. The primary finding was that 4R was indeed capable of reducing the neuronal damage caused by DFP when injected 24 hours after acute DFP intoxication. This finding is particularly relevant because, after the classical emergency treatment of victims of OP poisoning, there is no further treatment available to decrease the neurodegeneration that often follows. There is clearly a therapeutic gap in the medical treatment of the survivors of OP poisoning.
4.1 Experimental model
Rats were used here as a model of human exposure to nerve agents in combat or terrorist attack or to insecticides in industrial accidents. With this purpose in mind, the dose and vehicle of DFP were chosen to cause acute brain damage (Crawford et al., 2004, Deshpande et al., 2010).
The optimal neuroprotective dose of 4R in vivo is not known; however, at 6 mg/kg, 4R did not alter exploratory activity in naïve rats but abolished behavioral sensitization to nicotine (Ferchmin et al., 2001). In collaboration with SRI (Stanford Research International; Menlo Park, California), it was found that up to 98 mg/kg 4R was not toxic (manuscript in preparation). In vitro experiments suggest that even at lower doses, 4R could be effective (Ferchmin et al., 2005, Eterovic et al., 2011). On the basis of these in vivo and in vitro results, 6 mg/kg was used in this work.
As mentioned before, to study brain neurodegeneration caused by OPs, mortality has to be reduced with peripherally acting antidotes, including muscarinic antagonists such as methyl atropine. Methyl atropine was unavailable for the first three experiments; therefore, we resorted to its analogue ipratropium. Methyl atropine became available for the fourth experiment. Since we were the first to use ipratropium in this field, it was important to test methyl atropine in experiment 4 not only to compare the effect of both muscarinic antagonists in these experiments, but also to facilitate the comparison of our results with that of others. Methyl atropine and ipratropium are positively charged on the nitrogen and do not significantly penetrate the blood–brain barrier. We compared the efficacy of 4R neuroprotection using methyl atropine and ipratropium and found no significant difference. Likewise, Racine scores were equally affected by both drugs, suggesting that they are pharmacologically equivalent.
The effectiveness of DFP was tested by determining its capacity to inhibit AChE. Under our conditions, AChE was profoundly inhibited within minutes after DFP and recovered during the next 3 weeks, with different speeds in each brain area. Our data agree with the inhibition of AChE previously reported (Li et al., 2011) in those brain areas and times where comparison was possible. There is no direct correlation between inhibition of AChE and neurodegeneration. The inhibition of AChE by DFP was robust in cerebellum and pons and medulla, but those areas did not display neurodegeneration (data not shown). Therefore, it is reasonable to surmise that the return to normal AChE activity is not tightly correlated with recovery of function or morphology in the different brain areas. Under our conditions, DFP completely inhibited the activity of AChE and induced convulsions in all the subjects, but less than 50% had brain damage. This is explained by the finding that central seizures, not peripheral convulsions, correlate with neurodegeneration (Crawford et al., 2004). Recently, it was reported that the nerve agent soman failed to inhibit AChE in the basolateral amygdala (BLA) in a fraction of the animals. Status epilepticus and brain damage was observed only in those brains where AChE was inhibited in BLA (Prager et al., 2013). The explanation of why soman did not inhibit AChE in the BLA of some animals remains unknown. It is likely that, for the same reason, 9 mg/kg of DFP caused neurodegeneration in only about 50% of our rats.
