

Abstract
The purpose of this study was to identify functional genetic variants in the promoter of tumor necrosis factor superfamily member 15 (TNFSF15) and evaluate their effects on the risk of developing gastric adenocarcinoma. Forty DNA samples from healthy volunteers were sequenced to identify single nucleotide polymorphisms (SNPs) in the TNFSF15 promoter. Two TNFSF15 SNPs (−358T>C and −638A>G) were identified by direct sequencing. Next, genotypes and haplotypes of 470 gastric adenocarcinoma patients and 470 cancer-free controls were analyzed. Odds ratios (ORs) and 95% confidence intervals (CIs) were estimated by logistic regression. Serologic tests for Helicobacter pylori infection were measured by enzyme-linked immuno-sorbent assay (ELISA). Subjects carrying the TNFSF15 −358CC genotype were at an elevated risk for developing gastric adenocarcinoma, compared with those with the −358TT genotype (OR 1.42, 95% CI, 1.10 to 2.03). H. pylori infection was a risk factor for developing gastric adenocarcinoma (OR 2.31, 95% CI, 1.76 to 3.04). In the H. pylori infected group, subjects with TNFSF15 −358CC genotype were at higher risks for gastric adenocarcinoma compared with those carrying −358TT genotype (OR: 2.01, 95%CI: 1.65 to 4.25), indicating that H. pylori infection further influenced gastric adenocarcinoma susceptibility. The −358 T>C polymorphism eliminates a nuclear factor Y (NF-Y) binding site and the −358C containing haplotypes showed significantly decreased luciferase expression compared with −358T containing haplotypes. Collectively these findings indicate that functional genetic variants in TNFSF15 may play a role in increasing susceptibility to gastric adenocarcinoma.
Introduction
Gastric cancer is one of the most common cancers worldwide. According to the GLOBOCAN project, 952 000 new gastric cancer cases were estimated to have occurred in 2012, which attributed to 6.8% of newly diagnosed cancers [1]. Helicobacter pylori (H. pylori), a gram-negative bacterium, is one of the most important environmental risk factors for gastric cancer [2]–[4]. However, the mechanism underlying the development and progression of gastric cancer influenced by H. pylori infection is still unclear.
It has been suggested that inflammation is an important mediator of gastric cancer induced by H. Pylori and tumor necrosis factors (TNFs) is a key regulator of inflammation and has been indicated as a contributing factor for the development and progression of tumors [5]–[8]. Tumor necrosis factor superfamily member 15 (TNFSF15), also known as vascular endothelial growth inhibitor (VEGI) or TNF ligand related molecule 1 (TL1), is a unique cytokine that functions as a modulator of vascular homeostasis and inflammation [9]–[12]. The TNFSF15 gene is located on chromosome 9q32, and the full length of this gene is approximately 17 kb. TNFSF15 shares 20–30% identity (or similarity) in amino acid sequence with other TNF family members and has similar functions [13]–[15]. TNFSF15 is involved in numerous cellular processes including the suppression of neovascularization which is essential for tumor progression and spread [12], [16], [17]. Over-expression of TNFSF15 gene has been shown to inhibit the development of multiple tumor types [18], [19]. The TNFSF15 protein also inhibits the tumor growth in murine tumor models [13], [20]. Recently, TNFSF15 was also recognized as a valuable potential therapeutic target for cancer therapy [21], [22]. Collectively these findings suggest an important role of TNFSF15 in cancer suppression.
TNFSF15 gene is polymorphic, and single nucleotide polymorphisms (SNPs) in this gene were identified by genome-wide association study (GWAS) in Japanese cohort [23], but the frequency of these SNPs in other populations still have to be examined. Among these SNPs, several genetic variants in the promoter region of TNF15 have been documented to associate with increased risk to Crohn’s Disease in Japanese and European cohort, while the biological function was not reported [23], [24]. Moreover, little or nothing is known about the effects of these TNFSF15 polymorphisms on human cancer susceptibility.
In view of the importance of TNFSF15 in tumor progression and metastasis, we hypothesized that the TNFSF15 SNPs in promoter region may influence protein expression and function, which confers susceptibility to gastric cancer. Therefore, in this study we sought to identify the functional polymorphisms in the promoter of TNFSF15 gene and conduct a case-control study to investigate the frequency of these SNPs and the possible association with the risk for developing gastric cancer in the Chinese population.
