

Abstract
MicroRNAs (miRNAs), which are small and non-coding RNAs, are genome encoded from viruses to humans. They contribute to various developmental, physiological and pathological processes in living organisms. A huge amount of research results revealed that miRNAs regulate these processes also in the heart. miRNAs may have cell-type-specific or tissue-specific expression patterns or may be expressed ubiquitously. Primary studies of miRNA involvement in hypertrophy, heart failure and myocardial infarction analyzed miRNAs that are enriched in or specific for cardiomyocytes; however, growing evidence suggest that other miRNAs, not cardiac or muscle-specific, play a significant role in cardiovascular disease. Abnormal miRNA regulation has been shown to be involved in cardiac diseases, suggesting that miRNAs might affect cardiac structure and function. In this review, we focus on miRNAs that have been found to contribute to the pathogenesis of myocardial infarction (MI) and the response post-MI and characterized as diagnostic, prognostic and therapeutic targets. The majority of these studies were performed using mouse and rat models of MI, with a focus on the identification of basic cellular and molecular pathways involved in MI and in the response post-MI. Much research has also been performed on animal and human plasma samples from MI individuals to identify miRNAs that are possible prognostic and/or diagnostic targets of MI and other MI-related diseases. A large proportion of research is focused on miRNAs as promising therapeutic targets and biomarkers of drug responses and/or stem cell treatment approaches. However, only a few studies have described miRNA expression in human heart tissue following MI.
Keywords: MicroRNAs, Myocardial infarction, Human, Animal models, Biomarkers and targets
Core tip: MicroRNAs (miRNAs) contribute to various developmental, physiological and pathological processes in the heart. Cardiac diseases show abnormal miRNA regulation. Primary studies of miRNA involvement in cardiac disease analyzed mainly miRNAs that enriched in or specific for cardiomyocytes; however, growing evidence suggests that other cell-type-specific or ubiquitously expressed miRNAs are also involved in cardiovascular disease. miRNAs were found to contribute to the pathogenesis of myocardial infarction (MI) and post-MI. The majority of studies focused on miRNAs in animal models of MI, in human and animal plasma samples of MI (prognostic and diagnostic targets), and on miRNAs as promising therapeutic targets.
INTRODUCTION
MicroRNAs (miRNAs) are endogenously expressed small non-coding RNA molecules. Genes encoding miRNAs can be found in genomes of almost all organisms, including viruses. Their prime mechanism of action is post-transcriptional repression of gene expression[1]. It is suggested that the short length (22 nt) maximizes target-gene specificity and minimizes non-specific effects. It is estimated that miRNAs regulate approximately 30% of genes within the human genome[2]. There are over 2000 miRNAs known to be encoded by human genome. All sequenced and cloned miRNAs from humans as well as from other species are included in database miRBase (v20.0, June 2013, http://www.mirbase.org)[3].
Mechanism of action of miRNAs
Biogenesis (including genes encoding miRNAs), transcription and processing are beyond the scope of this review and are described elsewhere[1,2,4,5]. As mentioned above, miRNAs prime mechanism of action is repression of gene expression. By sequence-specific binding to the 3’-untranslated region (3’-UTR) of mRNAs, miRNA affects stability of the transcripts and cause mRNA degradation, which is the main mechanism in plants and happens when complementarity between miRNA and mRNA is perfect, or cause protein synthesis repression (translational repression), which happens when base pairing between these two molecules is incomplete and is the canonical mechanism in animals[1,2,4,5]. Due to incomplete base pairing in animals and humans, each miRNA could influences translation of many different mRNAs without degrading it (i.e., over 200 predicted target genes) and vice versa each mRNA may be influenced by different miRNA. It appears that the most efficient translational inhibition is provided through the multiplicity, which is the consequence of numerous target sites for the same miRNAs within 3’-UTR of the same mRNA (cooperative action of multiple identical miRNAs), and trough cooperativity, which is due to numerous target sites for the different miRNAs within 3’-UTR of the same mRNA. miRNA access to the UTRs could be on one hand restricted by proteins or mRNA secondary structures, and on the other hand these structures and protein binding may facilitate recognition of the mRNA targets[6]. Some miRNAs might also have other functions, although translational repression has been suggested to be the canonical one[2,4].
miRNA in regulating physiological functions
Different approaches in in vitro and in vivo experiments have been used to reveal function of majority of miRNA. Using mutated miRNA or its mutated complementary site within mRNA, consequently disrupting regulation of mRNA by miRNA, leads to the determination of the phenotypic consequence of this non-binding. Another possibility is use of transgenic constructs of either 3’-UTR or miRNA expressing vector and ectopic expression of the either miRNA or mRNA[1,7]. Perhaps the best evidence that miRNA are playing a significant role in normal physiological functions was established, when the components of the miRNA biogenesis pathway were depleted[8]. In normal cell conditions, miRNAs can repress translation in different ways: (1) as a switching-off the targets, when protein production is reduced to inconsequential levels in a cell type, where target mRNAs should not be expressed; (2) as fine-tuning expression of target gene, when protein output can be adjusted in a way, which provides customized expression in one cell type and uniformly expressed level within another cell type; and (3) as neutralizers of target gene expression, when mRNA downregulation by miRNAs is negated through feedback processes[1]. The role of miRNAs can be combinatorial (defined as cooperativity), different in different cell types, and either specific or housekeeping[9].
Through the studies of expression profiling of normal and disease tissues it has been shown that miRNAs are expressed in spatial as well as in temporal manner. miR-208 is a good example of expression in tissue-specific manner. Its expression can be detected specifically in the hearts, as well can be miR-122 found primarily in the liver. As an example of cell-type-specific miRNAs are miR-223, which is primarily expressed in granulocytes, and miR-1 and miR-133, which are believed to be myocyte-specific[10]. miRNAs are involved in a myriad of biological processes, including proliferation, apoptosis, metabolism, differentiation, epithelial-to-mesenchymal transition, regulation of insulin secretion, division of stem cells, embryonic development and pattering, fetal growth, immune system, including resistance to viral infection and vice versa viral production (in a case of HCV), etc[8]. miRNA activity is believed to have crucial role in regulatory role in maintaining tissue identity during embryogenesis as well as in adult life. Distinct miRNA expression profile, with completely different gene expression patterns might be observed in every cell type at each developmental stage[9-11].
Target prediction and bioinformatics
As mentioned above, miRBase is major database of all known miRNAs, which can also predict possible miRNA targets[3]. Predicting possible miRNA binding sites for specific mRNAs or potential targets for certain miRNA is usually the first step in target identification and for this purpose numerous computational methods have been developed. Main characteristics that are included in established programs are: evolutionarily conservation of the complementary 3’-UTR sequence, quality and stability of mRNA:miRNA pairing and involvement of “seed sequence”. It is believed that for base pairing the most important is “seed sequence” of the miRNA (2-8 nt at the 5’-end) and its interaction with seven consecutive nucleotides in the target mRNA[12]. However, all predicted targets have to be validated in vitro and in vivo since none of these programs can independently validate the targets[7,13]. Due the facts that 3’-UTR sites with perfect complementarity to the miRNA are not necessary functional and that mRNA sites with imperfect complementarity can themselves be very good miRNA targets, are bioinformatic analysis more prone to false positives[6]. Therefore, experimental demonstration that overexpression of the miRNA represses a luciferase reporter fused to the 3’-UTR of the predicted target and that this repression is not established by point mutations in the 3’-UTR target sequence is the gold standard for miRNA target identification[13,14]. Finally, association of mRNA:miRNA pairs with disease pathogenesis should be confirmed by expression profiling in human diseases by co-expression analyses[7,14]. Human MicroRNA Disease Database identifies all disease-related miRNAs with their tissue expression patterns[15]. Further, Tarbase lists experimentally validated miRNA targets for all organisms[12].
miRNAs AND DISEASE
Mutations, single-nucleotide polymorphisms and the epigenetics of miRNAs
There are several genetic and epigenetic abnormalities within miRNA genes that might contribute to a wide range of diseases. These abnormalities include small- and large-scale genomic alterations, as are rearrangements and chromosomal translocations, copy-number variation, nucleotide expansion, and single-nucleotide polymorphisms (SNPs) that beside protein-coding region also affect regions that code for non-coding RNAs. First, it has been shown that approximately 50% of the miRNA genes are encoded within fragile chromosomal sites or sites that are prone to cancer-associated rearrangements[10]. Second, although some SNPs are silent and cause no obvious functional consequence, other might cause disruption of binding between miRNAs and their targets, which can potentially lead to gain or loss of the function of miRNA or its target gene and consequently contribute to the disease state[16]. Variants identified in miRNA or their precursors (pri-miRNA or pre-miRNA) that beside targeting might also affect the processing and expression of miRNAs, are rarely observed. However, potential of variation in miRNA target sites is more huge[6,16]. Third, the aberrant DNA methylation of gene promoters has been shown to results in the inactivation of different genes, including miRNAs, and in parallel, miRNAs can also regulate proteins involved in DNA methylation[17].
Aberrant expression of miRNAs
Epigenetic mechanisms and genomic abnormalities frequently lead to abnormal miRNA expression profiles thus causing pathogenetic events in diseases. Numerous advances in miRNA research and numerous expressions profiling of diseased human tissue are suggesting that miRNAs are associated with various pathological conditions. miRNAs have been linked to wide range of diseases, including cancer genetic and immunological disorders, neurodegenerative and cardiovascular disorders[10].
THERAPEUTIC POTENTIAL OF miRNAs
miRNA expression patterns are dynamically regulated during various diseases and can also be used for pharmacological manipulation. Studies have demonstrated that the systemic use of antagomirs is well suited to block miRNA function in small animal models. For targeting a specific miRNA or disrupting binding between miRNA and its target mRNA the chemically modified oligonucleotides have been developed. miRNAs as small molecules of approximately 22 nt in length are more feasible delivered in vivo. Synthetic miRNAs can be therefore delivered systematically and may thus serve as therapeutic targets in the future[18].
Replenishing small RNAs
Underexpressed miRNA might be restored by reintroduction of the mature miRNA into the target tissue consequently restoring regulation of the miRNA target gene. miRNAs as potential therapeutic agents can be easily targeted and delivered to the appropriate tissue. Three major approaches are described below. Artificial miRNA or miRNA “mimics” enhance the expression of beneficial miRNAs. Artificial miRNAs are transient transfections of double-stranded miRNAs and possess the ability to bind to the homologous target site in various mRNAs. Another option is the introduction of a viral vector or plasmid expressing a specific miRNA from a short hairpin (sh) duplex (pre-miRNA-like shRNA). A high level of shRNA might lead to effective target knockdown; however, it may also saturate the miRNA biogenesis pathway and lead to off-target effects with fatal consequences. Therefore, another possibility arises, namely miRNA scaffolds. In scaffolds of endogenous pri-miRNA or pre-mRNA, siRNA is inserted and introduced to the target tissue leading to the degradation of homologous mRNA. This approach is advantageous in terms of specificity and stability over conventional shRNA because both siRNA and shRNA may trigger a non-specific interferon response in addition to off-target effects. As an example in cardiovascular disease, overexpression of miR-133 was used in a study of cardiac hypertrophy. By adenovirus delivery of a miRNA expression cassette, expression of miR-133 was restored, which results in protection of experimental animals from agonist-induced cardiac hypertrophy[18].
Inhibiting small RNAs
ASOs are short single-stranded antisense oligonucleotides, which are called anti-miRNA oligonucleotides, AMOs or antagomirs when talking about inhibition of miRNA. Antagomirs have been shown to efficiently and specifically silence endogenous miRNAs in mice. Overexpressed miRNA can also be downregulated by reducing the loop region of the miRNA precursor (pre-miRNA). The loop regions of different pre-miRNAs are not conserved and might therefore limit their application. Another approach is to use miRNA sponges, which are miRNA inhibitory transgenes containing multiple tandem binding sites for an endogenous miRNA and can inhibit several closely related miRNAs. miRNA sponges may be useful for sequestering a miRNA family with overlapping and redundant targets. miR-masks and miR-erasers have also been developed. Similarly to a miR-sponge, a miR-eraser sequesters more than one miRNA, except that there are only two copies of the antisense sequence. For masking the miRNA binding site on the target gene, miR-mask or miRNA masking antisense approach has been designed, which forms a duplex with target mRNA. They are also called antisense oligodeoxynucleotides (ODNs). Two approaches have been used in the context of studying cardiovascular diseases. A miRNA decoy using miRNA sponges was designed and used in research studying the effect of miR-133 in the pathogenesis of cardiac hypertrophy. Another approach used ODNs entirely complementary to the miRNA target motifs in the 3’-UTR of 2 cardiac pacemaker channel genes, HCN2 and HCN4[18,19].