4.2 Neuroprotection by 4R
We examined the window of therapeutic opportunity for 4R using cresyl violet, which estimates undamaged neurons and Fluoro-Jade B, which measures damaged neurons. The data from both methods supported the notion that 4R was neuroprotective when injected 1 hour before or 24 hours after DFP. Furthermore, the neuroprotective effects of 4R injected 1 hour before or 24 hours after DFP was confirmed in experiments 3 and 4 with Fluoro-Jade B and amino cupric silver staining. This report was focused on DFP-mediated neurotoxicity and 4R neuroprotection in the hippocampal area CA1; results from other brain areas will be presented in the future. A preliminary analysis of the damage and neuroprotection in other brain areas showed extensive damage in piriform cortex and amygdala. There was no damage in cerebellum and medulla. Our results were comparable to previously published data (Li et al., 2011). DFP inhibits the neuropathy target esterase (Ehrich et al., 1995, Qian et al., 2007), and therefore is a neuropathogenic OP that causes organophosphorous ester-induced delayed neurotoxicity (OPIDN). However, in this work OPIDN was not addressed, because rats are not a preferred model to study OPIDN, and hens are usually used for that purpose. In addition, the full expression of axonal neuropathy requires more time than the 48-hour period studied here (Ehrich et al., 1995, Sarin and Gill, 2000).
Two independent experiments showed that preinjection of 4R one hour before DFP did not alter Racine scores. This proves that 4R is not an anticonvulsant, in agreement with the report of the NIH-Anticonvulsant Screening Program (James Stables, 2007, personal communication). Most probably, the neuroprotection against DFP by 4R depends on the activation of antiapoptotic and anti-inflammatory pathways that protect against the delayed consequences of seizures, which include glutamatergic excitotoxicity and inflammation (Kadriu et al., 2009, Li et al., 2011). Therefore, it is not surprising that 4R is neuroprotective, despite its lack of anti-seizure activity. A similar mechanism was reported for neuroprotection by neuregulin-1 (Li et al., 2012).
Inflammation plays an important role not only in OP neurodegeneration but also in other neurodegenerative pathologies (Chapman et al., 2006, Johnson and Kan, 2010, Banks and Lein, 2012). DFP-mediated expression of GFAP confirms that neuroinflammation is a component of DFP pathology (Liu, 2012). In our model, nestin is co-expressed with GFAP; therefore, we used it as an index of neuroinflammation. The normal adult mammalian brain expresses very little nestin, mainly in endothelial and selected subventricular cells. However, a significant upregulation of nestin is observed after cerebral injury suggesting that nestin is involved in brain repair (Frisen et al., 1995, Holmin et al., 1997, Douen et al., 2004). There is abundant evidence in the literature that after brain injury nestin expression in mature brains is a marker of reactive astrocytes (Clarke et al., 1994, Lin et al., 1995, Duggal et al., 1997, Holmin et al., 1997, Wei et al., 2002, Douen et al., 2004, Moreels et al., 2008). The lack of nestin expression, in our conditions, in the absence of DFP is shown in supplementary data. However, 4R injected 1 hour before, or 24 hours after DFP robustly reduced the expression of nestin, showing that 4R is anti-inflammatory, with a window of opportunity of 24 hours.
The molecular mechanism of the 4R neuroprotection was not studied in vivo. However, in vitro work with hippocampal slices has shown that 4R, through a nicotinic mechanism, activates the pro-survival protein kinase Akt/PKB and inactivates the proapoptotic GSK3-β (Ferchmin et al., 1993, Ferchmin et al., 2013). It is likely that a similar mechanism acts in vivo. The brain damage caused by DFP is a dynamic process that involves necrosis, inflammation, and apoptosis in variable proportions, depending on the time after poisoning and the brain area. During the first 24 hours, DFP kills neurons by necrosis and apoptosis. This initial damage triggers NO-mediated glutamate release (Kim et al., 1997, Kim et al., 1999, Neitz et al., 2011) and further excitotoxic brain injury, resulting in additional neuronal death during the next 24 hours (Li et al., 2011, Li et al., 2012). We hypothesize that the long window of therapeutic opportunity of 4R is possible because 4R inhibits the second wave of neuronal death through its antiapoptotic and anti-inflammatory activity.
In conclusion, 4R is neuroprotective against the neurotoxicity of DFP by a mechanism that does not involve decreasing seizures. Instead, 4R seems to neuroprotect by inhibiting the apoptosis and inflammation caused by seizures. 4R appears to be a promising new neuroprotective drug that could be used by first responders as a prophylactic or administered to victims 24 hours after poisoning and treatment with classical antidotes.
