

Abstract
Gaucher disease is the prototypical lysosomal storage disease. It results from the accumulation of undegraded glucosylceramide in the reticuloendothelial system of the bone marrow, spleen and liver due to deficiency of the enzyme glucocerebrosidase. This leads to hematologic, visceral and skeletal maifestions. Build up of glucosylceramide in the liver and spleen results in hepatosplenomegaly. The normal bone marrow is replaced by the accumulating substrate leading to many of the hematologic signs including anemia. The visceral and skeletal manifestations can be visualized with various imaging modalities including radiography, computed tomography, magnetic resonance imaging (MRI) and radionuclide scanning. Prior to the development of enzyme replacement therapy, treatment was only supportive. However, once intravenous enzyme replacement therapy became available in the 1990s it quickly became the standard of care. Enzyme replacement therapy leads to improvement in all manifestations. The visceral and hematologic manifestations respond more quickly usually within a few months or years. The skeletal manifestations take much longer, usually several years, to show improvement. In recent years newer treatment strategies, such as substrate reduction therapy, have been under investigation. Imaging plays a key role in both initial diagnosis and routine monitoring of patient on treatment particularly volumetric MRI of the liver and spleen and MRI of the femora for evaluating bone marrow disease burden.
Keywords: Gaucher disease, Lysosomal storage disease, Enzyme replacement therapy, Genetics, Medical imaging, Magnetic resonance imaging, Bone marrow
Core tip: Gaucher disease is the most common lysosomal storage disease resulting from accumulation of undegraded glucosylceramide in the reticuloendothelial system of the bone marrow, spleen and liver. Although affecting all three organs, the bone manifestations lead to the most debilitation. Visceral and bone marrow infiltration respond to enzyme replacement therapy however, the bone marrow response typically takes much longer.
INTRODUCTION
Gaucher disease (GD) is the most common of the lysosomal storage diseases[1]. It results from accumulation of undegraded glucosylceramide in lysosomes within macrophages of the reticuloendothelial cell system due to a deficiency of the enzyme glucocerebrosidase. Consequently these macrophages, enlarged with a buildup of glycolipids, are called Gaucher cells and are most abundant in the bone marrow, spleen and liver. GD is inherited in an autosomal recessive manner[2].
Three clinical subtypes of GD have been described[3]. Type 1 does not have any involvement of the central nervous system and is the most common. It formerly was referred to as the “adult type”. However, this is a misnomer since type 1 can occur at any age and is currently know as the non-neuronopathic type. Although it is most common in the Ashkenazi Jewish population it can occur in all ethnic groups. Type 2 was formerly referred to as the “infantile type”. This type manifests with grave involvement of the central nervous system. It is rapidly progressive usually leading to death within 2 years. It is now known as the acute neuronopathic type. Type 3 also has central nervous system involvement but is less severe and is more indolent than type 2 leading to the current terminology, subacute neuronopathic type.
The clinical manifestations of GD are due to the accumulation of Gaucher cells in the reticuloendothelial system of the bone marrow, spleen and liver. There can be marked variability in the severity of symptoms and the course of the disease. This is particularly true for type 1 where some patients can remain asymptomatic through life. Although the visceral changes can be dramatic, the more debilitating symptoms arise from infiltration of the bone marrow and bone changes. Since type 1 is the most common and widely studied variant of Gaucher disease it will be the primary focus of this review.
GENETICS
Gaucher disease is inherited in an autosomal recessive manner. The diagnosis of GD is made by the demonstration of decreased glucocerebrosidase enzymatic activity in peripheral blood leukocytes or fibroblasts cultured from a skin biopsy. Generally there is a 70%-90% reduction in the enzyme activity when compared to normal[4].
Molecular testing by targeted mutation analysis is used for confirmation of diagnosis and may be helpful for genotype-phenotype correlations. There are more than 300 mutations in the glucocerebrosidase gene that cause Gaucher disease[2]. However, four common mutations - N370S, IVS2(+1), 84GG, L444P - account for approximately 96.5% of disease in Ashkenazi Jewish population in the western hemisphere and approximately 50%-60% in non-Jewish populations[5].
Genotyping is helpful to test at risk family members, for genetic counseling as well as for prognosis. However, genotype-phenotype correlations are limited due to the clinical heterogeneity of the disease. Moreover, the majority of the work on genotype-phenotype correlation was based on a heavily Ashkenazi Jewish population which could skew the results since many affected individuals with the N370S/N370S homozygous genotype may remain asymptomatic and not come to medical attention[2]. Never the less a few generalities can be made: (1) the presence of at least one N370S allele precludes development of neuronopathic disease; and (2) the presence of the L444P allele is strongly (but not exclusively) associated with neuronopathic involvement. In general, those homozygous for the N370S allele tend to have less severe manifestations of disease and compound heterozygotes with one copy of N370S and a second mutation being L444P, 84GG or IVS 2 + 1 tend to have more severe disease. In fact, adults homozygous for L444P mutation (L444P/L444P genotype) typically have the type 3 neuronopathic disease. However, these rules are not hard and fast due to the limited genotype-phenotype correlation. Some patients with the N370S/N370S genotype have profound symptomatic disease whereas a type 1 patient with N370S/L444P genotype may have mild symptoms.
HEMATOLOGIC MANIFESTATIONS
Hematologic abnormalities of GD are exceedingly common. Almost all patients with symptoms present with anemia and thrombocytopenia. The etiology can be explained by depressed hematopoiesis resulting from substitution of the bone marrow by Gaucher cells. However, hypersplenism or sequestration within the spleen can be a cause as well. Symptoms that arise due to the hematologic abnormalities include fatigue, easy bruising and frequent nosebleeds. Additional blood chemistries can be elevated in GD including angiotensin converting enzyme, chitotriosidase, and tartrate resistant acid phosphatase[6]. Changes towards normalization of the anemia, thrombocytopenia and blood chemistries can be used to monitor treatment response[7].
VISCERAL MANIFESTATIONS
The viscera most commonly involved with accumulation of Gaucher cells are the liver and spleen. The pulmonary system can be involved as well; although it is very rare. Current recommendation for evaluating and monitoring visceral involvement is volumetric MRI (preferred due to lack of ionizing radiation) or CT every 12 to 24 mo[6].
Gaucher cells accumulate in the Kupfer cells of the liver leading to hepatomegaly (Figure 1). Liver volumes in type 1 patients are typically approximately 2 times normal[8]. It is notable that glycolipid does not accumulate in the hepatocytes[8,9]. The Gaucher cells can conglomerate into nodules that can be seen with sonography or MRI. These nodules may be hypoechoic, hyperechoic, or mixed on sonography[10,11]. On MRI the nodules typically appear isointense or low signal intensity (SI) on T1 weighted imaging (WI) and high SI on T2 WI. Focal areas of extra-medullary hematopoiesis can have a similar appearance and can also be seen due the accompanying anemia. Hepatic infiltration can also lead to fibrosis and cirrhosis[12].