Delivering miRNAs or its inhibitor to the target tissue
Current limitations exist in the following areas and need improvement: the efficiency of delivery of miRNA therapeutics to target tissues; systemic administration of drug; the potential inhibition of non-target genes (off-target effects); redundancy in the efficacy of different miRNAs; potential toxicity; and immunogenic responses. Modifications have improved the stability of miRNAs or blocked their inhibition (i.e., nuclease resistance and pharmacokinetic properties such as half-life in serum and cellular uptake). Stabilization and facilitation of intravenous delivery of antagomirs could be improved by chemical modifications and cholesterol conjugations. However, toxicity due to chemical modifications should be taken into account. Local administration in easily accessible tissues has been used in the majority of the developed protocols; a major challenge remains for tissue- and cell-type-specific targeting. Viral and non-viral delivery systems have been developed in conjugation with homing signals for tissue- or cell-type-specific delivery, e.g., linked to lipids and/or proteins, cationic liposomes, cholesterol, bacterial phage, aptamers, etc[18,19].
miRNA IN MYOCARDIAL INFARCTION
MicroRNA in cardiovascular diseases
miRNAs contribute to the regulation of developmental, as well as physiological and pathological processes in the heart. Loss of cardiomyocyte renewal is a hallmark of numerous cardiac diseases, which might also influence miRNA expression patterns in the diseased heart[20]. Primary studies of miRNA involvement in hypertrophy, heart failure and myocardial infarction analyzed miRNAs that are enriched in or are specific for cardiomyocytes; however, growing evidence suggest that other miRNAs, not cardiac- or muscle-specific, play a significant role in cardiovascular disease[21-24]. Using experimental animals and human samples dysregulation of specific miRNAs has been shown that are distinct than those involved in other heart diseases as are hypertrophy and heart failure (HF). Cell lines and an animal models of different forms of myocardial ischemia, including myocardial infarction (MI), have been used to perform miRNA microarray expression profiling[20]. It has been shown that in response to limited amount of oxygen, numerous miRNAs are up- or downregulated. Many of dysregulated miRNAs are dependent on a transcription factor that plays an important role in response to low oxygen, hypoxia-inducible-factor. Further analyses showed that oxidative stress also activates other transcription factors that beside miRNA expression influence different homeostatic and physiological processes as are metabolism, angiogenesis, cell survival and oxygen delivery[25,26]. Numerous other pathways are activated in response to MI, including apoptosis and fibrosis, as well as numerous cell types such as cardiomyocytes, immune cells, fibroblasts and endothelial cells (ECs)[27]. However, the majority of studies were performed in terms of expression analysis and target gene identification, with only one publication focusing on MI, specifically, target site polymorphisms and the risk for MI.
Cardiac and muscle specific miRNAs in heart
There are five miRNAs recognized as muscle- and/or cardiac-specific or enriched, miR-1, miR-133, miR-206, miR-208 and miR-499. For miR-1 and miR-133 it is believed that are muscle-specific and that regulate heart development[28]. miR-208 has been identified as cardiac-specific and miR-499 as cardiac-enriched.
In the current review we have focused on miRNAs involved in MI pathogenesis and their diagnostic, prognostic and therapeutic potential regarding MI. The function of various miRNAs analyzed in cell lines, animal models of MI and patients with MI are presented. Table 1 summarizes all free-circulating miRNAs in experimental model of MI and human MI, describes their suggested function and predicted targets. We further overviewed the results of free-circulating miRNAs in different bodily fluids of patients and/or an animal model with MI. Table 2 summarizes all miRNAs as potential diagnostic and/or prognostic targets in MI. Lastly, therapeutic opportunities using miRNA strategies in the context of MI are also presented. More detailed description of all these miRNAs is below.
Table 1.
miRNAs with suggested role in experimental models of myocardial infarction and in myocardial infarction in humans
| miRNA | Role/function | Expression in MI | Target genes | Species | Ref. |
| miR-1 | After heat-shock up, protective against I/R | Nd | Repressing pro-apoptotic and up-regulating anti-apoptotic genes | Mouse | [40] |
| Nd | Down | Nd | Mouse | [59] | |
| Pro-apoptotic | Up | IGF1 | Rat | [45] | |
| Pro-arrhythmogenic | Up | Ion channels: Cx43, Kir2.1 | Rat | [46] | |
| Predictive | Down, up | Predictive | Human | [55,56] | |
| miR-15b | Anti-angiogenic | Down | Suggested VEGF and Ang2 | Endothelial cells | [30] |
| miR-21 | H2O2 induced cell injury | Up | PDCD4 | Cardiomyocytes | [29] |
| After heat-shock up, protective against I/R | Nd | Repressing pro-apoptotic and up-regulating anti-apoptotic genes | Mouse | [40] | |
| Response to I/R | Up in cardiac fibroblasts | PTEN | Mouse | [23] | |
| Anti-apoptotic | Down, up | PDCD4 | Rat | [47] | |
| Pro-arrhythmogenic | Up | Sprouty-1, collagen I, collagen III | Rat | [48] | |
| miR-24 | Anti-fibrotic | Down | Furin | Mouse | [35] |
| Anti-angiogenic, induce endothelial cell apoptosis | Down in cardiomyocytes and fibroblasts, up in endothelial cells | GATA2, PAK4 eNOS | Mouse Mouse | [36] [37] | |
| After heat-shock up, protective against I/R | Nd | Repressing pro-apoptotic and up-regulating anti-apoptotic genes | Mouse | [40] | |
| miR-29 family | Anti-fibrotic | Down | Proteins involved in fibrosis (COL1A1-2, COL3A1, FBN1, ELN) | Mouse, human | [34] |
| miR-29b | Anti-fibrotic | Down | Proteins involved in fibrosis (COL1A1, COL3A1, αSMA) | Rat | [52] |
| miR-34a | Pro-apoptotic | Up | ALDH2 | Rat | [49] |
| miR-92a | Anti-angiogenic | Up | ITGA5 | Mouse | [38] |
| miR-101a/b | Anti-fibrotic | Down | c-Fos | Rat | [50] |
| miR-106b | Anti-apoptotic | Up | p21 | Cardiomyocytes | [30] |
| miR-133a | Nd | Down | Nd | Mouse | [59] |
| Predictive | Down | Predictive | Human | [55,56] | |
| miR-133b | Predictive | Down | Predictive | Human | [55,56] |
| miR-146a | Predictive: inflammation and VR | Up | Predictive | Human | [57] |
| miR-150 | Predictive: inflammation and VR | Down | Predictive | Human | [57] |
| miR-155 | Predictive: inflammation and VR | Down | Predictive | Human | [57] |
| miR-206 | Pro-apoptotic | Up | IGF1 | Rat | [45] |
| miR-208a | Nd | Down | Nd | Mouse | [59] |
| Predictive | Up | Predictive | Human | [55] | |
| miR-320 | Pro-apoptotic | Down | HSP20 | Mouse | [41] |
| miR-494 | Activation of Akt pathway | Down | Pro- and anti-apoptotic proteins (PTEN, ROCK1, CaMKII; FGFR2, LIF) | Mouse | [42] |
| miR-499 | Nd | Down | Nd | Mouse | [59] |
| miR-711 | Involved in anti-fibrotic effect of pioglitazone | Down | SP1 | Rat | [51] |
| miR-874 | Regulated by Foxo3a in necrosis | Up | Caspase 8 | Mouse | [39] |
I/R: Ischemia/reperfusion; MI: Myocardial infarction; Nd: Not determinate; VR: Ventricular rupture.
Table 2.
miRNAs as potential diagnostic and prognostic biomarkers in myocardial infarction
| miRNAs as potential biomarker | Role of biomarker | Expression in body fluid | Species and body fluid | Ref. |
| let-7b | Potential diagnostic value | Up | Plasma; human | [72] |
| let-7f | Differentiating TTC and MI | Down | Plasma; human | [79] |
| miR-1 | Detection of AMI and AP | Up | Exosome, serum; human and mouse | [59] |
| Correlation with MI size | Up | Serum; rat and human | [60] | |
| Differentiating AMI and AP | Up | Serum; human | [61] | |
| Differentiating AMI and other cardiovascular diseases | Up | Plasma; rat and human | [67] | |
| Similar time course to cTnI and the same trend to cTnI concentration | Up | Plasma and tissue; human and mouse | [68,70] | |
| Differentiating AMI and non-AMI | Up | Plasma; human | [69] | |
| AMI biomarkers, not superior to cTnT | Up | Plasma; human | [71] | |
| Associated with various degree of cardiovascular damage (AMI, viral myocarditis, diastolic dysfunction, acute HF) | Up | Plasma; human | [76] | |
| No association with 30 d mortality post-MI and diagnosis of HF | Up | Plasma; human | [81] | |
| Biomarker for AMI, correlated with renal elimination | Up | Plasma, urine; human, pig | [85] | |
| Detected in urine | Up | Urine; rat | [86] | |
| miR-16 | Differentiating TTC and MI | Down | Plasma; human | [79] |
| Higher risk of impaired LV contractility | Up | Plasma; human | [83] | |
| miR-21 | Associated with various degree of cardiovascular damage (AMI, viral myocarditis, diastolic dysfunction, acute HF) | Nd | Plasma; human | [76] |
| Differentiating NSTEMI and CHF | Up | Plasma; human | [78] | |
| Time-dependent changes 2-90 d post-MI | Down, up | Plasma; human | [80] | |
| miR-26a | Differentiating TTC and MI | Down | Plasma; human | [79] |
| miR-27a | High risk of impaired LV contractility | Up | Plasma; human | [83] |
| miR-29a | Time-dependent changes 2-90 d post MI | Up | Plasma; human | [80] |
| miR-29b | Associated with various degree of cardiovascular damage (AMI, viral myocarditis, diastolic dysfunction, acute HF) | Nd | Plasma; human | [76] |
| miR-30a | Potential diagnostic value | Up | Plasma; human | [72] |
| miR-30c | Correlation with MI size | Up | Whole blood; human | [64] |
| miR-34a | Prognostic: correlated with LV end diastolic dimension | Up | Exosomes, serum; human | [62] |
| miR-101 | Higher risk of impaired LV contractility | Down | Plasma; human | [83] |
| miR-126 | The same trend to cTnI expression | Down | Plasma; human | [70] |
| Positive association to the risk for MI | Nd | Plasma; human | [84] | |
| miR-133a | Detection of AMI, AP: biomarker for cardiomyocyte death | Up | Exosome, serum; human and mouse | [59] |
| AMI biomarker, correlation to cTnI | Up | Plasma and whole blood; human | [66] | |
| Differentiating AMI and other cardiovascular diseases | Up | Plasma; rat and human | [67] | |
| Similar time-course to cTnI | Up | Plasma and tissue; human and mouse | [68] | |
| AMI biomarkers, not superior to cTnT | Up | Plasma; human | [71] | |
| Differentiating AMI and AP, positive correlation to severity of coronary stenosis | Up | Plasma; human | [75] | |
| Associated with various degree of cardiovascular damage (AMI, viral myocarditis, diastolic dysfunction, acute HF) | Up | Plasma; human | [76] | |
| Differentiating TCC and MI | Up | Plasma; human | [79] | |
| Biomarker for AMI, correlated with renal elimination | Up | Plasma, urine; human, pig | [85] | |
| miR-133b | Similar time-course to cTnI | Up | Plasma and tissue; human and mouse | [68] |
| miR-134 | Differentiating AMI and AP | Up | Serum; human | [61] |
| miR-145 | Correlation with MI size | Up | Whole blood; human | [64] |
| miR-146a | Associated with various degree of cardiovascular damage (AMI, viral myocarditis, diastolic dysfunction, acute HF) | Nd | Plasma; human | [76] |
| miR-150 | Associated with LV remodeling | Down | Plasma; human | [82] |
| Higher risk of impaired LV contractility | Down | Plasma; human | [83] | |
| miR-155 | Prognostic for cardiac death within 1 yr after MI | Up | Serum; human | [63] |
| Associated with various degree of cardiovascular damage (AMI, viral myocarditis, diastolic dysfunction, acute HF) | Nd | Plasma; human | [76] | |
| miR-181c* | Novel mirna dysregulated during MI | Nd | Whole blood; human | [65] |
| miR-186 | Differentiating AMI and AP | Up | Serum; human | [61] |
| miR-192 | Prognostic for development of ischemic HF | Up | Exosomes, serum; human | [62] |
| miR-194 | Prognostic: correlated with LV end diastolic dimension | Up | Exosomes, serum; human | [62] |
| miR-195 | Potential diagnostic value | Up | Plasma; human | [72] |
| miR-197 | Negative association to the risk for MI | Nd | Plasma; human | [84] |
| miR-208 | Differentiating AMI and AP | Up in AP compared to AMI | Serum; human | [61] |
| Differentiating AMI and other cardiovascular diseases | Up | Plasma; rat and human | [67] | |
| Time-dependent changes 2-90 d post MI | Up | Plasma; human | [80] | |
| Detected in urine | Up | Urine; rat | [86] | |
| miR-208b | AMI biomarkers, correlation to cTnT but not superior to cTnT | Up | Plasma; human | [71,76] |
| Differentiating STEMI and NSTEMI | Higher in STEMI | Plasma; human | [73] | |
| Associated with various degree of cardiovascular damage (AMI, viral myocarditis, diastolic dysfunction, acute HF) | Up | Plasma; human | [76] | |
| Higher risk for 30 d mortality post-MI and HF | Up | Plasma; human | [81] | |
| Biomarker for AMI, correlated with troponin | Up | Plasma, urine; human, pig | [85] | |
| miR-223 | Differentiating AMI and AP | Up | Serum; human | [61] |
| Associated with various degree of cardiovascular damage (AMI, viral myocarditis, diastolic dysfunction, acute HF) | Down | Plasma; human | [76] | |
| Negative association to the risk for MI | Nd | Plasma; human | [84] | |
| miR-328 | AMI biomarker, correlation to cTnI | Up | Plasma and whole blood; human | [66] |
| miR-380* | Prognostic for cardiac death within 1 yr after MI | Up | Serum; human | [63] |
| miR-423-5p | Before PCI compared to after | Up | Plasma; human | [77] |
| miR-499 | Differentiating AMI and AP | Up in AP compared to AMI | Serum; human | [61] |
| Differentiating AMI and other cardiovascular diseases | Up | Plasma; rat and human | [67] | |
| Similar time course to cTnI | Up | Plasma and tissue; human and mouse | [68] | |
| AMI biomarkers, correlation to cTnT but not superior to cTnT | Up | Plasma; human | [71,76] | |
| Differentiating STEMI and NSTEMI | Higher in STEMI | Plasma; human | [73] | |
| Differentiating MI, CHF and unstable AP | Up | Plasma; human | [74] | |
| Associated with various degree of cardiovascular damage (AMI, viral myocarditis, diastolic dysfunction, acute HF) | Up and also in acute HF | Plasma; human | [76] | |
| Differentiating NSTEMI and CHF | Up | Plasma; human | [78] | |
| Higher risk for 30 d mortality post MI and HF | Up | Plasma; human | [81] | |
| Biomarker for AMI, correlated with renal elimination | Up | Plasma, urine; human, pig | [85] | |
| miR-1915 | Novel miRNA dysregulated during MI | Nd | Whole blood; human | [65] |
| 11 miRNAs | Prognosis after MI | Up and down | Serum; human | [63] |
| 20 miRNAs | Predicting AMI (96% specificity; 90% sensitivity; 93% accuracy) | Up and down | Whole blood; human | [64] |
| A subset of miRNAs | Dysregulated during AMI course | Nd | Whole blood; human | [65] |
| 34 miRNAs | AMI biomarkers | 20 up, 14 down | Plasma and tissue; human and mouse | [68] |
| 19 candidate miRNAs | Prediction for risk of MI | Nd | Plasma; human | [84] |
AMI: Acute myocardial infarction; AP: Angina pectoris; cTnI: Cardiac troponin I; CHF: Chronic heart failure; cTnT: Cardiac troponin T; HF: Heart failure; LV: Left ventricle; MI: Myocardial infarction; Nd: Not determine; NSTEMI: Non-ST-elevation MI; PCI: Percutaneous coronary intervention; STEMI: ST-elevation MI; TTC: Takotsubo cardiomyopathy.
