

Abstract
The role of voltage-gated Ca2+ channels (VGCCs) in spontaneous miniature neurotransmitter release is incompletely understood. Here we show that stochastic opening of P/Q-, N-, and R-type VGCCs accounts for ~50% of all spontaneous glutamate release at rat cultured hippocampal synapses, and that R-type channels play a far greater role in spontaneous than in action potential-evoked exocytosis. VGCC-dependent ‘minis’ show similar sensitivity to presynaptic Ca2+ chelation as evoked release, arguing for direct triggering of spontaneous release by transient spatially localized Ca2+ domains. Experimentally constrained three-dimensional diffusion modeling of Ca2+ influx-exocytosis coupling is consistent with clustered distribution of VGCCs in the active zone of small hippocampal synapses, and shows that spontaneous VGCCs openings can account for the experimentally observed VGCC-dependent minis, although single channel openings trigger release with low probability. Uncorrelated stochastic VGCC opening is thus a major trigger for spontaneous glutamate release, with differential roles for distinct channel subtypes.
Introduction
Action potential-independent spontaneous miniature neurotransmitter release has important physiological functions including regulation of dendritic protein expression and maintenance of spines in glutamatergic synapses1, 2. Although ‘minis’ occur without extracellular Ca2+ the majority of spontaneous release under physiological conditions is Ca2+-dependent (e.g. refs.3, 4). Unlike action potential-evoked release, which is triggered by transient activation of presynaptic voltage-gated Ca2+ channels (VGCCs) and formation of local Ca2+-nano/microdomains in the immediate vicinity of release-ready synaptic vesicles5, Ca2+ regulation of miniature release remains incompletely understood. It has recently been shown that stochastic opening of presynaptic VGCCs at resting membrane potential (Vrest) is a major trigger of spontaneous release at GABAergic synapses with either tight (‘Ca2+-nanodomain’)6 or loose (‘Ca2+-microdomain’)7 coupling between VGCCs and vesicular Ca2+-release sensors. These findings are consistent with the low affinity Ca2+ sensor synaptotagmin-1 being the major Ca2+ sensor not only for evoked but also for spontaneous release4. In contrast, VGCCs have been reported not to be involved in triggering spontaneous glutamate release, because the broad non-specific inorganic VGCC blocker Cd2+ (50–100 μM) failed to consistently reduce the frequency of miniature excitatory postsynaptic currents (mEPSCs) (e.g. refs. 8, 9). This difference between spontaneous GABA and glutamate release is paradoxical, because the mechanisms of evoked release are broadly similar between inhibitory and excitatory central synapses4, 5. Evoked release of glutamate at central synapses is triggered by mixed populations of P/Q-type, N-type, and R-type VGCCs10-15. How these different VGCCs contribute to spontaneous release, and whether they do so through similar spatiotemporal domains as in evoked release, remains incompletely understood.
Here we use electrophysiological, optical, pharmacological and modeling approaches to understand the roles of different VGCC subtypes in spontaneous glutamate release at small hippocampal synapses. We show that stochastic uncorrelated activation of individual VGCCs at Vrest is a major source of spontaneous glutamate release. We further show that, consistent with a lower voltage activation threshold, R-type VGCCs are much more efficient in triggering spontaneous release than P/Q- and N-type channels. Finally, by comparing the effects of fast (BAPTA) and slow (EGTA) Ca2+ buffers on spontaneous and evoked release, complemented by experimentally constrained modeling of Ca2+ dynamics and activation of vesicular release sensors, we demonstrate that VGCC-dependent minis can be accounted for by fast, transient, 25–70 nm Ca2+-nano/microdomains around single VGCCs that open spontaneously at Vrest.
Results
Blockade of VGCCs inhibits spontaneous glutamate release
To determine the contribution of different presynaptic VGCCs to spontaneous miniature release at small excitatory synapses we tested the effects of specific VGCC blockers on mEPSC frequency in cultured hippocampal neurons. Acute blockade of P/Q-type (with 0.25 μM ω-Agatoxin-IVA, ω-Aga), N-type (with 5 μM ω-Conotoxin-GVIA, ω-Ctx), or R-type channels (with 0.5 μM SNX-482, SNX), each significantly decreased the frequency of mEPSCs. In contrast, blockade of T-type channels (with 1.0 μM TTA-P2) had no significant effect (Fig. 1a–d, and Supplementary Table 1). We also observed no evidence for non-linear summation of the effects of individual blockers (Fig. 1e).
Figure 1.

Differential triggering of spontaneous excitatory neurotransmission by P/Q-, N- , and R-type Ca2+ channels. (a–c) Representative experiment illustrating the effect of P/Q- and N-type VGCC blockade with ω-Aga and ω-Ctx on excitatory miniature release. (a) mEPSCs before (Control) and 10 min after application of both toxins, (b) time-course of mEPSC frequency, and (c) cumulative distributions of mEPSC amplitudes before (black) and after (gray) application of toxins, showing no effect of ω-Aga and ω-Ctx. (d) Summary graphs showing effects of the selective P/Q-, N-, R-, and T-type channel blockers on mEPSC frequency (mean ± s.e.m, ω-Aga, N = 14; ω-Ctx, N = 12; SNX N = 5; ω-Aga and ω-Ctx, N = 25; ω-Aga, ω-Ctx and SNX, N = 17; TTA-P2, N = 12 cells). See also Supplementary Table 1. (e) Linear summation of ω-Aga, ω-Ctx and SNX effects on mEPSC frequency (mean ± s.e.m). Sums of individual effects of the VGCC blockers (black bars) were calculated using data from (d). *** P < 0.001, ** P < 0.01, * P < 0.05, NS P > 0.75; single group mean (d) and unpaired comparison (e) t-tests.
A quantitative comparison of the different blockers revealed that simultaneous application of ω-Aga and ω-Ctx decreased mEPSC frequency by 27.7 ± 3.7%, implying that approximately one quarter of spontaneous glutamate release depends on P/Q- and N-type VGCCs, which underlie the majority of evoked Ca2+ influx and vesicular release10, 12, 14. Surprisingly, SNX reduced mEPSC frequency by a similar extent (23.1 ± 5.7%) as the combined application of ω-Aga and ω-Ctx, despite a much smaller contribution of R-type channels to evoked Ca2+ influx and exocytosis10, 12, 14 (and see below). A disproportionate role for SNX-sensitive VGCCs in spontaneous release is however consistent with the hypothesis that VGCC-mediated glutamatergic minis are triggered by stochastic VGCC opening: R-type channels have been shown to activate at more negative potentials, with slower gating kinetics, than P/Q- or N-type channels12, 16. This implies that, at physiological Vrest (between −80 mV and −60 mV) R-type channels should open more often and for a longer time than P/Q- and N-type channels, and therefore should be more efficient in triggering miniature release. Furthermore, in agreement with the hypothesis that spontaneous VGCC openings can trigger exocytosis, increasing the opening probability of VGCCs by depolarizing the presynaptic membrane with raised extracellular [K+]ext (20 mM) increased the frequency of VGCC-dependent minis (Supplementary Fig. 1).
Roles of different VGCCs in presynaptic Ca2+ dynamics
We next compared the relative contributions of different VGCCs to presynaptic Ca2+ dynamics and to spontaneous miniature release by examining the effects of VGCC blockers on presynaptic [Ca2+] at rest and following action potentials using fast fluorescence imaging of the Ca2+ indicator Fluo-4 (Fig. 2a–d, Online Methods). Consistent with previous reports10, 12, 15 ω-Aga and ω-Ctx profoundly reduced the evoked Ca2+ fluorescence transient, normalized by the fluorescence of a morphological indicator AP ΔGAP/R (by 74.3 ± 6.4%, Fig. 2b,e), confirming that P/Q-type and N-type VGCCs are the major source of spike-evoked Ca2+ entry. Simultaneous application of ω-Aga, ω-Ctx, and SNX almost completely abolished the evoked Ca2+ transient (by 90.0 ± 6.4%, Fig. 2c,e) arguing that the ω-Aga- and ω-Ctx-resistant Ca2+ influx is mainly mediated by R-type VGCCs. The inorganic non-specific VGCC blocker Cd2+ (100 μM) completely abolished the residual evoked Ca2+ fluorescence transient (Fig. 2d,e), implying that it was mediated by toxin-insensitive VGCCs.
Figure 2.

Relative contributions of P/Q-, N-, and R-type VGCCs to presynaptic Ca2+ dynamics assessed with fast Ca2+ fluorescence imaging. (a) Fluorescence image of a typical cultured hippocampal neuron loaded with AlexaFluor 568 and Fluo-4 (Alexa channel is shown). Bottom left: magnified image of axonal fragment containing several presynaptic boutons; arrows indicate position of the line-scan used to measure Ca2+ transients illustrated in (b). Scale bars 20 μm and 4 μm. (b–d) Line-scan fluorescence responses to a single action potential (average of 5 sweeps) in typical boutons from representative experiments before (left) and after (right) application of different VGCC blockers as indicated. (e, f) Summary graphs illustrating differential effects of organic VGCC blockers and Cd2+ on action potential-evoked (e) and resting (f) presynaptic Ca2+ fluorescence (mean ± s.e.m, N = 9 (Control), N = 7 (ω-Ctx), N = 18 (ω-Aga + ω-Ctx), N = 40 (ω-Aga + ω-Ctx + SNX) and N = 13 (Cd2+) boutons from 4–6 independent experiments for each condition. Stability of Ca2+ responses in control experiments was tested after the same duration as for VGCC blockers. * P < 0.05, *** P < 0.001, NS P > 0.3, Wilcoxon signed rank test for single group median.
Unexpectedly, the organic VGCC blockers and Cd2+ had very different effects on the resting Fluo-4 fluorescence (Grest/R). While ω-Aga, ω-Ctx and SNX did not change Grest/R, Cd2+ caused a several-fold increase of Grest/R (Fig. 2b–d,f). This could be due to a Cd2+-induced elevation of intracellular Ca2+ levels, or to a gradual accumulation of presynaptic Cd2+ binding to the Ca2+ indicator17, 18. In either case, the increase in Ca2+ indicator fluorescence suggests that Cd2+ may substantially alter resting presynaptic concentrations of divalent cations, which could affect miniature release through a mechanism separate from the direct blockade of VGCCs. We therefore only used organic VGCC blockers (ω-Aga, ω-Ctx and SNX) in the rest of the study.