Supplementary Material
4R decreases DFP-induced neuronal death.
DFP increases and 4R decreases the expression of nestin in activated astrocytes.
4R neuroprotects with a window of therapeutic opportunity of 24 hours.
Acknowledgments
We are grateful to Dr. Pamela Lein (Univ. of California Davis) for valuable advice on working with toxic organophosphates and for teaching the AChE assay. We wish to thank Dr. Byron Ford (Morehouse School of Medicine) for many interesting discussions and useful advice on the organophosphate field and training B. Cuadrado in the Fluoro-Jade B assay. We also want to thank Dr. Maria Braga (United States Army Medical Research Institute for Chemical Defense) for her counsel on the use of the Racine scale and other aspects of organophosphate research. We are very grateful for the important contributions of Dr. Rolando E. Dias Olivos, clinical neurologist (Universidad Central del Caribe School of Medicine), who revised the behavioral categories on the Racine scale on the basis of video recordings of our experimental subjects undergoing DFP-caused convulsions; Dr. José A. Quidgley, who trained our personnel in performing rat perfusions for histological analysis; and Dr. Aldo Perez, who performed a preliminary histological analysis of various brain regions that focused our attention on the CA1 hippocampal region. Ms. Dinely Perez contributed in all in vivo procedures, for which the authors are most grateful.
This work was supported by NIH grants NINDS5U01NS063555 to P.A. F.; 1U54NS083924-01 to AHM, and 8G12MD007583 to UCC. Dr. Jae Eun Lee's work on this project was supported by the National Institute on Minority Health and Health Disparities (NIMHD) under Award Number of U54MD008149.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Banks CN, Lein PJ. A review of experimental evidence linking neurotoxic organophosphorus compounds and inflammation. Neurotoxicology. 2012;33:575–584. doi: 10.1016/j.neuro.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical biochemistry. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Chapman S, Kadar T, Gilat E. Seizure duration following sarin exposure affects neuro-inflammatory markers in the rat brain. Neurotoxicology. 2006;27:277–283. doi: 10.1016/j.neuro.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Chen Y. Organophosphate-induced brain damage: mechanisms, neuropsychiatric and neurological consequences, and potential therapeutic strategies. Neurotoxicology. 2012;33:391–400. doi: 10.1016/j.neuro.2012.03.011. [DOI] [PubMed] [Google Scholar]
- Clarke SR, Shetty AK, Bradley JL, Turner DA. Reactive astrocytes express the embryonic intermediate neurofilament nestin. Neuroreport. 1994;5:1885–1888. doi: 10.1097/00001756-199410000-00011. [DOI] [PubMed] [Google Scholar]
- Craigie EH, Zeman W, Innes JRM. Neuroanatomy of the Rat. Academic Press; 1963. [Google Scholar]
- Crawford SM, Compton JR, Tetz LM, Ratcliffe RH, Steele KH, Gordon RK, Nambiar MP. Development Of A Rat Diisopropylfluorophosphate-Induced Seizure/Status Epilepticus Model For Screening Of Neuroprotectants Following Exposure To Chemical Warfare Agents. Paper presented at the Scientific Conference on Chemical and Biological Defense Research; Hunt Valley, MD. 2004. pp. 1–15. A976944. [Google Scholar]
- Damodaran TV, Attia MK, Abou-Donia MB. Early differential cell death and survival mechanisms initiate and contribute to the development of OPIDN: A study of molecular, cellular, and anatomical parameters. Toxicol Appl Pharmacol. 2011;256:348–359. doi: 10.1016/j.taap.2011.07.017. [DOI] [PubMed] [Google Scholar]