Figure 1.
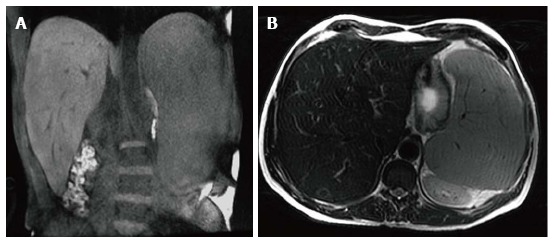
Hepatosplenomegaly. Coronal T1 WI (A) and axial T2 WI (B) images in a male type 1 GD patient with N370S/N370S genotype demonstrate marked hepatosplenomegaly. The liver volume measured 3235 cc. The spleen volume measured 2923 cc.
Splenomegaly results from accumulation of Gaucher cells within the spleen (Figure 1). Spleen volumes in type 1 GD are typically 5-15 times normal but the spleen size can be significantly enlarged in some cases and may be over 50 times normal[13]. Focal splenic masses are common and may represent clusters of Gaucher cells or extra-medullary hematopoiesis. They may be detected with sonography, CT or MRI. Similar to the liver, Gaucher masses in the spleen may be hypoechoic, hyperechoic, or mixed echogenicity[11,14]. On CT the masses are low density[15] and occasionally peripherally calcified (Figure 2). These masses are most commonly imaged with MRI. They typically are low SI or isointense on T1 WI and high SI on T2 WI[16] (Figure 3). Low SI on gradient recalled echo imaging in these masses is thought to be secondary to iron contained in the Gaucher cells[17]. Splenic infarcts can occur as well due to massive splenomegaly and can be detected with imaging as well (Figure 4).
Figure 2.
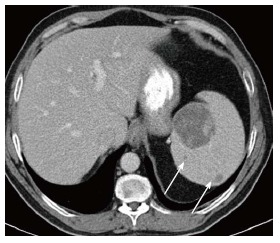
Splenic mass on computed tomography. Axial computed tomography image shows a large low density mass with patchy foci of soft tissue density within it in the medial aspect of the spleen. Additional smaller low density masses are present as well (arrows).
Figure 3.
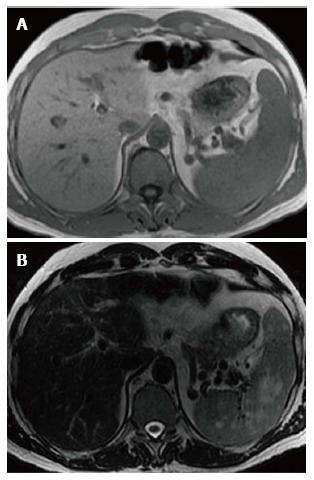
Splenic mass on magnetic resonance. Axial T1WI (A) image shows no apparent abnormality within the spleen consistent with isointense signal intensity (SI) masses. Axial T2WI (B) image at the same level reveals multiple masses in the spleen to be high SI.
Figure 4.
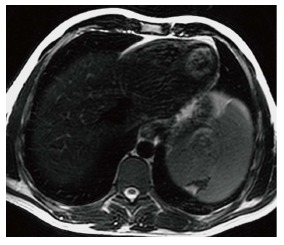
Splenic infarct. Axial T2 WI image demonstrates a wedge shaped defect in a subcapsular region of the spleen in its superior aspect. The defect has low signal intensity (SI) along the edges indicating fibrous tissue. In addition, high SI fat has filled the area left by the retracted capsule.
An infrequent manifestation of GD is pulmonary involvement which is more commonly seen in type 1 patients who have undergone splenectomy and those with type 3[18]. The lung findings are thought to be secondary to direct infiltration by Gaucher cells into the interstitial spaces, alveolar spaces and capillaries[19] as well as indirect causes secondary to hepatopulmonary syndrome related to the liver manifestations and/or aspiration associated with neurologic manifestations. Chest radiographs generally are normal or demonstrate a reticulo-nodular pattern. The findings are best imaged by high resolution CT and include interstitial thickening (both interlobular and intralobular), ground glass opacity, consolidation and bronchial wall thickening[20,21]. Pulmonary hypertension can be the result of lung involvement[22-24]. Symptomatic pulmonary involvement is generally seen in patients with more striking visceral and skeletal findings.
SKELETAL MANIFESTATIONS
The skeletal manifestations of GD lead to the most debilitating complications of the disease and significant morbidity. Gaucher cells infiltrate and accumulate in the bone marrow. The pathophysiology of how the infiltration leads to the bone changes is not well understood. Proposed mechanisms include altered bone formation and resorption, as well as increased intra-osseous pressure due to the infiltration leading to vascular occlusion[3,25].
An array of bone findings are seen in GD, including growth retardation in children, osteopenia, lytic lesions (Figure 5), pathologic fractures, bone pain, osteonecrosis (Figure 6), cortical and medullary infarcts (Figure 7) and evidence of bone crises[3,26]. The severity of bone findings in GD depend on the extent of medullary cavity substitution. Marrow replacement with Gaucher cells can lead to expansion of the medullary cavity with thinning of the cortex and endosteal scalloping (Figure 8) and consequent diffuse osteopenia. In addition, the medullary expansion leads to a failure of remodeling in the distal femurs resulting in the so called Erlenmeyer flask deformity (Figure 9). These manifestations can be imaged using a variety of modalities including radiography, MRI, dual energy X-ray absortiometry (DEXA) and radionuclide imaging. However, the mainstay of skeletal imaging in GD involves MRI.
Figure 5.
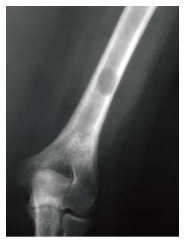
Lytic lesion. Frontal radiograph of the distal right humerus demonstrates a well demarcated lytic lesion that does not show sclerotic borders, endosteal erosion or associated expansion of the humeral shaft.
Figure 6.
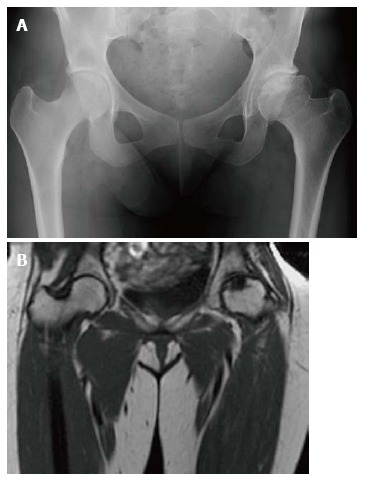
Osteonecrosis. Frontal radiograph of the pelvis (A) shows avascular necrosis of the left femoral head. The femoral head has a flattened contour with sclerosis in the subcapsular areas. Note that the joint space is maintained. Coronal T1 WI (B) in the same patient again demonstrated an abnormal shape of the left femoral head with flattening superiorly. In the same area there is a focus of low SI indicating the devasularized bone.