Distinct effects of BAPTA and EGTA on VGCC-mediated minis
How do presynaptic VGCCs trigger spontaneous glutamate release? One possibility is that VGCC-dependent minis are directly triggered by the formation of fast transient Ca2+-nano/microdomains around individual spontaneously opening channels, akin to those that underlie evoked exocytosis. Alternatively, stochastic opening of VGCCs may control the presynaptic resting [Ca2+]rest and thus affect the rate of miniature release on a slower timescale19. Ca2+ imaging however failed to detect a significant change in [Ca2+]rest upon blockade of VGCCs with the specific organic blockers (Fig. 2f). This argues against the hypothesis that stochastic opening of VGCCs facilitates spontaneous release simply by increasing [Ca2+]rest, and suggests instead that VGCC-dependent glutamatergic minis are directly triggered by the formation of transient Ca2+-nano/microdomains. If so, VGCC-dependent minis should be inhibited by intracellular Ca2+ chelation, because rapid Ca2+ transients are much more sensitive to exogenous Ca2+ buffers5 than [Ca2+]rest (which is determined by the balance of passive membrane Ca2+ fluxes and active pumping mechanisms20). To test this prediction we measured the effects of intracellular loading of either fast or slow membrane-permeable Ca2+ buffers (BAPTA-AM or EGTA-AM, respectively).
Both chelators applied at 20 μM significantly decreased the mEPSC frequency within 10–20 min: at steady state, BAPTA-AM by 42.6 ± 7.7% (Fig. 3a), and EGTA-AM by 37.8 ± 4.7% (Fig. 3b). At first sight the similar effects of BAPTA-AM and EGTA-AM argue that spontaneous glutamate release is triggered by relatively slow changes in intracellular [Ca2+]. However, the overall mini rate includes both VGCC-dependent minis and a residual population that persists in the presence of VGCC blockers (Fig. 1d). Moreover, the absolute concentrations of intracellular BAPTA and EGTA cannot be determined precisely in experiments where the AM esters are applied extracellularly21, so in principle BAPTA and EGTA may reach different steady-state concentrations in presynaptic terminals. Therefore, to compare the sensitivity of VGCC-dependent minis to BAPTA and EGTA, we measured the effects of ω-Aga, ω-Ctx and SNX on the mEPSC frequency in cultures pre-equilibrated with EGTA-AM or BAPTA-AM (Fig. 3c,d). Both Ca2+ chelators significantly reduced the effect of the combined VGCC blockers. However, while BAPTA-AM almost completely occluded the effect of the toxins (~7% inhibition, Fig. 3c, versus 52.1% in control conditions, Fig. 1d), EGTA-AM only reduced their effect by approximately half (28.6% inhibition, Fig. 3d, versus 52.1% in control conditions, Fig. 1d).
Figure 3.

Differential effects of slow (EGTA) and fast (BAPTA) exogenous Ca2+ buffers on VGCC-dependent minis. (a, b) Time-course of mEPSC frequency changes during incubation in BAPTA-AM (a) and EGTA-AM (b). Left, mEPSC traces from representative experiments before and after addition of the Ca2+ chelators. Right, average responses in N = 7 cells for BAPTA-AM and N = 9 cells for EGTA-AM. (c, d) Differential effects of VGCC blockers on mEPSC frequency in BAPTA-AM (c) and in EGTA-AM (d) pre-treated cultures (both in (c) and (d) N = 7 cells for ω-Aga and ω-Ctx, and N = 6 cells for ω-Aga, ω-Ctx, and SNX). To determine the remaining fraction of mEPSCs sensitive to VGCC blockade after BAPTA-AM and EGTA-AM treatment (shown on the right), the initial mEPSC frequencies in this set of experiments were normalized to the effects of BAPTA-AM or EGTA-AM determined in (a) and (b). In contrast to EGTA-AM, pretreatment with BAPTA-AM almost completely occluded the effect of toxins on miniature release. All data are mean ± s.e.m, *** P < 0.001 and ** P < 0.01, NS P > 0.3, single group mean t-test.
Distinct effects of BAPTA and EGTA on evoked exocytosis
The differential sensitivity of VGCC-dependent mEPSCs to BAPTA and EGTA observed here is similar to the effects of these buffers on evoked glutamate release at small central synapses in acute brain preparations5, 22. Indeed, in contrast to synapses with tight 10–20 nm Ca2+-nanodomain coupling, where EGTA fails to inhibit evoked release23, evoked release in small glutamatergic neocortical synapses is inhibited by intracellular EGTA in the millimolar range5, 22, and therefore is thought to be triggered by larger Ca2+-microdomains. Thus our data imply that VGCC-dependent glutamatergic minis in small hippocampal synapses are also mediated by the formation of transient Ca2+-microdomains in the vicinity of spontaneously opening VGCCs.
To test this model more directly we compared the effects of EGTA-AM and BAPTA-AM on VGCC-dependent mEPSCs to their effects on evoked release measured in the same preparation. Because whole-cell patch-clamp recordings would inevitably perturb cytoplasmic integrity in the presynaptic axon, and likely interfere with BAPTA-AM and EGTA-AM equilibration, we used an alternative approach based on imaging of evoked vesicular exocytosis in intact synapses with FM dyes24-26. We first labeled recycling vesicles with the fluorescent styryl dye SynaptoRedC1 (SRC1), and then measured the specific action potential-evoked SRC1 de-staining rate (kAP) during low frequency (0.5 Hz) stimulation, in control conditions or in the presence of BAPTA-AM or EGTA-AM (Fig. 4a–d and Online Methods). We have previously shown that kAP is proportional to the average release probability of individual release-ready vesicles25. The decrease of kAP in BAPTA- or EGTA-loaded boutons should thus report the specific reduction of evoked vesicular release by fast or slow Ca2+ chelation.
Figure 4.

FM dye imaging of action potential-evoked exocytosis reveals similar sensitivity of evoked and VGCC-dependent miniature release to presynaptic Ca2+ chelation. (a) Experimental paradigm (see also Online Methods). (b) Example of fluorescence loss in individual synaptic boutons during the time course of a typical control experiment. Arrows: individual boutons examined in (c). Scale bar 5 μm. (c) FM dye de-staining profiles in boutons 1 and 2; exponential fits of de-staining rates at rest (krest) and during 0.5 Hz action potential stimulation (kstim) are shown by red dashed lines. The effective specific action potential-evoked FM dye (SRC1) de-staining rate in each bouton was calculated as kAP = kstim – krest. (d) Effect of BAPTA-AM and EGTA-AM on SRC1 de-staining kinetics. Average de-staining profiles from three representative experiments: Control (black), BAPTA-AM (gray) and EGTA-AM (white) symbols. Each profile is an average of 100–200 boutons. (e) Comparison of BAPTA-AM and EGTA-AM relative effects on the rate of action potential-evoked vesicular exocytosis kAP (left 2 bars, BAPTA-AM N = 6 experiments, EGTA-AM N = 5 experiments) and on the frequency of VGCC-dependent minis (right 2 bars). The latter was calculated from electrophysiological mEPSC recordings in Figs. 1 and 3 as detailed in Online Methods. Data are mean ± s.e.m, * P < 0.05, *** P < 0.001 unpaired t-tests.
Both BAPTA-AM and EGTA-AM loading significantly decreased kAP in comparison to interleaved control cultures (Fig. 4d). BAPTA-AM almost completely blocked evoked exocytosis (by ~87%), while EGTA-AM inhibited it only by 33% (Fig. 4e, left pair of bars). A similar effect of EGTA-AM on evoked glutamate release in cultured hippocampal neurons has previously been reported by imaging vesicular release with vGLut-pHluorin27. Thus we conclude that, similar to small glutamatergic synapses in the brain, evoked release at synapses in hippocampal cultures is mediated by Ca2+-microdomains that are quenched by BAPTA and reduced by EGTA. Notably, the observed BAPTA-AM and EGTA-AM induced reductions in evoked release were quantitatively similar to the calculated specific effects of these chelators on VGCC-dependent mEPSCs (Fig. 4e, right pair of bars and Online Methods).
The conclusion that VGCC-dependent glutamatergic minis are triggered by transient Ca2+-microdomains is however difficult to reconcile with previous modeling studies that suggest that evoked release in synapses with loose Ca2+-microdomain coupling is likely to be controlled by several tens of VGCCs 5, 28 (although see ref.29). Indeed, the steep power relationship between vesicular release probability and presynaptic Ca2+ influx14, 22, 25, 28-31 implies that the release rate should be very low following stochastic opening of individual VGCCs. To address this paradox we assessed the properties of VGCC-dependent evoked and spontaneous glutamate release in small hippocampal boutons using quantitative modeling of presynaptic Ca2+ dynamics and activation of vesicular fusion.
Numbers of different VGCCs in an average synaptic bouton
To constrain the model parameters we first estimated the numbers of P/Q-, N-, and R-type VGCCs in an average presynaptic bouton. We simulated the stochastic behavior of different VGCC subtypes during action potentials using a six-state gating kinetic model describing VGCCs in hippocampal mossy fiber boutons12 (Fig. 5a, Supplementary Fig. 2 and Online Methods). By integrating the average Ca2+ current traces (including failures) (Fig. 5b) we estimated the number of Ca2+ ions entering the bouton during an action potential through single P/Q-type (nP/Q = 84), N-type (nN = 52), or R-type (nR = 248) channels.
Figure 5.

Estimation of the numbers of P/Q-, N-, and R-type VGCCs in an average presynaptic bouton. (a) Gating model for presynaptic VGCCs. (b) Average (500 simulations) action potential-evoked current traces (including failures) through P/Q-, N-, and R-type channels. Action potential waveform is shown above. Integration of the current traces (colored areas) yields estimates for the average number of Ca2+ ions entering the bouton following an action potential through single VGCCs of each subtype. (c) Representative Ca2+ imaging experiment. Top left, an axonal fragment loaded with Fluo-4 and AlexaFluor 568 (Alexa channel is shown), arrows indicate line-scan position, scale bar 2 μm. Top right, average of 5 line-scan Fluo-4 responses to a single spike followed by a saturating 100 Hz spike-train (brightness is color-coded). Bottom right, corresponding fluorescence time-course. Dashed lines, background (GBG) and maximal (Gm) Fluo-4 fluorescence. Brown curve, non-stationary single compartment model fit corresponding to Δ[Ca2+]total = 65 μM. (d, e) Distribution of [Ca2+]rest (d) and Δ[Ca2+]total (e) in 242 boutons from 12 axons. Insert in (e), the number of Ca2+ ions (NCa2+ ~3,650) entering a bouton of volume Vbout ~0.122 μm3 during action potential corresponding to the average Δ[Ca2+]total = 62.0 μM (see text for details). (f) Pie-chart illustrating the complement of presynaptic VGCCs in an average presynaptic bouton. The colored areas correspond to the fractions of spike-evoked Ca2+ influx mediated by ω-Aga-sensitive P/Q-type VGCCs (blue, 45%), ω-Ctx-sensitive N-type channels (green, 30%) and SNX-sensitive R-type channels (brown, 15%) as determined in Fig. 2e, together with the corresponding numbers of VGCCs.
