

Abstract
Bone homeostasis, which involves formation and resorption, is an important process for maintaining adequate bone mass in humans. Rheumatoid arthritis (RA) is an autoimmune disease characterized by inflammation and bone loss, leading to joint destruction and deformity, and is a representative disease of disrupted bone homeostasis. The bone loss and joint destruction are mediated by immunological insults by proinflammatory cytokines and various immune cells. The connection between bone and immunity has been intensely studied and comprises the emerging field of osteoimmunology. Osteoimmunology is an interdisciplinary science investigating the interplay between the skeletal and the immune systems. The main contributors in osteoimmunology are the bone effector cells, such as osteoclasts or osteoblasts, and the immune cells, particularly lymphocytes and monocytes. Physiologically, osteoclasts originate from immune cells, and immune cells regulate osteoblasts and vice versa. Pathological conditions such as RA might affect these interactions, thereby altering bone homeostasis, resulting in the unfavorable outcome of bone destruction. In this review, we describe the osteoclastogenic roles of the proinflammatory cytokines and immune cells that are important in the pathophysiology of RA.
1. Introduction
Rheumatoid arthritis (RA) is a devastating autoimmune disease characterized by progressive bone destruction. Under physiological conditions, bone remodeling occurs continually, as a coordinated process that results in the formation and degradation of bone. This process is a balance between bone formation, which is mediated by osteoblasts, and bone resorption, which is regulated by osteoclasts, and ensures bone homeostasis. In pathological conditions such as RA, bone homeostasis is disrupted, resulting in uncoordinated osteoclast formation.
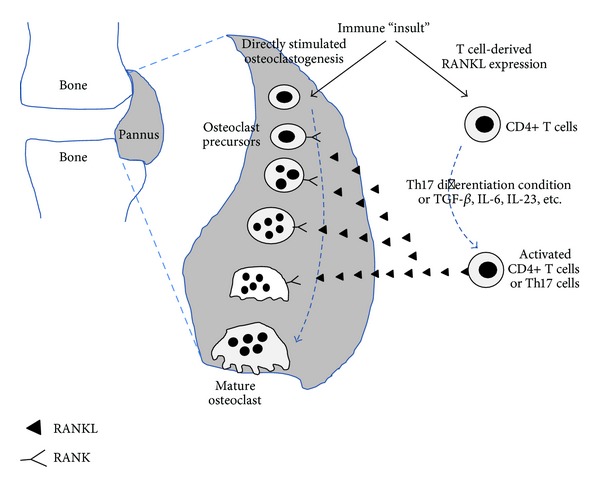
Osteoclasts are generated from precursor cells that are usually of the monocyte-macrophage lineage. Interactions between receptor activator of the nuclear factor kappa B (RANK) and its ligand (RANKL) are essential in osteoclastogenesis. RANK on monocyte binds to RANKL, initiating osteoclast differentiation. Under physiological conditions, the main source of RANKL is osteoblasts. However, immune cells and fibroblast-like synoviocytes (FLS) are the main source of RANKL in pathological conditions such as arthritic RA joints (Figure 1). Several systemic and local factors influence the process of osteoclastogenesis. In RA, excessive activation of the immune system could affect the formation and function of osteoclasts. Proinflammatory cytokines tend to be osteoclastogenic; however, the opposite has also been observed [1]. In our literature review, proinflammatory cytokines such as interleukin (IL)-1, IL-6, IL-8, IL-11, IL-17, and tumor necrosis factor (TNF)-α were frequently reported to be osteoclastogenic, and IL-4, IL-10, IL-13, IL-18, interferon (IFN)-γ, and IFN-β were anti-osteoclastogenic. T cell subpopulations have been studied for their contribution to osteoimmunology. T helper 17 cells (Th17 cells), a specific subtype of T helper cells that produce IL-17 and RANKL, were reported to be osteoclastogenic, whereas the classical Th1 and Th2 cells were generally reported to be anti-osteoclastogenic through their production of IFN-γ (Th1) and IL-4 (Th2) [2, 3].
Figure 1.
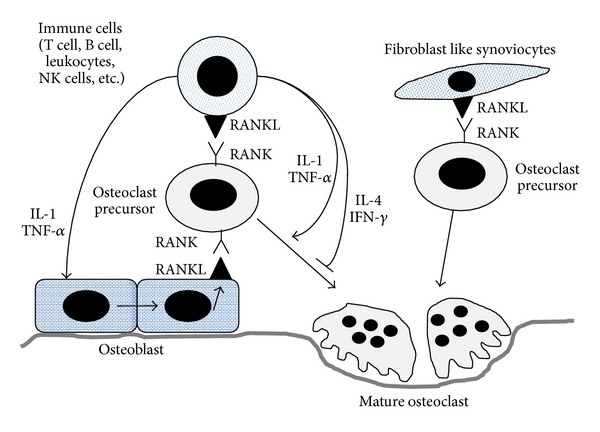
Osteoblast-derived RANKL binds to RANK on monocytes to differentiate them into mature osteoclasts. Osteoblast-derived RANKL plays important role in generating osteoclast in physiological condition. However, immune cell and FLS-derived RANKL play pathogenic role in RA. Proinflammatory cytokines such as IL-1 and TNFα effectively stimulate osteoblast to express RANKL. FLS-derived RANKL enhances osteoclastogenesis in RA joints. RANK: receptor activator of the nuclear factor kappa B; RANKL: receptor activator of the nuclear factor kappa B ligand; FLS: fibroblast like synoviocyte; RA: rheumatoid arthritis; IL-1: Interleukin-1; TNFα: tumor necrosis factor-alpha.
We could not draw uniform conclusions about the various factors involved in osteoclastogenesis. Some proinflammatory cytokines, such as IL-7, IL-12, IL-23, and TGF-β, possess dual osteoclastogenic and anti-osteoclastogenic properties. Their net effect depends on the specific pathophysiological conditions in in vivo models, whereas it depends on the developmental stage of the osteoclasts [4–6] in in vitro experiments. The determination of their exact role in the bone microenvironment is even more difficult because these cytokines can have synergistic or antagonistic effects on osteoclasts [7–11].
The joint structure is invaded and the bone is destroyed by the pannus, which contains a massive infiltration of immune cells, proliferative vessels, and increased numbers of osteoclasts (Figures 2(a) and 2(b)). These complicated structures are frequently observed in RA at the synovium-bone interface (Figure 2(c)). This review will address immune-mediated bone destruction in two sections. First, the osteoclastogenic role of proinflammatory cytokines will be discussed. In the following section, the osteoclastogenic role of inflammatory cells that play important roles in the pathogenesis of RA will be described.
Figure 2.
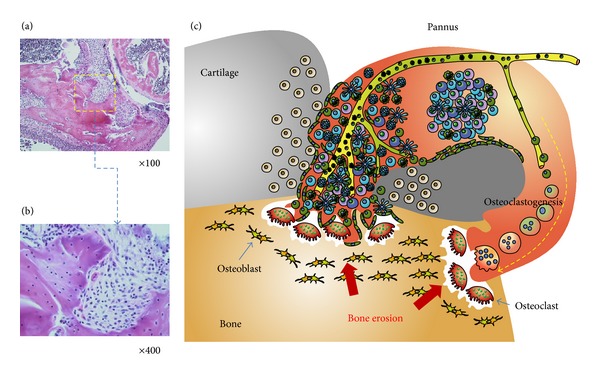
(a) Bone is destroyed by a proliferative and invasive synovium, which is called pannus. It originates from adjacent synovial tissue and invades the cartilages and bones. (b) Magnified view of the pannus-bone interface. The pannus-bone interface is lined with mature osteoclasts (arrows). Various inflammatory cells and stromal cells comprise the invading pannus. (c) Schematic depiction of the pannus-cartilage-bone structure. Inflammatory cells such as B cells, T cells, macrophages, monocytes, and fibroblast-like synoviocytes accumulate in the pannus. For metabolic support, intensive angiogenesis is usually followed. Excessive provision of RANKL from the accumulated cells in the pannus enhances osteoclastogenesis, resulting in the erosion of bone at the pannus-bone interface.
2. Cytokines and Bone: The Osteoclastogenic Effect of Proinflammatory Cytokines
Proinflammatory cytokines promote osteoclastogenesis via RANKL expression. Some researchers have shown that proinflammatory cytokines such as TNF-α, IL-1, and IL-6 are capable of inducing osteoclast differentiation independently of RANKL [12–14]. Others showed that a minimal level of RANKL is essential for TNF-α-induced osteoclastogenesis, revealing that TNF-α alone does not induce osteoclast formation [15]. To clarify this controversy, we adopted a simplified monocellular culture system instead of a co-culture system, which consists of osteoblasts and bone marrow cells [16]. In our experience, permissive levels of RANKL were required for cytokine-associated osteoclastogenesis. IL-1 increased and IL-6 decreased the number of mature osteoclasts in a dose-dependent manner. Treatment with IL-23, IL-17, or TNF-α resulted in various responses according to the exposure time and the cytokine concentration.
The effects of important cytokines on osteoclastogenesis in vitro and in vivo are summarized in Table 1. Based on laboratory observations, cytokine-targeting therapies were tested in bone resorptive conditions. The results of experimental and clinical trials are presented in Table 2.
Table 1.