Figure 7.
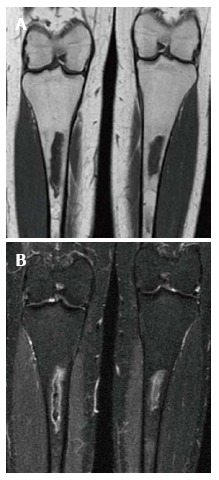
Medullary infarction. Coronal T1 WI (A) image shows irregularly bordered areas of low SI within the medullary cavity of both tibiae. The same areas show peripheral serpigenous high SI on the coronal short tau inversion recovery (STIR) (B) image. The appearance is typical of an infarct.
Figure 8.
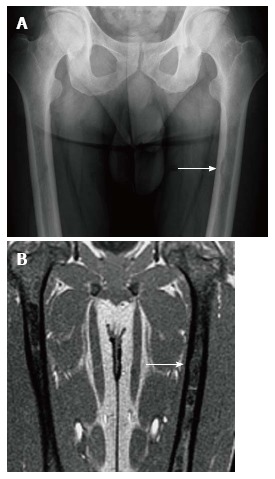
Endosteal scalloping. Frontal radiograph (A) of the femurs shows an area of rounded thinning of the medial cortex of the left femur (arrow). Coronal out of phase image of the femurs in the same patient (B) shows low signal intensity in that same area due to expansion of the medullary cavity due to infiltration. The thinning of the cortex is less apparent on magnetic resonance than on radiography.
Figure 9.

Erlenmeyer flask deformity. Frontal radiograph of the distal femurs demonstrates flaring of the bone and thinning of the cortex due to under-tubulation of the metadiaphysis.
Bone crises are most common in childhood and adolescence presenting as episodes of severe bone pain associated with fever and leucocytosis. The signs and symptoms are indistinguishable from osteomyelitis, however no infection exists. The terms “pseudo-osteomyelitis” and “aseptic osteomyelitis” have been historically used to characterize this condition[3,27] (Figure 10).
Figure 10.
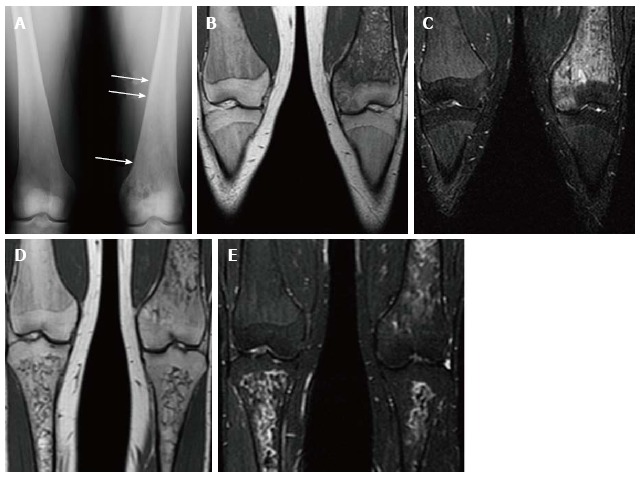
Psuedo-osteomyelitis. Frontal radiograph of both distal femurs in 2009 (A) demonstrates an irregular area of patchy lucency in the medial condyle of the left femur. There is periosteal reaction in this area as well as more superiorly (arrows). Coronal T1 WI (B) image at the same time in 2009 demonstrate low SI in the medial condyle of the femur extending into the medial epiphysis. There is high SI in these areas on coronal STIR (C) image which extends into the adjacent soft tissues where the periosteal reaction is seen on the radiograph. There is no joint effusion. The patient presented with left knee pain and the imaging was suspicious for osteomyelitis involving the medial distal femur. However, the patient has no fever and cultures were negative. Coronal T1 WI (D) of the same area in 2011 shows resolution of the low SI in the medial condyle and epiphysis. The corresponding high SI on the STIR image (E) has resolved as well.
Osteonecrosis, otherwise known as avascular necrosis, is due to lack of blood supply and consequent bone death. It is most commonly seen in the femoral heads (Figure 6), proximal humeri and vertebral bodies. The vertebral body can “cave in” leading to the “H-shaped” vertebra, i.e., Reynolds phenomenon, similar to sickle cell disease (Figure 11). While the end result is similar in both diseases the mechanism of formation is different. In GD the entire vertebral body collapses followed by peripheral regrowth while in sickle cell disease the deformity is secondary to central growth arrest[28]. The necrotic bone crumples and leads to malformation and/or fracture, sometimes requiring treatment with a bone prosthesis or joint replacement.
Figure 11.
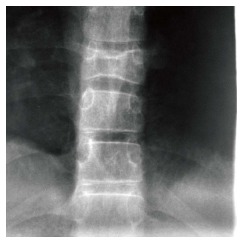
H-shaped vertebra. Cone down frontal radiograph of the lower thoracic spine demonstrates collapse of a lower thoracic vertebral body with bi-concave upper and lower end plates giving the Reynolds phenomenon of Gaucher disease.
Radiography is used primarily to image cortical bone. It can detect lytic (Figure 5) or sclerotic lesions within bones. Fractures, both traumatic and pathologic, are readily detected on radiographs. In addition, endosteal scalloping (Figure 8) and the Erlenmeyer flask deformity (Figure 9) due to marrow expansion are also detected with radiography. Although changes in cortical bone secondary to marrow infiltration can be detected with this modality, the marrow space itself is cannot be evaluated by radiography.
Osteopenia is near universal in GD as a representation of decreased bone mineral density. A significant decrease in bone density must occur before osteopenia is perceived on radiography leading to its poor sensitivity for detecting this abnormality. DEXA is the current modality of choice for evaluation of osteopenia and bone mineral density. However, care must be taken to avoid areas of osteonecrosis during DEXA evaluations.
The bone marrow itself is best assessed with MRI. Normal yellow (fatty) marrow is seen as high signal on T1 WI and T2 WI. The infiltration of the marrow by Gaucher cells replaces the normal yellow marrow. Marrow infiltration generally follows the distribution of cellular read marrow progressing from the axial to the peripheral skeleton and from the proximal to the distal aspects of the long bones with a tendency to spare the epiphyses[29].This is recognized as a change to low SI on both T1 and T2 WI[29,30] (Figure 12). On short tau inversion recovery (STIR) images the infiltration appears slightly high SI[31]. High SI within the marrow on T2 or STIR images suggests edema within the marrow and the presence of an “active” process such as a bone crisis or infection[32]. Evaluation of bone marrow infiltration in children is complicated by the fact that normal red marrow which is seen in this age group manifests with low SI on both T1 and T2 WI.