We next imaged Fluo-4 Ca2+ fluorescence transients triggered by single spikes in individual boutons, followed by 100 Hz spike-trains to define the saturated fluorescence level (Fig. 5c and Online Methods). We have previously shown that fast presynaptic Ca2+ dynamics are well approximated by a non-stationary single compartment model25. Using direct fitting of individual Ca2+ traces (Fig. 5c and Online Methods) we obtained an estimate of [Ca2+]rest = 53.9 ± 2.2 nM and of the total action potential-evoked Ca2+ concentration change Δ[Ca2+]total = 62.0 ± 1.3 μM (Fig. 5d,e). The latter value allowed us to estimate the average number of Ca2+ ions entering the bouton during an action potential as Ca2+ ions entering the bouton during an action potential as NCa2+ = Δ[Ca2+]total · λfree · Vbout · NA ~3.650 (where Vbout ~0.122 μm3 is the average bouton volume in cultures32,33, λfree is the fraction of intra-terminal volume free of synaptic vesicles, mitochondria, other presynaptic organelles, and cytomatrix32, 34, 35, and NA is Avogadro’s number). By multiplying NCa2+ by the proportions of spike-evoked Ca2+ influx mediated by each channel subtype (P/Q ~0.45, N ~0.3, and R ~0.15, Fig. 2e) we estimated the average number of Ca2+ ions entering the bouton through P/Q-, N-, and R-type channels (NP/Q ~1,640, NN ~1,100, and NR ~550). Finally by dividing these values by the number of Ca2+ ions entering through individual VGCCs (Fig. 5b) we estimated that an average presynaptic bouton contains ~20 P/Q-type, ~21 N-type, and ~2 R-type VGCCs (Fig. 5f).
Modeling of action potential-evoked glutamate release
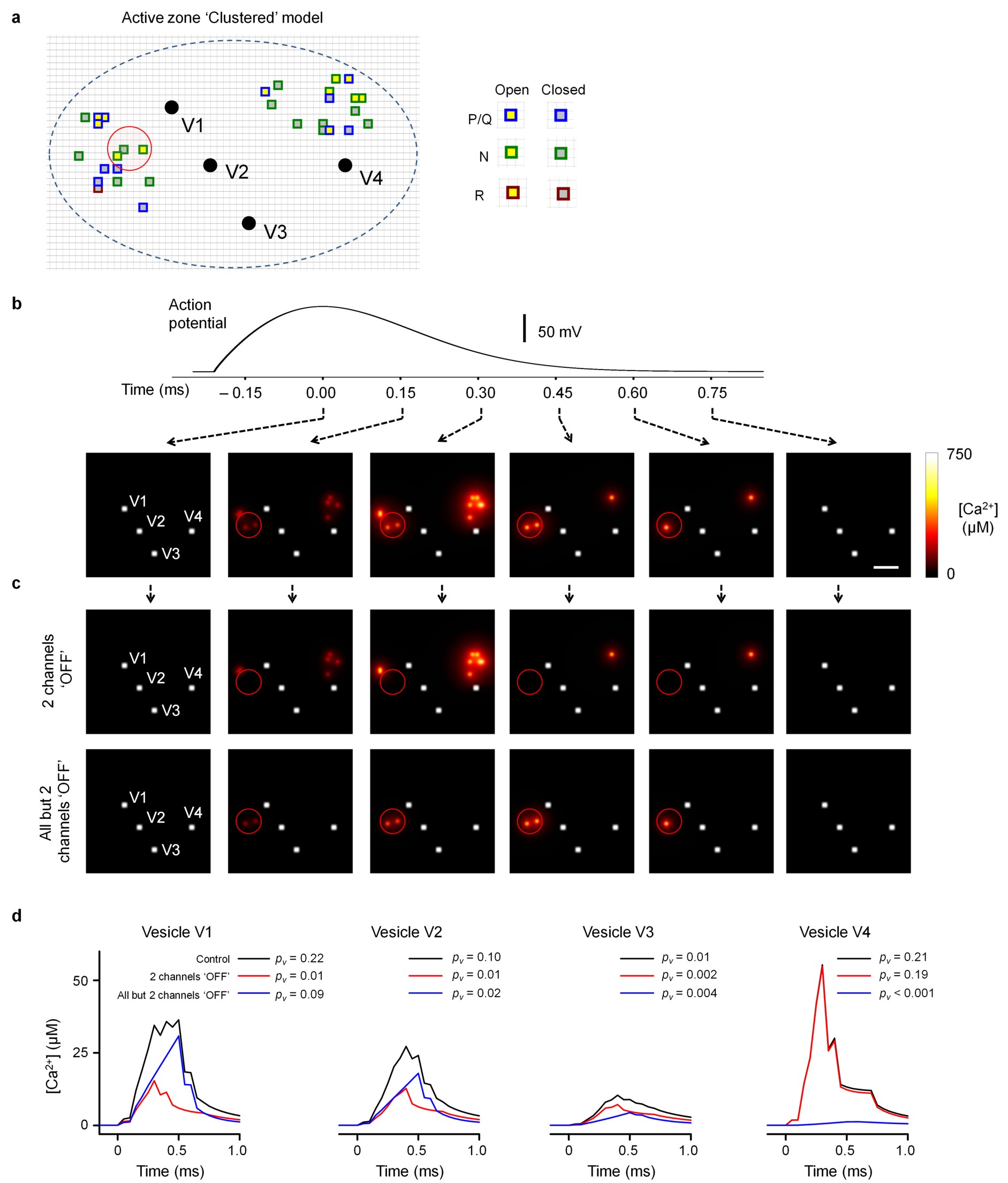
To model VGCC-glutamate release coupling in small hippocampal synapses we used an allosteric model of Ca2+ activation of vesicle fusion developed in the calyx of Held19 (Fig. 6a). Guided by available ultrastructural data15, 32 we considered a typical active zone as an elliptical disk (with area SAZ = 0.04 μm2) located within the truncated surface of a sphere corresponding to a synaptic bouton of radius Rbout = 0.35 μm (Fig. 6b). Each active zone contained 4 docked release-ready vesicles. Accumulating experimental data demonstrate that presynaptic VGCCs in central synapses are almost exclusively located in the active zone11, 15, 23, 33. Because small hippocampal boutons contain on average 1.3 active zones15, 32 we adjusted the numbers of VGCCs in a whole bouton by this factor and considered that a typical active zone contains: ~15 P/Q-type (equivalent to ~375 μm−2), ~16 N-type (~400 μm−2), and ~1.5 R-type VGCCs (~37.5 μm−2). This result is consistent with an estimate of the P/Q-type channel density in the active zones of small CA3 synapses obtained with immunogold electron microscopy (~396 μm−2 or ~16 P/Q-type channels)15. The exact distribution of different VGCCs subtypes within the active zone however remains largely unknown. Most P/Q-type VGCCs are not distributed uniformly in the active zones of small CA3 glutamatergic synapses, but instead occur in oval clusters with characteristic dimensions of 50–100 nm15. In some active zones however the spatial distribution of P/Q-type VGCCs could not be distinguished from a random distribution. Therefore we initially considered both limiting cases in parallel: ‘Clustered’ and ‘Random’ VGCC distributions in the active zone (Fig. 6c). To account for the EGTA sensitivity of VGCC-dependent release (Fig. 4e) we also assumed that the minimal distance between VGCCs and vesicular release sensors was 25 nm (ref. 5).
Figure 6.

Modeling action potential-evoked release in small hippocampal synapses. (a) Allosteric model of Ca2+ activation of vesicle fusion19. (b) Presynaptic bouton geometry used in VCell simulations. Scale bar 0.5 μm. (c) Representative distributions of VGCCs and vesicles in the active zone for Clustered (left) and Random (right) models. Top, XY cross-sections 2.5 nm above the active zone; bottom, XZ cross-sections corresponding to black dashed lines in the XY plane. Blue dashed lines, active zone borders; brown dots, VGCCs; black dots, space occupied by vesicles; gray circles, vesicle projections on the XY plane; green dots, locations of Ca2+-release sensors, grid 5 nm. (d) Simulation results corresponding to geometries in (c). Top, action potential waveform; middle, average [Ca2+] transients at Ca2+-release sensors; bottom, corresponding release rates; legends, resulting fusion probabilities pv. (e) Cumulative probability plots of pv for Clustered and Random models. (n = 28 vesicles from 7 simulated synapses for each model). (f) Cumulative probability plots showing the average number of VGCCs located within a given distance from the vesicular Ca2+-release sensors (n = 240 vesicles from 60 simulated synapses). (g) Model predictions for inhibition of evoked release by BAPTA and EGTA. Dotted lines show the experimental effects of BAPTA-AM and EGTA-AM as determined in Fig. 4e. (h) Dependency of pv on Δ[Ca2+]total simulated by progressive deletion of active VGCCs. Data are from 5 simulated synapses, each point represents average pv for 4 release-ready vesicles. Data on both axes are normalized to the corresponding maximal values at basal conditions. Dotted lines, fitted power function, with the slope corresponding to Ca2+ current cooperativity mICa = 2.46.
The results of two typical simulations for the Clustered and Random models are illustrated in Fig. 6c,d. Each run consisted of the following steps (see Online Methods for details): (1) generate the spatial distribution of docked synaptic vesicles and VGCCs within the active zone (Fig. 6c); (2) simulate the action potential-evoked Ca2+ influx and three-dimensional buffered diffusion using the Virtual Cell (VCell) environment, and estimate [Ca2+] transients at vesicular release sensors (Fig. 6d middle traces); and (3) calculate vesicular release rates (Fig. 6d bottom traces) and corresponding vesicle fusion probabilities pv (Fig. 6d top) using the allosteric model of the Ca2+ activation of vesicle fusion19 (Fig. 6a).
For each model we simulated exocytosis in seven active zones with different VGCC-vesicle distributions and different realizations of stochastic VGCC behavior during an action potential (i.e. in total 28 vesicles for each model). Although the overall VGCC density in the active zone and the total evoked Ca2+ influx were the same in the Clustered and Random models, the average peak [Ca2+]peak at the vesicular release sensors and the corresponding vesicular fusion probability pv were substantially lower in the Clustered model ([Ca2+]peak = 27.7 μM, coefficient of variation CV = 0.648; pv = 0.09, CV = 0.82) than in the Random model ([Ca2+]peak = 49.4 μM, CV = 0.61; pv = 0.29, CV = 0.88) (Fig. 6d,e). This difference was a direct consequence of a lower number of VGCCs located in the immediate vicinity of docked vesicles (Fig. 6f) in the Clustered than in the Random model. In agreement with this, evoked release in the Clustered model was more sensitive to Ca2+ chelation than in the Random model, and as expected for synapses with loose VGCC-release coupling, both models revealed differential effects of BAPTA and EGTA on evoked vesicle fusion (Fig. 6g). Notably, whilst the average pv predicted by the Clustered model (0.09) is within the range of experimentally determined average pv values at hippocampal synapses (0.05–0.1)24, 25, 30, the average pv predicted by the Random model (0.29) is several-fold higher. Thus our modeling results are consistent with the hypothesis that the majority of presynaptic VGCCs within the active zone are indeed clustered15.