Roles of cytokines on osteoclastogenesis.
| In Vitro | In Vivo | |
|---|---|---|
| TNF-α | Osteoclastogenic (i) Upregulates the expression of RANKL and osteoclast activators (ii) Enhances osteoclast differentiation synergistically with RANKL or independently of RANKL (iii) Inhibits osteoclast apoptosis |
Osteoclastogenic (i) Upregulates the expression of RANKL and osteoclast activators (ii) Induces osteoclastogenesis in the presence or absence of RANKL (iii) Plays a critical role in inflammatory arthritis (iv) Associated with estrogen-deficient osteoporosis and joint destruction in RA |
| References: [12, 21, 110–116] | References: [15, 24, 36, 117–119] | |
|
| ||
| IL-1 | Osteoclastogenic (i) Upregulates the expression of RANKL and osteoclast activators (ii) Enhances osteoclast differentiation synergistically with RANKL or independently of RANKL |
Osteoclastogenic (i) Induces osteoclastogenesis in the presence or absence of RANKL (ii) Mediates TNF-α-induced osteoclastogenesis (iii) Participates in physiological bone metabolism (iv) Associated with estrogen-deficient osteoporosis |
| References: [33, 35, 116, 120–122] | References: [23, 29–32, 36, 37] | |
|
| ||
| IL-6 | Osteoclastogenic (i) Upregulates the expression of RANKL and osteoclast activators (ii) Induces RANKL-dependent osteoclastogenesis |
Osteoclastogenic (i) Enhances osteoclastogenesis in the prepubertal stage (ii) Supports osteoclastogenesis in callus formation during fracture healing (iii) Associated with bone loss from inflammatory arthritis and estrogen deficiency |
| References: [10, 44, 123–129] | References: [46, 48, 49, 130–132] | |
| Antiosteoclastogenic (i) Suppresses the RANK signaling pathway (ii) Diverts cells into the macrophage lineage |
Antiosteoclastogenic (i) Suppresses the differentiation of early osteoclast precursor cells (ii) Decreases osteoclast formation, leading to reduced bone turnover |
|
| References: [6, 133, 134] | References: [46, 47, 135] | |
|
| ||
| IL-17 | Osteoclastogenic (i) Induces the expression of RANKL and proinflammatory cytokines (ii) Increases sensitivity to RANKL (iii) Enhances osteoclastogenesis via prostaglandin E2 (PGE2) in osteoblasts |
Osteoclastogenic (i) Induces the expression of RANKL and proinflammatory cytokines (ii) Mediates estrogen-deficient osteoporosis |
| References: [1, 53, 136–141] | References: [52, 142–144] | |
| Anti-osteoclastogenic (i) Suppresses osteoclast formation at high concentrations (ii) Inhibits osteoclastogenesis by induction of GM-CSF |
||
| References: [145, 146] | ||
|
| ||
| IL-23 | Osteoclastogenic Induces osteoclastogenesis via IL-17 |
Osteoclastogenic (i) Induces the expression of RANKL (ii) Expands myeloid-lineage osteoclast precursors |
| References: [56] | References: [65, 147–150] | |
| Antiosteoclastogenic Inhibits osteoclast formation via T cells |
Antiosteoclastogenic Limits the resorption of immature bone below the growth plate |
|
| References: [57, 151] | References: [57] | |
Table 2.
Effects of biologic therapies on bone.
| Mice | Human | |
|---|---|---|
| TNF-α blockers | Bone-protective in inflammatory arthritis and estrogen deficiency | Bone-protective in inflammatory disease Changes in bone turnover markers in postmenopause (small observational study) |
| References: [152–158] | References: [17, 19, 20, 159] | |
|
| ||
| IL-1 blockers | Bone-protective in inflammatory arthritis and estrogen deficiency | Bone-protective in RA (not usually recommended; less effective than other biologic agents) Changes in bone turnover markers in postmenopause (small observational study) |
| References: [28, 155, 157] | References: [19, 160, 161] | |
|
| ||
| IL-6 blockers | Bone-protective in inflammatory arthritis No effects in estrogen deficiency |
Bone-protective in RA |
| References: [155, 156, 162, 163] | References: [41, 42, 164, 165] | |
|
| ||
| IL-17 blockers | Bone-protective in inflammatory arthritis and estrogen deficiency | No data in bone metabolism |
| References: [55, 138, 144, 166, 167] | ||
|
| ||
| IL-23 blockers | Bone-protective in inflammatory arthritis | No data |
| References: [56] | ||
2.1. TNF-α
TNF-α has received attention from immunologists and rheumatologists because several TNF-α inhibitors show enormous pharmaceutical success in treating RA. TNF-α is produced by activated T cells and is involved in inflammation- and cancer-induced bone loss [17]. Treatment with TNF-α inhibitors results in decreased inflammation and bone protection in RA patients [18]. In vivo blockade of TNF-α reduces bone resorption in postmenopausal osteoporosis [19]. Thus, TNF-α is regarded as a major contributor to bone destruction and osteoclast formation.
TNF-α promotes bone destruction by upregulating the production of RANKL and macrophage colony-stimulating factor (M-CSF) from osteoblasts and stromal cells, and by augmenting differentiation into osteoclasts independently of RANK-RANKL signaling [20]. In addition, TNF-α and RANKL synergistically upregulate the expression of RANK [21]. This osteoclastogenic effect of TNF-α is closely associated with other inflammatory cytokines, including IL-1 and M-CSF [22–24]. Although osteoclastogenesis is a more dominant mechanism in the bone erosion of inflammatory disease, osteoblast formation is also affected by TNF-α. TNF-α inhibits osteoblast differentiation primarily through TNF-receptor 1 signaling [25, 26].
2.2. IL-1
IL-1, a proinflammatory cytokine, is highly expressed in patients with RA [27]. An earlier study showed a prominent protective effect of IL-1 blockade against structural damage in an arthritis animal model, suggesting a crucial effect of IL-1 on bone metabolism [28]. Animal models with a deficiency of IL-1 signaling present with reduced osteoclastogenesis, leading to significantly increased levels of bone density, trabecular bone mass, and cortical thickness [29, 30]. IL-1 also plays an important role in the bone loss induced by estrogen deficiency; the level of IL-1 increases after menopause and decreases with estrogen replacement [31, 32]. Bone resorption is suppressed by blockade of IL-1 in postmenopausal women [19].
IL-1 induces RANKL to promote osteoclastogenesis through the production of prostaglandin E in periodontal tissue [33, 34]. Furthermore, IL-1 might exert a bone resorptive effect via an alternative pathway independent of the RANK/RANKL signal [35, 36]. IL-1 is essential for TNF-α-induced osteoclastogenesis. Human TNF-α transgenic mice lacking IL-1β were protected from systemic bone loss regardless of sustained inflammation [37]. The activation of p38 mitogen-activated proteinase kinase is involved in TNF-α- and IL-1-mediated osteoclastogenesis by upregulating RANKL expression in stromal cells and stimulating osteoclast precursor differentiation [23].
2.3. IL-6
Dysregulation of IL-6 is frequently observed in RA patients [38–40]. IL-6 is responsible for synovial inflammation as well as the structural damage of RA. An IL-6 receptor antagonist, a new immunotherapeutic, reduced bone turnover, favoring bone protection in RA patients [41, 42]. IL-6 is also involved in other diseases associated with accelerated bone turnover, such as multiple myeloma and Paget's disease of bone [43].
The previous data indicate the dual functions of IL-6 on bone remodeling. The addition of IL-6 and the soluble IL-6 receptor into bone tissue cultures stimulates bone resorption through increased RANKL expression on osteoblasts [44] via activation of the STAT3 pathway [45]. However, IL-6 exhibits a direct inhibitory effect on RANK signaling in osteoclast progenitor cells in the absence of other supporting cells [6].
In vivo studies also suggest that the role of IL-6 varies in a context-dependent manner. IL-6 transgenic mice with a high level of circulating IL-6 exhibited enhanced osteoclastogenesis, leading to impaired skeletal growth at the prepubertal stage [46] but decreased osteoclast formation at the adult stage [46, 47]. Under physiological conditions, IL-6 deficiency resulted in no detectable change in osteoclast number [48]. However, IL-6 knockout mice were protected against ovariectomy-induced bone loss [48]. IL-6 knockout mice with experimental arthritis showed significantly decreased osteoclastogenic activity and impaired osteoclast recruitment to inflammatory sites [49]. These results indicate that IL-6 is associated with bone loss from inflammation and estrogen deprivation. IL-6, along with TGF-β, induces the differentiation of naïve T cells into Th17 cells, which are typically osteoclastogenic [50].
2.4. IL-17
IL-17 is predominantly expressed by Th17 cells, a specific type of human T helper cells [51]. It is hypothesized that this cytokine plays a crucial role in inflammation and the development of autoimmune diseases, including RA. There is evidence that IL-17 enhances osteoclastogenesis by a RANKL-RANK dependent mechanism. Studies of an arthritis animal model indicate that IL-17 induces the expression of RANKL and proinflammatory cytokines such as IL-1 and TNF-α [52]. These inflammatory mediators (IL-17, IL-1, TNF-α, and RANKL) interact with each other in the progression of RA. IL-17A also upregulates the expression of RANK on osteoclast precursors and increases their sensitivity to RANKL [53]. Similarly, treatment with an IL-17 neutralizing antibody inhibited bone destruction in collagen-induced arthritis [54, 55]. However, the mechanisms of action of IL-17 in bone erosion remain to be determined, particularly in association with other osteoclastogenic cytokines such as IL-1, TNF-α, and RANKL.
2.5. IL-23
One of the most important stimuli for IL-17 synthesis is IL-23 produced by activated dendritic cells and macrophages [50]. IL-23 is implicated in inflammatory diseases, in association with IL-17. Accordingly, the IL-23/IL-17 axis plays a critical role in controlling inflammatory bone loss. Recent work suggests that osteoclastogenesis is promoted by IL-23 and inhibited by an anti-IL-23 antibody [56]. By contrast, another study shows the indirect inhibition of osteoclast differentiation by IL-23 in vitro. Under physiological conditions, IL-23 promotes higher bone mass in long bones by limiting bone resorption near the growth plate in vivo [57]. These conflicting data suggest different roles for this cytokine in physiological or inflammatory bone turnover.
3. Immune Cells and Bone: The Osteoclastogenic Effect of Inflammatory Cells
Various immune cells play important roles in the pathogenesis of RA. These cells comprise the rheumatoid synovium that is continuously inflamed and invades adjacent tissue, resulting in joint destruction (Figure 2). Although osteoclasts are the final effectors of bone erosion, osteoclastogenesis is regulated by various cells in the RA synovium. FLS are the main cellular component of the matrix that is involved in bone turnover. Monocytes, T cells, B cells, and neutrophils also infiltrate the RA synovium and interact with each other. These cells vigorously contribute to osteoclast formation under inflammatory conditions by producing osteoclastogenic cytokines or RANKL (Figure 3).
Figure 3.