Figure 12.
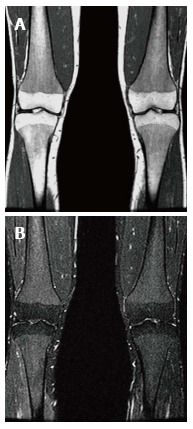
Bone marrow infiltration. Coronal T1 WI (A) of the distal femora and proximal tibiae in a type 1 gaucher disease patient shows low signal intensity (SI) in the bone marrow which spares the epiphyses that demonstrate the normal fatty marrow SI. Coronal short tau inversion recover (STIR) (B) image demonstrates that the low SI on T1 becomes slightly high SI on STIR.
Radionuclide imaging is useful for evaluating bone changes in GD. Bone scintigraphy utilizing Technicium 99m-methylene diphosphonate (99mTc-MDP) can be used to evaluate for fractures that are not readily apparent on radiography. In addition, this tracer can be used to help differentiate a bone crisis (aseptic infarction) from osteomyelitis. In a bone crisis bone scintigraphy performed within 1-3 d of the onset of pain will demonstrate decreased tracer uptake at the involved site unlike infection that shows increased uptake[33,34]. The same agent can be used to help evaluate for complication related to joint prostheses such as loosening[31]. Scintigraphy using leucocytes labeled with Indium-111 is commonly used to image areas of suspected infection including osteomyelitis and around joint prostheses.
Prior to the advent of MRI, radionuclide imaging was also used for evaluation of the bone marrow. Technicium 99m sulfur colloid (99mTC-SC) accumulates in normal bone marrow. Therefore in marrow infiltrated and replaced by Gaucher cells there will be decreased uptake or an abnormal pattern of uptake compared to normal[34]. This gives an indirect sign of infiltration. Another tracer, Technicium 99m sestamibi, has the advantage of being accumulated in areas of Gaucher cell deposition[35,36]. Mariani et al[35] imaged 74 Italian patients with Gaucher disease using technetium 99m sestamibi and showed 71 of 74 demonstrated uptake predominantly in the distal femur. An undisclosed number of these patients had MR imaging performed at the same approximate time revealing low SI in the same regions. Therefore it is a method of direct visualization of infiltration. Bone marrow sestamibi imaging can be advantageous when imaging children and trying to differentiate Gaucher infiltration from normal red marrow in children. Positron emission tomography has become widely available in recent years. Imaging with the most common radiotracer, fluoride-18 fluorodeoxyglucose, has not proven beneficial for detecting marrow involvement in GD. However, a newer tracer, fluoride-18 L-thymidine, shows promise for imaging bone marrow[37]. Since it is not yet FDA approved its use is limited to clinical trials and its role if any in GD has not been established. Therefore, MRI remains the modality of choice for imaging bone marrow due the poor spatial resolution of scintigraphy as well as the associated radiation dose of radionuclide imaging.
An essential problem of imaging is that it only gives a qualitative assessment of bone marrow infiltration. An MRI shows decreased SI on T1 WI but there is no way to measure the “amount” of signal. A visual assessment of improvement or worsening can be made on the basis of the MR image but that is qualitative and not very useful to clinicians. One way of directly measuring bone marrow disease has been developed, Dixon’s quantitative chemical shift imaging[38]. Chemical shift imaging leverages the difference in resonance frequencies between water and fat molecules thereby defining the amount of fat or fat fraction within bone marrow. The fat fraction of normal marrow decreases as the amount of infiltration replaces the triglyceride rich fat cells of normal marrow. Studies have shown a low marrow fat fraction as measured by quantitative chemical sift imaging to correspond to worse clinical disease and more bone complications[39-42]. However, the technique is complex and not widely used outside academic centers. To overcome this problem several semi-quantitative methods have been developed including the Rosenthal staging system[29], the Dusseldorf score[43], the Terk classification[44] and the bone marrow burden (BMB) score[45]. All use conventional MR imaging technology and assign points based on changes in marrow signal intensity at different anatomic locations.
The BMB score is the most widely used and validated[45,46]. The BMB score[45] incorporates both the visual interpretation of SI and the geographic location of the disease on conventional MR images of the lumbar spine and femora. The SI of the bone marrow in the femora is compared to the subcutaneous fat on both T1 and T2 WI sequences. The SI of the bone marrow in the lumbar spine is compared to a non-diseased intervertebral disc on both T1 and T2 WI sequences. The SI is scored as hyper-intense, slightly hyper-intense, iso-intense, slightly hypo-intense, and hypo-intense. A numeric value ranging from 0-2 is assigned based on the SI. Point values are also assigned based on the location/distribution of the marrow infiltration. In the femora, sites of involvement including the diaphysis, proximal epiphysis/apophysis and distal epiphysis are evaluated. In the lumbar spine, the distribution of infiltration is evaluated as patchy or diffuse with special attention given to absence of fat in basivertebral vein region. The score for the femora (0-8) and the lumbar spine (0-8) are added together for a total score which can range from 0 to 16. A higher score indicates more severe the bone marrow involvement. In their study of 12 patients with Gaucher disease[45], Maas et al[45] demonstrated good correlation of the BMB score with fat fraction determination by the Dixon technique. That study also showed a decrease in the BMB score in patients on enzyme replacement therapy (ERT), however, it was less sensitive for detection of marrow improvement than Dixon method.
TREATMENT
Only supportive treatment or surgical intervention including splenectomy and joint replacement was available for patients with Gaucher disease into the early 1990s. In 1991 ERT with placentally derived enzyme alglucerase (Ceredase®, Genzyme Corporation, Cambridge, Mass.) came into existence. Following this, in 1994, recombinant mannose-terminated human glucocerebrosidase (Cerezyme®, Genzyme Corporation) received FDA approval. The goal of ERT is to treat the symptoms of the disease as well as to prevent complications particularly the skeletal complications[6].
The visceral and hematologic manifestations of GD respond relatively quickly to ERT[47,48] (Figure 13). The anemia and thrombocytopenia can improve within 6 mo to one year of initialing ERT. The liver and spleen volumes generally can decrease by approximately 50% within the first 2 years but rarely ever return to a normal volume even with long term treatment. Although some improvement in pulmonary involvement has been reported with ERT, response is generally slow, and sometimes no improvement is seen[23,49]. The marrow infiltration responds to ERT as well but takes much longer to be seen[50,51] (Figures 14 and 15). Some skeletal manifestations, such as osteonecrosis, osteosclerosis and vertebral body collapse, remain irreversible. The neurologic manifestations of GD do not respond to ERT since it does not cross the blood-brain barrier[52].