How many VGCCs from each cluster contribute to the release of a single vesicle during an action potential? In other words, what is VGCC cooperativity (mCh) of triggering glutamate release at small hippocampal synapses? The six-state VGCC gating model12 provides estimates for the opening probabilities of P/Q-type (Popen_P/Q = 0.5), N-type (Popen_N = 0.4), and R-type VGCCs (Popen_R = 0.32) during an action potential (Supplementary Fig. 2). Thus on average ~14 VGCCs are open in the whole active zone during an action potential, which yields an upper bound for mCh. To estimate the lower bound of mCh we simulated the dependency of pv on the magnitude of total calcium influx Δ[Ca2+]total as the number of open channels during an action potential decreased (without changing the currents through the channels that remained open) (Fig. 6h). This model corresponded to experiments where evoked Ca2+ influx was progressively reduced with slowly dissociating VGCC blockers29, 36. By fitting the obtained dependency with a power function we thus obtained a slope value for Ca2+ current cooperativity mICa = 2.5, which is close to the lower bound of VGCC cooperativity mCh37.
Furthermore our modeling showed that the apparent mCh differed among vesicles in the active zone. For vesicles with high pv which were located close to VGCC clusters (e.g. vesicles V1 and V4 in Fig. 6c Clustered model and Supplementary Fig. 3) release was almost completely controlled by the VGCCs from the nearest cluster and [Ca2+] transients around such vesicles were largely determined by the 2–3 closest VGCCs. This effect was most prominent at the end of the action potential repolarization phase when the channel open probability was low but the current through individual VGCCs was high because of the increased Ca2+ driving force. In contrast, in the other limiting case, for vesicles with low pv that were located further away from VGCC clusters (e.g. vesicle V3 in Fig. 6c Clustered model and Supplementary Fig. 3) release was jointly controlled by all the VGCCs that opened during the action potential.
Modeling of VGCC-dependent miniature glutamate release
To model VGCC-dependent miniature release we simulated [Ca2+] transients at vesicular release sensors produced by spontaneous openings of a single VGCC for different VGCC open-channel durations (Δt) and at different VGCC-vesicle distances (d) (Fig. 7a,b). Here we assumed a constant single channel current of 0.34 pA, corresponding to Vrest = −70 mV (Online Methods and ref. 38). Using the same allosteric model (Fig. 6a) and the simulated [Ca2+] transients corresponding to different (Δt,d) pairs we then calculated a pv (Δt,d) map, which showed that stochastic opening of a single VGCC can indeed trigger vesicular fusion, with a steep dependence on both the VGCC-distance and the open-channel duration (Fig. 7c).
Figure 7.

Modeling VGCC-dependent glutamate miniature release. (a) Schematics illustrating VCell simulations. As in Fig. 6c the black dot indicate space taken up by the vesicle in the XY plane 2.5 nm above the active zone; gray circle, vesicle projection on the active zone plane; green dots, assumed positions of Ca2+-release sensors, grid 5 nm. (b) Examples of average [Ca2+] transients at release sensors produced by single VGCC openings (for 0.33 ms) for 4 different VGCC-release sensor distances. Insert, corresponding vesicle fusion probabilities. (c) Color-coded map showing dependency of vesicle fusion probability pv(Δt,d) on VGCC-vesicle distance d and VGCC open-channel duration Δt. (d) Frequency histograms φ(Δt) for the durations of spontaneous P/Q-, N-, and R-type channel opening at Vrest obtained using the VGCC gating model12 (Fig. 5a). (e) Dependencies of vesicle fusion probability pv(d) on VGCC-vesicle distance for different VGCC subtypes. (f) Frequency histogram ψ(d) for the relative VGCC-vesicle distances in the Clustered model (n = 60 simulated active zones). (g) Dependency of spontaneous P/Q-, N-, and R-type channel opening on Vrest, calculated using the six-state VGCC gating model12 (Fig. 5a). (h) Distribution of Vrest in cultured hippocampal neurons (mean 71.9 ± 0.7 mV, n = 98 neurons). (i) Cumulative fractions of VGCC-mediated mEPSCs and VGCC numbers plotted as functions of the distance from the vesicular release sensor. ~90% of all VGCC-dependent minis are mediated by only ~20% of all VGCCs present in the active zone located within 70 nm of docked vesicles. (j) Model predictions for the effects of BAPTA and EGTA on VGCC-dependent mEPSC frequency. Dotted lines, experimental effects of BAPTA-AM and EGTA-AM as estimated in Fig. 4e.
We next determined the relative efficiency of different VGCCs in triggering spontaneous miniature release. For each channel subtype we multiplied the pv(Δt,d) map by the probability density function for open-channel duration φ(Δt) (Fig. 7d). By integrating the products over the entire range of possible open-channel durations we thus obtained pv(d)=∫pv(Δt,d)·φ(Δt)·d(Δt) dependencies of the vesicular fusion probability on VGCC-release sensor distance for P/Q-, N-, and R-type VGCCs (Fig. 7e). Consistent with a longer duration of spontaneous channel openings, R-type VGCCs were ~30 fold more efficient in triggering vesicular fusion than P/Q- and N-type channels (e.g. at d = 50 nm, for R-type VGCCs pv(d) ~0.03, whilst for P/Q- and N-type VGCCs pv(d) ~0.001, Fig. 7e). Finally, by multiplying pv(d) by the probability density function ψ(d) for the distribution of VGCC-release sensor distances in the Clustered model (Fig. 7f) and by integrating the product pv(d)·ψ(d) for each channel subtype over the entire range of distances, we estimated the average probabilities 〈pv〉 of vesicle fusion in response to single P/Q-, N-, and R-type VGCC openings (Table 1, 〈pv〉 ~0.0006 for P/Q- and N-type channels, and 〈pv〉 ~0.009 for R-type channels).
Table 1. Triggering of miniature glutamate release by spontaneous opening of P/Q-, N-, and R-type VGCCs. Model results.
| 〈pv〉 Average vesicle fusion probability in response to a single VGCC opening | ν Average single channel opening frequency (Hz) | NCh_type Number of VGCCs in an average active zone | VGCC-dependent mEPSC rate at a single synapse* (Hz) | |
|---|---|---|---|---|
| P/Q-type | 0.0006 | 0.067 | 15 | 0.0029 |
| N-type | 0.0006 | 0.069 | 16 | 0.0035 |
| R-type | 0.0089 | 0.115 | 1.5 | 0.0080 |
| Total | 32.5 | 0.0144 |
By taking into account that on average there are 4 release ready vesicles per active zone, and that there are ~ 1.3 active zones per synaptic bouton we considered the readily releasable pool size RRP = 5.2 vesicles.
To estimate VGCC-mediated miniature release rates in a typical bouton we modeled the stochastic opening of single P/Q-, N-, and R-type VGCCs at different Vrest (Fig. 7g, Supplementary Fig. 4 and Online Methods) and calculated the average frequencies of spontaneous channel opening (ν, Table 1) for each channel subtype corresponding to the experimentally determined distribution of Vrest in hippocampal neurons in culture (Fig. 7h). This allowed us to estimate the frequencies of VGCC-dependent minis mediated by each channel subtype in a typical synapse as: , where NCh_type stands for the average number of each VGCC subtype in the active zone, and RRP is the average size of the readily releasable pool of vesicles (Table 1).
Consistent with our experimental data (Fig. 1d) the modeling results confirmed that R-type VGCCs have a privileged role in triggering spontaneous glutamate release (~50% of VGCC-dependent minis). The frequency of all VGCC-dependent mEPSCs in an average synapse predicted by the model was fVGCC ~0.014 Hz (or ~0.85 vesicles per minute). Taking into account that this subset of minis accounts for ~50% of all mEPSCs (Fig. 1d), the model predicts the overall mEPSC rate at a single synapse ftotal ~0.028 Hz (or 1.7 vesicles per minute). This value is consistent with experimentally determined spontaneous vesicular release rates at individual synaptic boutons (0.7–2 vesicles per minute) 39-41. Furthermore, the average mEPSC frequency across all cells recorded in standard conditions was 5.3 ± 0.5 Hz (n = 110 recordings). Dividing this value by the model-predicted mEPSC rate in a single bouton yields a plausible estimate (~190) for the number of synapses whose activity can be readily monitored by somatic patch clamp in a typical neuron in culture. Thus the modeling results demonstrate that uncorrelated spontaneous opening of VGCCs at Vrest can fully account for the experimentally observed VGCC-dependent mEPSC rates.
What fraction of VGCCs contributes to spontaneous miniature release? To answer this we simulated VGCC-vesicle distributions according to the Clustered model (n = 60 synapses) and then calculated VGCC-dependent mEPSC rates during progressive removal of VGCCs that were more distant from the docked synaptic vesicles in the active zone. Direct comparison of the obtained cumulative fractions of VGCC-mediated minis and of VGCC number (plotted as functions of VGCC-vesicle distance, Fig. 7i) revealed that 90% of all VGCC-dependent mEPSCs were triggered by only 20% of all VGCCs that are located within 70 nm from the docked vesicles.