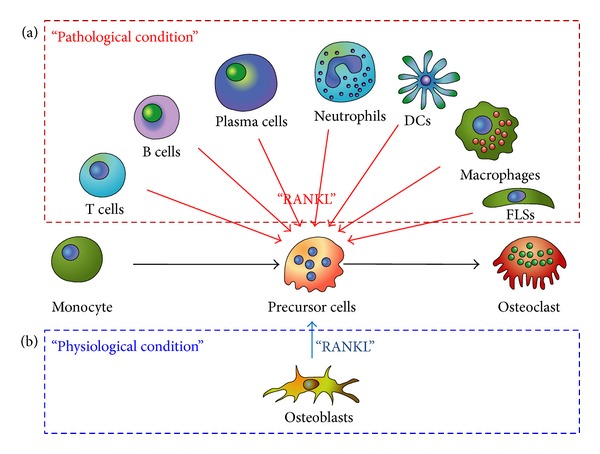
(a) Monocytes are differentiated into mature osteoclasts by the aid of RANKL. In pathological conditions such as inflammation, cancer, and hypermetabolism, various cells extraordinarily provide RANKL to the monocytes, resulting in overweighed osteoclastogenesis. In this condition, the osteoclasts outnumber the osteoblasts, disrupting the bone homeostasis. Bone erosion or osteoporosis is the major outcome of disrupted homeostasis. (b) In normal physiological conditions, a few cells, predominantly osteoblasts, express RANKL. A similar number of osteoclasts and osteoblasts maintain the bone mass by homeostatic equilibrium.
3.1. Fibroblast-Like Synoviocytes (FLS)
Under physiological conditions, the synovium secretes synovial fluid and provides mechanical stability to the joint. However, pathological conditions such as RA render the synovium more aggressive. The synovium forms a pannus with inflammatory cells, enabling invasion into the bone [58, 59]. Histopathology demonstrates increased bone resorption at the bone-pannus interface in the joints of patients with RA. Thus, FLS play an active role in the pathogenesis of RA [59].
The bone and cartilage destruction in RA patients is partly mediated by metalloproteinases secreted by activated synoviocytes and chondrocytes [60, 61]. More importantly, bone destruction is further exacerbated by osteoclasts induced by the RA synovium [62, 63]. We reported that RANKL is produced by FLS from RA patients (RA-FLS) and that osteoclasts are formed in cocultures of RA-FLS and human monocytes [64]. Consistent with a previous report [62], this result indicates that RA-FLS have the capability to support osteoclast differentiation. In RA, FLS upregulate the expression of RANKL and osteoclastogenic cytokines. Earlier studies show that RANKL in RA-FLS can be increased by IL-23 [65], IL-22 [66], and SDF-1 [67]. Furthermore, FLS produce osteoclastogenic cytokines such as IL-6 in response to IL-17 and IL-23 [68, 69]. These inflammatory mediators from stimulated RA-FLS act on stromal cells to upregulate RANKL expression and on osteoclast precursor cells to promote differentiation into osteoclasts (Figure 4).
Figure 4.
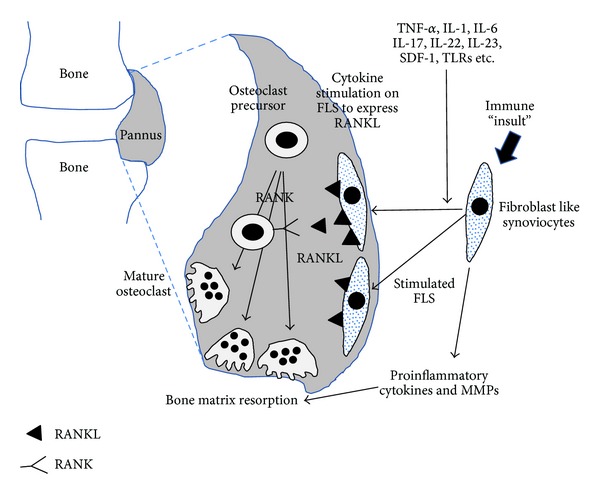
FLS could express RANKL in response to various stimuli. FLS-expressed RANKL enhances osteoclastogenesis and results in bone erosion in RA. Inflammatory and immune stimulation induce the FLS to produce proinflammatory cytokines and matrix metalloproteinase. These cytokines and enzymes aid osteoclasts to destroy the bone matrix.
3.2. Monocyte and Dendritic Cells
Bloodstream monocytes migrate into inflammatory tissue where they differentiate into resident macrophages and dendritic cells (DCs) [70]. Macrophages and DCs express a variety of inflammatory cytokines involved in the pathogenesis of RA [71].
Synovial macrophages play a central role in rheumatoid inflammation. TNF-α, IL-1, and IL-6 are largely produced by activated macrophages and synovial fibroblasts in the RA synovium [71, 72]. As discussed above, these cytokines directly exert osteoclastogenic effects, either synergistically with RANKL or independently of the RANKL signaling pathway. Moreover, macrophages in the RA synovium also secrete TGF-β, IL-21, and IL-23 to differentiate CD4+ T cells into Th17 cells, which are typically referred to as osteoclastogenic T cells.
DC, highly differentiated antigen-presenting cells, interact with T cells and B cells in RA. The physiological function of DC in bone remodeling appears to be modest, as DCs are not frequently observed in bone or the adjacent stroma under normal conditions. By contrast, active lesions of RA and periodontitis retain mature and immature DCs [73–78]. At these sites, DCs contact and interact with T cells to elicit inflammatory processes that involve RANK-RANKL signaling [77]. In multiple myeloma, DCs promote osteoclastogenesis, leading to bone destruction, possibly by activation of RANK-RANKL signaling [79] and the overproduction of IL-17 [80].
DCs can also affect bone metabolism in a more direct manner. Rivollier and colleagues showed that human monocyte-derived DCs transdifferentiate into osteoclasts in the presence of M-CSF and RANKL in vitro, suggesting that DCs might directly contribute to osteoclastogenesis [81]. Alnaeeli et al. tested whether the interaction between DCs and T cells supports osteoclast development using an in vitro co-culture system of bone marrow-derived CD11c+ DC and CD4+ T cells. Murine CD11c+ DC developed into functional osteoclasts after interactions with CD4+ T cells and stimulation with microbial or protein antigens. Adoptive transfer of DC-derived osteoclasts could induce bone resorption in NOD/SCID mice calvarias in vivo [82]. The differentiation of DCs into osteoclasts is frequently reported in the pathogenesis of multiple myeloma [79, 83].
3.3. T Cells
T cells are one of the key regulators of synovial inflammation in RA, having both stimulatory and inhibitory roles [71]. T cells can also play a destructive or a protective role in bone metabolism in a context- and subtype-dependent manner.
In the resting state, T cells seem to have a positive effect on bone mineral density, as T cell depletion increased osteoclastogenesis in vitro [84] and accelerated bone resorption in vivo [85]. T cell-deficient nude mice have significantly higher numbers of osteoclasts and reduced bone density compared to controls [85].
In response to antigenic stimuli, CD4+ T cells differentiate into distinct effector subsets, Th1 and Th2 cells, which are classically defined on the basis of cytokine production profiles [86]. Th1 cells are characterized by the secretion of IFN-γ, IL-2, IL-12, TNF-α, and TNF-β, and are involved in the elimination of intracellular pathogens [87]. Th2 cells produce IL-4, IL-5, IL-6, IL-9, and IL-13, and are responsible for parasite eradication and allergic disorders [87, 88]. In one comprehensive study, Th1 and Th2 cells were shown to inhibit osteoclastogenesis through IFN-γ and IL-4, respectively [89]. However, the bone-preserving effects of Th1 and Th2 cells are not certain, because contradictory responses have been observed in inflammatory conditions. Infection and inflammation could activate T cells to produce osteoclastogenic cytokines such as TNF-α and RANKL. In the pathogenic state, lymphocytes express significantly higher levels of RANKL and have the capacity to induce RANKL-dependent osteoclast differentiation, unlike in healthy conditions [90]. In addition, IFN-γ exerts a bone resorptive effect instead of a bone-protective effect in an animal model with ovariectomy, infection, and inflammation [4, 91]. Thus, further research is required to understand the net effect of Th1/Th2 cells in disease states such as RA.
Th17 cells, a more recently characterized subset of CD4+ T cells, have been shown to be more osteoclastogenic. Th17 cells are produced when naïve T cells are activated by TGF-β and IL-6 in mice or TGF-β and inflammatory cytokines in humans [50, 92]. Th17 cell play a pivotal role in the pathogenesis of RA through the production of Th17 signature cytokines [50]. Since IL-17 is predominantly produced by Th17 cells and is closely associated with osteoclastogenesis, Th17 cells are likely to affect bone metabolism primarily through IL-17. IL-17 directly induces the expression of RANKL from surrounding cells and facilitates the recruitment of inflammatory cells, leading to an abundance of inflammatory cytokines such as TNF-α and IL-1. Moreover, Th17 cells drive RA-FLS to produce IL-6, IL-8, and matrix metalloproteinases, which potentiate structural damage [93]. A prominent role for Th17 cells has been demonstrated in bone destructive diseases such as RA and multiple myeloma [94, 95] (Figure 5).
Figure 5.
T cells are activated to produce RANKL or osteoclastogenic cytokines by various stimuli. RANKL and activated T cell-cytokines have the potential to induce osteoclastogenesis. With T cells, the outnumbered osteoclasts destroy the bone in RA.
3.4. B Cells
Multiple myeloma is a malignant B cell disease characterized by multiple bone lesions. These are caused by plasma cells expressing RANKL, which stimulate osteoclast formation, leading to osteolysis [96]. This phenomenon indicates that B cells could affect bone metabolism via RANKL expression. In RA, the pathophysiologic role of B cells is highlighted by the therapeutic success of B cell-depleting therapy with an anti-CD20 monoclonal antibody (rituximab) [97, 98]. B cells play an important role in producing autoantibodies. Although the role of autoantibodies such as rheumatoid factor (RF) and anti-citrullinated protein antibody is not fully understood, these autoantibodies are associated with more severe bone destruction [99]. Treatment with rituximab reduced bone destruction as well as joint inflammation. Taken together, these findings indicate that B cells contribute to bone destruction through RANKL expression and the production of autoantibodies in cooperation with other immune cells.