Cell line models
miR-21: miR-21 was upregulated after inducing injury of cardiac myocytes using H2O2, and H2O2-induced cardiac cell death and apoptosis were increased by a miR-21 inhibitor. Programmed cell death 4 (PDCD4) has been identified as a target of miR-21, and activator protein 1 (AP-1) has been identified as a downstream signaling molecule of PDCD4[29]. All miRNAs with suggested role in apoptosis in MI are summarized in Figure 1.
Figure 1.
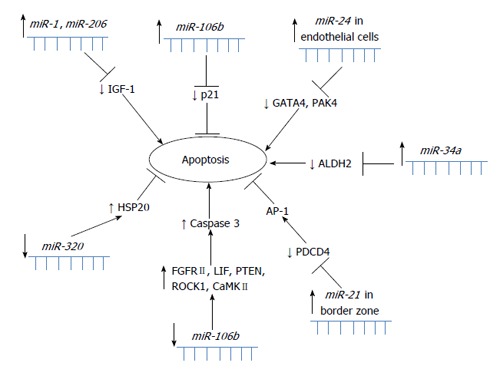
Schematic overview of miRNAs involved in apoptosis in myocardial infarction.
miR-15b and miR-106b: After retrieving 119 MI-related miRNAs from publications, GO and pathway analyses for their predicted gene targets demonstrated that these dysregulated miRNAs were enriched in cardiovascular-related phenotypes. By highlighting miRNA-gene networks, overall relationships between miRNAs and gene targets were discovered, particularly in apoptosis and angiogenesis. Experimental data identified miR-106b as an anti-apoptotic modulator through inhibition of p21 expression and miR-15b as an anti-angiogenic miRNA with the possible targets vascular endothelial growth factor and Ang2 (angiopoietin 2)[30]. All miRNAs with suggested role in angiogenesis in MI are summarized in Figure 2.
Figure 2.
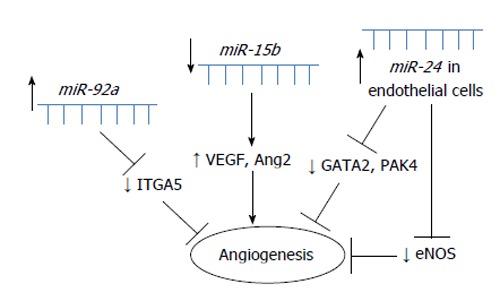
Schematic overview of miRNAs involved in angiogenesis in myocardial infarction.
To investigate the possible release of miRNAs from activated platelets, the miRNA content of platelets was screened from control patients and patients with MI. Nine miRNAs found to be differentially expressed in MI patients compared with healthy controls were screened, and 8 of these were decreased in MI patients. Of these, miR-22, miR-185, miR-320b and miR-423-5p increased after aggregation in the supernatant of platelets and were depleted in thrombi aspirated from MI patients. Platelets from patients with MI exhibit a loss of specific miRNAs, and activated platelets shed miRNAs that can regulate EC gene expression[31].
Mouse models
Whole genome microarray analysis: Genome-wide mRNA and miRNA expression profiles were performed at three time points post-MI: 2 d, 2 wk and 2 mo. The majority of differentially expressed miRNAs were uniquely regulated at each of the time points analyzed. Bioinformatic analysis demonstrated that several genes and miRNAs in various pathways are regulated in a temporal or phenotype-specific manner[32]. In another study, a mouse MI was induced and one week after MI, a set of 29 upregulated miRNAs was found in the left ventricle originating from the Dlk1-deiodinase type 3 gene (Dio3) genomic imprinted region, which has been identified as a hallmark of pluripotency and proliferation. This miRNA signature was associated with an increase in expression of the Dio3 located in this region. Dio3 is a fetally expressed enzyme associated with cell proliferation, which was shown to be upregulated in cardiomyocytes. These data suggest that a regenerative process is initiated, but not completed, in adult cardiomyocytes after MI[33].
miR-29 and fibrosis: One of the first studies regarding MI was comparing expression profiles of miRNA from mouse border zone of the infarcted region as well as from the remote myocardium 3 and 14 d after MI. The miR-29 family was downregulated in the region of the heart adjacent to the infarct. It has been shown that downregulation of miR-29b with anti-miRs induces the expression of collagen and that overexpression of miR-29 reduces collagen. Three days after the MI, in the infarcted region, miR-29 downregulation correlated with upregulation of collagen types I and III (COL1A1, COL1A2, COL3A1) and fibrillin, and in the remote myocardium expression of elastin was increased. The miR-29 family was thus identified as a regulator of fibrosis[34]. All miRNAs with suggested role in fibrosis in MI are summarized in Figure 3.
Figure 3.
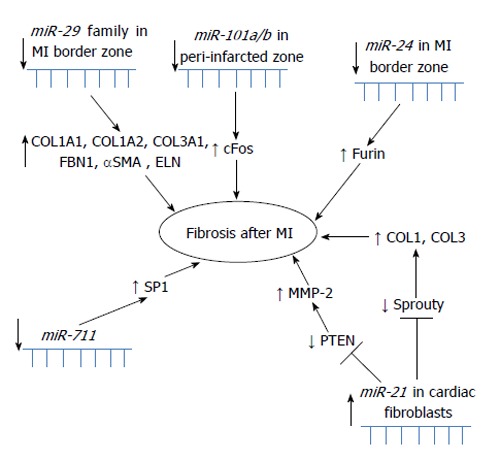
Schematic overview of miRNAs involved in fibrosis after myocardial infarction.
miR-24, fibrosis and angiogenesis: The downregulation of miR-24 in a mouse MI model was closely related to extracellular matrix remodeling. Intra-myocardial injection of miR-24 was able to improve heart function and attenuate fibrosis in the infarct border zone. In vitro experiments suggested that the upregulation of miR-24 could reduce fibrosis and decrease the differentiation and migration of cardiac fibroblasts (CFs). Transforming growth factor β (TGF-β) increased miR-24 expression, and overexpression of miR-24 reduced TGF-β secretion and Smad2/3 phosphorylation in CFs. Furin was found to be a potential target for miR-24 in fibrosis and both protein and mRNA levels of furin were regulated by miR-24 in CFs[35]. miR-24 is markedly upregulated after cardiac ischemia and it has been also shown to be enriched in cardiac ECs. miR-24 has been reported to induce apoptosis in ECs and abolishes endothelial capillary network formation by targeting the endothelium-enriched transcription factor GATA2 and the p21-activated kinase PAK4. MI size in mice has been limited by blocking endothelial miR-24. Reduced MI size as well as preserved cardiac function and survival were probably due to prevention of endothelial apoptosis and enhancement of vascularity as a consequence of blocked miR-24[36]. Another mouse model showed that after a MI induction, miR-24 expression was lower in the peri-infarct tissue and its resident cardiomyocytes and fibroblasts, while it increased in ECs. Local adenovirus-mediated miR-24 decoy delivery increased angiogenesis and blood perfusion in the peri-infarct myocardium, reduced infarct size, induced fibroblast apoptosis and overall improved cardiac function. The miR-24 decoy increased apoptosis in cardiomyocytes. In vitro miR-24 inhibition enhanced EC survival and proliferation and induced cardiomyocyte and fibroblast apoptosis. Endothelial nitric oxide synthase has been identified as a novel direct target of miR-24 in human cultured ECs and in vivo[37].
miR-92a and angiogenesis: miR-92a has been shown to control the growth of new blood vessels (angiogenesis). Systemic administration of antagomir-92a led to enhanced blood vessel growth and functional recovery of damaged tissue. Overexpression of miR-92a blocked angiogenesis and vessel formation. miR-92a was shown to be upregulated after induction of acute MI (AMI). Antagomir-92a treatment reduced the infarct size, suppressed the number of apoptotic cells and augmented the number of in vivo perfused vessels in the infarct border zone. Among its targets are several pro-angiogenic proteins, including integrin subunit alpha5[38].
miR-874 and necrosis: Another study revealed that in response to H2O2 treatment, miR-874 was substantially increased. Knockdown of miR-874 attenuated necrosis in the cellular model and also MI in the mouse model. As downstream mediator and target of miR-874 was identified caspase-8. Caspase-8 was able to antagonize necrosis. When suppressed by miR-874, caspase-8 lost the ability to repress the necrotic program. Foxo3a was identified as a transcriptional repressor of miR-874 expression. This study determined a novel myocardial necrotic regulatory model consisting of Foxo3a, miR-874 and caspase-8[39].
miR-1, miR-21, miR-24, miR-320 and ischemia-reperfusion: Heat-shock treatment protects the heart against ischemia-reperfusion (I/R) injury. A significant induction and increase of miR-1, miR-21 and miR-24 has been observed in hearts of mice, which were subject to cytoprotective heat-shock (HS). miRNAs isolation from HS mice and injection into non-HS mice, resulted in significantly reduction of the infarct size in the heart following global I/R injury. Further analysis showed that reduction in MI size is accompanied by downregulation of expression of genes that induce apoptosis and upregulation of those that reduce apoptosis. These results showed that in the non-heat-shocked mice, miRNA function in heat-shock-like protection against I/R. Proposed mechanism of miRNAs action is through repression of pro-apoptotic genes (caspases 1, 2, 8, and 14, Bid, Bcl-10, Cidea, Ltbr, Trp53, and Fasl) and induction of anti-apoptotic genes (Bag-3 and Prdx2). Through administration of miR-21, it has been shown that chemically synthesized miRNA can reduce MI size, an outcome that was blocked with a miR-21 inhibitor[40]. Another miRNA in the mouse hearts with I/R has been shown to be dysregulated, miR-320. miR-320 was shown to be significantly decreased after MI and to target heat-shock protein 20. Experiments involving cardiac-specific overexpression of miR-320 in transgenic mice resulted in increased apoptosis and infarct size in the hearts with I/R, and treatment with antagomir-320 reduced the infarct size[41].
miR-21, I/R and fibrosis: Further research on I/R models led to the identification of miRNAs with significant expression changes on days 2 and 7 post-I/R. Elevated miR-21 levels were observed on day 2 as well as on day 7; however, miR-21 induction in response to I/R was limited to CFs. CFs were shown to be the major cell type in the infarct zone. A marked decrease in phosphatase and tensin homolog (PTEN), a target of miR-21, has also been observed in the infarct zone. This decrease has been associated with increased matrix metalloproteinase-2 (MMP-2) expression, suggesting a miR-21-PTEN-Akt-MMP-2 pathway in CFs after MI[23].
miR-494 and apoptosis in I/R: A mouse model with cardiac-specific miR-494 overexpression showed improved recovery of contractile performance during the reperfusion period. This was accompanied by a reduction of apoptosis in transgenic mice and reduced MI in I/R. Cultured adult cardiomyocytes with short-term overexpression of miR-494 showed an inhibition of caspase-3 activity and reduced cell death after stimulated I/R. miR-494 inhibited three pro-apoptotic (PTEN, ROCK1, CaMKII) as well as two anti-apoptotic proteins (FGFR2 and LIF). miR-494 targets both pro- and anti-apoptotic proteins and was downregulated in human infarcted hearts. Divergent targets of a miRNA may work unequally to balance a common signaling pathway and eventually affect its functional consequences[42].