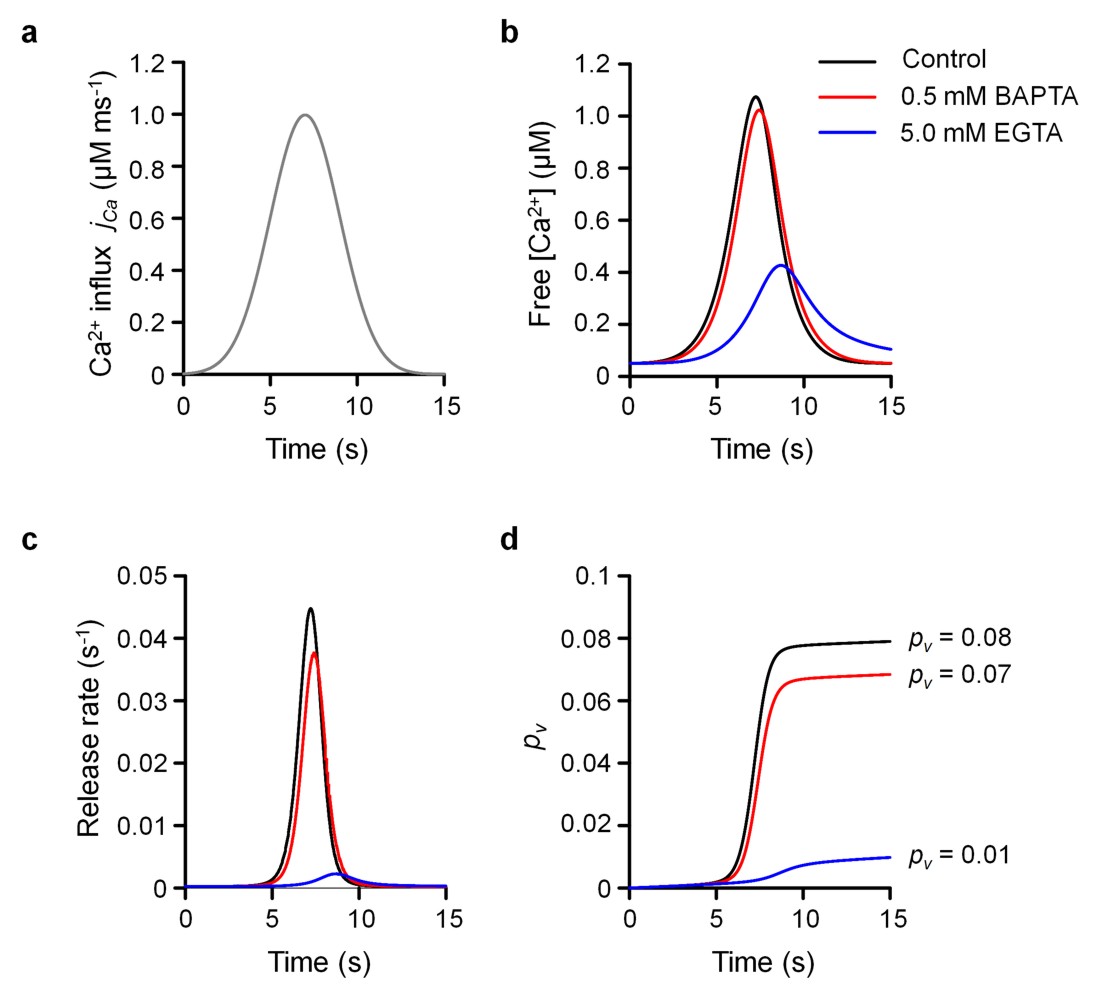
Similar to evoked release, model simulations revealed differential effects of BAPTA and EGTA on VGCC-dependent miniature release (Fig. 7j). Comparing the modeling results with the experimentally determined effects of BAPTA-AM and EGTA-AM (see also Fig. 6g) allowed us to make an estimate of the unknown concentrations of BAPTA (~0.5 mM) and EGTA (~5 mM) after AM-ester loading, implying that EGTA-AM in our experimental conditions was taken up by cultured hippocampal neurons ~10 fold more efficiently than BAPTA-AM. This provides a plausible explanation for the paradoxical finding that VGCC-dependent mEPSCs were much more sensitive to BAPTA-AM than EGTA-AM loading, whilst the overall mEPSC frequency was reduced to the same extent by both chelators (Fig. 3). Indeed, it is likely that at least some of the ~50% of mEPSCs that remain in the presence of VGCC blockers depend on slow global intracellular [Ca2+] changes as might occur from intracellular stores42, 43. Then according to this model 0.5 mM BAPTA should be much less efficient than 5 mM EGTA in inhibiting such minis, as demonstrated by modeling of vesicular release (using the same allosteric model) triggered by small (~1 μM) and slow (~2 s) elevation of presynaptic [Ca2+] from [Ca2+]rest = 50 nM (Supplementary Fig. 5).
Discussion
We have shown that presynaptic P/Q-, N-, and R-type VGCCs directly trigger approximately half of spontaneous miniature glutamate release events at small hippocampal synapses. The finding that the relative sensitivity of VGCC-dependent mEPSCs to EGTA and BAPTA was similar to that of action potential-evoked neurotransmitter release argues that both forms of exocytosis are triggered by similar Ca2+-nano/microdomains resulting from VGCC opening, which in turn are sensed by the same low-affinity Ca2+ sensor synaptotagmin-1 (ref. 4). The results of experimentally constrained modeling of Ca2+ influx-exocytosis coupling are consistent with this hypothesis, and show that spontaneous VGCCs openings can fully account for the experimentally observed VGCC-dependent minis, although single channel openings trigger release with low probability. Taken together, our results argue that stochastic VGCC opening can engage the same signaling cascade that underlies fast evoked neurotransmitter release, and thus challenge the view that action potential-evoked and action potential-independent release of glutamate are mediated by non-overlapping sources of Ca2+.
We have also identified an unexpectedly large role for R-type VGCCs in miniature release. This finding suggests a potential mechanism for differential regulation of evoked and spontaneous exocytosis at the level of individual presynaptic boutons. Indeed, the complement of presynaptic VGCCs varies substantially among small glutamatergic boutons both in cultures and the brain10, 13-15. Synapses with higher proportions of R-type channels are therefore expected to have significantly higher rates of spontaneous miniature release relative to evoked release.
It has been proposed that spontaneous release of GABA is triggered by synchronized activation of multiple VGCCs of different types6. Indeed, similar to evoked release, which is normally triggered by several VGCCs, ω-Aga and ω-Ctx caused non-additive supralinear reductions of GABAergic minis in cultured neocortical neurons6. In contrast, we did not observe such non-additive effects of VGCC blockers: simultaneous application of specific VGCC blockers reduced mEPSCs frequency to the same extent as the sum of the effects of the individual blockers (Fig. 1e). Furthermore, assuming independent operation of individual VGCCs, our modeling predicts that the baseline probability of coincident opening of more than one channel in the active zone is low (Supplementary Fig. 4). Thus our results argue that VGCC-dependent glutamatergic minis are triggered by uncorrelated opening of individual VGCCs.
At a first sight this finding contradicts to the prevailing view that several VGCCs are required to trigger exocytosis at synapses with loose Ca2+-microdomain VGCC-Ca2+ sensor coupling, where neurotransmitter release is sensitive to the slow Ca2+ buffer EGTA in the millimolar concentration range5, 22, 28. The detailed modeling of presynaptic Ca2+ dynamics and vesicular release performed here provides a plausible explanation for the apparent inconsistency. In line with previous modeling of the calyx of Held28 our model revealed a large heterogeneity of vesicular fusion probabilities within the same active zone. This was mainly a consequence of variable distances between docked vesicles and VGCC clusters. Furthermore, our simulations showed that the number of VGCCs that control vesicular fusion (i.e. VGCC cooperativity, mCh 37) varies several-fold depending on the position of an individual docked vesicle in the active zone. Release of ‘distal’ vesicles with low pv (located >100 nm away from VGCC clusters) was controlled by overlapping Ca2+ domains from all VGCCs in the active zone that open during an action potential (~14). In contrast, in the other limiting case, release of ‘proximal’ vesicles with high pv (located within ~30–40 nm of the nearest cluster) was mainly controlled by the 2–3 closest VGCCs. In a full agreement with this, our model demonstrated that 90% of all VGCC-dependent mEPSCs could be accounted for by stochastic opening of single VGCCs located within 25–70 nm of the docked vesicles.
It should be noted that although our results are consistent with non-uniform (Clustered) VGCC distribution in the active zone, the real distributions of different VGCCs subtypes and docked synaptic vesicles in active zones of central synapses remain largely unknown. Evidence exists for a direct interaction between presynaptic VGCCs and the release machinery44, 45, implying that some channels are likely to be directly tethered to the vesicular release sites. This subpopulation of VGCCs should therefore have a disproportionately important role in triggering both evoked and spontaneous release.
Previous studies have reported that blockade of VGCCs with the non-selective inorganic channel blocker Cd2+ either failed to reduce the rate of spontaneous glutamate release (e.g. at cortical8 and hippocampal9 synapses), or even increased the mEPSC frequency (e.g. in the spinal cord46). This apparent contradiction with the present evidence that VGCCs contribute to triggering miniature release can be explained by other presynaptic actions of Cd2+ than simple blockade of VGCCs. Indeed, there is accumulating evidence that Cd2+ applied extracellularly disrupts intracellular divalent cation homeostasis and leads to an elevation of intracellular Ca2+ and/or Cd2+ levels (e.g. refs. 17, 18 and Fig. 2f). An alternative possible explanation for previous evidence that Cd2+ fails to inhibit minis is that Vrest (the parameter that sets the rate of spontaneous VGCC opening) differed among studies. In our experimental conditions the average Vrest was approximately −72 mV (ranging from −55 to −80 mV), similar to previous estimates of Vrest in excitatory hippocampal neurons in acute brain preparations47-49. This argues that estimates of VGCC-dependent miniature release rates in cultured hippocampal neurons can be extrapolated to small excitatory synapses in the brain.
Does VGCC-mediated miniature release have a physiological significance, or is it simply a consequence of the inevitable spontaneous openings of VGCCs that are arranged within the active zone to optimize evoked release? Because the opening probability of VGCCs depends steeply on the membrane potential, spontaneous exocytosis is likely to be modulated by subthreshold axonal voltage fluctuations, including those propagating passively from the cell body50. Thus, VGCC-dependent minis potentially provide a mechanism for synaptic homeostasis1, 2 to be regulated by activity in the parent axon. This complements other forms of modulation of spontaneous exocytosis by neurotransmitters that act via presynaptic receptors and activate signaling cascades linked to intracellular Ca2+ stores (e.g. refs. 42, 43). It is tempting to speculate that the co-existence of VGCC-dependent and VGCC-independent minis provides for two forms of homeostasis, one reflecting the degree of depolarization of the presynaptic neuron, and the other reflecting the synaptic microenvironment, allowing complementary processes to control the excitability and development of neuronal networks.
Online Methods
Cell cultures and solutions
Animal care and use protocols were approved by the UK Home Office. Hippocampal neurons were isolated from P0–P1 Sprague Dawley rat pups and cultured in Neurobasal A / B27-based medium on a rat astrocyte feeder layer. All experiments were conducted in a custom-made open field-stimulation chamber (0.35 ml) at ambient temperature (23°C–25°C) 14–19 days after plating. The standard extracellular solution contained (in mM) 125 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 30 glucose, 25 HEPES (pH 7.4). Drugs were directly added to the recirculating perfusion system (total volume 10 ml) to achieve the following final concentrations: 0.25 μM ω-Aga (Bachem, Germany), 5 μM ω-Ctx, 0.5 μM SNX 482 (Abcam, UK), 1.0 μM TTA-P251 (Merck, USA), 100 μM CdCl2 (Sigma), 20 μM EGTA-AM and 20 μM BAPTA-AM (Invitrogen, USA). The time required for complete equilibration of the added drugs throughout the perfusion system (~3–4 min) was determined using the soluble fluorescence tracer Alexa Fluor 568 (Alexa 568, Invitrogen, USA).