3.5. Neutrophils
The neutrophil is the most abundant type of white blood cell in mammals, and comprises an essential part of the innate immune system. Neutrophils normally circulate in the bloodstream and migrate to the site of inflammation in response to inflammatory stimuli. In the RA synovium, neutrophils regulate inflammation through the secretion of inflammatory mediators [100]. Histological analysis of bony lesions in humans and animal models indicates the involvement of neutrophils in pathogenic bone remodeling. Infiltration of neutrophils is observed in human periodontitis and experimental arthritis [101–103]. The RANKL-RANK-osteoprotegerin pathway is upregulated in activated neutrophils from inflammatory sites [104]. Membrane RANKL on neutrophils is strongly overexpressed after stimulation with lipopolysaccharide and thus mediates osteoclastic bone resorption through the interactions between neutrophils and osteoclasts [105]. The osteoclastogenic effect of neutrophils could be reproduced with purified neutrophil membranes, but not with culture supernatants from activated neutrophils. Thus, the effect of RANKL in activated neutrophils is predominantly mediated by the membrane-bound form, in contrast to activated T cells, where RANKL signaling is mediated by both cell surface and soluble RANKL [106, 107]. In addition, neutrophils affect the function of osteoblasts in children on chronic glucocorticoid therapy and in patients with tophaceous gout, resulting in altered bone remodeling [108, 109].
4. Conclusions
The human body attempts to maintain bone mass in order to maintain skeletal strength. Bone mass is not static but dynamic, and results from the formation or resorbtion of the bony matrix by osteoblasts or osteoclasts. In pathological states such as RA, in which bone resorption is favored over bone formation, osteoblasts are outnumbered by osteoclasts. Osteoclastogenesis is also favored over osteoblastogenesis by the inflammatory milieu. Recent studies have shown that numerous cytokines and immune cells have osteoclastogenic effects, although their exact roles in pathological states are difficult to determine because of the complexity of immune networks in the human body. Proinflammatory cytokines such as TNF-α, IL-1, IL-6, and IL-17 tend to be osteoclastogenic. The immune cells that participate in the pathogenesis of RA often enhance osteoclastogenesis by upregulating RANKL directly or by secreting proinflammatory cytokines that influence RANKL expression indirectly. Understanding the precise mechanisms of immune-mediated bone destruction would increase opportunities for target-specific inhibition of bone erosion or osteoporosis. Therapeutic interventions specifically targeting osteoclastogenesis might enable clinicians to spare bone mass in RA patients.
Acknowledgments
This work was supported by Grants from the Korea Healthcare Technology R&D Project of the Ministry for Health, Welfare, and Family Affairs, Republic of Korea (A092258); from the Basic Science Research Program of the Ministry of Science, ICT, and Future Planning of the National Research Foundation of Korea (2013R1A1A1076125); and from the Advanced Production Technology Development Program of the Ministry for Food, Agriculture, Forestry, and Fisheries (312037-05).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publishing of this paper.
Authors' Contribution
Seung Min Jung and Kyoung Woon Kim equally contributed.
References
- 1.Nakashima T, Kobayashi Y, Yamasaki S, et al. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-κB ligand: modulation of the expression by osteotropic factors and cytokines. Biochemical and Biophysical Research Communications. 2000;275(3):768–775. doi: 10.1006/bbrc.2000.3379. [DOI] [PubMed] [Google Scholar]
- 2.Takayanagi H, Kim S, Taniguchi T. Signaling crosstalk between RANKL and interferons in osteoclast differentiation. Arthritis Research & Therapy. 2002;4(supplement 3):S227–S232. doi: 10.1186/ar581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kotake S, Nanke Y, Yago T, Kawamoto M, Yamanaka H. Human osteoclastogenic T cells and human osteoclastology. Arthritis and Rheumatism. 2009;60(11):3158–3163. doi: 10.1002/art.24886. [DOI] [PubMed] [Google Scholar]
- 4.Gao Y, Grassi F, Ryan MR, et al. IFN-γ stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. The Journal of Clinical Investigation. 2007;117(1):122–132. doi: 10.1172/JCI30074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janssens K, ten Dijke P, Janssens S, van Hul W. Transforming growth factor-β1 to the bone. Endocrine Reviews. 2005;26(6):743–774. doi: 10.1210/er.2004-0001. [DOI] [PubMed] [Google Scholar]
- 6.Yoshitake F, Itoh S, Narita H, Ishihara K, Ebisu S. Interleukin-6 directly inhibits osteoclast differentiation by suppressing receptor activator of NF-κB signaling pathways. Journal of Biological Chemistry. 2008;283(17):11535–11540. doi: 10.1074/jbc.M607999200. [DOI] [PubMed] [Google Scholar]
- 7.Franchimont N, Rydziel S, Canalis E. Transforming growth factor-β increases interleukin-6 transcripts in osteoblasts. Bone. 2000;26(3):249–253. doi: 10.1016/s8756-3282(99)00275-6. [DOI] [PubMed] [Google Scholar]
- 8.Horwood NJ, Elliott J, Martin TJ, Gillespie MT. IL-12 alone and in synergy with IL-18 inhibits osteoclast formation in vitro. Journal of Immunology. 2001;166(8):4915–4921. doi: 10.4049/jimmunol.166.8.4915. [DOI] [PubMed] [Google Scholar]
- 9.Huang W, Drissi MH, O'Keefe RJ, Schwarz EM. A rapid multiparameter approach to study factors that regulate osteoclastogenesis: demonstration of the combinatorial dominant effects of TNF-α and TGF-β in RANKL-mediated osteoclastogenesis. Calcified Tissue International. 2003;73(6):584–593. doi: 10.1007/s00223-003-0059-8. [DOI] [PubMed] [Google Scholar]
- 10.Liu X, Kirschenbaum A, Yao S, Levine AC. Cross-talk between the interleukin-6 and prostaglandin E2 signaling systems results in enhancement of osteoclastogenesis through effects on the osteoprotegerin/receptor activator of nuclear factor-κB (RANK) ligand/RANK system. Endocrinology. 2005;146(4):1991–1998. doi: 10.1210/en.2004-1167. [DOI] [PubMed] [Google Scholar]
- 11.Ragab AA, Nalepka JL, Bi Y, Greenfield EM. Cytokines synergistically induce osteoclast differentiation: support by immortalized or normal calvarial cells. American Journal of Physiology-Cell Physiology. 2002;283(3):C679–C687. doi: 10.1152/ajpcell.00421.2001. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi K, Takahashi N, Jimi E, et al. Tumor necrosis factor α stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. Journal of Experimental Medicine. 2000;191(2):275–285. doi: 10.1084/jem.191.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim JH, Jin HM, Kim K, et al. The mechanism of osteoclast differentiation induced by IL-1. Journal of Immunology. 2009;183(3):1862–1870. doi: 10.4049/jimmunol.0803007. [DOI] [PubMed] [Google Scholar]
- 14.Kudo O, Sabokbar A, Pocock A, Itonaga I, Fujikawa Y, Athanasou NA. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone. 2003;32(1):1–7. doi: 10.1016/s8756-3282(02)00915-8. [DOI] [PubMed] [Google Scholar]
- 15.Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. Journal of Clinical Investigation. 2000;106(12):1481–1488. doi: 10.1172/JCI11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moon SJ, Ahn IE, Jung H, et al. Temporal differential effects of proinflammatory cytokines on osteoclastogenesis. International Journal of Molecular Medicine. 2013;31(4):769–777. doi: 10.3892/ijmm.2013.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Osta B, Benedetti G, Miossec P. Classical and paradoxical effects of TNF-alpha on bone homeostasis. Frontiers in Immunology. 2014;5, article 48 doi: 10.3389/fimmu.2014.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scott DL, Kingsley GH. Tumor necrosis factor inhibitors for rheumatoid arthritis. The New England Journal of Medicine. 2006;355(7):704–712. doi: 10.1056/NEJMct055183. [DOI] [PubMed] [Google Scholar]
- 19.Charatcharoenwitthaya N, Khosla S, Atkinson EJ, McCready LK, Riggs BL. Effect of blockade of TNF-α and interleukin-1 action on bone resorption in early postmenopausal women. Journal of Bone and Mineral Research. 2007;22(5):724–729. doi: 10.1359/jbmr.070207. [DOI] [PubMed] [Google Scholar]
- 20.Kawai VK, Stein CM, Perrien DS, Griffin MR. Effects of anti-tumor necrosis factor α agents on bone. Current Opinion in Rheumatology. 2012;24(5):576–585. doi: 10.1097/BOR.0b013e328356d212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Komine M, Kukita A, Kukita T, Ogata Y, Hotokebuchi T, Kohashi O. Tumor necrosis factor-α cooperates with receptor activator of nuclear factor κB ligand in generation of osteoclasts in stromal cell-depleted rat bone marrow cell culture. Bone. 2001;28(5):474–483. doi: 10.1016/s8756-3282(01)00420-3. [DOI] [PubMed] [Google Scholar]
- 22.Kudo O, Fujikawa Y, Itonaga I, Sabokbar A, Torisu T, Athanasou NA. Proinflammatory cytokine (TNFα/IL-Iα) induction of human osteoclast formation. The Journal of Pathology. 2002;198(2):220–227. doi: 10.1002/path.1190. [DOI] [PubMed] [Google Scholar]
- 23.Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL. IL-1 mediates TNF-induced osteoclastogenesis. Journal of Clinical Investigation. 2005;115(2):282–290. doi: 10.1172/JCI23394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitaura H, Zhou P, Kim H, Novack DV, Ross FP, Teitelbaum SL. M-CSF mediates TNF-induced inflammatory osteolysis. Journal of Clinical Investigation. 2005;115(12):3418–3427. doi: 10.1172/JCI26132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abbas S, Zhang YH, Clohisy JC, Abu-Amer Y. Tumor necrosis factor-α inhibits pre-osteoblast differentiation through its type-1 receptor. Cytokine. 2003;22(1-2):33–41. doi: 10.1016/s1043-4666(03)00106-6. [DOI] [PubMed] [Google Scholar]