Methods
TNFSF15 SNPs identification
Forty DNA samples derived from peripheral blood of unrelated healthy Han Chinese individuals were used to search for SNPs within the promoter of TNFSF15 (1274 bp). The PCR primers were as follows: Primer F (5′-TCC AAC ACC ACC TCT TTC TCC AG −3′) and Primer R (5′-TAT GTG GTG AGT CCT GCA AGG −3′). PCR products were bidirectionally sequenced to identify the genetic variants in the TNFSF15 promoter (373 Automated DNA Sequencer, Applied Biosystems, Foster City, USA). Finally, we used the Mutation Explorer program (Todaysoft Inc, Beijing, China) to identify SNP candidates that were further confirmed by re-amplifying and re-sequencing SNP sites from the opposite DNA strand.
By re-sequencing the TNFSF15 promoter of 40 healthy subjects, we identified two genetic variants (−358 T>C, rs6478109 and −638 A>G, rs7848647), which were located at −358 bp and −638 bp upstream of the translation start site, ATG.
Study participants
This hospital-based, case-control study consisted of 470 patients with histopathologically verified primary gastric adenocarcinoma and 470 healthy individuals. All subjects were unrelated and ethnically classified as Han Chinese. The cases were consecutively recruited (90% recruitment rate) from January 1997 to January 2004, at the Cancer Hospital, Chinese Academy of Medical Sciences (Beijing). The exclusion criteria included previous cancer and previous chemotherapy or radiotherapy. The stage was evaluated according to the UICC Tumor-Node-Metastasis (TNM) classification for gastric carcinoma at diagnosis on the basis of postoperative pathological examination of specimens. The control subjects were randomly selected from a database consisting of 2500 individuals based on a physical examination. The selection criteria included no history of cancer and matched the frequencies of study cases for age and sex. The detailed recruitment of patients and controls was described previously [25]. At recruitment, written informed consent was obtained from each subject and this study was approved by the Institutional Review Board of the Cancer Institute, Chinese Academy of Medical Sciences.
TNFSF15 Genotyping Assays
Genotypes were determined by PCR-based restriction fragment length polymorphism (PCR-RFLP), using −358F(5′-AAA TGT GAT TTC CGT TTC CCC A -3′)/−358R (5′-TGG GTG GGG CAA AAT ATA CC-3′) and −638F (5′-AGT CAC CTC GAT CTG TGG CCT C-3′)/−638R (5′-AAT CAC GGC TTG GAG TTG TAA CCT C-3′) as PCR primer sets for −358T>C (rs6478109) or −638A>G (rs7848647) sites, respectively. The PCR profile consisted of an initial denaturation for 2 min at 95°C, followed by 35 cycles of 25 s at 94°C, 25 s at 59°C, 25 s at 72°C, and a final elongation step of 5 min at 72°C. The PCR products were then digested with BccI (for −358 T>C site) or RsaI (for −638 T>C site) (New England BioLabs, Inc., Beverly, USA) and separated on a 3% agarose gel. TNFSF15 genotypes detected by PCR-RFLP analysis were further confirmed by DNA sequencing. Genotyping was performed without knowledge of subjects’ case/control status. To ensure the quality control, 10% random samples of cases and controls were tested twice by different persons and results were in 100% concordance.
Serologic Tests for H. Pylori Infection
Serologic assay was performed for the H. pylori infection status as previously described [26]. In brief, H. pylori stains cultured from gastric biopsies of five patients in a former study were used to provide a local antigen preparation for serology. Serum levels of anti- H. pylori IgG and IgA were measured separately in duplicate with ELISA procedures. Quality-control samples were assayed at Vanderbit University, Nashville, Tennessee. An individual was determined to be positive for H. pylori infection if the mean optical density for either anti- H. pylori IgG or anti- H. pylori IgA was >0.1, a cut-off value from the examination of a group of H. pylori -negative persons and reference sera. Serum levels of sCD14 were measured by commercially available ELISA kits (JingMei Biotech, Shenzhen, China). The concentration of each sample (unknown) was determined by extrapolation from a standard curve estimated from a panel of standards of known concentrations.