Electrophysiology
Somatic whole-cell voltage-clamp recordings of spontaneous mEPSCs were performed in extracellular solution supplemented with 1 μM tetrodotoxin, 50 μM AP5, 50 μM picrotoxin and 5 μM CGP52432, using 3-6 MΩ pipettes filled either with a K gluconate based intracellular solution containing (in mM): 130 K gluconate, 10 KCl, 1 EGTA, 4 Mg-ATP; 0.5 Na2GTP, 10 phosphocreatine, 10 HEPES (pH 7.36), or with a CsCl based solution containing (in mM): 120 CsCl, 8 NaCl, 2 EGTA, 2 Mg-ATP, 0.3 Na-GTP, 5 QX314-Br, 10 phosphocreatine, 10 HEPES (pH 7.36). In some experiments 10 μM cyclothiazide was added to the extracellular solution to facilitate detection of mEPSCs. Recordings were obtained using a MultiClamp 700B amplifier (Molecular Devices, USA), filtered at 4 kHz, digitized at 10 kHz. Data acquisition and off-line analysis were performed using either Strathclyde Electrophysiology Windows Electrophysiology Disk Recorder (WinEDR v3.0.1, Dr. John Dempster, University of Strathclyde, Glasgow, UK) or LabView 8.0 (National Instruments, Austin, TX, USA). For mEPSC recordings neurons were held at −70 mV and the series resistance was monitored throughout the experiment using a −5 mV step command. All mEPSC recordings were performed between 10 min and 45 min after establishing the whole-cell configuration to allow for stable recording conditions. Cells showing a > 25% change in series resistance, or an unstable holding current, were rejected. The acute effects of organic VGCC blockers were determined by comparing mEPSC frequencies averaged over 5–10 min of continuous recording, immediately before and 10 min after toxin application. Individual mEPSCs were detected using a peak detection algorithm of MiniAnalysis 6.0 software (Synaptosoft Inc., Decatur, GA, USA), which measured the peak amplitude, and rise and decay times. Automated search parameters were selected based on the typical mEPSC kinetics. Amplitude threshold values were set at three times the root mean square of the baseline noise amplitude. All undetected events and false positives were corrected by visual inspection. Vrest was measured in the whole-cell current clamp mode (immediately after of breaking in, <1 min) with either K gluconate-based intracellular solution (used to record mEPSCs) or KMeSO3-based intracellular solution (used for Fluo-4 and Alexa 568 loading in imaging experiments, see below) and corrected for the liquid junction potentials (−15.5 mV for K gluconate and −10.2 mV for KMeSO3) calculated using Clampex software (Molecular Devices).
Estimation of the specific effects of BAPTA-AM and EGTA-AM on VGCC-dependent component of mEPSCs
The specific effects of BAPTA-AM and EGTA-AM treatment on VGCC-dependent minis (Fig. 4e) were estimated as follows. Let us denote: ftotal, average overall mEPSCs frequency in control conditions; fVGCC, average frequency of VGCC-dependent mEPSCs in control conditions; and , average mEPSCs frequencies after BAPTA-AM or EGTA-AM pre-incubation; and , average frequencies of VGCC-dependent mEPSCs after EGTA-AM or BAPTA-AM pre-incubation.
Then the proportions of VGCC-dependent mEPSCs remaining after BAPTA-AM or EGTAAM loading were calculated as and , using the experimentally determined values for the ratios , outlined in Supplementary Table 1. The error bars (s.e.m.) were calculated using the standard formula for error propagation: .
Imaging of presynaptic Ca2+ dynamics
Fluorescence imaging experiments were performed on an inverted LSM 510 confocal microscope (Zeiss) using a 63x (1.4 NA) oil immersion objective as previously described25. The high affinity calcium indicator Fluo-4 (Invitrogen, USA) was excited using the 488 nm line of an argon laser and its emission was recorded using a band-pass 505–550 nm filter. AlexaFluor 568 (Invitrogen, USA) was excited by a 543 nm He-Ne laser and its emission was recorded using a band-pass 560–615 nm filter.
To image presynaptic Ca2+ dynamics a neuron was loaded, via a whole-cell pipette, with 200 μM Fluo-4 and 200 μM AlexaFluor 568 in a solution containing (mM): 135 KMeSO3, 10 HEPES, 10 Na-phosphocreatine, 4 MgCl2, 4 Na2ATP, 0.4 Na2GTP. Five minutes after breaking in, the patch pipette was slowly withdrawn to minimize cytosol dialysis. Experiments were terminated if Vrest was above −55 mV, or if the gigaseal was lost during pipette withdrawal. The fluorophores were allowed to equilibrate throughout the neuron for at least 30 min after retracting the patch pipette, before Ca2+ fluorescence recordings were started. Action potentials were evoked by field stimulation via platinum bath electrodes separated by 1 cm (12.5–15 V, 1 ms pulses). To avoid recurrent activity, synaptic transmission was blocked in all imaging experiments by supplementing the extracellular solution with 25 μM NBQX, 50 μM AP5 and 50 μM picrotoxin.
Axons were identified using Alexa fluorescence by their thin shaft, tortuous trajectory, and occurrence of varicosities with diameter ~1 μm (Fig. 2a). Fluorescence transients in identified boutons were recorded in fast line-scan mode (~500 Hz, 5 trials averaged for analysis). The background fluorescence values in Fluo-4 (GBG) and Alexa (RBG) channels were determined in the image areas adjacent to the loaded axon and subtracted from the recorded fluorescence transients. At the beginning of each experiment the [Ca2+]rest in individual boutons was estimated using the formula (1): [Ca2+]rest = Kd · (Grest/Gm – 1/γ)/(1 – Grest/Gm) (ref. 52), where Kd = 350 nM and γ = 100 are the Ca2+ dissociation constant and dynamic range of Fluo-4 respectively52, and Gm is the maximal fluorescence of Fluo-4 determined during a saturating 100 Hz action potential train, as previously described25, 52. Experiments where the initial [Ca2+]rest was higher than 100 nM were rejected. To minimize optical artifacts during application of VGCC blockers the Fluo-4 fluorescence was normalized to the average AlexaFluor 568 fluorescence determined in each sweep (G/R ratio). The effect of VGCC blockers was measured 10–20 min after application. Stability of the baseline (Grest/R) and action potential-evoked Ca2+ responses (ΔGAP/R) during this time course were confirmed in control experiments (Fig. 2e,f). The sensitivity of the method to estimate the changes in [Ca2+]rest was principally limited by variability in basal Fluo-4 fluorescence during the time course of the experiment (Fig. 2f, Grest/R, coefficient of variation in control conditions CV ~30%). Formula (1) then yields an upper limit for the error in the [Ca2+]rest estimate ~20–40 nM.
FM dye imaging
FM dye imaging of vesicular release rates was performed as previously described25. Because hippocampal cultures are dominated by glutamatergic neurons (~94%)53 the FM-dye approach mainly reports vesicular release in glutamatergic synapses. The experimental paradigm is illustrated in Fig. 4a. Recycling vesicles were first labeled with 200 μM SRC1 by saturating high-frequency field stimulation (4 trains of 120 action potentials at 30 Hz delivered at 20 sec intervals) followed by dye washout for 15 min. SRC1 de-staining kinetics at rest and during low frequency stimulation (450 action potentials at 0.5 Hz) were imaged in a ~150 μm × 150 μm region of interest (ROI, 1024 × 1024 pixels) containing several hundred boutons. The background SRC1 fluorescence was determined by applying 3 rounds of high frequency stimulation used for loading. Images were analyzed using ImageJ (National Institutes of Health, Bethesda). Following the X-Y alignment of consecutive recorded SRC1 images, active boutons were identified by subtracting the five-frame background average from the five-frame average immediately after completion of SRC1 washout. SRC1 de-staining rates were measured in circular ROIs of 1.6 μm diameter centered at the fluorescence maxima of individual boutons. Boutons with overlapping ROIs were excluded from the analysis. The de-staining rates at rest (krest) and during 0.5 Hz stimulations (kstim) were calculated by fitting monoexponential functions to the fluorescence time course in each selected ROI, after subtracting the background value. Boutons with low signal to noise ratio (goodness of the fit χ2 > 0.025) were excluded from the analysis. To correct for spontaneous SRC1 vesicular release and for the non-specific loss of SRC1 fluorescence, the effective specific action potential-evoked release rate in each bouton was calculated as kAP = kstim – krest.
Modeling of VGCC gating
The stochastic behavior of VGCCs at rest and during action potentials was modeled in the NEURON simulation environment54 using the six-state channel gating kinetics previously used to describe P/Q-, N-, and R-type VGCCs in hippocampal mossy fiber boutons at room temperature12.
The model consisted of five closed and one open states:
Voltage-dependent forward and backward rates were calculated as follows: αi(V) = αi,0exp(V/ki), βi(V) = βi,0exp(V/ki). The values for forward and backward rates at 0 mV, αi,0 and βi,0, and for the slope factors ki were taken for each channel type from Table 2 in ref.12. We assumed the same action potential waveform as in the previous modeling study in the Calyx of Held28 but with action potential half-width of ~0.37 ms. This estimate (for recording temperature 23–25°C) was obtained by using the action potential half-width of ~0.2–0.25 ms recorded in the axons of CA3 pyramidal cells in slice hippocampal cultures at 32°C55, and the Q10 temperature coefficient for action potential duration ~1.8 (estimated from data in ref.56). Ca2+ currents were calculated as iCa2+(V) = g(V – ECa2+), where ECa2+ = 55 mV is the reversal potential for Ca2+ and g = 2.7 pS is VGCC conductance at 2 mM extracellular [Ca2+]ext 38 (we assumed the same conductance for all three VGCC subtypes11).
Non-stationary single compartment model of presynaptic Ca2+ dynamics
The total magnitude of action potential-evoked presynaptic Ca2+ influx was estimated using a non-stationary single compartment model, which was previously developed for quantification of fast action potential-evoked Ca2+ fluorescence transients(e.g. refs. 25, 57). The model assumes spatial homogeneity of Ca2+ concentration in the nerve terminal, and the presynaptic Ca2+ dynamics governed by the Ca2+ entry and removal rates and binding-unbinding reactions with the endogenous and exogenous buffers as well as the fluorescent Ca2+ indicator. These considerations are reflected in the system of equations below, where the square brackets denote concentrations, and the superscript indices of the reaction rate constants denote endogenous Ca2+ buffers Bi or the indicator I.