Rat models
Microarray analysis: Using genome-wide expression profiling of miRNAs in an ischemic myocardium from rat, seventeen miRNAs were shown to be significantly dysregulated during the AMI progression. Expression was analyzed 2, 7 and 14 d after AMI. On day 2, four miRNAs were upregulated (miR-31, miR-223, miR-18a and miR-18b) and two were downregulated (miR-451 and miR-499-5p). On day 7, four miRNAs were upregulated (miR-31, miR-214, miR-199a-5p and miR-199a-3p) and seven were downregulated (miR-181c, miR-181d, miR-499-5p, miR-29b, miR-26b, miR-126 and miR-1). On day 14, five miRNAs were upregulated (miR-214, miR-923, miR-711, miR-199a-3p and miR-31). Some of these dysregulated miRNAs were related to processes included in response to low oxygen as are hypoxia, inflammation, and fibrosis[43]. In another study, propanolol was chronically administered to induce reversal of the MI. A long-term MI model in rats was established and microarray data analysis showed that long-term propranolol administration resulted in 18 of 31 dysregulated miRNAs undergoing reversed expression. miR-1, miR-29b and miR-98 were suggested to play predominant roles in MI. Bioinformatic analysis suggested that miR-1 regulates myocyte growth, miR-29b regulates fibrosis and miR-98 regulates inflammation[44].
miR-1 and miR-206 and apoptosis: The potential roles of muscle-specific miR-1 and miR-206 and their expression in a rat model of MI have been analyzed. Both miRNAs were significantly increased, while insulin-like growth factor 1 (IGF-1) protein levels were markedly reduced. Caspase-3 activity was increased in cells transfected with either miR-1 or siRNA against IGF-1. Enhanced apoptosis could be therefore induced in cardiomyocytes with a low level of IGF-1 mediated by the post-transcriptional repression caused by miR-1/miR-206[45].
miR-1 and arrhythmia: Propranolol was shown to reduce the incidence of arrhythmias in a rat model of MI. Increased expression of miR-1 was observed in an ischemic myocardium. Administration of propranolol reversed the upregulation of miR-1 to near control levels, significantly diminishing the incidence of arrhythmias in the first 12 h after MI. The suggested targets for miR-1 were the cardiac ion channels Cx43 and Kir2.1[46].
miR-21, apoptosis and atrial fibrillation: The miRNA expression profiling has been performed 6 h after AMI induction in rats. Thirty-eight miRNAs were dysregulated when infarcted area has been compared to non-infarcted heart tissue and 33 in the border zone of the MI when compared to non-infarcted area. miR-21 was significantly downregulated in the infarcted area but was upregulated in the border zone (6 and 24 h after MI). miR-21 had a protective effect on ischemia-induced cell apoptosis by targeting PDCD4 and AP-1, which might play critical roles in the early phase of AMI. Importantly, some miRNAs in the non-infarcted area were also differentially expressed 6 h after AMI, suggesting that in addition to dysregulated miRNAs in infarcted tissue and border zone, some miRNAs dysregulated in the remote myocardium might also contribute in the pathophysiological response to AMI[47]. Another potential role of miR-21 in the atrial fibrillation (AF) resulted from experimental HF after MI. miR-21 was upregulated in atrial tissues following MI, along with the dysregulation of target genes sprouty-1, collagen-I, and collagen-III. Anti-miR-21 treatment reduced atrial miR-21 expression, decreased AF duration, and reduced atrial fibrous tissue[48].
miR-34a and apoptosis: In an experimental rat model of MI, the expression of miR-34a was highly increased while the expression of aldehyde dehydrogenase 2 (ALDH2) was decreased. Overexpression of miR-34a in neonatal rat cardiomyocytes significantly enhanced apoptosis and downregulated ALDH2, suggesting that ALDH2 is a direct target of miR-34a. Serum miR-34a levels in AMI patients and rats were significantly higher than those in controls[49].
miR-101, miR-711, miR-29b and fibrosis: Four weeks after MI induction in rats, examination of miRNAs expression in the peri-infarct area revealed down-regulation of miR-101a/b. In rat neonatal CFs, enforced expression of miR-101a/b lead to suppression of collagen production and proliferation. These effects were abrogated by co-transfection with antisense inhibitors of miR-101a/b. The fibroblast proto-oncogene c-Fos was suggested as a target of miR-101a. Anti-fibrotic action of miR-101a was mimicked by silencing c-Fos using siRNA, whereas effect of miR-101a in cultured CFs was cancelled by enforced expression of the c-Fos. In rats with chronic MI, four weeks after overexpression of miR-101a using adenovirus, remarkable improvement in cardiac performance was observed as well as reduction in interstitial fibrosis and inhibition of c-Fos and TGF-β1 expression[50]. Pioglitazone was further shown to increase miR-711 expression and significantly reduce collagen-I levels similar to CFs, and overexpression of miR-711 suppressed collagen-I levels. Therefore, pioglitazone may upregulate miR-711 to reduce collagen-I levels in rats with MI. The miR-711-transcription factor SP1-collagen-I pathway may be involved in the anti-fibrotic effects of pioglitazone[51]. Another fibrosis study has been performed showing that carvedilol protected against myocardial injury induced by AMI. In male rats, cardiac remodeling and impaired heart function were observed 4 wk after MI; the upregulation of COL1A1, COL3A1, and α-smooth muscle actin (α-SMA) mRNA was observed as well as the downregulation of miR-29b. COL1A1, COL3A1, and α-SMA were downregulated and miR-29b was upregulated by carvedilol in a dose-dependent manner in rat CFs. Enforced expression of miR-29b significantly suppressed COL1A1, COL3A1, and α-SMA expression[52]. An alternative strategy has also been hypothesized that overexpression of miR-29b, which would inhibit mRNAs that encode CF proteins involved in fibrosis, would similarly facilitate progenitor cell migration into the infarcted rat myocardium. The number of GFP-positive cells, capillary density, and heart function were significantly increased in hearts overexpressing miR-29b, and downregulation of miR-29b with anti-miR-29b induced interstitial fibrosis and cardiac remodeling[53].
Human MI
Microarray analysis: Our group performed genome-wide miRNA expression profiling of human MI (7 d post-MI and 4 wk post-MI) comparing fetal hearts to healthy adult hearts. A number of novel miRNAs were identified as well as some similar expression patterns between human MI and fetal hearts, suggesting involvement of cardiac gene reprogramming also in response after MI. Seven miRNAs were confirmed as dysregulated, including miR-1, miR-133a/b, miR-150, miR-186, miR-210 and miR-451[54].
miR-29: Several miRNAs were shown to be dysregulated in the murine MI model, including miR-29. Similarly dysregulation has been observed in human MI, after obtaining border zone of the infarcted cardiac tissue from the patients that received a cardiac transplant[34].
miR-1, miR-133a/b, miR-208a: Our group further showed that miR-1, miR-133a/b and miR-208 were differentially expressed in human MI and fetal hearts when compared to healthy adults. Time-course changes were observed in human MI, with miR-208 upregulated across all time points and miR-1 and miR-133a/b downregulated 2-7 d after MI. All four miRNAs were downregulated in fetal hearts in comparison to healthy adults. We have also observed some similar patterns of miRNA expression between fetal hearts and MI[55]. The remote myocardium was also analyzed and compared to healthy adult hearts and the infarcted area. Whereas miR-1 expression was similar in MI and healthy adults, it was upregulated in the remote myocardium. Downregulation of both miR-133a and miR-133b was observed in the infarcted tissue as well as in the remote myocardium of patients with MI when compared to healthy adult hearts[56].
miRNAs and ventricular rupture: Evidence suggests that an intense inflammatory reaction after a MI might contribute to the development of ventricular rupture (VR). In 50 patients with MI (with or without VR), we showed an altered expression of miR-146a, miR-150 and miR-155 compared to healthy adult hearts. miR-146a showed upregulation and miR-150 and miR-155 showed downregulation in patients with VR compared to those without. These miRNAs are involved in the regulation of innate immunity and the inflammatory response, providing further evidence that innate immunity resulting in an intense inflammatory reaction plays an important role in the pathogenesis of VR after a MI in humans[57].
miRNAs and SERCA2: In another study our group also showed 43 dysregulated miRNAs and decreased expression of the protein SERCA2 when infarcted tissue was compared to the corresponding remote myocardium. The prediction of miRNA binding to SERCA2 identified 213 putative miRNAs. miRNA annotation of dysregulated miRNAs revealed 18 functional and 21 disease states that are linked to the cardiovascular diseases. Half of the dysregulated miRNAs were associated with SERCA2. Free-energy binding and flanking regions were defined for 10 upregulated miRNAs (miR-122, miR-320a/b/c/d, miR-574-3p/-5p, miR-199a, miR-140 and miR-483). The dysregulation of 9 miRNAs was confirmed (miR-21, miR-122, miR-126, miR-1, miR-133, miR-125a/b and miR-98)[58].
Comparison of the number of differentially expressed miRNAs from microarray studies performed on human MI[54,58], on mouse[32,33] and rat model of MI[43,44] is summarized in Figure 4. Only a small proportion of differentially expressed miRNAs overlaps between three different species, these are let-7b, let-7f, miR-26b, miR-126-3p, miR-126-5p, miR-195, miR-199a-3p, miR-214 and miR-451. All these microarray analyses were performed at different time point post-MI. However, comparison has been performed including any dysregulated miRNA at any time point post-MI within species.
Figure 4.
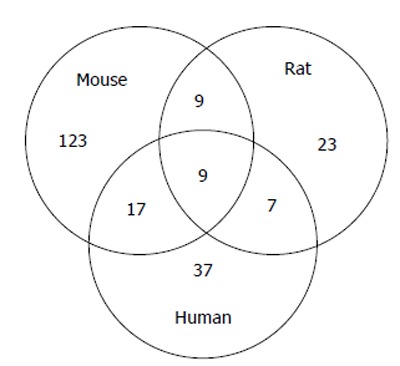
Venn’s diagram of number of differentially expressed miRNAs in human myocardial infarction compared to mouse and rat model of myocardial infarction. Experimental data were obtained from microarray analysis performed on mouse model of MI[32,33], rat model of MI[43,44] and human MI[54,58]. MI: Myocardial infarction.
Circulating miRNAs
Serum and exosome miRNAs as AMI biomarkers: In patients with AMI as well as in patients with angina pectoris (AP), significant increase in serum levels was observed for miR-1 and miR-133a. miR-133a has been recognized as a circulating marker for cardiomyocyte death, because its elevated expression is observed in patients with an injured myocardium. Using an experimental mouse model, it was further identified that significant reduction in levels of miR-1, miR-133a, miR-208a and miR-499 occur in the infarcted myocardium. After stimulation of cardiomyoblasts, exosome fraction of the culture medium was obtained. The measurement of miR-133a was performed. Significant elevation of miR-133a was observed upon the detection of cell death[59]. Using an in vitro cardiac cell necrosis model, it was shown that cardiac miR-1 was released into the culture media 20 min after induction, where it is stable for at least 24 h. The amount of miR-1 released was related to the number of necrotic cardiac myocytes. Furthermore, a time-course study of serum miR-1 in a rat model included time points at 1, 3, 6, 12, 24 h, and 3, 7, 14, 21 and 28 d after AMI. Serum miR-1 levels were increased after AMI with a peak at 6 h, returning to the basal level 3 d after AMI, and showed a strong positive correlation with MI size. Research in humans has shown that in 31 patients with AMI, miR-1 was significantly increased within 24 h after AMI and showed a positive correlation with serum creatine kinase-MB, suggesting its relationship to MI size also occurs in humans. At days 3 and 7, the serum levels returned to baseline[60]. In another study, serum samples were taken from 117 patients with AMI, 182 patients with AP and 100 age- and gender-matched controls. Six serum miRNAs, miR-1, miR-134, miR-186, miR-208, miR-223 and miR-499, were identified as AMI biomarkers and presented significant differences between the AMI and AP cases. miR-208 and miR-499 showed higher expression in the AP cases than in the AMI cases[61].
Serum and exosome miRNAs and prognosis after MI: Using sera collected a median of 18 d after AMI onset, miRNAs were screened in 21 patients who experienced development of HF within 1 year after AMI and in 65 matched controls. miR-192, miR-194 and miR-34a, all p53-responsive miRNAs, were coordinately increased, particularly in exosomes. The serum level of miR-192 was significantly upregulated in AMI patients with development of ischemic HF. miR-194 and miR-34a expression levels were significantly correlated with the left ventricular (LV) end-diastolic dimension 1 year after AMI[62]. The prognostic impact of circulating miRNAs in patients who survived AMI was also analyzed by a high-throughput array consisting of 667 miRNAs. Eleven miRNAs were differentially expressed in the serum from patients at high-risk for cardiac death, and a subset of circulating miRNAs might be predictive for cardiac death in post-AMI patients. Serum levels of miR-155 and miR-380* were higher in patients who experienced cardiac death within 1 year after discharge[63].
Whole blood miRNAs as AMI biomarkers: After performing miRNA expression profiling in peripheral whole-blood samples of patients with AMI, 121 dysregulated miRNAs have been identified. These miRNAs possess a unique signature of 20 miRNAs predicting AMI with 96% specificity, 90% sensitivity and 93% accuracy. miR-30c and miR-145 levels were expressed in correlation with infarct size, which was estimated by release of Troponin T (TnT). Identification of miRNAs that is not based solely on the release of miRNAs from a necrotic myocardium is important for understanding active processes involved in the pathogenesis of MI (inflammation, plaque, rupture and vascular injury). Dysregulated miRNAs in AMI might be equally derived from other cellular populations that play an active role in AMI pathophysiology[64]. To characterize temporal expression patterns of miRNAs in MI, another study was performed with miRNA expression levels measured at multiple time points (0, 2, 4, 12, 24 h after the initial presentation) in patients with acute MI. A subset of miRNAs was found to be significantly dysregulated both at the initial presentation and during the course of AMI. Novel miRNAs that are dysregulated early during MI were identified (miR-1915 and miR-181c*)[65].
Whole blood and plasma miRNAs as AMI biomarkers: The whole blood and plasma samples were obtained from 51 AMI patients and compared with 28 control subjects. Sample collection from AMI patients was performed within 24 h and 7 d after the onset of AMI. In plasma as well as in whole blood from AMI patients, elevated miR-133 and miR-328 levels was observed. Seven days after onset of AMI symptoms increased circulating miR-133 and miR-328 levels returned to control levels. There has also been observed a correlation between cardiac Troponin I (cTnI) and circulating miR-133 or miR-328[66].