Figure 13.
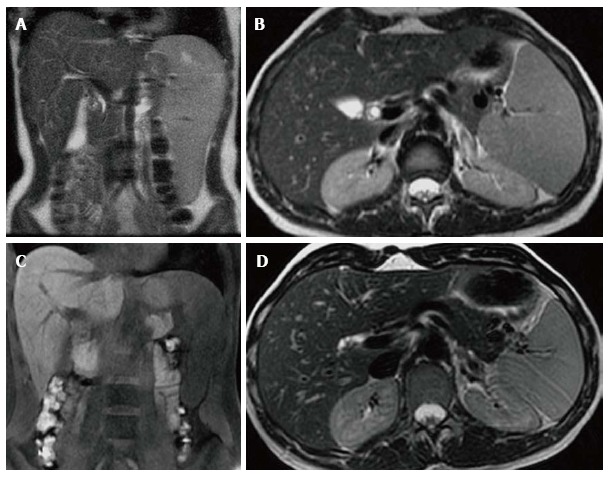
Visceral improvement on enzyme replacement therapy. Female type 3 Gaucher disease patient who began enzyme replacement therapy (ERT) in 2007. Initial evaluation in 2007 before starting ERT shows that the spleen extends down to the iliac crest on the T2 WI coronal image (A). On the axial T2 WI (B) the left lobe of the liver extends across the midline posterior to the lateral aspect of the left rectus abdominus muscle and anterior to the stomach. At that time the liver volume measured 1702 cc and the spleen volume measured 769 cc. After 6 years on treatment, repeat imaging in 2013 reveals the inferior edge of the spleen is now above the iliac crest on the coronal T1 WI (C) and the left lobe of the liver now extends only slightly across the midline to end posterior to the medial aspect of the rectus abdominus muscle and the stomach is now lateral to the liver on the axial T2 WI (D). In 2013, the liver volume measured 1163 cc and the spleen volume measured368 cc.
Figure 14.
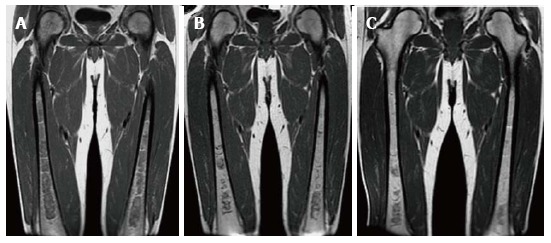
Bone marrow improvement on enzyme replacement therapy. Type 1 GD male patient with N370S/N370S genotype who began enzyme replacement therapy (ERT) in October of 2007 shows relatively rapid improvement of the bone marrow infiltration. Initial coronal T1 WI of the femora (A) demonstrates diffuse low SI throughout the medullary cavity consistent with marked infiltration. Coronal T1 WI in 2009 (B) after only 2 years of treatment shows significant improvement in the infiltration manifest by decreased low SI in the medullary cavity. In 2011, coronal T1 WI (C) shows continued slight decrease in the amount of low SI in the bone marrow.
Figure 15.
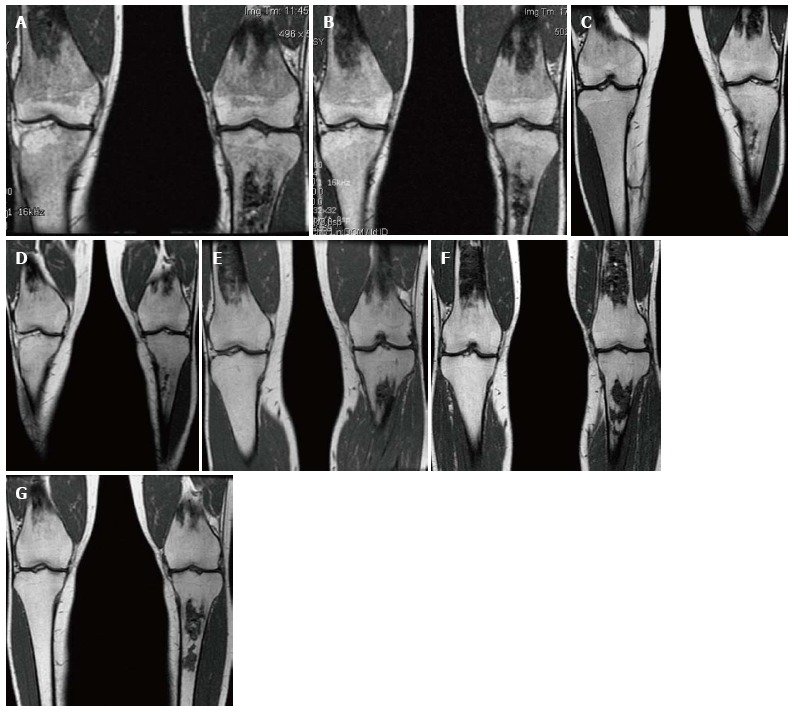
Bone marrow improvement. Gaucher Type 1 patient with N370S/N370S genotype who started enzyme replacement therapy (ERT) in 1997. Coronal T1 WI of the distal femora and proximal tibiae in 1998 (A), 1999 (B), 2002 (C), 2003 (D), 2006 (E), 2008 (F) and 2011 (G). Medullary infarcts are partially seen in both distal femurs and the left tibia. The low T1 SI significantly improves between 1998 and 2002 with near normal marrow signal seen in the noninfarcted areas in 2008 and years later. Although this patient has the same genotype as the patient in Figure 14, there is a longer time to improvement indicating the limited genotype/phenotype correlation.
An alternative to ERT for treatment of GD is substrate reduction therapy. Whereas ERT works by replacing the deficient enzyme, substrate reduction works by decreasing the production of the substrate glucosylceramide. Since most type 1 GD patients have some residual enzyme activity, reducing the amount of substrate may allow the native enzyme to succeed. The first medication of this kind was N-butyldeoxynojirimycin (miglustat) approved by the FDA in 2003. It is an oral medication for use in mild to moderate type 1 GD patients who cannot tolerate ERT. Studies have shown improvement of anemia, platelet count, liver volume and spleen volume alone or in combination with ERT[53-55]. One study also showed improvement of bone disease on miglustat[56,57]. However, side effects particularly diarrhea, weight loss and tremors were significant[52] and its use is limited in the United States Unlike ERT miglustat can cross the blood-brain barrier[52] and has shown promising results in treating neurologic manifestations in combination with ERT[58,59]. A study also shows improvement of pulmonary manifestations with single drug treatment[60]. Currently there is a newer oral substrate reduction therapy agent, eliglustat tartrate, which has demonstrated efficacy in GD type 1 patients with a more favorable side effect profile[61]. It is currently undergoing phase III trials.