Plasmid Construction
A pGL3-basic reporter plasmid encompassing −1079/−1 bp of TNFSF15 was constructed, using 5′-ATG CGG TAC CAG AAG CCA GCA GCC AGC CT-3′ and 5′-ATG CGC TAG CGC TCC TGC TGC TCC TGG AGG -3′ as PCR primers. The resulting construct was named as pA−638-C−358 according to sequence analysis. Subsequently, site-specific mutagenesis was performed to generate other constructs, pA−638-T−358, pG−638-C−358, and pG−638-T−358, by using pA−638-C−358 as templates. All constructs used in this study were restriction mapped and sequenced to confirm their authenticity.
Luciferase Reporter Gene Assays
Human gastric carcinoma cell lines (MGC-803, AGS, and BGC-823), originally purchased from Cell Resource Center, Chinese Academy of Medical Sciences (CAMS, Beijing, China), were cultured in RPMI 1640 (Gibco, Grand Island, USA) (for MGC-803 and AGS cells) or F12 medium (Gibco, Grand Island, USA) (for BGC-823 cells) supplemented with 10% fetal bovine serum, in a humidified, 5% CO2 incubator at 37°C. Next, 1×104 cells were plated in 48-multiwell plates (Corning, NY, USA) and grown to 80–90% confluence for transient transfection using Lipofectamine 2000 Reagent (Life Technologies, Inc., Rockville, USA) according to the manufacturer’s protocol. Cells were then co-transfected with 500 ng of pA−638-C−358, pA−638-T−358, pG−638-C−358, and pG−638-T−358, or PGL3-basic plasmids and 1.0 ng of Renilla luciferase reporter plasmid pRL-SV40 (Luciferase Assay System, Promega, Madison, USA) for standardization of the transfection efficiencies. Luciferase activity was measured using the Dual-Luciferase Reporter Assay system (Promega, Madison, USA) on a TD-20/20n Luminometer (Turner Designs, Promega, Madison, USA) according to the manufacturer’s protocol. Results were normalized for Renilla activity and were expressed as relative luciferase activity (RLA). Three independent transfection experiments were performed, and each was done in triplicate.
Electrophoretic Mobility Shift Assay (EMSA)
Synthetic biotin labeled double-stranded oligonucleotides were −358T probe (5′-biotin-ATT TCC GTT TCC CAA TCT GCA AAC CAC ACA-3′/5′-biotin-TGT GTG GTT TGC AGA TTG GGA AAC GGA AAT-3′) and −358C probe (5′-biotin-ATT TCC GTT TCC CAA CCT GCA AAC CAC ACA-3′/5′-biotin-TGT GTG GTT TGC AGG TTG GGA AAC GGA AAT-3′). The gel shift assay was accomplished using the LightShift Chemiluminescent EMSA Kit (Pierce, Rockford, IL, USA) according to the manufacturer’s instructions. For each gel shift reaction, an aliquot of labeled oligonucleotide (10 fmoles) was incubated with 3 µg of nuclear extract from HeLa cells (Promega). For competition experiments, a 100-fold molar excess of unlabeled −358T or −358C oligonucleotide, a NF-Y recognition element (NF-Y cons, 5′-AGT TCA TCA GCC AAT CAG AGC ACA GG-3′/5′-CCT GTG CTC TGA TTG GCT GAT GAA CT-3′), or a mutant NF-Y recognition element (NF-Y mut, 5′-AGC TGT AGA GTG TGA GTG ATG AAG ACA T-3′/5′-ATG TCT TCA TCA CTC ACA CTC TAC AGC T-3′) was pre-incubated for 2 minutes at room temperature with the nuclear extracts before the addition of the labeled probe. Samples were then run on a non-denaturing 6% polyacrylamide gel and the electrophoresed binding reactions were transferred to positively charged nylon membranes (Pierce) in a Mini Trans-Blot Cell (BioRad, Hercules, USA). Cross-link was performed in a GS Gene Linker UV chamber (BioRad, Hercules, USA). Detection of biotin-labeled DNA was performed using stabilized streptavidin/horseradish peroxidase conjugate (Pierce) according to the manufacturer’s instructions.