Plasma miRNAs as AMI biomarkers: In AMI rats, plasma samples were taken at 1, 3, 6, 12 and 24 h. At these time points, measurement of levels of miR-1, miR-133a, miR-499 and miR-208a has been performed. All these miRNAs were significantly increased, at least at one time point. miR-208a was undetectable at time 0 h, increased 1 h after AMI and reached its peak at 3 h. At time point 6-12 h, it began decreasing and at 24 h it was undetectable. At time point 1-3 h, miR-1, miR-133a and miR-499 were elevated, at 3-12 h reached their peak and at 12-24 h finally decreased. miR-1, miR-133a, miR-499 and miR-208a were present at very low levels or were absent in the plasma of healthy people, but were substantially higher in the plasma of 33 AMI patients compared with that of patients with other cardiovascular diseases, whereas miR-208a remained undetectable in patients with non-AMI heart diseases[67]. In another study, miRNAs were analyzed in human plasma, mouse plasma and mouse cardiac muscle. A microarray analysis showed 20 upregulated and 14 downregulated miRNAs in 17 healthy donors compared with 33 patients with AMI. miR-1, miR-133a/b and miR-499-5p were upregulated and miR-122 and miR-375 were downregulated 6-12 h after MI onset. Five days later, all miRNAs were back to basal plasma levels, except that miR-122 was lower than in controls through day 30. Compared to cTnI, peak expression was observed at a similar time in MI patients for miR-1 and miR-133a/b, but miR-499-5p showed a slower time course. In mice, the pattern of upregulated miRNAs was similar to that in MI patients, but reciprocal expression was observed in cardiac tissue 3-6 h after MI[68]. miR-1 level was measured in a larger cohort of patients (159) with or without AMI. In the plasma from AMI patients, miR-1 was significantly increased when compared with non-AMI patients. Its levels decreased to normal after medication. Statistical analysis revealed that elevated levels of circulating miR-1 were not in correlation to patient characteristics (established biomarkers for AMI, concurrent disease as are blood pressure and diabetes mellitus or either age or gender)[69]. Increased miR-1 and decreased miR-126 expression were consistently observed in the plasma from 17 patients with AMI compared with 25 healthy subjects. cTnI, miR-1 and miR-126 expression levels showed the same trend[70]. miR-1, miR-133a, miR-208b and miR-499 were further compared to cTnT for diagnostic value. Study has been performed on 67 patients with AMI and 32 healthy volunteers. The levels of all plasma miRNAs were significantly higher in AMI patients than in healthy volunteers. At the time of hospital discharge of AMI patients, expression of the cardiac-specific miRNAs was reduced to near baseline levels. However, it has turned out that for the diagnosis of AMI, the four plasma miRNAs were not superior to cTnT[71]. In another study, plasma samples were obtained from 18 patients with AMI and 30 healthy adults. In this cohort of samples, miR-30a, miR-195 and let-7b levels were examined. At time points 4 h, 8 h and 12 h after the onset of AMI, circulating miR-30a was highly elevated. In AMI patients, miR-195 was also highly expressed, when compared to control, but only at time points 8 h and 12 h. Through all the time points, let-7b was lower in AMI patients when compared to control samples. All three investigated circulating miRNAs, miR-30a, miR-195 and let-7b, showed the peak expression at 8 h and were of significant diagnostic value for AMI[72].
Plasma miRNAs for differentiating MI: Plasma concentrations of cardiac-enriched miR-208b and miR-499 were measured in a case-control study of 510 MI patients and 87 healthy controls. miR-208b and miR-499 showed elevated expression in patients with MI and were nearly undetectable in healthy controls. In 397 patients with ST-elevation MI (STEMI), miRNAs had higher concentrations than in 113 patients with non-STEMI (NSTEMI)[73].
Plasma miRNAs for differentiating MI from other cardiovascular diseases: In all individuals with AMI, the concentration of plasma miR-499 was shown to be increased; however it was below the detection limit in other groups of patient [control, chronic HF (CHF), and unstable AP][74]. The expression level of plasma miR-133a has been analyzed in 13 AMI patients, 176 AP patients and 127 control subjects for its relationship to the severity of coronary stenosis. The results showed that circulating miR-133a levels were significantly increased in AMI patients in a time-dependent manner, achieving a peak at 21.6 ± 4.5 h after the onset of AMI symptoms and showed a similar trend as the level of plasma cTnI. Importantly, the levels of circulating miR-133a positively correlated with the severity of coronary artery stenosis[75]. Another study showed that plasma levels of miR-1, miR-133a, miR-208b and miR-499 (muscle- or cardiac-specific or enriched miRNAs), miR-21 and miR-29b (fibrosis-related miRNAs) miR-146, miR-155, miR-223 (leukocyte-associated miRNAs) are associated with different degrees of cardiac injury as are AMI, acute HF, diastolic dysfunction and even viral myocarditis. In the plasma of 32 patients with AMI, miR-208b and miR-499 were highly elevated compared with control subjects and both correlated with plasma cTnT levels. Both miRNAs also showed significant but milder elevation in viral myocarditis. However, in patients with acute HF, only miR-499 showed significant elevation, whereas no significant change was observed in diastolic dysfunction[76]. Another study group consisted of 17 patients with AMI, 4 with stable coronary artery disease (CAD) and 5 with no history of CAD. Expression of miR-423-5p, miR-208 and miR-1 was measured in plasma before percutaneous coronary intervention (PCI), at 6, 12 and 24 h. In stable CAD, the expression of miR-1, miR-208a and miR-423-5p did not show any significant differences at any time point. There was a higher number of miR-423-5p copies in patients with AMI before the PCI. However, 6, 12 and 24 h after PCI, the expression levels were similar to the control group and significantly lower than the baseline level. The expression levels of miR-1 and miR-208a were not significantly different from the control group[77]. In another study, the increased expression levels of miR-1, miR-21, miR-133a, miR-423-5p and miR-499-5p has been showed in plasma of 92 patients with NSTEMI compared to 99 age-matched healthy control subjects. miR-499-5p and miR-21 showed increased expression in NSTEMI compared to 81 patients with CHF. mir-499-5p also showed good diagnostic accuracy in differentiating patients with NSTEMI and CHF[78]. Takotsubo cardiomyopathy (TTC) is clinically indistinguishable from AMI, and no established biomarkers are available for the early diagnosis of TTC and differentiation from AMI. After miRNA profiling, eight miRNAs were selected for verification in 36 patients with TTC, 27 patients with AMI and 28 healthy controls. Upregulation of miR-16 and miR-26a was confirmed in patients with TTC compared with healthy subjects, and upregulation of miR-16, miR-26a and let-7f was observed in TTC compared with MI patients. Compared with healthy controls, miR-1 and miR-133a showed upregulation in patients with MI, and miR-133a was substantially increased in patients with MI when compared with TTC. A unique signature comprising miR-1, miR-16, miR-26a and miR-133a differentiated TTC from healthy subjects and from MI patients[79].
Plasma miRNAs and prognosis after MI: Plasma miR-1, miR-21, miR-29a, miR-133a and miR-208 were measured in 12 age-matched reference controls and 12 post-MI patients from day 2 through day 90 post-MI. After MI, a progressive increase of LV end-diastolic volume was accompanied by time-dependent changes in specific miRNAs. Two days post-MI, miR-21 decreased and 5 d post-MI increased. At later time points its expression level reached the control values. Similarly, at day 5 post-MI, miR-29a increased and then decreased to the control level at later time points. miR-208 showed elevated expression at day 5 post-MI and did not show any decrease up to day 90 post-MI[80]. miR-1, miR-208b and miR-499-5p were further measured in plasma samples from 424 patients for discrimination of a clinical diagnosis of MI and for association with 30-d mortality and for diagnosis of HF. Discrimination of MI was accurate for miR-208b and miR-499-5p but was considerably lower than for TnT. Increased miRNA levels were strongly associated with an increased risk of mortality or heart failure within 30 d for miR-208b and miR-499-5p, but the association was lost when adjusting for TnT[81]. In another study, circulating miRNAs were measured in 90 patients after AMI and several miRNAs were identified as potentially involved in LV remodeling. miR-150 was downregulated in patients with remodeling compared with patients without. miR-150 outperformed B-type natriuretic peptide (BNP) to predict remodeling and reclassified 54% of patients misclassified by BNP and 59% of patients misclassified by a multi-parameter clinical model[82]. Furthermore, plasma samples from 150 patients with AMI were obtained for determination of the levels of miR-16, miR-27a, miR-101 and miR-150. A combination of the four miRNAs improved the prediction of LV contractility based on clinical variables. Patients with low levels of miR-150 or miR-101 and elevated levels of miR-16 were at high risk for impaired LV contractility. The four-miRNA panel reclassified a significant proportion of patients, with a net reclassification improvement of 66%[83].
Plasma miRNAs and prospective study for MI: The association between baseline levels of miRNAs, the incidence of MI, and the cellular origin of miRNAs was analyzed in 820 participants with 19 candidate miRNAs. Three miRNAs were consistently and significantly related to the incidence of MI; miR-126 showed a positive association, miR-223 and miR-197 were negatively related to the risk of disease. Control group consisted of healthy volunteers, in who limb I/R was performed by thigh cuff inflation. After obtaining plasma samples at baseline, 10 min, 1 h, 5 h, 2 d, and 7 d, miRNA expression was analyzed. Six distinct miRNA clusters were identified by computational analysis, and one of them consisted of all miRNAs that were related to the risk of a future MI. This cluster included miRNAs predominantly expressed in platelets and its characteristic was activation 1 h post-I/R (early) and activation 7 d post-I/R (sustained). Platelets were suggested as being a major contributor to this miRNA expression pattern, since in subjects with a subsequent MI, dysregulated patterns of circulating miRNAs occurred with endothelium-enriched miR-126[84].
Plasma and urine miRNAs: In a pig I/R model, miR-1, miR-133a and miR-208b increased rapidly in plasma with a peak at 120 min, while miR-499-5p remained elevated longer. In humans, 25 patients with MI revealed that all four miRNAs were increased in plasma, with a peak at 12 h. Peak values of miR-208b correlated with peak troponin levels. miR-1 and miR-133a both correlated strongly with renal elimination, which was confirmed by detection of miR-1 and miR-133a, but not miR-208b or miR-499-5p, in the urine[85].
Urine miRNAs: Blood protein MI biomarkers (creatine phosphokinase-muscle band, TnT and TnI) are not typically filtered into urine. Urine miR-1 was quickly increased in rats with a peak at 24 h after AMI and returned to the basal level 7 d after AMI. No miR-208 was observed in normal urine; however, miR-208 was easily detected in urine from rats with AMI. Serum exosomes from rats after AMI were isolated and injected into the circulating blood of normal rats; urine miR-1 was significantly increased in the exosome-injected animals. The levels of urine miR-1 were also significantly increased in patients with AMI[86].
In summary, it has been shown that miRNAs may be useful circulating biomarkers for the diagnosis of AMI, differentiating them from other cardiovascular diseases and prognoses after MI. However, two studies have shown that miRNAs are not useful circulating biomarkers for some aspects of MI, (1) for prognosis of patients with STEMI; or (2) for an incidence of LV remodeling 1 year after anterior AMI[87,88].
Therapeutic opportunities
All miRNAs as potential therapeutic targets were tested in mouse or rat models of MI. miRNAs and different therapeutic approaches analyzed in mouse model of MI are summarized in Figure 5 and miRNAs and different therapeutic approaches analyzed in rat model of MI are overviewed Figure 6.
Figure 5.
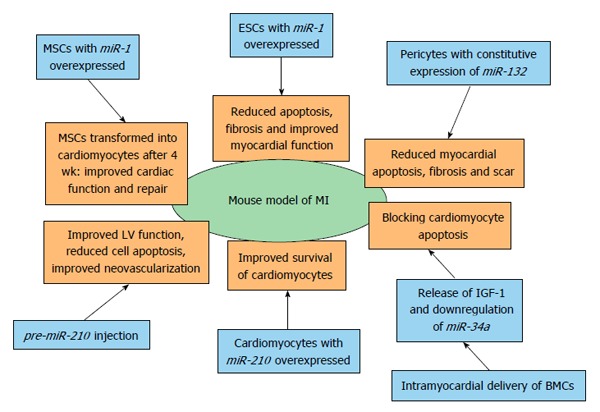
Therapeutic opportunities of miRNAs in myocardial infarction identified by using mouse model of myocardial infarction. BMC: Bone marrow cell; ESC: Embryonic stem cell; LV: Left ventricle; MSC: Mesenchymal stem cell; MI: Myocardial infarction; IGF-1: Insulin-like growth factor 1.
Figure 6.