- 26.Gilbert LC, Rubin J, Nanes MS. The p55 TNF receptor mediates TNF inhibition of osteoblast differentiation independently of apoptosis. The American Journal of Physiology—Endocrinology and Metabolism. 2005;288(5):E1011–E1018. doi: 10.1152/ajpendo.00534.2004. [DOI] [PubMed] [Google Scholar]
- 27.Symons JA, McDowell TL, Di Giovine FS, Wood NC, Capper SJ, Duff GW. Interleukin 1 in rheumatoid arthritis: potentiation of immune responses with the joint. Lymphokine Research. 1989;8(3):365–372. [PubMed] [Google Scholar]
- 28.Joosten LA, Helsen MM, Saxne T, van De Loo FA, Heinegård D, van Den Berg WB. IL-1αβ blockade prevents cartilage and bone destruction in murine type II collagen-induced arthritis, whereas TNF-α blockade only ameliorates joint inflammation. Journal of Immunology. 1999;163(9):5049–5055. [PubMed] [Google Scholar]
- 29.Lee Y, Fujikado N, Manaka H, Yasuda H, Iwakura Y. IL-1 plays an important role in the bone metabolism under physiological conditions. International Immunology. 2010;22(10):805–816. doi: 10.1093/intimm/dxq431. [DOI] [PubMed] [Google Scholar]
- 30.Simsa-Maziel S, Zaretsky J, Reich A, Koren Y, Shahar R, Monsonego-Ornan E. IL-1RI participates in normal growth plate development and bone modeling. American Journal of Physiology—Endocrinology and Metabolism. 2013;305(1):E15–E21. doi: 10.1152/ajpendo.00335.2012. [DOI] [PubMed] [Google Scholar]
- 31.Rogers A, Eastell R. Effects of estrogen therapy of postmenopausal women on cytokines measured in peripheral blood. Journal of Bone and Mineral Research. 1998;13(10):1577–1586. doi: 10.1359/jbmr.1998.13.10.1577. [DOI] [PubMed] [Google Scholar]
- 32.Pacifici R, Brown C, Puscheck E, et al. Effect of surgical menopause and estrogen replacement on cytokine release from human blood mononuclear cells. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(12):5134–5138. doi: 10.1073/pnas.88.12.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fukushima H, Jimi E, Okamoto F, Motokawa W, Okabe K. IL-1-induced receptor activator of NF-κB ligand in human periodontal ligament cells involves ERK-dependent PGE2 production. Bone. 2005;36(2):267–275. doi: 10.1016/j.bone.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 34.Nukaga J, Kobayashi M, Shinki T, et al. Regulatory effects of interleukin-1β and prostaglandin E2 on expression of receptor activator of nuclear factor-κB ligand in human periodontal ligament cells. Journal of Periodontology. 2004;75(2):249–259. doi: 10.1902/jop.2004.75.2.249. [DOI] [PubMed] [Google Scholar]
- 35.Bloemen V, Schoenmaker T, de Vries TJ, Everts V. IL-1β favors osteoclastogenesis via supporting human periodontal ligament fibroblasts. Journal of Cellular Biochemistry. 2011;112(7):1890–1897. doi: 10.1002/jcb.23109. [DOI] [PubMed] [Google Scholar]
- 36.Kim N, Kadono Y, Takami M, et al. Osteoclast differentiation independent of the TRANCE-RANK-TRAF6 axis. Journal of Experimental Medicine. 2005;202(5):589–595. doi: 10.1084/jem.20050978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Polzer K, Joosten L, Gasser J, et al. Interleukin-1 is essential for systemic inflammatory bone loss. Annals of the Rheumatic Diseases. 2010;69(1):284–290. doi: 10.1136/ard.2008.104786. [DOI] [PubMed] [Google Scholar]
- 38.Okamoto H, Yamamura M, Morita Y, Harada S, Makino H, Ota Z. The synovial expression and serum levels of interleukin-6, interleukin- 11, leukemia inhibitory factor, and oncostatin M in rheumatoid arthritis. Arthritis and Rheumatism. 1997;40(6):1096–1105. doi: 10.1002/art.1780400614. [DOI] [PubMed] [Google Scholar]
- 39.Kotake S, Sato K, Kim KJ, et al. Interleukin-6 and soluble interleukin-6 receptors in the synovial fluids from rheumatoid arthritis patients are responsible for osteoclast-like cell formation. Journal of Bone and Mineral Research. 1996;11(1):88–95. doi: 10.1002/jbmr.5650110113. [DOI] [PubMed] [Google Scholar]
- 40.Wood NC, Symons JA, Dickens E, Duff GW. In situ hybridization of IL-6 in rheumatoid arthritis. Clinical and Experimental Immunology. 1992;87(2):183–189. doi: 10.1111/j.1365-2249.1992.tb02972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garnero P, Thompson E, Woodworth T, Smolen JS. Rapid and sustained improvement in bone and cartilage turnover markers with the anti-interleukin-6 receptor inhibitor tocilizumab plus methotrexate in rheumatoid arthritis patients with an inadequate response to methotrexate: results from a substudy of the multicenter double-blind, placebo-controlled trial of tocilizumab in inadequate responders to methotrexate alone. Arthritis and Rheumatism. 2010;62(1):33–43. doi: 10.1002/art.25053. [DOI] [PubMed] [Google Scholar]
- 42.Karsdal MA, Schett G, Emery P, et al. IL-6 receptor inhibition positively modulates bone balance in rheumatoid arthritis patients with an inadequate response to anti-tumor necrosis factor therapy: biochemical marker analysis of bone metabolism in the tocilizumab RADIATE study ( NCT00106522) Seminars in Arthritis and Rheumatism. 2012;42(2):131–139. doi: 10.1016/j.semarthrit.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 43.Ehrlich LA, Roodman GD. The role of immune cells and inflammatory cytokines in Paget's disease and multiple myeloma. Immunological Reviews. 2005;208:252–266. doi: 10.1111/j.0105-2896.2005.00323.x. [DOI] [PubMed] [Google Scholar]
- 44.Palmqvist P, Persson E, Conaway HH, Lerner UH. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-κB ligand, osteoprotegerin, and receptor activator of NF-κB in mouse calvariae. The Journal of Immunology. 2002;169(6):3353–3362. doi: 10.4049/jimmunol.169.6.3353. [DOI] [PubMed] [Google Scholar]
- 45.O'Brien CA, Lin SC, Bellido T, Manolagas SC. Expression levels of gp130 in bon e marrow stromal cells determine the magnitude of osteoclastogenic signals generated by IL-6-type cytokines. Journal of Cellular Biochemistry. 2000;79(4):532–541. doi: 10.1002/1097-4644(20001215)79:4<532::aid-jcb20>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 46.de Benedetti F, Rucci N, Del Fattore A, et al. Impaired skeletal development in interleukin-6-transgenic mice: a model for the impact of chronic inflammation on the growing skeletal system. Arthritis and Rheumatism. 2006;54(11):3551–3563. doi: 10.1002/art.22175. [DOI] [PubMed] [Google Scholar]
- 47.Kitamura H, Kawata H, Takahashi F, Higuchi Y, Furuichi T, Ohkawa H. Bone marrow neutrophilia and suppressed bone turnover in human interleukin-6 transgenic mice: a cellular relationship among hematopoietic cells, osteoblasts, and osteoclasts mediated by stromal cells in bone marrow. The American Journal of Pathology. 1995;147(6):1682–1692. [PMC free article] [PubMed] [Google Scholar]
- 48.Poli V, Balena R, Fattori E, et al. Interleukin-6 deficient mice are protected from bone loss caused by estrogen depletion. EMBO Journal. 1994;13(5):1189–1196. doi: 10.1002/j.1460-2075.1994.tb06368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong PKK, Quinn JMW, Sims NA, van Nieuwenhuijze A, Campbell IK, Wicks IP. Interleukin-6 modulates production of T lymphocyte-derived cytokines in antigen-induced arthritis and drives inflammation-induced osteoclastogenesis. Arthritis and Rheumatism. 2006;54(1):158–168. doi: 10.1002/art.21537. [DOI] [PubMed] [Google Scholar]
- 50.Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. New England Journal of Medicine. 2009;361(9):848–898. doi: 10.1056/NEJMra0707449. [DOI] [PubMed] [Google Scholar]
- 51.Yao Z, Painter SL, Fanslow WC, et al. Human IL-17: a novel cytokine derived from T cells. Journal of Immunology. 1995;155(12):5483–5486. [PubMed] [Google Scholar]
- 52.Lubberts E, Koenders M, van den Berg WB. The role of T cell interleukin-17 in conducting destructive arthritis: lessons from animal models. Arthritis Research and Therapy. 2005;7(1):29–37. doi: 10.1186/ar1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adamopoulos IE, Chao C, Geissler R, et al. Interleukin-17A upregulates receptor activator of NF-κB on osteoclast precursors. Arthritis Research and Therapy. 2010;12, article R29 doi: 10.1186/ar2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chao CC, Chen SJ, Adamopoulos IE, et al. Anti-IL-17A therapy protects against bone erosion in experimental models of rheumatoid arthritis. Autoimmunity. 2011;44(3):243–252. doi: 10.3109/08916934.2010.517815. [DOI] [PubMed] [Google Scholar]
- 55.Lubberts E, Koenders MI, Oppers-Walgreen B, et al. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis and Rheumatism. 2004;50(2):650–659. doi: 10.1002/art.20001. [DOI] [PubMed] [Google Scholar]
- 56.Yago T, Nanke Y, Kawamoto M, et al. IL-23 induces human osteoclastogenesis via IL-17 in vitro, and anti-IL-23 antibody attenuates collagen-induced arthritis in rats. Arthritis Research and Therapy. 2007;9(5, article no. R96) doi: 10.1186/ar2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Quinn JMW, Sims NA, Saleh H, et al. IL-23 inhibits osteoclastogenesis indirectly through lymphocytes and is required for the maintenance of bone mass in mice. Journal of Immunology. 2008;181(8):5720–5729. doi: 10.4049/jimmunol.181.8.5720. [DOI] [PubMed] [Google Scholar]