Both ERT and substrate reduction therapy can lead to improvement in the signs and symptoms of GD. However, there is a significant cost to the treatment ranging from US$100000 to $250000 per year[62]. Thus judicious use is warranted. Imaging plays a significant role in determining which patients need treatment and in surveillance of those on treatment. Current recommendations call for abdominal MRI for determination of liver and spleen volume, MRI of the bilateral femora, radiography of the spine and DEXA of the hips and lumbar spine in the initial assessment[6]. For those patients not on treatment being followed and those on treatment, the same imaging protocol is recommended every 12-24 mo or at the time of a dosage change or significant clinical complication[6].
CONCLUSION
Gaucher disease is the most common lysomal storage disease which affects all ethnic groups. The accumulation of glycolipids in the reticuloendothelial system leads to symptoms. Anemia, thrombocytopenia, and hepatosplenomegaly are commonly seen and respond relatively rapidly to treatment with ERT. The skeletal manifestations are due to build up of the glycolipids in the bone marrow and lead to the most debilitating aspects of GD. Treatment results in improvement of marrow infiltration however it takes much longer than the visceral and hematologic manifestations. Imaging plays a key role in both initial diagnosis and treatment monitoring. MRI of the abdomen is used to monitor liver and spleen volumes. MRI of the femora and lumbar spine is used for evaluation of bone marrow infiltration burden.
Footnotes
P- Reviewer: Palumbo B S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
References
- 1.Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- 2.Grabowski GA, Petsko GA, Kolodny EH. Gaucher Disease. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, eds. Scriver’s Online Metabolic and Molecular Bases of Inherited Disease. Published January 2006. Accessed November 29; 2013. [Google Scholar]
- 3.Wenstrup RJ, Roca-Espiau M, Weinreb NJ, Bembi B. Skeletal aspects of Gaucher disease: a review. Br J Radiol. 2002;75 Suppl 1:A2–12. doi: 10.1259/bjr.75.suppl_1.750002. [DOI] [PubMed] [Google Scholar]
- 4.Beutler E, Saven A. Misuse of marrow examination in the diagnosis of Gaucher disease. Blood. 1990;76:646–648. [PubMed] [Google Scholar]
- 5.Beutler E, Gelbart T, Kuhl W, Zimran A, West C. Mutations in Jewish patients with Gaucher disease. Blood. 1992;79:1662–1666. [PubMed] [Google Scholar]
- 6.Weinreb NJ, Aggio MC, Andersson HC, Andria G, Charrow J, Clarke JT, Erikson A, Giraldo P, Goldblatt J, Hollak C, et al. Gaucher disease type 1: revised recommendations on evaluations and monitoring for adult patients. Semin Hematol. 2004;41:15–22. doi: 10.1053/j.seminhematol.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 7.Charrow J, Esplin JA, Gribble TJ, Kaplan P, Kolodny EH, Pastores GM, Scott CR, Wappner RS, Weinreb NJ, Wisch JS. Gaucher disease: recommendations on diagnosis, evaluation, and monitoring. Arch Intern Med. 1998;158:1754–1760. doi: 10.1001/archinte.158.16.1754. [DOI] [PubMed] [Google Scholar]
- 8.James SP, Stromeyer FW, Chang C, Barranger JA. LIver abnormalities in patients with Gaucher’s disease. Gastroenterology. 1981;80:126–133. [PubMed] [Google Scholar]
- 9.James SP, Stromeyer FW, Stowens DW, Barranger JA. Gaucher disease: hepatic abnormalities in 25 patients. Prog Clin Biol Res. 1982;95:131–142. [PubMed] [Google Scholar]
- 10.Patlas M, Hadas-Halpern I, Abrahamov A, Elstein D, Zimran A. Spectrum of abdominal sonographic findings in 103 pediatric patients with Gaucher disease. Eur Radiol. 2002;12:397–400. doi: 10.1007/s003300101031. [DOI] [PubMed] [Google Scholar]
- 11.Neudorfer O, Hadas-Halpern I, Elstein D, Abrahamov A, Zimran A. Abdominal ultrasound findings mimicking hematological malignancies in a study of 218 Gaucher patients. Am J Hematol. 1997;55:28–34. doi: 10.1002/(sici)1096-8652(199705)55:1<28::aid-ajh5>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 12.Lee RE. The pathology of Gaucher disease. Prog Clin Biol Res. 1982;95:177–217. [PubMed] [Google Scholar]
- 13.Grabowski GA, Kolodny EH, Weinreb NJ, Rosenbloom BE, Prakash-Cheng A, Kaplan P, Charrow J, Pastores GM, Mistry PK Gaucher disease: Phenotypic and Genetic Variation,. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, eds. Scriver’s Online Metabolic and Molecular Bases of Inherited Disease. Accessed November 29, 2013. New York: McGraw-Hill; 2006. [Google Scholar]
- 14.Hill SC, Reinig JW, Barranger JA, Fink J, Shawker TH. Gaucher disease: sonographic appearance of the spleen. Radiology. 1986;160:631–634. doi: 10.1148/radiology.160.3.3526400. [DOI] [PubMed] [Google Scholar]
- 15.Poll LW, Koch JA, vom Dahl S, Sarbia M, Häussinger D, Mödder U. Gaucher disease of the spleen: CT and MR findings. Abdom Imaging. 2000;25:286–289. doi: 10.1007/s002610000010. [DOI] [PubMed] [Google Scholar]
- 16.Hill SC, Damaska BM, Ling A, Patterson K, Di Bisceglie AM, Brady RO, Barton NW. Gaucher disease: abdominal MR imaging findings in 46 patients. Radiology. 1992;184:561–566. doi: 10.1148/radiology.184.2.1620865. [DOI] [PubMed] [Google Scholar]
- 17.Terk MR, Esplin J, Lee K, Magre G, Colletti PM. MR imaging of patients with type 1 Gaucher’s disease: relationship between bone and visceral changes. AJR Am J Roentgenol. 1995;165:599–604. doi: 10.2214/ajr.165.3.7645477. [DOI] [PubMed] [Google Scholar]
- 18.Hill SC, Damaska BM, Tsokos M, Kreps C, Brady RO, Barton NW. Radiographic findings in type 3b Gaucher disease. Pediatr Radiol. 1996;26:852–860. doi: 10.1007/BF03178036. [DOI] [PubMed] [Google Scholar]