- de Olmos JS, Beltramino CA, de Olmos de Lorenzo S. Use of an amino-cupric-silver technique for the detection of early and semiacute neuronal degeneration caused by neurotoxicants, hypoxia, and physical trauma. Neurotoxicology and teratology. 1994;16:545–561. doi: 10.1016/0892-0362(94)90033-7. [DOI] [PubMed] [Google Scholar]
- Deshpande LS, Carter DS, Blair RE, DeLorenzo RJ. Development of a prolonged calcium plateau in hippocampal neurons in rats surviving status epilepticus induced by the organophosphate diisopropylfluorophosphate. Toxicol Sci. 2010;116:623–631. doi: 10.1093/toxsci/kfq157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douen AG, Dong L, Vanance S, Munger R, Hogan MJ, Thompson CS, Hakim AM. Regulation of nestin expression after cortical ablation in adult rat brain. Brain research. 2004;1008:139–146. doi: 10.1016/j.brainres.2003.08.070. [DOI] [PubMed] [Google Scholar]
- Duggal N, Schmidt-Kastner R, Hakim AM. Nestin expression in reactive astrocytes following focal cerebral ischemia in rats. Brain research. 1997;768:1–9. doi: 10.1016/s0006-8993(97)00588-x. [DOI] [PubMed] [Google Scholar]
- Ehrich M, Jortner BS, Padilla S. Comparison of the relative inhibition of acetylcholinesterase and neuropathy target esterase in rats and hens given cholinesterase inhibitors. Fundamental and applied toxicology : official journal of the Society of Toxicology. 1995;24:94–101. doi: 10.1006/faat.1995.1011. [DOI] [PubMed] [Google Scholar]
- El Sayed KA, Laphookhieo S, Baraka HN, Yousaf M, Hebert A, Bagaley D, Rainey FA, Muralidharan A, Thomas S, Shah GV. Biocatalytic and semisynthetic optimization of the anti-invasive tobacco (1S,2E,4R,6R,7E,11E)-2,7,11-cembratriene-4,6-diol. Bioorg Med Chem. 2008;16:2886–2893. doi: 10.1016/j.bmc.2007.12.056. [DOI] [PubMed] [Google Scholar]
- Ellman GL, Courtney KD, Andres V, Jr, Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Eterovic VA, Del Valle-Rodriguez A, Perez D, Carrasco M, Khanfar MA, El Sayed KA, Ferchmin PA. Protective activity of (1S,2E,4R,6R,7E,11E)-2,7,11-cembratriene-4,6-diol analogues against diisopropylfluorophosphate neurotoxicity: preliminary structure-activity relationship and pharmacophore modeling. Bioorganic & medicinal chemistry. 2013;21:4678–4686. doi: 10.1016/j.bmc.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eterovic VA, Perez D, Martins AH, Cuadrado BL, Carrasco M, Ferchmin PA. A cembranoid protects acute hippocampal slices against paraoxon neurotoxicity. Toxicol In Vitro. 2011;25:1468–1474. doi: 10.1016/j.tiv.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferchmin PA, Hao J, Perez D, Penzo M, Maldonado HM, Gonzalez MT, Rodriguez AD, de Vellis J. Tobacco cembranoids protect the function of acute hippocampal slices against NMDA by a mechanism mediated by alpha4beta2 nicotinic receptors. J Neurosci Res. 2005;82:631–641. doi: 10.1002/jnr.20666. [DOI] [PubMed] [Google Scholar]
- Ferchmin PA, Lukas RJ, Hann RM, Fryer JD, Eaton JB, Pagan OR, Rodriguez AD, Nicolau Y, Rosado M, Cortes S, Eterovic VA. Tobacco cembranoids block behavioral sensitization to nicotine and inhibit neuronal acetylcholine receptor function. J Neurosci Res. 2001;64:18–25. doi: 10.1002/jnr.1049. [DOI] [PubMed] [Google Scholar]