- 58.Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nature Reviews Rheumatology. 2013;9(1):24–33. doi: 10.1038/nrrheum.2012.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tolboom TC, Pieterman E, van der Laan WH, et al. Invasive properties of fibroblast-like synoviocytes: correlation with growth characteristics and expression of MMP-1, MMP-3, and MMP-10. Annals of the Rheumatic Diseases. 2002;61(11):975–980. doi: 10.1136/ard.61.11.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pap T, Meinecke I, Müller-Ladner U, Gay S. Are fibroblasts involved in joint destruction? Annals of the Rheumatic Diseases. 2005;64(4):iv52–iv54. doi: 10.1136/ard.2005.042424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takayanagi H, Oda H, Yamamoto S, et al. A new mechanism of bone destruction in rheumatoid arthritis: synovial fibroblasts induce osteoclastogenesis. Biochemical and Biophysical Research Communications. 1997;240(2):279–286. doi: 10.1006/bbrc.1997.7404. [DOI] [PubMed] [Google Scholar]
- 62.Gravallese EM, Harada Y, Wang J, Gorn AH, Thornhill TS, Goldring SR. Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. The American Journal of Pathology. 1998;152(4):943–951. [PMC free article] [PubMed] [Google Scholar]
- 63.Harris ED., Jr. Rheumatoid arthritis. Pathophysiology and implications for therapy. The New England Journal of Medicine. 1990;322(18):1277–1289. doi: 10.1056/NEJM199005033221805. [DOI] [PubMed] [Google Scholar]
- 64.Kim KW, Cho ML, Lee SH, et al. Human rheumatoid synovial fibroblasts promote osteoclastogenic activity by activating RANKL via TLR-2 and TLR-4 activation. Immunology Letters. 2007;110(1):54–64. doi: 10.1016/j.imlet.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 65.Li X, Kim K, Cho M, et al. IL-23 induces receptor activator of NF-κB ligand expression in fibroblast-like synoviocytes via STAT3 and NF-κB signal pathways. Immunology Letters. 2010;127(2):100–107. doi: 10.1016/j.imlet.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 66.Kim KW, Kim HR, Park JY, et al. Interleukin-22 promotes osteoclastogenesis in rheumatoid arthritis through induction of RANKL in human synovial fibroblasts. Arthritis and Rheumatism. 2012;64(4):1015–1023. doi: 10.1002/art.33446. [DOI] [PubMed] [Google Scholar]
- 67.Kim HR, Kim KW, Kim BM, Jung HG, Cho ML, Lee SH. Reciprocal activation of CD4+ T cells and synovial fibroblasts by stromal cell-derived factor 1 promotes RANKL expression and osteoclastogenesis in rheumatoid arthritis. Arthritis and Rheumatism. 2014;66(3):538–548. doi: 10.1002/art.38286. [DOI] [PubMed] [Google Scholar]
- 68.Liu FL, Chen CH, Chu SJ, et al. Interleukin (IL)-23 p19 expression induced by IL-1β in human fibroblast-like synoviocytes with rheumatoid arthritis via active nuclear factor-κB and AP-1 dependent pathway. Rheumatology. 2007;46(8):1266–1273. doi: 10.1093/rheumatology/kem055. [DOI] [PubMed] [Google Scholar]
- 69.Hwang SY, Kim JY, Kim KW, et al. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-κB- and PI3-kinase/Akt-dependent pathways. Arthritis Research & Therapy. 2004;6(2):R120–128. doi: 10.1186/ar1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nature Reviews Immunology. 2011;11(11):762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. The New England Journal of Medicine. 2011;365(23):2205–2219. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 72.Li J, Hsu H, Mountz JD. Managing macrophages in rheumatoid arthritis by reform or removal. Current Rheumatology Reports. 2012;14(5):445–454. doi: 10.1007/s11926-012-0272-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cirrincione C, Pimpinelli N, Orlando L, Romagnoli P. Lamina propria dendritic cells express activation markers and contact lymphocytes in chronic periodontitis. Journal of Periodontology. 2002;73(1):45–52. doi: 10.1902/jop.2002.73.1.45. [DOI] [PubMed] [Google Scholar]
- 74.Highton J, Kean A, Hessian PA, Thomson J, Rietveld J, Hart DNJ. Cells expressing dendritic cell markers are present in the rheumatoid nodule. The Journal of Rheumatology. 2000;27(2):339–346. [PubMed] [Google Scholar]
- 75.Thomas R, MacDonald KPA, Pettit AR, Cavanagh LL, Padmanabha J, Zehntner S. Dendritic cells and the pathogenesis of rheumatoid arthritis. Journal of Leukocyte Biology. 1999;66(2):286–292. doi: 10.1002/jlb.66.2.286. [DOI] [PubMed] [Google Scholar]
- 76.Page G, Lebecque S, Miossec P. Anatomic localization of immature and mature dendritic cells in an ectopic lymphoid organ: correlation with selective chemokine expression in rheumatoid synovium. Journal of Immunology. 2002;168(10):5333–5341. doi: 10.4049/jimmunol.168.10.5333. [DOI] [PubMed] [Google Scholar]
- 77.Page G, Miossec P. RANK and RANKL expression as markers of dendritic cell-T cell interactions in paired samples of rheumatoid synovium and lymph nodes. Arthritis and Rheumatism. 2005;52(8):2307–2312. doi: 10.1002/art.21211. [DOI] [PubMed] [Google Scholar]
- 78.Cutler CW, Jotwani R. Dendritic cells at the oral mucosal interface. Journal of Dental Research. 2006;85(8):678–689. doi: 10.1177/154405910608500801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tucci M, Stucci S, Strippoli S, Dammacco F, Silvestris F. Dendritic cells and malignant plasma cells: an alliance in multiple myeloma tumor progression? Oncologist. 2011;16(7):1040–1048. doi: 10.1634/theoncologist.2010-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dhodapkar KM, Barbuto S, Matthews P, et al. Dendritic cells mediate the induction of polyfunctional human IL17-producing cells (Th17-1 cells) enriched in the bone marrow of patients with myeloma. Blood. 2008;112(7):2878–2885. doi: 10.1182/blood-2008-03-143222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rivollier A, Mazzorana M, Tebib J, et al. Immature dendritic cell transdifferentiation into osteoclasts: a novel pathway sustained by the rheumatoid arthritis microenvironment. Blood. 2004;104(13):4029–4037. doi: 10.1182/blood-2004-01-0041. [DOI] [PubMed] [Google Scholar]
- 82.Alnaeeli M, Penninger JM, Teng YA. Immune interactions with CD4+ T cells promote the development of functional osteoclasts from murine CD11c+ dendritic cells. Journal of Immunology. 2006;177(5):3314–3326. doi: 10.4049/jimmunol.177.5.3314. [DOI] [PubMed] [Google Scholar]
- 83.Kukreja A, Radfar S, Sun B, Insogna K, Dhodapkar MV. Dominant role of CD47-thrombospondin-1 interactions in myeloma-induced fusion of human dendritic cells: implications for bone disease. Blood. 2009;114(16):3413–3421. doi: 10.1182/blood-2009-03-211920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.John V, Hock JM, Short LL, Glasebrook AL, Galvin RJS. A role for CD8+ T lymphocytes in osteoclast differentiation in vitro. Endocrinology. 1996;137(6):2457–2463. doi: 10.1210/endo.137.6.8641199. [DOI] [PubMed] [Google Scholar]
- 85.Li Y, Toraldo G, Li A, et al. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood. 2007;109(9):3839–3848. doi: 10.1182/blood-2006-07-037994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mosmann TR, Cherwinski H, Bond MW. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. Journal of Immunology. 1986;136(7):2348–2357. [PubMed] [Google Scholar]
- 87.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383(6603):787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 88.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annual Review of Immunology. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 89.Sato K, Suematsu A, Okamoto K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. Journal of Experimental Medicine. 2006;203(12):2673–2682. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kawai T, Matsuyama T, Hosokawa Y, et al. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. The American Journal of Pathology. 2006;169(3):987–998. doi: 10.2353/ajpath.2006.060180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kotake S, Nanke Y, Mogi M, et al. IFN-γ-producing human T cells directly induce osteoclastogenesis from human monocytes via the expression of RANKL. European Journal of Immunology. 2005;35(11):3353–3363. doi: 10.1002/eji.200526141. [DOI] [PubMed] [Google Scholar]
- 92.Volpe E, Servant N, Zollinger R, et al. A critical function for transforming growth factor-β, interleukin 23 and proinflammatory cytokines in driving and modulating human TH-17 responses. Nature Immunology. 2008;9(6):650–657. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 93.van Hamburg JP, Asmawidjaja PS, Davelaar N, et al. Th17 cells, but not Th1 cells, from patients with early rheumatoid arthritis are potent inducers of matrix metalloproteinases and proinflammatory cytokines upon synovial fibroblast interaction, including autocrine interleukin-17A production. Arthritis and Rheumatism. 2011;63(1):73–83. doi: 10.1002/art.30093. [DOI] [PubMed] [Google Scholar]
- 94.Noonan K, Marchionni L, Anderson J, Pardoll D, Roodman GD, Borrello I. A novel role of IL-17-producing lymphocytes in mediating lytic bone disease in multiple myeloma. Blood. 2010;116(18):3554–3563. doi: 10.1182/blood-2010-05-283895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. The American Journal of Pathology. 2012;181(1):8–18. doi: 10.1016/j.ajpath.2012.03.044. [DOI] [PubMed] [Google Scholar]
- 96.Matsumoto T, Abe M. Bone destruction in multiple myeloma. Annals of the New York Academy of Sciences. 2006;1068(1):319–326. doi: 10.1196/annals.1346.035. [DOI] [PubMed] [Google Scholar]
- 97.Edwards JCW, Cambridge G. Sustained improvement in rheumatoid arthritis following a protocol designed to deplete B lymphocytes. Rheumatology. 2001;40(2):205–211. doi: 10.1093/rheumatology/40.2.205. [DOI] [PubMed] [Google Scholar]
- 98.Edwards JCW, Szczepański L, Szechiński J, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. The New England Journal of Medicine. 2004;350(25):2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 99.Nakken B, Munthe LA, Konttinen YT, et al. B-cells and their targeting in rheumatoid arthritis—current concepts and future perspectives. Autoimmunity Reviews. 2011;11(1):28–34. doi: 10.1016/j.autrev.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 100.Cascão R, Rosário HS, Souto-Carneiro MM, Fonseca JE. Neutrophils in rheumatoid arthritis: more than simple final effectors. Autoimmunity Reviews. 2010;9(8):531–535. doi: 10.1016/j.autrev.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 101.Kantarci A, Oyaizu K, van Dyke TE. Neutrophil-mediated tissue injury in periodontal disease pathogenesis: findings from localized aggressive periodontitis. Journal of Periodontology. 2003;74(1):66–75. doi: 10.1902/jop.2003.74.1.66. [DOI] [PubMed] [Google Scholar]