- 19.Amir G, Ron N. Pulmonary pathology in Gaucher’s disease. Hum Pathol. 1999;30:666–670. doi: 10.1016/s0046-8177(99)90092-8. [DOI] [PubMed] [Google Scholar]
- 20.McHugh K, Olsen E ØE, Vellodi A. Gaucher disease in children: radiology of non-central nervous system manifestations. Clin Radiol. 2004;59:117–123. doi: 10.1016/j.crad.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 21.Aydin K, Karabulut N, Demirkazik F, Arat A. Pulmonary involvement in adult Gaucher’s disease: high resolution CT appearance. Br J Radiol. 1997;70:93–95. doi: 10.1259/bjr.70.829.9059303. [DOI] [PubMed] [Google Scholar]
- 22.Theise ND, Ursell PC. Pulmonary hypertension and Gaucher’s disease: logical association or mere coincidence? Am J Pediatr Hematol Oncol. 1990;12:74–76. doi: 10.1097/00043426-199021000-00014. [DOI] [PubMed] [Google Scholar]
- 23.Mistry PK, Sirrs S, Chan A, Pritzker MR, Duffy TP, Grace ME, Meeker DP, Goldman ME. Pulmonary hypertension in type 1 Gaucher’s disease: genetic and epigenetic determinants of phenotype and response to therapy. Mol Genet Metab. 2002;77:91–98. doi: 10.1016/s1096-7192(02)00122-1. [DOI] [PubMed] [Google Scholar]
- 24.Pastores GM, Miller A. Pulmonary hypertension in Gaucher’s disease. Lancet. 1998;352:580. doi: 10.1016/S0140-6736(05)79295-3. [DOI] [PubMed] [Google Scholar]
- 25.Sims KB, Pastores GM, Weinreb NJ, Barranger J, Rosenbloom BE, Packman S, Kaplan P, Mankin H, Xavier R, Angell J, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet. 2008;73:430–440. doi: 10.1111/j.1399-0004.2008.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hermann G, Goldblatt J, Levy RN, Goldsmith SJ, Desnick RJ, Grabowski GA. Gaucher’s disease type 1: assessment of bone involvement by CT and scintigraphy. AJR Am J Roentgenol. 1986;147:943–948. doi: 10.2214/ajr.147.5.943. [DOI] [PubMed] [Google Scholar]
- 27.Yossipovitch ZH, Herman G, Makin M. Aseptic osteomyelitis in Gaucher’s disease. Isr J Med Sci. 1965;1:531–536. [PubMed] [Google Scholar]
- 28.Schwartz AM, Homer MJ, McCauley RG. „Step-off“ vertebral body: Gaucher‘s disease versus sickle cell hemoglobinopathy. AJR Am J Roentgenol. 1979;132:81–85. doi: 10.2214/ajr.132.1.81. [DOI] [PubMed] [Google Scholar]
- 29.Rosenthal DI, Scott JA, Barranger J, Mankin HJ, Saini S, Brady TJ, Osier LK, Doppelt S. Evaluation of Gaucher disease using magnetic resonance imaging. J Bone Joint Surg Am. 1986;68:802–808. [PubMed] [Google Scholar]
- 30.Lanir A, Hadar H, Cohen I, Tal Y, Benmair J, Schreiber R, Clouse ME. Gaucher disease: assessment with MR imaging. Radiology. 1986;161:239–244. doi: 10.1148/radiology.161.1.3763873. [DOI] [PubMed] [Google Scholar]
- 31.Katz R, Booth T, Hargunani R, Wylie P, Holloway B. Radiological aspects of Gaucher disease. Skeletal Radiol. 2011;40:1505–1513. doi: 10.1007/s00256-010-0992-3. [DOI] [PubMed] [Google Scholar]
- 32.Hermann G, Shapiro RS, Abdelwahab IF, Grabowski G. MR imaging in adults with Gaucher disease type I: evaluation of marrow involvement and disease activity. Skeletal Radiol. 1993;22:247–251. doi: 10.1007/BF00197668. [DOI] [PubMed] [Google Scholar]
- 33.Katz K, Mechlis-Frish S, Cohen IJ, Horev G, Zaizov R, Lubin E. Bone scans in the diagnosis of bone crisis in patients who have Gaucher disease. J Bone Joint Surg Am. 1991;73:513–517. [PubMed] [Google Scholar]
- 34.Mikosch P, Kohlfürst S, Gallowitsch HJ, Kresnik E, Lind P, Mehta AB, Hughes DA. Is there a role for scintigraphic imaging of bone manifestations in Gaucher disease? A review of the literature. Nuklearmedizin. 2008;47:239–247. [PubMed] [Google Scholar]
- 35.Mariani G, Filocamo M, Giona F, Villa G, Amendola A, Erba P, Buffoni F, Copello F, Pierini A, Minichilli F, et al. Severity of bone marrow involvement in patients with Gaucher’s disease evaluated by scintigraphy with 99mTc-sestamibi. J Nucl Med. 2003;44:1253–1262. [PubMed] [Google Scholar]
- 36.Mariani G, Molea N, La Civita L, Porciello G, Lazzeri E, Ferri C. Scintigraphic findings on 99mTc-MDP, 99mTc-sestamibi and 99mTc-HMPAO images in Gaucher’s disease. Eur J Nucl Med. 1996;23:466–470. doi: 10.1007/BF01247378. [DOI] [PubMed] [Google Scholar]
- 37.Agool A, Schot BW, Jager PL, Vellenga E. 18F-FLT PET in hematologic disorders: a novel technique to analyze the bone marrow compartment. J Nucl Med. 2006;47:1592–1598. [PubMed] [Google Scholar]
- 38.Dixon WT. Simple proton spectroscopic imaging. Radiology. 1984;153:189–194. doi: 10.1148/radiology.153.1.6089263. [DOI] [PubMed] [Google Scholar]
- 39.Maas M, Hollak CE, Akkerman EM, Aerts JM, Stoker J, Den Heeten GJ. Quantification of skeletal involvement in adults with type I Gaucher’s disease: fat fraction measured by Dixon quantitative chemical shift imaging as a valid parameter. AJR Am J Roentgenol. 2002;179:961–965. doi: 10.2214/ajr.179.4.1790961. [DOI] [PubMed] [Google Scholar]
- 40.Johnson LA, Hoppel BE, Gerard EL, Miller SP, Doppelt SH, Zirzow GC, Rosenthal DI, Dambrosia JM, Hill SC, Brady RO. Quantitative chemical shift imaging of vertebral bone marrow in patients with Gaucher disease. Radiology. 1992;182:451–455. doi: 10.1148/radiology.182.2.1732964. [DOI] [PubMed] [Google Scholar]
- 41.Maas M, Poll LW, Terk MR. Imaging and quantifying skeletal involvement in Gaucher disease. Br J Radiol. 2002;75 Suppl 1:A13–A24. doi: 10.1259/bjr.75.suppl_1.750013. [DOI] [PubMed] [Google Scholar]
- 42.Maas M, Akkerman EM, Venema HW, Stoker J, Den Heeten GJ. Dixon quantitative chemical shift MRI for bone marrow evaluation in the lumbar spine: a reproducibility study in healthy volunteers. J Comput Assist Tomogr. 2001;25:691–697. doi: 10.1097/00004728-200109000-00005. [DOI] [PubMed] [Google Scholar]