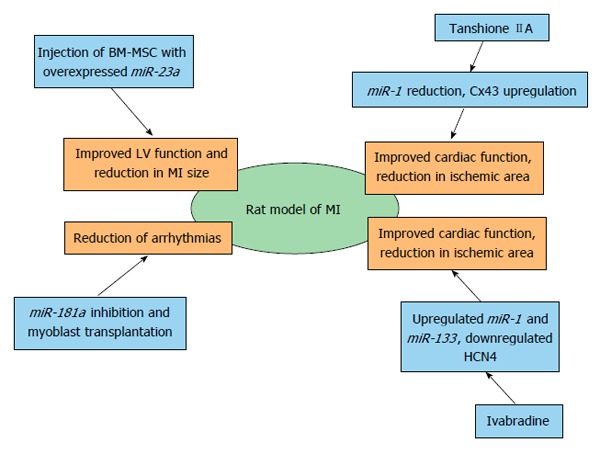
Therapeutic opportunities of miRNAs in myocardial infarction identified by using rat model of myocardial infarction. BM-MSC: Bone marrow-mesenchymal stem cell; LV: Left ventricle; MI: Myocardial infarction; Cx43: Gap junction alpha-1 protein; HCN4: Potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 4.
miR-181a and skeletal myoblast transplantation in rats with MI: A lentiviral siRNA against the loop region of miR-181a was shown to upregulate the skeletal myoblast (SKM) differentiation repressor Hox-A11 and reduce arrhythmias following SKM transplantation into ischemic myocardium of rats. Engraftments of SKMs with miR-181a knockdown improved cardiac function and significantly decreased the arrhythmogenic effect of SKM transplantation in rats with experimental MI[89].
miR-210 and treatment of ischemic heart disease: miR-210 was highly expressed in mouse cardiomyocytes that survived 48 h after hypoxia exposure compared with apoptotic cardiomyocytes. Mice receiving a miR-210 precursor showed significant improvement of LV fractional shortening after 8 wk. Histological analysis showed decreased cellular apoptosis and increased neovascularization. Two target genes involved in inhibition of angiogenesis/vascular remodeling and induction of apoptosis, Ephrin-A3 and Ptp1 (non-receptor phospho-tyrosine protein phosphatase), were confirmed. It has been shown that miR-210 can improve angiogenesis, inhibit apoptosis and improve cardiac function in a mouse model of MI[90].
Phosphorinositide-3-kinase-regulated miRNA and mRNA: Activation of phosphorinositide-3-kinase (PIK3) is considered a new strategy for the treatment of heart failure and MI. To identify cardiac-selective miRNAs and mRNAs that mediate the protective properties of PIK3, experimental mice were used and identified growth factor receptor-bound protein (Grb14) gene expression that positively correlated with cardiac function. Grb14 is highly expressed in the mouse heart compared with other tissues. Three miRNAs were also highly correlated with Grb14, namely miR-210, miR-34a and miR-222[91].
Tanshinone and miR-1: Accumulating evidence suggests that tanshinone IIA can reduce the ischemic area and improve cardiac function and has been shown to suppress miR-1 expression. Using a rat model of MI, tanshinone IIA was administered daily for 7 d before MI and lasted for 3 mo following MI. Tanshinone IIA was shown to relieve ischemia-induced injury, decrease the elevated miR-1 levels in ischemic and hypoxic cardiomyocytes, and consequently restored the normal level of the miR-1 target Cx43. In ischemic and hypoxic cardiomyocytes, tanshinone IIA also inhibited activated p38 MAPK, SRF and MEF2[92].
Ivabradine and miR-1 and miR-133a: Ivabradine is a selective inhibitor of the hyperpolarization-activated, cyclic nucleotide-gated pacemaker current. Its effect on electrophysiological remodeling of myocytes from post-MI rats was observed as a decrease in the transcription of HCN4, a target of miR-1 and miR-133a. Both, miR-1 and miR-133 were significantly elevated in myocytes. The beneficial effects of ivabradine may be due to the reversal of electrophysiological cardiac remodeling by reducing the overexpressed HCN channels in post-MI rats[93].
Embryonic stem cells and miRNAs: Embryonic stem cells (ESC) with overexpressed miR-1 were transplanted into the infarcted myocardium of experimental animals, and reduced apoptosis was subsequently observed 4 wk post-MI. A significant elevation in p-Akt levels and diminished PTEN levels were also observed. The mice also had a significant improvement in some physiological cardiac functions[94]. The same author investigated whether overexpression of miR-1 in ESCs would enhance cardiac myocyte differentiation following transplantation into the infarcted myocardium. Two weeks after transplantation into the border zone of the infarcted heart, cardiac myocyte differentiation, adverse ventricular remodeling, and cardiac function were assessed. Overexpression of miR-1 in transplanted ESCs protected the host myocardium from MI-induced apoptosis. A significant reduction in interstitial and vascular fibrosis was observed as well as significantly improved heart function[95].
Mesenchymal-stem cells and miRNAs: One week after MI, mice were intramyocardially injected at the heart infarcted zone with miR-1-transduced mesenchymal-stem cells (MSCs). At 4 wk post-transplantation, transplanted MSCs were able to differentiate into cardiomyocytes in the infarcted zone. Cardiac function with the miR-1-transduced MSCs was significantly improved, and treatment of MSCs expressing miR-1 was more effective for cardiac repair, most likely by enhancing cell survival and cardiac myocyte differentiation compared with the MSCs without miR-1[96]. In vitro co-culture between cardiomyocytes and MSCs has been established to test whether MSCs deliver miR-210 to host cardiomyocytes; this showed co-localization of miR-210 with the gap-junction protein Cx43. miR-210 has been proposed to be transferred through gap junctions. Higher survival rates of cardiomyocytes co-cultured with MSCs was observed with concomitant expression of caspase-8 associated protein 2 (CASP8AP2) suggesting that miR-210 translocates from MSCs to protect host cardiomyocytes. Direct transfer of pro-survival miR-210 from MSCs to host cardiomyocytes led to a functional recovery of the ischemic hearts of the experimental animals[97]. The clinical application of MSC-based therapy is restricted because of the poor survival of implanted cells. Using a tumor necrosis factor α-(TNF-α)-induced bone marrow (BM)-MSC injury model and a rat MI, it has been shown that miR-23a was involved in TNF-α-induced BM-MSC apoptosis through regulating caspase-7 and that the injection of BM-MSCs overexpressing miR-23a could improve LV function and reduce the infarct size in the rat MI model[98].
Bone marrow cells and miR-34a: Cell therapy with bone marrow cells (BMC) can improve the recovery of cardiac function after ischemia. I intra-myocardial delivery of BMCs in infarcted mice has been shown to regulate the expression of miRNAs in the heart and downregulate the expression of miR-34a, a pro-apoptotic miRNA. Transplanted BMCs regulate cardiac miRNAs by paracrine mode and thus contribute to the protective effects. IGF-1 inhibits the miR-34a processing and is released by BMCs, thereby blocking apoptosis in cardiomyocytes[99].
Pericytes and miR-132: Pericytes are key regulators of vascular maturation and therapeutic activity, and mechanistic targets of saphenous vein-derived pericyte progenitor cells (SVPs) have been investigated using a mouse MI model. Transplantation of SVPs into the peri-infarct zone of mice attenuated LV dilatation, reduced myocardial scar, cardiomyocyte apoptosis and interstitial fibrosis, and blood flow and neovascularization. miR-132 was constitutively expressed and secreted by SVPs and markedly upregulated. Ras-GTPase activating protein and methyl-CpG-binding protein 2 were shown to be targets of miR-132. miR-132 inhibition decreased SVP capacity to improve contractility, reparative angiogenesis, and interstitial fibrosis in infarcted hearts[100].
Telocytes and miRNAs: Telocytes (TCs) are a novel type of interstitial cell recently discovered in the myocardium. Rat experimental MI was investigated using electron microscopy, immunocytochemistry and analysis of several pro-angiogenic miRNAs that provided evidence for TC involvement in neo-angiogenesis after MI. TCs contain measurable quantities of angiogenic miRNAs (let-7e, miR-10a, miR-21, miR-27b, miR-100, miR-126-3p, miR-130a, miR-143, miR-155 and miR-503)[101].
Bioinformatics analysis
Rationally designed bioinformatic analysis combined with experimental approaches to screen key therapeutic members of the IUPHAR database was conducted, following establishment of the whole genome protein interaction network and a comprehensive topological assessment. The number of validated and confidently predicted miRNAs regulating each gene encoding an ion channel or a gap junction protein was counted. Cx43 showed more intensive miRNA regulation compared with other ion channel and gap junction proteins[102].
My-Inflamome: One of crucial processes in cardiac repair after MI is inflammation. In a study, a network has been established that enhances understanding of the inflammatory responses and its interaction network in human MI. The network is called My-Inflamome and it assembles protein interactions that are associated to inflammation and related to prognosis after MI. Classification models were established based on microarray data of blood samples from patients after MI with various disease consequences. Significant associations were experimentally verified. Different biological processes included in the heart repair are organized into modules. Small set of miRNAs is also included in modules that are significantly associated with transcriptional regulation[103].
My-DTome: Another computational approach has been performed. It is based on different drug and protein interaction and it is called My-DTome (it is assembling the MI drug-target). It is also consisted of modules, which are related to the important molecular processes and pathways and to potential therapeutic approaches in MI that might be miRNAs-regulated. Non-cardiovascular drugs may also possess the cardiovascular effects and this systemic insight was established. This network might represent the basis for an investigation of new multidrug treatment and new targets MI[104].
Polymorphisms in miRNA binding sites
After searching across dbSNP and TargetScan, 10 SNPs in potential miRNA binding sites of 8 RAAS-related genes were identified and genotyped for risk for MI and blood pressure. It was found that nine SNPs in seven genes were prevalent. Of the nine SNPs, four in three genes were associated with blood pressure. The rare allele of the mineralocorticoid receptor (NR3C2) (SNP rs5534) was associated with a twofold increased risk of MI in men younger than 50 years of age. The reduction in miR-induced repression of gene expression was demonstrated[105].
CONCLUSION
In recent years miRNAs have been recognized as promising therapeutic, diagnostic and prognostic factors in the field of cardiovascular diseases. The usefulness of circulating miRNAs in the diagnosis and prognosis of MI has been established through numerous studies. Moreover, the therapeutic potential of miRNA has been established, especially in field of stem cell research. Heart tissue expression patterns examined in numerous experimental animals still need to be confirmed on human MI. Much more work is necessary before establishing routine use of miRNAs in clinical diagnosis, prognosis and therapy; however, the current findings are encouraging.