- 102.Hasturk H, Kantarci A, Ohira T, et al. RvE1 protects from local inflammation and osteoclast-mediated bone destruction in periodontitis. The FASEB Journal. 2006;20(2):401–403. doi: 10.1096/fj.05-4724fje. [DOI] [PubMed] [Google Scholar]
- 103.Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. Journal of Immunology. 2001;167(3):1601–1608. doi: 10.4049/jimmunol.167.3.1601. [DOI] [PubMed] [Google Scholar]
- 104.Poubelle PE, Chakravarti A, Fernandes MJ, Doiron K, Marceau A. Differential expression of RANK, RANK-L, and osteoprotegerin by synovial fluid neutrophils from patients with rheumatoid arthritis and by healthy human blood neutrophils. Arthritis Research and Therapy. 2007;9(2, article R25) doi: 10.1186/ar2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chakravarti A, Raquil M, Tessier P, Poubelle PE. Surface RANKL of Toll-like receptor 4-stimulated human neutrophils activates osteoclastic bone resorption. Blood. 2009;114(8):1633–1644. doi: 10.1182/blood-2008-09-178301. [DOI] [PubMed] [Google Scholar]
- 106.Walsh NC, Alexander KA, Manning CA, et al. Activated human T cells express alternative mRNA transcripts encoding a secreted form of RANKL. Genes and Immunity. 2013;14(5):336–345. doi: 10.1038/gene.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kanamaru F, Iwai H, Ikeda T, Nakajima A, Ishikawa I, Azuma M. Expression of membrane-bound and soluble receptor activator of NF-κB ligand (RANKL) in human T cells. Immunology Letters. 2004;94(3):239–246. doi: 10.1016/j.imlet.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 108.Brunetti G, Faienza MF, Piacente L, et al. High dickkopf-1 levels in sera and leukocytes from children with 21-hydroxylase deficiency on chronic glucocorticoid treatment. The American Journal of Physiology: Endocrinology and Metabolism. 2013;304(5):E546–E554. doi: 10.1152/ajpendo.00535.2012. [DOI] [PubMed] [Google Scholar]
- 109.Allaeys I, Rusu D, Picard S, Pouliot M, Borgeat P, Poubelle PE. Osteoblast retraction induced by adherent neutrophils promotes osteoclast bone resorption: implication for altered bone remodeling in chronic gout. Laboratory Investigation. 2011;91(6):905–920. doi: 10.1038/labinvest.2011.46. [DOI] [PubMed] [Google Scholar]
- 110.Boyce BF, Schwarz EM, Xing L. Osteoclast precursors: Cytokine-stimulated immunomodulators of inflammatory bone disease. Current Opinion in Rheumatology. 2006;18(4):427–432. doi: 10.1097/01.bor.0000231913.32364.32. [DOI] [PubMed] [Google Scholar]
- 111.Fuller K, Murphy C, Kirstein B, Fox SW, Chambers TJ. TNFalpha potently activates osteoclasts, through a direct action independent of and strongly synergistic with RANKL. Endocrinology. 2002;143(3):1108–1118. doi: 10.1210/endo.143.3.8701. [DOI] [PubMed] [Google Scholar]
- 112.Glantschnig H, Fisher JE, Wesolowski G, Rodan GA, Reszka AA. M-CSF, TNFα and RANK ligand promote osteoclast survival by signaling through mTOR/S6 kinase. Cell Death and Differentiation. 2003;10(10):1165–1177. doi: 10.1038/sj.cdd.4401285. [DOI] [PubMed] [Google Scholar]
- 113.Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A. Tumor necrosis factor-α induces differentiation of and bone resorption by osteoclasts. Journal of Biological Chemistry. 2000;275(7):4858–4864. doi: 10.1074/jbc.275.7.4858. [DOI] [PubMed] [Google Scholar]
- 114.Yarilina A, Xu K, Chen J, Ivashkiv LB. TNF activates calcium-nuclear factor of activated T cells (NFAT)c1 signaling pathways in human macrophages. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(4):1573–1578. doi: 10.1073/pnas.1010030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lader CS, Flanagan AM. Prostaglandin E2, interleukin and tumor necrosis factor-α increase human osteoclast formation and bone resorption in vitro. Endocrinology. 1998;139(7):3157–3164. doi: 10.1210/endo.139.7.6085. [DOI] [PubMed] [Google Scholar]
- 116.O' Gradaigh D, Ireland D, Bord S, Compston JE. Joint erosion in rheumatoid arthritis: Interactions between tumour necrosis factor α, interleukin 1, and receptor activator of nuclear factor κB ligand (RANKL) regulate osteoclasts. Annals of the Rheumatic Diseases. 2004;63(4):354–359. doi: 10.1136/ard.2003.008458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kitaura H, Kimura K, Ishida M, Kohara H, Yoshimatsu M, Takano-Yamamoto T. Immunological reaction in TNF-α-mediated osteoclast formation and bone resorption in vitro and in vivo . Clinical and Developmental Immunology. 2013;2013:8 pages. doi: 10.1155/2013/181849.181849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Li P, Schwarz EM, O'Keefe RJ, Ma L, Boyce BF, Xing L. RANK signaling is not required for TNFα-mediated increase in CD11bhi osteoclast precursors but is essential for mature osteoclast formation in TNFα-mediated inflammatory arthritis. Journal of Bone and Mineral Research. 2004;19(2):207–213. doi: 10.1359/JBMR.0301233. [DOI] [PubMed] [Google Scholar]
- 119.Neidel J, Schulze M, Lindschau J. Association between degree of bone-erosion and synovial fluid-levels of tumor necrosis factor α in the knee-joints of patients with rheumatoid arthritis. Inflammation Research. 1995;44(5):217–221. doi: 10.1007/BF01782262. [DOI] [PubMed] [Google Scholar]
- 120.Ma T, Miyanishi K, Suen A, et al. Human interleukin-1-induced murine osteoclastogenesis is dependent on RANKL, but independent of TNF-α. Cytokine. 2004;26(3):138–144. doi: 10.1016/j.cyto.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 121.Lee S, Gardner AE, Kalinowski JF, Jastrzebski SL, Lorenzo JA. RANKL-stimulated osteoclast-like cell formation in vitro is partially dependent on endogenous interleukin-1 production. Bone. 2006;38(5):678–685. doi: 10.1016/j.bone.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 122.Tanabe N, Maeno M, Suzuki N, et al. IL-1α stimulates the formation of osteoclast-like cells by increasing M-CSF and PGE2 production and decreasing OPG production by osteoblasts. Life Sciences. 2005;77(6):615–626. doi: 10.1016/j.lfs.2004.10.079. [DOI] [PubMed] [Google Scholar]
- 123.Heymann D, Rousselle A. gp130 cytokine family and bone cells. Cytokine. 2000;12(10):1455–1468. doi: 10.1006/cyto.2000.0747. [DOI] [PubMed] [Google Scholar]
- 124.Liu XH, Kirschenbaum A, Yao S, Levine AC. The role of the interleukin-6/gp130 signaling pathway in bone metabolism. Vitamins and Hormones. 2006;74:341–355. doi: 10.1016/S0083-6729(06)74014-6. [DOI] [PubMed] [Google Scholar]
- 125.Wong PKK, Campbell IK, Egan PJ, Ernst M, Wicks IP. The role of the interleukin-6 family of cytokines in inflammatory arthritis and bone turnover. Arthritis and Rheumatism. 2003;48(5):1177–1189. doi: 10.1002/art.10943. [DOI] [PubMed] [Google Scholar]
- 126.Steeve KT, Marc P, Sandrine T, Dominique H, Yannick F. IL-6, RANKL, TNF-alpha/IL-1: interrelations in bone resorption pathophysiology. Cytokine and Growth Factor Reviews. 2004;15(1):49–60. doi: 10.1016/j.cytogfr.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 127.Kim S, Yamazaki M, Shevde NK, Pike JW. Transcriptional control of receptor activator of nuclear factor-κB ligand by the protein kinase A activator forskolin and the transmembrane glycoprotein 130-activating cytokine, oncostatin M, is exerted through multiple distal enhancers. Molecular Endocrinology. 2007;21(1):197–214. doi: 10.1210/me.2006-0315. [DOI] [PubMed] [Google Scholar]
- 128.O'Brien CA, Gubrij I, Lin S, Saylors RL, Manolagas SC. STAT3 activation in stromal/osteoblastic cells is required for induction of the receptor activator of NF-κB ligand and stimulation of osteoclastogenesis by gp130-utilizing cytokines or interleukin-1 but not 1,25-dihydroxyvitamin D3 or parathyroid hormone. The Journal of Biological Chemistry. 1999;274(27):19301–19308. doi: 10.1074/jbc.274.27.19301. [DOI] [PubMed] [Google Scholar]
- 129.Yokota K, Sato K, Miyazaki T, Kitaura H, Kayama H, Miyoshi F. Combination of tumor necrosis factor alpha and interleukin-6 induces mouse osteoclast-like cells with bone resorption activity both in vitro and in vivo. Arthritis & Rheumatology. 2014;66(1):121–129. doi: 10.1002/art.38218. [DOI] [PubMed] [Google Scholar]
- 130.Wallace A, Cooney TE, Englund R, Lubahn JD. Effects of interleukin-6 ablation on fracture healing in mice. Journal of Orthopaedic Research. 2011;29(9):1437–1442. doi: 10.1002/jor.21367. [DOI] [PubMed] [Google Scholar]
- 131.Yang X, Ricciardi BF, Hernandez-Soria A, Shi Y, Pleshko Camacho N, Bostrom MPG. Callus mineralization and maturation are delayed during fracture healing in interleukin-6 knockout mice. Bone. 2007;41(6):928–936. doi: 10.1016/j.bone.2007.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rozen N, Lewinson D, Bick T, Jacob ZC, Stein H, Soudry M. Fracture repair: modulation of fracture-callus and mechanical properties by sequential application of IL-6 following PTH 1-34 or PTH 28-48. Bone. 2007;41(3):437–445. doi: 10.1016/j.bone.2007.04.193. [DOI] [PubMed] [Google Scholar]
- 133.Duplomb L, Baud'huin M, Charrier C, et al. Interleukin-6 inhibits receptor activator of nuclear factor κB ligand-induced osteoclastogenesis by diverting cells into the macrophage lineage: key role of serine727 phosphorylation of signal transducer and activator of transcription 3. Endocrinology. 2008;149(7):3688–3697. doi: 10.1210/en.2007-1719. [DOI] [PubMed] [Google Scholar]