The action potential-dependent Ca2+ influx time course jCa was approximated by the Gaussian function where tAP denotes the time of peak of action potential-evoked Ca2+ current, and Δ[Ca2+]total denotes the total time integral of the volume averaged presynaptic Ca2+ entry. We assumed σ ~0.4 ms (Fig. 5b). The mass conservation rules for this system are: [I]total = [I]+[CaI], [Bi]total = [Bi]+[CaBi], Ca2+ removal was approximated by a first-order reaction Prem = krem ([Ca2+]–[Ca2+]rest)57.
We assumed that a typical small hippocampal excitatory presynaptic bouton contains two types of mobile endogenous buffers (ATP5, 28, 58 and calbindin-D28K 5, 59) and a single immobile endogenous buffer (calmodulin58, which is immobilized at rest due to its binging to neuromodulin60). The complete set of model parameters and Ca2+ binding reactions is specified in Supplementary Table 2. To estimate Δ[Ca2+]total in individual synaptic boutons the above model was numerically solved using the adaptive step-size Runge-Kutta algorithm, and the Fluo-4 fluorescence profile normalized to maximal fluorescence of the saturated indicator was calculated as , where γ ~100 is the dynamic range of Fluo-4. As was previously shown the model operated with only two adjustable (free) parameters Δ[Ca2+]total and krem which have virtually independent effects on Ca2+ fluorescence signal57. Thus both of these parameters were constrained by a straightforward fitting procedure that would match the calculated and experimental fluorescence profiles. The model predicts that the fast high affinity buffer calbindin-D28K has only limited effects on the peak amplitude of action potential-evoked Ca2+ fluorescence (10%–20% reduction for 100–200 uM of total calbindin binding sites)25, and that the low affinity buffers ATP and calmodulin have even smaller effects on the amplitude of Ca2+ fluorescence transients. Thus the error in determining of Δ[Ca2+]total due to possible variations in concentration of endogenous Ca2+ buffers was limited to 20%.
Modeling of action potential-evoked and spontaneous glutamate release
Modeling of Ca2+ influx and buffered diffusion was performed in the VCell simulation environment (http://vcell.org) using the fully implicit finite volume regular grid solver and a 5 nm mesh. In line with electron microscopy data15, 32, 34 the synaptic bouton was considered as a truncated sphere of radius Rbout = 0.35 μm (described by the equation [x2 + y2 + z2 < 0.352]·[z < 0.25], all distances are in μm). Readily releasable vesicles and VGCCs were placed within an ellipse shaped active zone of area SAZ = 0.04 μm2 (described by the equation [(x/0.146)2 + (y/0.089)2 <1]) situated in the centre of the truncated plane z = 0.25 according to either the Clustered or Random distribution models (as described below). Readily releasable vesicles docked at the active zone were described by the equations (x – xv)2 + (y – yv)2 + (z – 0.228)2 < Rv2, where xv and yv denote x and y coordinates of the vesicle centre and Rv = 0.02 μm is the outer vesicle radius. Ca2+ vesicular release sensors were assumed to be evenly distributed around the vesicle periphery within a 5 nm thick z-slice directly above the active zone (i.e. 12 voxels for each vesicle highlighted in green in Fig. 6c). The concentrations and properties of endogenous and exogenous Ca2+ buffers used in VCell simulations are specified in Supplementary Table 2. Ca2+ removal was approximated by a first-order reaction at the bouton surface (excluding the active zone): , where krem ~3600 s−1 was estimated by fitting experimental data with non-stationary single compartment model (described above), and the ratio of bouton volume to its surface area was = 0.104 μm. After performing several test simulations we restricted the computations to a dome described by the equation [x2 + y2 + (z – 0.35)2 < 0.352]·[z < 0.25] . This modification did not significantly affect Ca2+ dynamics calculated near the docked vesicles (less than 1% difference with the original model), but substantially increased the computation speed.
The spatial distributions of VGCCs and vesicles within the active zone used in VCell simulations were obtained from Monte Carlo simulations performed in MathCad 15.0 (Parametric Technology Corporation, USA). In the Clustered model we first randomly simulated positions of 2 ellipse shaped VGCC clusters (each 100 nm long and 50 nm wide) and then randomly distributed 32–33 VGCCs within these two clusters (i.e. VGCC density 4000 μm−2 within the clusters and 800 μm−2 in the whole active zone). We next randomly simulated position of 4 release-ready vesicles (vesicle centers were separated by a minimal distance of 45 nm to prevent them from overlapping). To account for the EGTA-sensitivity of action potential-evoked release the minimal distance between VGCC clusters and docked synaptic vesicles was set to 25 nm. For the Random model we fist simulated positions of 4 docked vesicles and then randomly distributed 32–33 VGCCs the whole active zone. Again, the minimal distance between VGCCs and docked synaptic vesicles was set to 25 nm. The subtype of each VGCC was randomly simulated according to the relative occurrence frequency: 20/43 (P/Q-type), 21/43 (N-type), and 2/43 (R-type) (Fig. 5f). Thus on average there were 15 P/Q-type, 16 N-type, and 1.5 R-type VGCCs in the active zone. Action potential-evoked Ca2+ currents through each of the VGCCs were simulated in NEURON simulation environment as described above.
Vesicular release rates were calculated using a previously published six-state allosteric model of Ca2+ activation of vesicle fusion in the calyx of Held (Fig. 6a)19. All the model parameters were as in the original calyx of Held model: kon = 1 × 108 M−1 s−1, koff = 4,000 s−1, b = 0.5, f = 31.3, and I+ = 2 × 10−4 s−1. At the beginning of each simulation we assumed that the relative occupancy of different model states corresponded to [Ca2+]rest = 50 nM.
The model was solved using a variable-order stiff multistep method based on the numerical differentiation formulas (function ode15s in MATLAB, MathWorks USA) for action potential-evoked Ca2+ concentration profiles obtained in VCell simulations at each of the 12 voxels surrounding the vesicle (assumed positions for Ca2+ release sensors, green pixels Fig. 6c). The average occupancies for the different model states 〈Vi(t)〉 were calculated by averaging the obtained 12 solutions. Finally the time dependency of vesicular release probability was calculated as and the release rate as Rrel = dpv(t)/dt. The three dimensional pv(Δt,d) maps (Fig. 7c) were obtained by piecewise cubic Hermite polynomial interpolation (pchip function in MATLAB) of the pv(Δt,d) array calculated on the grid corresponding to VGCC open times (ms) 0.03, 0.1, 0.33, 1.0, 2.0, and 3.0 and to VGCC-vesicle distances (nm) 20, 30, 40, 50, 60, 70, 80, 90, 100, 120, and 140.
Statistical analysis
All data are presented as mean ± s.e.m. The distribution of data in each set of experiments was first tested for normality using a Kolmogorov-Smirnov test. The similarity of variances between each group of data was tested using the F-test. For normally distributed data Student’s t-tests for single group mean, unpaired and paired comparisons were used as indicated. For the data that failed the normality test Wilcoxon singed rank tests for single group median, unpaired and paired comparisons were used. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications in the field6-8, 25. For normally distributed data, standard post hoc power calculations for t-test were performed using the experimentally determined standard deviations, to verify that the sample sizes were sufficient for detecting with 90% power 5–15% changes in the mean values at P < 0.05. Data collection and analysis were not performed blind to the conditions of the experiments. The effects of different VGCC blockers were tested in randomly interleaved experiments. All statistical tests were performed using SigmaPlot 11 (Systat Software, Inc.).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We are grateful to Leonid Savtchenko for help with stochastic VGCC modeling and to Yuri Ushkaryov, Dmitri Rusakov, Christian Henneberger, and Matthew Walker for critical reading of the manuscript, to Marife Cano for help with neuronal cultures, and to Victor Uebele (Merck, USA) for the gift of TTA-P2. The study was supported by the Medical Research Council, the Wellcome Trust, the Biotechnology and Biological Sciences Research Council, the German Research Foundation, the Brain Research Trust, the European Research Council, the Special Trustees of the UCLH NHS Foundation Trust, Epilepsy Research UK, and The Worshipful Company of Pewterers.