Footnotes
P- Reviewer: Rassaf T, Zamilpa R S- Editor: Song XX L- Editor: A E- Editor: Liu SQ
References
- 1.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Pillai RS, Bhattacharyya SN, Filipowicz W. Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol. 2007;17:118–126. doi: 10.1016/j.tcb.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 3.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ying SY, Chang DC, Lin SL. The microRNA (miRNA): overview of the RNA genes that modulate gene function. Mol Biotechnol. 2008;38:257–268. doi: 10.1007/s12033-007-9013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pillai RS. MicroRNA function: multiple mechanisms for a tiny RNA? RNA. 2005;11:1753–1761. doi: 10.1261/rna.2248605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnes MR, Deharo S, Grocock RJ, Brown JR, Sanseau P. The micro RNA target paradigm: a fundamental and polymorphic control layer of cellular expression. Expert Opin Biol Ther. 2007;7:1387–1399. doi: 10.1517/14712598.7.9.1387. [DOI] [PubMed] [Google Scholar]
- 7.Krützfeldt J, Poy MN, Stoffel M. Strategies to determine the biological function of microRNAs. Nat Genet. 2006;38 Suppl:S14–S19. doi: 10.1038/ng1799. [DOI] [PubMed] [Google Scholar]
- 8.Soifer HS, Rossi JJ, Saetrom P. MicroRNAs in disease and potential therapeutic applications. Mol Ther. 2007;15:2070–2079. doi: 10.1038/sj.mt.6300311. [DOI] [PubMed] [Google Scholar]
- 9.Perera RJ, Ray A. MicroRNAs in the search for understanding human diseases. BioDrugs. 2007;21:97–104. doi: 10.2165/00063030-200721020-00004. [DOI] [PubMed] [Google Scholar]
- 10.Williams AE. Functional aspects of animal microRNAs. Cell Mol Life Sci. 2008;65:545–562. doi: 10.1007/s00018-007-7355-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 12.Min H, Yoon S. Got target? Computational methods for microRNA target prediction and their extension. Exp Mol Med. 2010;42:233–244. doi: 10.3858/emm.2010.42.4.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuhn DE, Martin MM, Feldman DS, Terry AV, Nuovo GJ, Elton TS. Experimental validation of miRNA targets. Methods. 2008;44:47–54. doi: 10.1016/j.ymeth.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Rooij E. The art of microRNA research. Circ Res. 2011;108:219–234. doi: 10.1161/CIRCRESAHA.110.227496. [DOI] [PubMed] [Google Scholar]
- 15.Lu M, Zhang Q, Deng M, Miao J, Guo Y, Gao W, Cui Q. An analysis of human microRNA and disease associations. PLoS One. 2008;3:e3420. doi: 10.1371/journal.pone.0003420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borel C, Antonarakis SE. Functional genetic variation of human miRNAs and phenotypic consequences. Mamm Genome. 2008;19:503–509. doi: 10.1007/s00335-008-9137-6. [DOI] [PubMed] [Google Scholar]
- 17.Chuang JC, Jones PA. Epigenetics and microRNAs. Pediatr Res. 2007;61:24R–29R. doi: 10.1203/pdr.0b013e3180457684. [DOI] [PubMed] [Google Scholar]
- 18.Liu Z, Sall A, Yang D. MicroRNA: An emerging therapeutic target and intervention tool. Int J Mol Sci. 2008;9:978–999. doi: 10.3390/ijms9060978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sayed D, Rane S, Abdellatif M. MicroRNAs challenge the status quo of therapeutic targeting. J Cardiovasc Transl Res. 2009;2:100–107. doi: 10.1007/s12265-008-9052-y. [DOI] [PubMed] [Google Scholar]
- 20.D’Alessandra Y, Pompilio G, Capogrossi MC. MicroRNAs and myocardial infarction. Curr Opin Cardiol. 2012;27:228–235. doi: 10.1097/HCO.0b013e3283522052. [DOI] [PubMed] [Google Scholar]
- 21.Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–710. doi: 10.1038/nature08195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, Vatner DE, Vatner SF, Abdellatif M. Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res. 2009;104:879–886. doi: 10.1161/CIRCRESAHA.108.193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roy S, Khanna S, Hussain SR, Biswas S, Azad A, Rink C, Gnyawali S, Shilo S, Nuovo GJ, Sen CK. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res. 2009;82:21–29. doi: 10.1093/cvr/cvp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song XW, Li Q, Lin L, Wang XC, Li DF, Wang GK, Ren AJ, Wang YR, Qin YW, Yuan WJ, et al. MicroRNAs are dynamically regulated in hypertrophic hearts, and miR-199a is essential for the maintenance of cell size in cardiomyocytes. J Cell Physiol. 2010;225:437–443. doi: 10.1002/jcp.22217. [DOI] [PubMed] [Google Scholar]
- 25.Kulshreshtha R, Ferracin M, Negrini M, Calin GA, Davuluri RV, Ivan M. Regulation of microRNA expression: the hypoxic component. Cell Cycle. 2007;6:1426–1431. [PubMed] [Google Scholar]
- 26.Kulshreshtha R, Davuluri RV, Calin GA, Ivan M. A microRNA component of the hypoxic response. Cell Death Differ. 2008;15:667–671. doi: 10.1038/sj.cdd.4402310. [DOI] [PubMed] [Google Scholar]
- 27.Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res. 2008;58:88–111. doi: 10.1016/j.phrs.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niu Z, Iyer D, Conway SJ, Martin JF, Ivey K, Srivastava D, Nordheim A, Schwartz RJ. Serum response factor orchestrates nascent sarcomerogenesis and silences the biomineralization gene program in the heart. Proc Natl Acad Sci USA. 2008;105:17824–17829. doi: 10.1073/pnas.0805491105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng Y, Liu X, Zhang S, Lin Y, Yang J, Zhang C. MicroRNA-21 protects against the H(2)O(2)-induced injury on cardiac myocytes via its target gene PDCD4. J Mol Cell Cardiol. 2009;47:5–14. doi: 10.1016/j.yjmcc.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Z, Yang D, Xie P, Ren G, Sun G, Zeng X, Sun X. MiR-106b and MiR-15b modulate apoptosis and angiogenesis in myocardial infarction. Cell Physiol Biochem. 2012;29:851–862. doi: 10.1159/000258197. [DOI] [PubMed] [Google Scholar]
- 31.Gidlöf O, van der Brug M, Ohman J, Gilje P, Olde B, Wahlestedt C, Erlinge D. Platelets activated during myocardial infarction release functional miRNA, which can be taken up by endothelial cells and regulate ICAM1 expression. Blood. 2013;121:3908–3917, S1-26. doi: 10.1182/blood-2012-10-461798. [DOI] [PubMed] [Google Scholar]
- 32.Port JD, Walker LA, Polk J, Nunley K, Buttrick PM, Sucharov CC. Temporal expression of miRNAs and mRNAs in a mouse model of myocardial infarction. Physiol Genomics. 2011;43:1087–1095. doi: 10.1152/physiolgenomics.00074.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janssen R, Zuidwijk M, Muller A, Mulders J, Oudejans CB, Simonides WS. Cardiac expression of deiodinase type 3 (Dio3) following myocardial infarction is associated with the induction of a pluripotency microRNA signature from the Dlk1-Dio3 genomic region. Endocrinology. 2013;154:1973–1978. doi: 10.1210/en.2012-2017. [DOI] [PubMed] [Google Scholar]
- 34.van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Huang W, Xu R, Nie Y, Cao X, Meng J, Xu X, Hu S, Zheng Z. MicroRNA-24 regulates cardiac fibrosis after myocardial infarction. J Cell Mol Med. 2012;16:2150–2160. doi: 10.1111/j.1582-4934.2012.01523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fiedler J, Jazbutyte V, Kirchmaier BC, Gupta SK, Lorenzen J, Hartmann D, Galuppo P, Kneitz S, Pena JT, Sohn-Lee C, et al. MicroRNA-24 regulates vascularity after myocardial infarction. Circulation. 2011;124:720–730. doi: 10.1161/CIRCULATIONAHA.111.039008. [DOI] [PubMed] [Google Scholar]
- 37.Meloni M, Marchetti M, Garner K, Littlejohns B, Sala-Newby G, Xenophontos N, Floris I, Suleiman MS, Madeddu P, Caporali A, et al. Local inhibition of microRNA-24 improves reparative angiogenesis and left ventricle remodeling and function in mice with myocardial infarction. Mol Ther. 2013;21:1390–1402. doi: 10.1038/mt.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, Burchfield J, Fox H, Doebele C, Ohtani K, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–1713. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
- 39.Wang K, Liu F, Zhou LY, Ding SL, Long B, Liu CY, Sun T, Fan YY, Sun L, Li PF. miR-874 regulates myocardial necrosis by targeting caspase-8. Cell Death Dis. 2013;4:e709. doi: 10.1038/cddis.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin C, Wang X, Kukreja RC. Endogenous microRNAs induced by heat-shock reduce myocardial infarction following ischemia-reperfusion in mice. FEBS Lett. 2008;582:4137–4142. doi: 10.1016/j.febslet.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ren XP, Wu J, Wang X, Sartor MA, Qian J, Jones K, Nicolaou P, Pritchard TJ, Fan GC. MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation. 2009;119:2357–2366. doi: 10.1161/CIRCULATIONAHA.108.814145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Zhang X, Ren XP, Chen J, Liu H, Yang J, Medvedovic M, Hu Z, Fan GC. MicroRNA-494 targeting both proapoptotic and antiapoptotic proteins protects against ischemia/reperfusion-induced cardiac injury. Circulation. 2010;122:1308–1318. doi: 10.1161/CIRCULATIONAHA.110.964684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi B, Guo Y, Wang J, Gao W. Altered expression of microRNAs in the myocardium of rats with acute myocardial infarction. BMC Cardiovasc Disord. 2010;10:11. doi: 10.1186/1471-2261-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu W, Yang L, Shan H, Zhang Y, Zhou R, Su Z, Du Z. MicroRNA expression analysis: clinical advantage of propranolol reveals key microRNAs in myocardial infarction. PLoS One. 2011;6:e14736. doi: 10.1371/journal.pone.0014736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shan ZX, Lin QX, Fu YH, Deng CY, Zhou ZL, Zhu JN, Liu XY, Zhang YY, Li Y, Lin SG, et al. Upregulated expression of miR-1/miR-206 in a rat model of myocardial infarction. Biochem Biophys Res Commun. 2009;381:597–601. doi: 10.1016/j.bbrc.2009.02.097. [DOI] [PubMed] [Google Scholar]
- 46.Lu Y, Zhang Y, Shan H, Pan Z, Li X, Li B, Xu C, Zhang B, Zhang F, Dong D, et al. MicroRNA-1 downregulation by propranolol in a rat model of myocardial infarction: a new mechanism for ischaemic cardioprotection. Cardiovasc Res. 2009;84:434–441. doi: 10.1093/cvr/cvp232. [DOI] [PubMed] [Google Scholar]
- 47.Dong S, Cheng Y, Yang J, Li J, Liu X, Wang X, Wang D, Krall TJ, Delphin ES, Zhang C. MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. J Biol Chem. 2009;284:29514–29525. doi: 10.1074/jbc.M109.027896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cardin S, Guasch E, Luo X, Naud P, Le Quang K, Shi Y, Tardif JC, Comtois P, Nattel S. Role for MicroRNA-21 in atrial profibrillatory fibrotic remodeling associated with experimental postinfarction heart failure. Circ Arrhythm Electrophysiol. 2012;5:1027–1035. doi: 10.1161/CIRCEP.112.973214. [DOI] [PubMed] [Google Scholar]
- 49.Fan F, Sun A, Zhao H, Liu X, Zhang W, Jin X, Wang C, Ma X, Shen C, Zou Y, et al. MicroRNA-34a promotes cardiomyocyte apoptosis post myocardial infarction through down-regulating aldehyde dehydrogenase 2. Curr Pharm Des. 2013;19:4865–4873. doi: 10.2174/13816128113199990325. [DOI] [PubMed] [Google Scholar]
- 50.Pan Z, Sun X, Shan H, Wang N, Wang J, Ren J, Feng S, Xie L, Lu C, Yuan Y, et al. MicroRNA-101 inhibited postinfarct cardiac fibrosis and improved left ventricular compliance via the FBJ osteosarcoma oncogene/transforming growth factor-β1 pathway. Circulation. 2012;126:840–850. doi: 10.1161/CIRCULATIONAHA.112.094524. [DOI] [PubMed] [Google Scholar]
- 51.Zhao N, Yu H, Yu H, Sun M, Zhang Y, Xu M, Gao W. MiRNA-711-SP1-collagen-I pathway is involved in the anti-fibrotic effect of pioglitazone in myocardial infarction. Sci China Life Sci. 2013;56:431–439. doi: 10.1007/s11427-013-4477-1. [DOI] [PubMed] [Google Scholar]
- 52.Zhu JN, Chen R, Fu YH, Lin QX, Huang S, Guo LL, Zhang MZ, Deng CY, Zou X, Zhong SL, et al. Smad3 inactivation and MiR-29b upregulation mediate the effect of carvedilol on attenuating the acute myocardium infarction-induced myocardial fibrosis in rat. PLoS One. 2013;8:e75557. doi: 10.1371/journal.pone.0075557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang W, Dai B, Wen Z, Millard RW, Yu XY, Luther K, Xu M, Zhao TC, Yang HT, Qi Z, et al. Molecular strategy to reduce in vivo collagen barrier promotes entry of NCX1 positive inducible pluripotent stem cells (iPSC(NCX¹+)) into ischemic (or injured) myocardium. PLoS One. 2013;8:e70023. doi: 10.1371/journal.pone.0070023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bostjancic E, Zidar N, Glavac D. MicroRNA microarray expression profiling in human myocardial infarction. Dis Markers. 2009;27:255–268. doi: 10.3233/DMA-2009-0671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bostjancic E, Zidar N, Stajer D, Glavac D. MicroRNAs miR-1, miR-133a, miR-133b and miR-208 are dysregulated in human myocardial infarction. Cardiology. 2010;115:163–169. doi: 10.1159/000268088. [DOI] [PubMed] [Google Scholar]
- 56.Bostjancic E, Zidar N, Stajner D, Glavac D. MicroRNA miR-1 is up-regulated in remote myocardium in patients with myocardial infarction. Folia Biol (Praha) 2010;56:27–31. doi: 10.14712/fb2010056010027. [DOI] [PubMed] [Google Scholar]
- 57.Zidar N, Boštjančič E, Glavač D, Stajer D. MicroRNAs, innate immunity and ventricular rupture in human myocardial infarction. Dis Markers. 2011;31:259–265. doi: 10.3233/DMA-2011-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boštjančič E, Zidar N, Glavač D. MicroRNAs and cardiac sarcoplasmic reticulum calcium ATPase-2 in human myocardial infarction: expression and bioinformatic analysis. BMC Genomics. 2012;13:552. doi: 10.1186/1471-2164-13-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuwabara Y, Ono K, Horie T, Nishi H, Nagao K, Kinoshita M, Watanabe S, Baba O, Kojima Y, Shizuta S, et al. Increased microRNA-1 and microRNA-133a levels in serum of patients with cardiovascular disease indicate myocardial damage. Circ Cardiovasc Genet. 2011;4:446–454. doi: 10.1161/CIRCGENETICS.110.958975. [DOI] [PubMed] [Google Scholar]