- 134.Honda K. Interleukin-6 and soluble interleukin-6 receptor suppress osteoclastic differentiation by inducing PGE(2) production in chondrocytes. Journal of Oral Science. 2011;53(1):87–96. doi: 10.2334/josnusd.53.87. [DOI] [PubMed] [Google Scholar]
- 135.Darowish M, Rahman R, Li P, et al. Reduction of particle-induced osteolysis by interleukin-6 involves anti-inflammatory effect and inhibition of early osteoclast precursor differentiation. Bone. 2009;45(4):661–668. doi: 10.1016/j.bone.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kotake S, Udagawa N, Takahashi N, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. Journal of Clinical Investigation. 1999;103(9):1345–1352. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Van Bezooijen RL, Papapoulos SE, Löwik CWGM. Effect of interleukin-17 on nitric oxide production and osteoclastic bone resorption: is there dependency on nuclear factor-κB and receptor activator of nuclear factor κB (RANK)/RANK ligand signaling? Bone. 2001;28(4):378–386. doi: 10.1016/s8756-3282(00)00457-9. [DOI] [PubMed] [Google Scholar]
- 138.Kelchtermans H, Schurgers E, Geboes L, et al. Effector mechanisms of interleukin-17 in collagen-induced arthritis in the absence of interferon-γ and counteraction by interferon-γ . Arthritis Research and Therapy. 2009;11(4, article R122) doi: 10.1186/ar2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Van Bezooijen RL, Farih-Sips HCM, Papapoulos SE, Löwik CWGM. Interleukin-17: a new bone acting cytokine in vitro. Journal of Bone and Mineral Research. 1999;14(9):1513–1521. doi: 10.1359/jbmr.1999.14.9.1513. [DOI] [PubMed] [Google Scholar]
- 140.Yago T, Nanke Y, Ichikawa N, et al. IL-17 induces osteoclastogenesis from human monocytes alone in the absence of osteoblasts, which is potently inhibited by anti-TNF-α antibody: a novel mechanism of osteoclastogenesis by IL-17. Journal of Cellular Biochemistry. 2009;108(4):947–955. doi: 10.1002/jcb.22326. [DOI] [PubMed] [Google Scholar]
- 141.Oostlander AE, Everts V, Schoenmaker T, et al. T cell-mediated increased osteoclast formation from peripheral blood as a mechanism for crohn's disease-associated bone loss. Journal of Cellular Biochemistry. 2012;113(1):260–268. doi: 10.1002/jcb.23352. [DOI] [PubMed] [Google Scholar]
- 142.Lubberts E, van den Bersselaar L, Oppers-Walgreen B, et al. IL-17 promotes bone erosion in murine collagen-induced arthritis through loss of the receptor activator of NF-κB ligand/osteoprotegerin balance1. Journal of Immunology. 2003;170(5):2655–2662. doi: 10.4049/jimmunol.170.5.2655. [DOI] [PubMed] [Google Scholar]
- 143.Deselm CJ, Takahata Y, Warren J, et al. IL-17 mediates estrogen-deficient osteoporosis in an Act1-dependent manner. Journal of Cellular Biochemistry. 2012;113(9):2895–2902. doi: 10.1002/jcb.24165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tyagi AM, Srivastava K, Mansoori MN, Trivedi R, Chattopadhyay N, Singh D. Estrogen deficiency induces the differentiation of IL-17 secreting Th17 cells: a new candidate in the pathogenesis of osteoporosis. PloS one. 2012;7(9) doi: 10.1371/journal.pone.0044552.e44552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Balani D, Aeberli D, Hofstetter W, Seitz M. Interleukin-17A stimulates granulocyte-macrophage colony-stimulating factor release by murine osteoblasts in the presence of 1,25-dihydroxyvitamin D 3 and inhibits murine osteoclast development in vitro. Arthritis and Rheumatism. 2013;65(2):436–446. doi: 10.1002/art.37762. [DOI] [PubMed] [Google Scholar]
- 146.Kitami S, Tanaka H, Kawato T, et al. IL-17A suppresses the expression of bone resorption-related proteinases and osteoclast differentiation via IL-17RA or IL-17RC receptors in RAW264.7 cells. Biochimie. 2010;92(4):398–404. doi: 10.1016/j.biochi.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 147.Adamopoulos IE, Tessmer M, Chao C, et al. IL-23 is critical for induction of arthritis, osteoclast formation, and maintenance of bone mass. Journal of Immunology. 2011;187(2):951–959. doi: 10.4049/jimmunol.1003986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ji HJ, Cho M, Moon Y, et al. IL-23 induces receptor activator of NF-κB ligand expression on CD4+ T cells and promotes osteoclastogenesis in an autoimmune arthritis model. Journal of Immunology. 2008;181(2):1507–1518. doi: 10.4049/jimmunol.181.2.1507. [DOI] [PubMed] [Google Scholar]
- 149.Hu Y, Xiong H, Peng B. Expression of a cytokine, interleukin-23, in experimental periapical lesions. International Endodontic Journal. 2013;46(10):896–903. doi: 10.1111/iej.12077. [DOI] [PubMed] [Google Scholar]
- 150.Chen L, Wei X, Evans B, Jiang W, Aeschlimann D. IL-23 promotes osteoclast formation by up-regulation of receptor activator of NF-κB (RANK) expression in myeloid precursor cells. European Journal of Immunology. 2008;38(10):2845–2854. doi: 10.1002/eji.200838192. [DOI] [PubMed] [Google Scholar]
- 151.Kamiya S, Nakamura C, Fukawa T, et al. Effects of IL-23 and IL-27 on osteoblasts and osteoclasts: inhibitory effects on osteoclast differentiation. Journal of Bone and Mineral Metabolism. 2007;25(5):277–285. doi: 10.1007/s00774-007-0766-8. [DOI] [PubMed] [Google Scholar]
- 152.Binder NB, Puchner A, Niederreiter B, et al. Tumor necrosis factor-inhibiting therapy preferentially targets bone destruction but not synovial inflammation in a tumor necrosis factor-driven model of rheumatoid arthritis. Arthritis and Rheumatism. 2013;65(3):608–617. doi: 10.1002/art.37797. [DOI] [PubMed] [Google Scholar]
- 153.Redlich K, Görtz B, Hayer S, et al. Repair of local bone erosions and reversal of systemic bone loss upon therapy with anti-tumor necrosis factor in combination with osteoprotegerin or parathyroid hormone in tumor necrosis factor-mediated arthritis. The American Journal of Pathology. 2004;164(2):543–555. doi: 10.1016/S0002-9440(10)63144-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Saidenberg-Kermanac'h N, Corrado A, Lemeiter D, Devernejoul MC, Boissier MC, Cohen-Solal ME. TNF-α antibodies and osteoprotegerin decrease systemic bone loss associated with inflammation through distinct mechanisms in collagen-induced arthritis. Bone. 2004;35(5):1200–1207. doi: 10.1016/j.bone.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 155.Kitazawa R, Kimble RB, Vannice JL, Kung VT, Pacifici R. Interleukin-1 receptor antagonist and tumor necrosis factor binding protein decrease osteoclast formation and bone resorption in ovariectomized mice. The Journal of Clinical Investigation. 1994;94(6):2397–2406. doi: 10.1172/JCI117606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kimble RB, Bain S, Pacifici R. The functional block of TNF but not of IL-6 prevents bone loss in ovariectomized mice. Journal of Bone and Mineral Research. 1997;12(6):935–941. doi: 10.1359/jbmr.1997.12.6.935. [DOI] [PubMed] [Google Scholar]
- 157.Feige U, Hu YL, Gasser J, Campagnuolo G, Munyakazi L, Bolon B. Anti-interleukin-1 and anti-tumor necrosis factor-α synergistically inhibit adjuvant arthritis in lewis rats. Cellular and Molecular Life Sciences. 2000;57(10):1457–1470. doi: 10.1007/PL00000629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shealy DJ, Wooley PH, Emmell E, et al. Anti-TNF-alpha antibody allows healing of joint damage in polyarthritic transgenic mice. Arthritis Research. 2002;4(5):p. R7. doi: 10.1186/ar430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Smolen JS, Aletaha D, Koeller M, Weisman MH, Emery P. New therapies for treatment of rheumatoid arthritis. The Lancet. 2007;370(9602):1861–1874. doi: 10.1016/S0140-6736(07)60784-3. [DOI] [PubMed] [Google Scholar]
- 160.Mertens M, Singh JA. Anakinra for rheumatoid arthritis. Cochrane Database of Systematic Reviews. 2009;(1) doi: 10.1002/14651858.CD005121.pub3.CD005121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bresnihan B, Alvaro-Gracia JM, Cobby M. Treatment of rheumatoid arthritis with recombinant human interleukin-1 receptor antagonist. Arthritis and Rheumatism. 1998;41(12):2196–2204. doi: 10.1002/1529-0131(199812)41:12<2196::AID-ART15>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 162.Tanaka K, Hashizume M, Mihara M, Yoshida H, Suzuki M, Matsumoto Y. Anti-interleukin-6 receptor antibody prevents systemic bone mass loss via reducing the number of osteoclast precursors in bone marrow in a collagen-induced arthritis model. Clinical and Experimental Immunology. 2014;175(2):172–180. doi: 10.1111/cei.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Axmann R, Böhm C, Krönke G, Zwerina J, Smolen J, Schett G. Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis and Rheumatism. 2009;60(9):2747–2756. doi: 10.1002/art.24781. [DOI] [PubMed] [Google Scholar]
- 164.Finzel S, Rech J, Schmidt S, Engelke K, Englbrecht M, Schett G. Interleukin-6 receptor blockade induces limited repair of bone erosions in rheumatoid arthritis: a micro CT study. Annals of the Rheumatic Diseases. 2013;72(3):396–400. doi: 10.1136/annrheumdis-2011-201075. [DOI] [PubMed] [Google Scholar]
- 165.Terpos E, Fragiadaki K, Konsta M, Bratengeier C, Papatheodorou A, Sfikakis PP. Early effects of IL-6 receptor inhibition on bone homeostasis: a pilot study in women with rheumatoid arthritis. Clinical and Experimental Rheumatology. 2011;29(6):921–925. [PubMed] [Google Scholar]
- 166.Zwerina K, Koenders M, Hueber A, et al. Anti IL-17A therapy inhibits bone loss in TNF-α-mediated murine arthritis by modulation of the T-cell balance. European Journal of Immunology. 2012;42(2):413–423. doi: 10.1002/eji.201141871. [DOI] [PubMed] [Google Scholar]
- 167.Koenders MI, Lubberts E, Oppers-Walgreen B, et al. Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. The American Journal of Pathology. 2005;167(1):141–149. doi: 10.1016/S0002-9440(10)62961-6. [DOI] [PMC free article] [PubMed] [Google Scholar]