- Ferchmin PA, Pagan OR, Ulrich H, Szeto AC, Hann RM, Eterovic VA. Actions of octocoral and tobacco cembranoids on nicotinic receptors. Toxicon. 2009;54:1174–1182. doi: 10.1016/j.toxicon.2009.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferchmin PA, Perez D, Castro Alvarez W, Penzo MA, Maldonado HM, Eterovic VA. gamma-Aminobutyric acid type a receptor inhibition triggers a nicotinic neuroprotective mechanism. J Neurosci Res. 2013;91:416–425. doi: 10.1002/jnr.23155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferchmin PA, Rivera E, Eterovic VA. alpha-Difluoromethylornithine does not antagonize the behavioral effects of putrescine. Pharmacology, biochemistry, and behavior. 1993;45:967–971. doi: 10.1016/0091-3057(93)90149-n. [DOI] [PubMed] [Google Scholar]
- Frisen J, Johansson CB, Torok C, Risling M, Lendahl U. Rapid, widespread, and longlasting induction of nestin contributes to the generation of glial scar tissue after CNS injury. The Journal of cell biology. 1995;131:453–464. doi: 10.1083/jcb.131.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmin S, Almqvist P, Lendahl U, Mathiesen T. Adult nestin-expressing subependymal cells differentiate to astrocytes in response to brain injury. Eur J Neurosci. 1997;9:65–75. doi: 10.1111/j.1460-9568.1997.tb01354.x. [DOI] [PubMed] [Google Scholar]
- Johnson EA, Kan RK. The acute phase response and soman-induced status epilepticus: temporal, regional and cellular changes in rat brain cytokine concentrations. J Neuroinflammation. 2010;7:40. doi: 10.1186/1742-2094-7-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadriu B, Guidotti A, Costa E, Auta J. Imidazenil, a non-sedating anticonvulsant benzodiazepine, is more potent than diazepam in protecting against DFP-induced seizures and neuronal damage. Toxicology. 2009;256:164–174. doi: 10.1016/j.tox.2008.11.021. [DOI] [PubMed] [Google Scholar]
- Kim YB, Hur GH, Shin S, Sok DE, Kang JK, Lee YS. Organophosphate-induced brain injuries: delayed apoptosis mediated by nitric oxide. Environmental Toxicology and Pharmacology. 1999;7:147–152. doi: 10.1016/s1382-6689(99)00006-x. [DOI] [PubMed] [Google Scholar]
- Kim YB, Hur GH, Lee YS, Han BG, Shin S. A role of nitric oxide in organophosphate-induced convulsions. Environmental Toxicology and Pharmacology. 1997;3:53–56. doi: 10.1016/s1382-6689(96)00139-1. [DOI] [PubMed] [Google Scholar]
- Li Y, Lein PJ, Liu C, Bruun DA, Giulivi C, Ford GD, Tewolde T, Ross-Inta C, Ford BD. Neuregulin-1 is neuroprotective in a rat model of organophosphate-induced delayed neuronal injury. Toxicol Appl Pharmacol. 2012;262:194–204. doi: 10.1016/j.taap.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lein PJ, Liu C, Bruun DA, Tewolde T, Ford G, Ford BD. Spatiotemporal pattern of neuronal injury induced by DFP in rats: a model for delayed neuronal cell death following acute OP intoxication. Toxicol Appl Pharmacol. 2011;253:261–269. doi: 10.1016/j.taap.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RC, Matesic DF, Marvin M, McKay RD, Brustle O. Re-expression of the intermediate filament nestin in reactive astrocytes. Neurobiol Dis. 1995;2:79–85. doi: 10.1006/nbdi.1995.0008. [DOI] [PubMed] [Google Scholar]
- Liu C, Yonggang Li, Lein Pamela J, Ford Byron D. Spatiotemporal patterns of GFAP upregulation in rat brain following acute intoxication with diisopropylfluorophosphate (DFP) Current Neurobiology. 2012;3:90–97. [PMC free article] [PubMed] [Google Scholar]
- Miyaki K, Nishiwaki Y, Maekawa K, Ogawa Y, Asukai N, Yoshimura K, Etoh N, Matsumoto Y, Kikuchi Y, Kumagai N, Omae K. Effects of sarin on the nervous system of subway workers seven years after the Tokyo subway sarin attack. Journal of occupational health. 2005;47:299–304. doi: 10.1539/joh.47.299. [DOI] [PubMed] [Google Scholar]