- 43.Poll LW, Koch JA, vom Dahl S, Willers R, Scherer A, Boerner D, Niederau C, Häussinger D, Mödder U. Magnetic resonance imaging of bone marrow changes in Gaucher disease during enzyme replacement therapy: first German long-term results. Skeletal Radiol. 2001;30:496–503. doi: 10.1007/s002560100375. [DOI] [PubMed] [Google Scholar]
- 44.Terk MR, Dardashti S, Liebman HA. Bone marrow response in treated patients with Gaucher disease: evaluation by T1-weighted magnetic resonance images and correlation with reduction in liver and spleen volume. Skeletal Radiol. 2000;29:563–571. doi: 10.1007/s002560000276. [DOI] [PubMed] [Google Scholar]
- 45.Maas M, van Kuijk C, Stoker J, Hollak CE, Akkerman EM, Aerts JF, den Heeten GJ. Quantification of bone involvement in Gaucher disease: MR imaging bone marrow burden score as an alternative to Dixon quantitative chemical shift MR imaging--initial experience. Radiology. 2003;229:554–561. doi: 10.1148/radiol.2292020296. [DOI] [PubMed] [Google Scholar]
- 46.DeMayo RF, Haims AH, McRae MC, Yang R, Mistry PK. Correlation of MRI-Based bone marrow burden score with genotype and spleen status in Gaucher’s disease. AJR Am J Roentgenol. 2008;191:115–123. doi: 10.2214/AJR.07.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weinreb NJ, Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, Rosenbloom BE, Scott CR, Wappner RS, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med. 2002;113:112–119. doi: 10.1016/s0002-9343(02)01150-6. [DOI] [PubMed] [Google Scholar]
- 48.Andersson H, Kaplan P, Kacena K, Yee J. Eight-year clinical outcomes of long-term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics. 2008;122:1182–1190. doi: 10.1542/peds.2007-2144. [DOI] [PubMed] [Google Scholar]
- 49.Goitein O, Elstein D, Abrahamov A, Hadas-Halpern I, Melzer E, Kerem E, Zimran A. Lung involvement and enzyme replacement therapy in Gaucher’s disease. QJM. 2001;94:407–415. doi: 10.1093/qjmed/94.8.407. [DOI] [PubMed] [Google Scholar]
- 50.Poll LW, Maas M, Terk MR, Roca-Espiau M, Bembi B, Ciana G, Weinreb NJ. Response of Gaucher bone disease to enzyme replacement therapy. Br J Radiol. 2002;75 Suppl 1:A25–A36. doi: 10.1259/bjr.75.suppl_1.750025. [DOI] [PubMed] [Google Scholar]
- 51.Hermann G, Pastores GM, Abdelwahab IF, Lorberboym AM. Gaucher disease: assessment of skeletal involvement and therapeutic responses to enzyme replacement. Skeletal Radiol. 1997;26:687–696. doi: 10.1007/s002560050313. [DOI] [PubMed] [Google Scholar]
- 52.Ficicioglu C. Review of miglustat for clinical management in Gaucher disease type 1. Ther Clin Risk Manag. 2008;4:425–431. doi: 10.2147/tcrm.s6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cox T, Lachmann R, Hollak C, Aerts J, van Weely S, Hrebícek M, Platt F, Butters T, Dwek R, Moyses C, et al. Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet. 2000;355:1481–1485. doi: 10.1016/S0140-6736(00)02161-9. [DOI] [PubMed] [Google Scholar]
- 54.Elstein D, Dweck A, Attias D, Hadas-Halpern I, Zevin S, Altarescu G, Aerts JF, van Weely S, Zimran A. Oral maintenance clinical trial with miglustat for type I Gaucher disease: switch from or combination with intravenous enzyme replacement. Blood. 2007;110:2296–2301. doi: 10.1182/blood-2007-02-075960. [DOI] [PubMed] [Google Scholar]
- 55.Pastores GM, Barnett NL, Kolodny EH. An open-label, noncomparative study of miglustat in type I Gaucher disease: efficacy and tolerability over 24 months of treatment. Clin Ther. 2005;27:1215–1227. doi: 10.1016/j.clinthera.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 56.Pastores GM, Elstein D, Hrebícek M, Zimran A. Effect of miglustat on bone disease in adults with type 1 Gaucher disease: a pooled analysis of three multinational, open-label studies. Clin Ther. 2007;29:1645–1654. doi: 10.1016/j.clinthera.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 57.Mikosch P, Reed M, Baker R, Holloway B, Berger L, Mehta AB, Hughes DA. Changes of bone metabolism in seven patients with Gaucher disease treated consecutively with imiglucerase and miglustat. Calcif Tissue Int. 2008;83:43–54. doi: 10.1007/s00223-008-9143-4. [DOI] [PubMed] [Google Scholar]
- 58.Capablo JL, Franco R, de Cabezón AS, Alfonso P, Pocovi M, Giraldo P. Neurologic improvement in a type 3 Gaucher disease patient treated with imiglucerase/miglustat combination. Epilepsia. 2007;48:1406–1408. doi: 10.1111/j.1528-1167.2007.01074.x. [DOI] [PubMed] [Google Scholar]
- 59.Cox-Brinkman J, van Breemen MJ, van Maldegem BT, Bour L, Donker WE, Hollak CE, Wijburg FA, Aerts JM. Potential efficacy of enzyme replacement and substrate reduction therapy in three siblings with Gaucher disease type III. J Inherit Metab Dis. 2008;31:745–752. doi: 10.1007/s10545-008-0873-2. [DOI] [PubMed] [Google Scholar]
- 60.Schiffmann R, Fitzgibbon EJ, Harris C, DeVile C, Davies EH, Abel L, van Schaik IN, Benko W, Timmons M, Ries M, et al. Randomized, controlled trial of miglustat in Gaucher’s disease type 3. Ann Neurol. 2008;64:514–522. doi: 10.1002/ana.21491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lukina E, Watman N, Arreguin EA, Dragosky M, Iastrebner M, Rosenbaum H, Phillips M, Pastores GM, Kamath RS, Rosenthal DI, et al. Improvement in hematological, visceral, and skeletal manifestations of Gaucher disease type 1 with oral eliglustat tartrate (Genz-112638) treatment: 2-year results of a phase 2 study. Blood. 2010;116:4095–4098. doi: 10.1182/blood-2010-06-293902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet. 2008;372:1263–1271. doi: 10.1016/S0140-6736(08)61522-6. [DOI] [PubMed] [Google Scholar]