Reference List
- 1.Sutton MA, Wall NR, Aakalu GN, Schuman EM. Regulation of dendritic protein synthesis by miniature synaptic events. Science. 2004;304:1979–1983. doi: 10.1126/science.1096202. [DOI] [PubMed] [Google Scholar]
- 2.McKinney RA, Capogna M, Durr R, Gahwiler BH, Thompson SM. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat. Neurosci. 1999;2:44–49. doi: 10.1038/4548. [DOI] [PubMed] [Google Scholar]
- 3.Groffen AJ, et al. Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science. 2010;327:1614–1618. doi: 10.1126/science.1183765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu J, Pang ZP, Shin OH, Sudhof TC. Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat. Neurosci. 2009;12:759–766. doi: 10.1038/nn.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eggermann E, Bucurenciu I, Goswami SP, Jonas P. Nanodomain coupling between Ca(2+) channels and sensors of exocytosis at fast mammalian synapses. Nat. Rev. Neurosci. 2011;13:7–21. doi: 10.1038/nrn3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams C, et al. Coactivation of multiple tightly coupled calcium channels triggers spontaneous release of GABA. Nat. Neurosci. 2012;15:1195–1197. doi: 10.1038/nn.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goswami SP, Bucurenciu I, Jonas P. Miniature IPSCs in Hippocampal Granule Cells Are Triggered by Voltage-Gated Ca2+ Channels via Microdomain Coupling. J. Neurosci. 2012;32:14294–14304. doi: 10.1523/JNEUROSCI.6104-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vyleta NP, Smith SM. Spontaneous glutamate release is independent of calcium influx and tonically activated by the calcium-sensing receptor. J. Neurosci. 2011;31:4593–4606. doi: 10.1523/JNEUROSCI.6398-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scanziani M, Capogna M, Gahwiler BH, Thompson SM. Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus. Neuron. 1992;9:919–927. doi: 10.1016/0896-6273(92)90244-8. [DOI] [PubMed] [Google Scholar]
- 10.Wu LG, Saggau P. Pharmacological identification of two types of presynaptic voltage-dependent calcium channels at CA3-CA1 synapses of the hippocampus. J. Neurosci. 1994;14:5613–5622. doi: 10.1523/JNEUROSCI.14-09-05613.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheng J, et al. Calcium-channel number critically influences synaptic strength and plasticity at the active zone. Nat. Neurosci. 2012;15:998–1006. doi: 10.1038/nn.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li L, Bischofberger J, Jonas P. Differential gating and recruitment of P/Q-, N-, and R-type Ca2+ channels in hippocampal mossy fiber boutons. J. Neurosci. 2007;27:13420–13429. doi: 10.1523/JNEUROSCI.1709-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reid CA, Bekkers JM, Clements JD. N- and P/Q-type Ca2+ channels mediate transmitter release with a similar cooperativity at rat hippocampal autapses. J. Neurosci. 1998;18:2849–2855. doi: 10.1523/JNEUROSCI.18-08-02849.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mintz IM, Sabatini BL, Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- 15.Holderith N, et al. Release probability of hippocampal glutamatergic terminals scales with the size of the active zone. Nat. Neurosci. 2012;15:988–997. doi: 10.1038/nn.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soong TW, et al. Structure and functional expression of a member of the low voltage-activated calcium channel family. Science. 1993;260:1133–1136. doi: 10.1126/science.8388125. [DOI] [PubMed] [Google Scholar]
- 17.Regehr WG, Atluri PP. Calcium transients in cerebellar granule cell presynaptic terminals. Biophys. J. 1995;68:2156–2170. doi: 10.1016/S0006-3495(95)80398-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu B, et al. Calcium Signaling Is Involved in Cadmium-Induced Neuronal Apoptosis via Induction of Reactive Oxygen Species and Activation of MAPK/mTOR Network. PLoS ONE. 2011;6:e19052. doi: 10.1371/journal.pone.0019052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lou X, Scheuss V, Schneggenburger R. Allosteric modulation of the presynaptic Ca2+ sensor for vesicle fusion. Nature. 2005;435:497–501. doi: 10.1038/nature03568. [DOI] [PubMed] [Google Scholar]
- 20.Neher E. Usefulness and limitations of linear approximations to the understanding of Ca2+ signals. Cell Calcium. 1998;24:345–357. doi: 10.1016/s0143-4160(98)90058-6. [DOI] [PubMed] [Google Scholar]
- 21.Atluri PP, Regehr WG. Determinants of the time course of facilitation at the granule cell to Purkinje cell synapse. J. Neurosci. 1996;16:5661–5671. doi: 10.1523/JNEUROSCI.16-18-05661.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohana O, Sakmann B. Transmitter release modulation in nerve terminals of rat neocortical pyramidal cells by intracellular calcium buffers. J. Physiol. 1998;513:135–148. doi: 10.1111/j.1469-7793.1998.135by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bucurenciu I, Kulik A, Schwaller B, Frotscher M, Jonas P. Nanodomain coupling between Ca2+ channels and Ca2+ sensors promotes fast and efficient transmitter release at a cortical GABAergic synapse. Neuron. 2008;57:536–545. doi: 10.1016/j.neuron.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 24.Murthy VN, Schikorski T, Stevens CF, Zhu Y. Inactivity produces increases in neurotransmitter release and synapse size. Neuron. 2001;32:673–682. doi: 10.1016/s0896-6273(01)00500-1. [DOI] [PubMed] [Google Scholar]
- 25.Ermolyuk YS, et al. Independent Regulation of Basal Neurotransmitter Release Efficacy by Variable Ca2+ Influx and Bouton Size at Small Central Synapses. PLoS Biol. 2012;10:e1001396. doi: 10.1371/journal.pbio.1001396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaffield MA, Betz WJ. Imaging synaptic vesicle exocytosis and endocytosis with FM dyes. Nat. Protoc. 2006;1:2916–2921. doi: 10.1038/nprot.2006.476. [DOI] [PubMed] [Google Scholar]
- 27.Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA. alpha2delta expression sets presynaptic calcium channel abundance and release probability. Nature. 2012;486:122–125. doi: 10.1038/nature11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meinrenken CJ, Borst JG, Sakmann B. Calcium secretion coupling at calyx of held governed by nonuniform channel-vesicle topography. J. Neurosci. 2002;22:1648–1667. doi: 10.1523/JNEUROSCI.22-05-01648.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scimemi A, Diamond JS. The Number and Organization of Ca2+ Channels in the Active Zone Shapes Neurotransmitter Release from Schaffer Collateral Synapses. J. Neurosci. 2012;32:18157–18176. doi: 10.1523/JNEUROSCI.3827-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ariel P, Ryan TA. Optical mapping of release properties in synapses. Front Neural Circuits. 2010;4 doi: 10.3389/fncir.2010.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rozov A, Burnashev N, Sakmann B, Neher E. Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J. Physiol. 2001;531:807–826. doi: 10.1111/j.1469-7793.2001.0807h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schikorski T, Stevens CF. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J. Neurosci. 1997;17:5858–5867. doi: 10.1523/JNEUROSCI.17-15-05858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Novak P, et al. Nanoscale-targeted patch-clamp recordings of functional presynaptic ion channels. Neuron. 2013;79:1067–1077. doi: 10.1016/j.neuron.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shepherd GM, Harris KM. Three-dimensional structure and composition of CA3-->CA1 axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. J. Neurosci. 1998;18:8300–8310. doi: 10.1523/JNEUROSCI.18-20-08300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siksou L, et al. Three-dimensional architecture of presynaptic terminal cytomatrix. J. Neurosci. 2007;27:6868–6877. doi: 10.1523/JNEUROSCI.1773-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bucurenciu I, Bischofberger J, Jonas P. A small number of open Ca2+ channels trigger transmitter release at a central GABAergic synapse. Nat. Neurosci. 2010;13:19–21. doi: 10.1038/nn.2461. [DOI] [PubMed] [Google Scholar]
- 37.Matveev V, Bertram R, Sherman A. Calcium cooperativity of exocytosis as a measure of Ca(2)+ channel domain overlap. Brain Res. 2011;1398:126–38. doi: 10.1016/j.brainres.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weber AM, et al. N-type Ca2+ channels carry the largest current: implications for nanodomains and transmitter release. Nat. Neurosci. 2010;13:1348–1350. doi: 10.1038/nn.2657. [DOI] [PubMed] [Google Scholar]
- 39.Atasoy D, et al. Spontaneous and evoked glutamate release activates two populations of NMDA receptors with limited overlap. J. Neurosci. 2008;28:10151–10166. doi: 10.1523/JNEUROSCI.2432-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geppert M, et al. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- 41.Murthy VN, Stevens CF. Reversal of synaptic vesicle docking at central synapses. Nat. Neurosci. 1999;2:503–507. doi: 10.1038/9149. [DOI] [PubMed] [Google Scholar]
- 42.Sharma G, Vijayaraghavan S. Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron. 2003;38:929–939. doi: 10.1016/s0896-6273(03)00322-2. [DOI] [PubMed] [Google Scholar]
- 43.Emptage NJ, Reid CA, Fine A. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron. 2001;29:197–208. doi: 10.1016/s0896-6273(01)00190-8. [DOI] [PubMed] [Google Scholar]
- 44.Mochida S, et al. Requirement for the synaptic protein interaction site for reconstitution of synaptic transmission by P/Q-type calcium channels. Proc. Natl. Acad. Sci. U. S. A. 2003;100:2819–2824. doi: 10.1073/pnas.262787699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaeser PS, et al. RIM Proteins Tether Ca(2+) Channels to Presynaptic Active Zones via a Direct PDZ-Domain Interaction. Cell. 2011;144:282–295. doi: 10.1016/j.cell.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bao J, Li JJ, Perl ER. Differences in Ca2+ channels governing generation of miniature and evoked excitatory synaptic currents in spinal laminae I and II. J. Neurosci. 1998;18:8740–8750. doi: 10.1523/JNEUROSCI.18-21-08740.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pavlov I, Scimemi A, Savtchenko L, Kullmann DM, Walker MC. I(h)-mediated depolarization enhances the temporal precision of neuronal integration. Nat. Commun. 2011;2:199. doi: 10.1038/ncomms1202. doi: 10.1038/ncomms1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Banke TG, McBain CJ. GABAergic input onto CA3 hippocampal interneurons remains shunting throughout development. J. Neurosci. 2006;26:11720–11725. doi: 10.1523/JNEUROSCI.2887-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruiz A, Campanac E, Scott RS, Rusakov DA, Kullmann DM. Presynaptic GABAA receptors enhance transmission and LTP induction at hippocampal mossy fiber synapses. Nat. Neurosci. 2010;13:431–438. doi: 10.1038/nn.2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alle H, Geiger JR. Analog signalling in mammalian cortical axons. Curr. Opin. Neurobiol. 2008;18:314–320. doi: 10.1016/j.conb.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 51.Dreyfus FM, et al. Selective T-type calcium channel block in thalamic neurons reveals channel redundancy and physiological impact of I(T)window. J. Neurosci. 2010;30:99–109. doi: 10.1523/JNEUROSCI.4305-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maravall M, Mainen ZF, Sabatini BL, Svoboda K. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys. J. 2000;78:2655–2667. doi: 10.1016/S0006-3495(00)76809-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benson DL, Watkins FH, Steward O, Banker G. Characterization of GABAergic neurons in hippocampal cell cultures. J. Neurocytol. 1994;23:279–295. doi: 10.1007/BF01188497. [DOI] [PubMed] [Google Scholar]
- 54.Hines ML, Carnevale NT. The NEURON simulation environment. Neural Comput. 1997;9:1179–1209. doi: 10.1162/neco.1997.9.6.1179. [DOI] [PubMed] [Google Scholar]
- 55.Sasaki T, Matsuki N, Ikegaya Y. Action-potential modulation during axonal conduction. Science. 2011;331:599–601. doi: 10.1126/science.1197598. [DOI] [PubMed] [Google Scholar]
- 56.Borst JG, Helmchen F, Sakmann B. Pre- and postsynaptic whole-cell recordings in the medial nucleus of the trapezoid body of the rat. J. Physiol. 1995;489:825–840. doi: 10.1113/jphysiol.1995.sp021095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scott R, Rusakov DA. Main determinants of presynaptic Ca2+ dynamics at individual mossy fiber-CA3 pyramidal cell synapses. J. Neurosci. 2006;26:7071–7081. doi: 10.1523/JNEUROSCI.0946-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Faas GC, Raghavachari S, Lisman JE, Mody I. Calmodulin as a direct detector of Ca2+ signals. Nat. Neurosci. 2011;14:301–304. doi: 10.1038/nn.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nagerl UV, Novo D, Mody I, Vergara JL. Binding kinetics of calbindin-D(28k) determined by flash photolysis of caged Ca(2+) Biophys. J. 2000;79:3009–3018. doi: 10.1016/S0006-3495(00)76537-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xia Z, Storm DR. The role of calmodulin as a signal integrator for synaptic plasticity. Nat. Rev. Neurosci. 2005;6:267–276. doi: 10.1038/nrn1647. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.