- Moreels M, Vandenabeele F, Dumont D, Robben J, Lambrichts I. Alpha-smooth muscle actin (alpha-SMA) and nestin expression in reactive astrocytes in multiple sclerosis lesions: potential regulatory role of transforming growth factor-beta 1 (TGF-beta1) Neuropathol Appl Neurobiol. 2008;34:532–546. doi: 10.1111/j.1365-2990.2007.00910.x. [DOI] [PubMed] [Google Scholar]
- Neitz A, Mergia E, Eysel UT, Koesling D, Mittmann T. Presynaptic nitric oxide/cGMP facilitates glutamate release via hyperpolarization-activated cyclic nucleotide-gated channels in the hippocampus. The European journal of neuroscience. 2011;33:1611–1621. doi: 10.1111/j.1460-9568.2011.07654.x. [DOI] [PubMed] [Google Scholar]
- Pope C, Karanth S, Liu J. Pharmacology and toxicology of cholinesterase inhibitors: uses and misuses of a common mechanism of action. Environ Toxicol Pharmacol. 2005;19:433–446. doi: 10.1016/j.etap.2004.12.048. [DOI] [PubMed] [Google Scholar]
- Prager EM, Aroniadou-Anderjaska V, Almeida-Suhett CP, Figueiredo TH, Apland JP, Braga MF. Acetylcholinesterase inhibition in the basolateral amygdala plays a key role in the induction of status epilepticus after soman exposure. Neurotoxicology. 2013;38:84–90. doi: 10.1016/j.neuro.2013.06.006. [DOI] [PubMed] [Google Scholar]
- Qian Y, Venkatraj J, Barhoumi R, Pal R, Datta A, Wild JR, Tiffany-Castiglioni E. Comparative non-cholinergic neurotoxic effects of paraoxon and diisopropyl fluorophosphate (DFP) on human neuroblastoma and astrocytoma cell lines. Toxicol Appl Pharmacol. 2007;219:162–171. doi: 10.1016/j.taap.2006.11.030. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Ray DE, Richards PG. The potential for toxic effects of chronic, low-dose exposure to organophosphates. Toxicol Lett. 2001;120:343–351. doi: 10.1016/s0378-4274(01)00266-1. [DOI] [PubMed] [Google Scholar]
- Sarin S, Gill KD. Biochemical characterization of dichlorvos-induced delayed neurotoxicity in rat. IUBMB Life. 2000;49:125–130. doi: 10.1080/15216540050022449. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade: novel fluorochromes for detecting toxicant-induced neuronal degeneration. Toxicol Pathol. 2000;28:91–99. doi: 10.1177/019262330002800111. [DOI] [PubMed] [Google Scholar]
- Sparenborg S, Brennecke LH, Beers ET. Pharmacological dissociation of the motor and electrical aspects of convulsive status epilepticus induced by the cholinesterase inhibitor soman. Epilepsy Res. 1993;14:95–103. doi: 10.1016/0920-1211(93)90014-x. [DOI] [PubMed] [Google Scholar]
- Terry AV., Jr Functional consequences of repeated organophosphate exposure: potential non-cholinergic mechanisms. Pharmacol Ther. 2012;134:355–365. doi: 10.1016/j.pharmthera.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry AV, Jr, Buccafusco JJ, Gearhart DA, Beck WD, Middlemore-Risher ML, Truan JN, Schwarz GM, Xu M, Bartlett MG, Kutiyanawala A, Pillai A. Repeated, intermittent exposures to diisopropylfluorophosphate in rats: protracted effects on cholinergic markers, nerve growth factor-related proteins, and cognitive function. Neuroscience. 2011;176:237–253. doi: 10.1016/j.neuroscience.2010.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei LC, Shi M, Chen LW, Cao R, Zhang P, Chan YS. Nestin-containing cells express glial fibrillary acidic protein in the proliferative regions of central nervous system of postnatal developing and adult mice. Brain Res Dev Brain Res. 2002;139:9–17. doi: 10.1016/s0165-3806(02)00509-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.