- 60.Cheng Y, Tan N, Yang J, Liu X, Cao X, He P, Dong X, Qin S, Zhang C. A translational study of circulating cell-free microRNA-1 in acute myocardial infarction. Clin Sci (Lond) 2010;119:87–95. doi: 10.1042/CS20090645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li C, Fang Z, Jiang T, Zhang Q, Liu C, Zhang C, Xiang Y. Serum microRNAs profile from genome-wide serves as a fingerprint for diagnosis of acute myocardial infarction and angina pectoris. BMC Med Genomics. 2013;6:16. doi: 10.1186/1755-8794-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matsumoto S, Sakata Y, Suna S, Nakatani D, Usami M, Hara M, Kitamura T, Hamasaki T, Nanto S, Kawahara Y, et al. Circulating p53-responsive microRNAs are predictive indicators of heart failure after acute myocardial infarction. Circ Res. 2013;113:322–326. doi: 10.1161/CIRCRESAHA.113.301209. [DOI] [PubMed] [Google Scholar]
- 63.Matsumoto S, Sakata Y, Nakatani D, Suna S, Mizuno H, Shimizu M, Usami M, Sasaki T, Sato H, Kawahara Y, et al. A subset of circulating microRNAs are predictive for cardiac death after discharge for acute myocardial infarction. Biochem Biophys Res Commun. 2012;427:280–284. doi: 10.1016/j.bbrc.2012.09.039. [DOI] [PubMed] [Google Scholar]
- 64.Meder B, Keller A, Vogel B, Haas J, Sedaghat-Hamedani F, Kayvanpour E, Just S, Borries A, Rudloff J, Leidinger P, et al. MicroRNA signatures in total peripheral blood as novel biomarkers for acute myocardial infarction. Basic Res Cardiol. 2011;106:13–23. doi: 10.1007/s00395-010-0123-2. [DOI] [PubMed] [Google Scholar]
- 65.Vogel B, Keller A, Frese KS, Kloos W, Kayvanpour E, Sedaghat-Hamedani F, Hassel S, Marquart S, Beier M, Giannitis E, et al. Refining diagnostic microRNA signatures by whole-miRNome kinetic analysis in acute myocardial infarction. Clin Chem. 2013;59:410–418. doi: 10.1373/clinchem.2011.181370. [DOI] [PubMed] [Google Scholar]
- 66.Wang R, Li N, Zhang Y, Ran Y, Pu J. Circulating microRNAs are promising novel biomarkers of acute myocardial infarction. Intern Med. 2011;50:1789–1795. doi: 10.2169/internalmedicine.50.5129. [DOI] [PubMed] [Google Scholar]
- 67.Wang GK, Zhu JQ, Zhang JT, Li Q, Li Y, He J, Qin YW, Jing Q. Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur Heart J. 2010;31:659–666. doi: 10.1093/eurheartj/ehq013. [DOI] [PubMed] [Google Scholar]
- 68.D’Alessandra Y, Devanna P, Limana F, Straino S, Di Carlo A, Brambilla PG, Rubino M, Carena MC, Spazzafumo L, De Simone M, et al. Circulating microRNAs are new and sensitive biomarkers of myocardial infarction. Eur Heart J. 2010;31:2765–2773. doi: 10.1093/eurheartj/ehq167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ai J, Zhang R, Li Y, Pu J, Lu Y, Jiao J, Li K, Yu B, Li Z, Wang R, et al. Circulating microRNA-1 as a potential novel biomarker for acute myocardial infarction. Biochem Biophys Res Commun. 2010;391:73–77. doi: 10.1016/j.bbrc.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 70.Long G, Wang F, Duan Q, Chen F, Yang S, Gong W, Wang Y, Chen C, Wang DW. Human circulating microRNA-1 and microRNA-126 as potential novel indicators for acute myocardial infarction. Int J Biol Sci. 2012;8:811–818. doi: 10.7150/ijbs.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li YQ, Zhang MF, Wen HY, Hu CL, Liu R, Wei HY, Ai CM, Wang G, Liao XX, Li X. Comparing the diagnostic values of circulating microRNAs and cardiac troponin T in patients with acute myocardial infarction. Clinics (Sao Paulo) 2013;68:75–80. doi: 10.6061/clinics/2013(01)OA12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Long G, Wang F, Duan Q, Yang S, Chen F, Gong W, Yang X, Wang Y, Chen C, Wang DW. Circulating miR-30a, miR-195 and let-7b associated with acute myocardial infarction. PLoS One. 2012;7:e50926. doi: 10.1371/journal.pone.0050926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Devaux Y, Vausort M, Goretti E, Nazarov PV, Azuaje F, Gilson G, Corsten MF, Schroen B, Lair ML, Heymans S, et al. Use of circulating microRNAs to diagnose acute myocardial infarction. Clin Chem. 2012;58:559–567. doi: 10.1373/clinchem.2011.173823. [DOI] [PubMed] [Google Scholar]
- 74.Adachi T, Nakanishi M, Otsuka Y, Nishimura K, Hirokawa G, Goto Y, Nonogi H, Iwai N. Plasma microRNA 499 as a biomarker of acute myocardial infarction. Clin Chem. 2010;56:1183–1185. doi: 10.1373/clinchem.2010.144121. [DOI] [PubMed] [Google Scholar]
- 75.Wang F, Long G, Zhao C, Li H, Chaugai S, Wang Y, Chen C, Wang DW. Plasma microRNA-133a is a new marker for both acute myocardial infarction and underlying coronary artery stenosis. J Transl Med. 2013;11:222. doi: 10.1186/1479-5876-11-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Corsten MF, Dennert R, Jochems S, Kuznetsova T, Devaux Y, Hofstra L, Wagner DR, Staessen JA, Heymans S, Schroen B. Circulating MicroRNA-208b and MicroRNA-499 reflect myocardial damage in cardiovascular disease. Circ Cardiovasc Genet. 2010;3:499–506. doi: 10.1161/CIRCGENETICS.110.957415. [DOI] [PubMed] [Google Scholar]
- 77.Nabiałek E, Wańha W, Kula D, Jadczyk T, Krajewska M, Kowalówka A, Dworowy S, Hrycek E, Włudarczyk W, Parma Z, et al. Circulating microRNAs (miR-423-5p, miR-208a and miR-1) in acute myocardial infarction and stable coronary heart disease. Minerva Cardioangiol. 2013;61:627–637. [PubMed] [Google Scholar]
- 78.Olivieri F, Antonicelli R, Lorenzi M, D’Alessandra Y, Lazzarini R, Santini G, Spazzafumo L, Lisa R, La Sala L, Galeazzi R, et al. Diagnostic potential of circulating miR-499-5p in elderly patients with acute non ST-elevation myocardial infarction. Int J Cardiol. 2013;167:531–536. doi: 10.1016/j.ijcard.2012.01.075. [DOI] [PubMed] [Google Scholar]
- 79.Jaguszewski M, Osipova J, Ghadri JR, Napp LC, Widera C, Franke J, Fijalkowski M, Nowak R, Fijalkowska M, Volkmann I, et al. A signature of circulating microRNAs differentiates takotsubo cardiomyopathy from acute myocardial infarction. Eur Heart J. 2014;35:999–1006. doi: 10.1093/eurheartj/eht392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zile MR, Mehurg SM, Arroyo JE, Stroud RE, DeSantis SM, Spinale FG. Relationship between the temporal profile of plasma microRNA and left ventricular remodeling in patients after myocardial infarction. Circ Cardiovasc Genet. 2011;4:614–619. doi: 10.1161/CIRCGENETICS.111.959841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gidlöf O, Smith JG, Miyazu K, Gilje P, Spencer A, Blomquist S, Erlinge D. Circulating cardio-enriched microRNAs are associated with long-term prognosis following myocardial infarction. BMC Cardiovasc Disord. 2013;13:12. doi: 10.1186/1471-2261-13-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Devaux Y, Vausort M, McCann GP, Zangrando J, Kelly D, Razvi N, Zhang L, Ng LL, Wagner DR, Squire IB. MicroRNA-150: a novel marker of left ventricular remodeling after acute myocardial infarction. Circ Cardiovasc Genet. 2013;6:290–298. doi: 10.1161/CIRCGENETICS.113.000077. [DOI] [PubMed] [Google Scholar]
- 83.Devaux Y, Vausort M, McCann GP, Kelly D, Collignon O, Ng LL, Wagner DR, Squire IB. A panel of 4 microRNAs facilitates the prediction of left ventricular contractility after acute myocardial infarction. PLoS One. 2013;8:e70644. doi: 10.1371/journal.pone.0070644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zampetaki A, Willeit P, Tilling L, Drozdov I, Prokopi M, Renard JM, Mayr A, Weger S, Schett G, Shah A, et al. Prospective study on circulating MicroRNAs and risk of myocardial infarction. J Am Coll Cardiol. 2012;60:290–299. doi: 10.1016/j.jacc.2012.03.056. [DOI] [PubMed] [Google Scholar]
- 85.Gidlöf O, Andersson P, van der Pals J, Götberg M, Erlinge D. Cardiospecific microRNA plasma levels correlate with troponin and cardiac function in patients with ST elevation myocardial infarction, are selectively dependent on renal elimination, and can be detected in urine samples. Cardiology. 2011;118:217–226. doi: 10.1159/000328869. [DOI] [PubMed] [Google Scholar]
- 86.Cheng Y, Wang X, Yang J, Duan X, Yao Y, Shi X, Chen Z, Fan Z, Liu X, Qin S, et al. A translational study of urine miRNAs in acute myocardial infarction. J Mol Cell Cardiol. 2012;53:668–676. doi: 10.1016/j.yjmcc.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eitel I, Adams V, Dieterich P, Fuernau G, de Waha S, Desch S, Schuler G, Thiele H. Relation of circulating MicroRNA-133a concentrations with myocardial damage and clinical prognosis in ST-elevation myocardial infarction. Am Heart J. 2012;164:706–714. doi: 10.1016/j.ahj.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 88.Bauters C, Kumarswamy R, Holzmann A, Bretthauer J, Anker SD, Pinet F, Thum T. Circulating miR-133a and miR-423-5p fail as biomarkers for left ventricular remodeling after myocardial infarction. Int J Cardiol. 2013;168:1837–1840. doi: 10.1016/j.ijcard.2012.12.074. [DOI] [PubMed] [Google Scholar]
- 89.Li YG, Zhang PP, Jiao KL, Zou YZ. Knockdown of microRNA-181 by lentivirus mediated siRNA expression vector decreases the arrhythmogenic effect of skeletal myoblast transplantation in rat with myocardial infarction. Microvasc Res. 2009;78:393–404. doi: 10.1016/j.mvr.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 90.Hu S, Huang M, Li Z, Jia F, Ghosh Z, Lijkwan MA, Fasanaro P, Sun N, Wang X, Martelli F, et al. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation. 2010;122:S124–S131. doi: 10.1161/CIRCULATIONAHA.109.928424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lin RC, Weeks KL, Gao XM, Williams RB, Bernardo BC, Kiriazis H, Matthews VB, Woodcock EA, Bouwman RD, Mollica JP, et al. PI3K(p110 alpha) protects against myocardial infarction-induced heart failure: identification of PI3K-regulated miRNA and mRNA. Arterioscler Thromb Vasc Biol. 2010;30:724–732. doi: 10.1161/ATVBAHA.109.201988. [DOI] [PubMed] [Google Scholar]
- 92.Zhang Y, Zhang L, Chu W, Wang B, Zhang J, Zhao M, Li X, Li B, Lu Y, Yang B, et al. Tanshinone IIA inhibits miR-1 expression through p38 MAPK signal pathway in post-infarction rat cardiomyocytes. Cell Physiol Biochem. 2010;26:991–998. doi: 10.1159/000324012. [DOI] [PubMed] [Google Scholar]
- 93.Suffredini S, Stillitano F, Comini L, Bouly M, Brogioni S, Ceconi C, Ferrari R, Mugelli A, Cerbai E. Long-term treatment with ivabradine in post-myocardial infarcted rats counteracts f-channel overexpression. Br J Pharmacol. 2012;165:1457–1466. doi: 10.1111/j.1476-5381.2011.01627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Glass C, Singla DK. MicroRNA-1 transfected embryonic stem cells enhance cardiac myocyte differentiation and inhibit apoptosis by modulating the PTEN/Akt pathway in the infarcted heart. Am J Physiol Heart Circ Physiol. 2011;301:H2038–H2049. doi: 10.1152/ajpheart.00271.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Glass C, Singla DK. ES cells overexpressing microRNA-1 attenuate apoptosis in the injured myocardium. Mol Cell Biochem. 2011;357:135–141. doi: 10.1007/s11010-011-0883-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huang F, Li ML, Fang ZF, Hu XQ, Liu QM, Liu ZJ, Tang L, Zhao YS, Zhou SH. Overexpression of MicroRNA-1 improves the efficacy of mesenchymal stem cell transplantation after myocardial infarction. Cardiology. 2013;125:18–30. doi: 10.1159/000347081. [DOI] [PubMed] [Google Scholar]
- 97.Kim HW, Jiang S, Ashraf M, Haider KH. Stem cell-based delivery of Hypoxamir-210 to the infarcted heart: implications on stem cell survival and preservation of infarcted heart function. J Mol Med (Berl) 2012;90:997–1010. doi: 10.1007/s00109-012-0920-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mao J, Lv Z, Zhuang Y. MicroRNA-23a is involved in tumor necrosis factor-α induced apoptosis in mesenchymal stem cells and myocardial infarction. Exp Mol Pathol. 2014;97:23–30. doi: 10.1016/j.yexmp.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 99.Iekushi K, Seeger F, Assmus B, Zeiher AM, Dimmeler S. Regulation of cardiac microRNAs by bone marrow mononuclear cell therapy in myocardial infarction. Circulation. 2012;125:1765–173, 1765-173. doi: 10.1161/CIRCULATIONAHA.111.079699. [DOI] [PubMed] [Google Scholar]
- 100.Katare R, Riu F, Mitchell K, Gubernator M, Campagnolo P, Cui Y, Fortunato O, Avolio E, Cesselli D, Beltrami AP, et al. Transplantation of human pericyte progenitor cells improves the repair of infarcted heart through activation of an angiogenic program involving micro-RNA-132. Circ Res. 2011;109:894–906. doi: 10.1161/CIRCRESAHA.111.251546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Manole CG, Cismaşiu V, Gherghiceanu M, Popescu LM. Experimental acute myocardial infarction: telocytes involvement in neo-angiogenesis. J Cell Mol Med. 2011;15:2284–2296. doi: 10.1111/j.1582-4934.2011.01449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhou R, Hang P, Zhu W, Su Z, Liang H, Du Z. Whole genome network analysis of ion channels and connexins in myocardial infarction. Cell Physiol Biochem. 2011;27:299–304. doi: 10.1159/000327956. [DOI] [PubMed] [Google Scholar]
- 103.Azuaje FJ, Rodius S, Zhang L, Devaux Y, Wagner DR. Information encoded in a network of inflammation proteins predicts clinical outcome after myocardial infarction. BMC Med Genomics. 2011;4:59. doi: 10.1186/1755-8794-4-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Azuaje FJ, Zhang L, Devaux Y, Wagner DR. Drug-target network in myocardial infarction reveals multiple side effects of unrelated drugs. Sci Rep. 2011;1:52. doi: 10.1038/srep00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nossent AY, Hansen JL, Doggen C, Quax PH, Sheikh SP, Rosendaal FR. SNPs in microRNA binding sites in 3’-UTRs of RAAS genes influence arterial blood pressure and risk of myocardial infarction. Am J Hypertens. 2011;24:999–1006. doi: 10.1038/ajh.2011.92. [DOI] [PubMed] [Google Scholar]