Haplotype Construction and Statistical Analysis
Unconditional logistic regression was used to assess the association between genotypes and risk of gastric cancer using the Statistical Analysis System software (version 6.12; SAS Institute, Cary, USA). Odds ratios (ORs) were adjusted for age and sex where it was appropriate. HaploStats software package (Mayo Clinic/Foundation, Rochester, USA, http://mayoresearch.mayo.edu/mayo/research/schaid_lab/software.cfm) developed using the R language was employed to estimate and test haplotype-environment interactions in the general linear model framework. Simulations were run 1000 times for empirical P-values. Haploview 3.2 software (http://www.broad.mit.edu/mpg/haploview/) was also used to construct the haplotypes, evaluate the linkage disequilibrium of two SNPs and test the association of single markers and haplotypes with gastric carcinomas, correcting for multiple testing bias by permutation tests.
Bioinformatics
The genetic variants identified were compared with the Ensemble database (ENSG00000181634, http://asia.ensembl.org). The potential transcription factor binding capability between two different alleles of TNFSF15 −358 T>C and −638 A>G were predicted by TRANSFAC program and the accompanying MATCH at http://www.gene-regulation.com.
Results
Identification of SNPs in the TNFSF15 promoter
By resequencing the TNFSF15 promoter of 40 healthy subjects, we identified two genetic variants (−358 T>C, rs6478109 and −638 A>G, rs7848647), which were located at −358 bp and −638 bp upstream of the translation start site, ATG. The allele frequencies for the −358C and −638G were 0.498 and 0.485 in healthy controls, respectively. No novel SNP were identified.
Next, we estimated the degree of linkage disequilibrium between the two SNPs, using two marker expectation maximizations (EMs) to estimate the maximum-likelihood values of the four-gamete frequencies. Results indicated that these two polymorphisms were in linkage disequilibrium, with the D′ = 0.932, LOD = 142.15, r2 = 0.826 in our study population.
Subject Characteristics
As shown in Table 1, no statistically significant difference was found between 470 cases and 470 controls in terms of age (P = 0.553) and sex (P = 0.385). However, the percentage of H. pylori infection in patients was higher than in controls (73.2% versus 54.3%). Among 470 patients, 383 (81.5%) had detailed tumor stage information, while this information was unavailable for the remaining patients (18.5%).
Table 1. Distribution of selected characteristics of gastric cancer patients (cases) and controls.
| Cases (n = 470) | Controls (n = 470) | ||||
| No. | (%) | No. | (%) | P-value* | |
| Sex | 0.385 | ||||
| Male | 330 | (70.2) | 342 | (72.8) | |
| Female | 140 | (29.8) | 128 | (27.2) | |
| Age (years) | 0.553 | ||||
| ≤57 | 253 | (53.8) | 242 | (51.5) | |
| >57 | 217 | (46.2) | 228 | (48.5) | |
| H pyroli infection | <0.0001 | ||||
| Negative | 126 | (26.8) | 215 | (45.7) | |
| Positive | 344 | (73.2) | 255 | (54.3) | |
| Tumor stage† | |||||
| 0 | 6 | (1.3) | |||
| I | 69 | (14.7) | |||
| II | 63 | (13.4) | |||
| III | 117 | (24.8) | |||
| IV | 128 | (27.2) | |||
| Unknown | 87 | (18.5) | |||
*Two-sided χ2 test.
According to the UICC Tumor-Node-Metastasis classification for gastric carcinoma (1997).
Association of TNFSF15 Promoter Polymorphisms with Gastric Adenocarcinoma
The frequencies of TNFSF15 −358 T>C and −638 A>G genotypes in cases and controls and their association with gastric adenocarcinoma are shown in Table 2. The observed genotype distribution of −358 T>C and −638 A>G polymorphisms within the control population did not deviate significantly from those expected from the Hardy-Weinberg equilibrium (P = 0.871 and 0.715, respectively). The frequencies of TNFSF15 −358 T>C genotypes in infected cases (TT, 22.8%; TC, 45.5%; CC, 31.7%) were significantly different from those in controls (TT, 24.9%; CT, 50.6%; CC, 24.5%) (P = 0.047). Logistic regression analysis showed an elevated risk of gastric adenocarcinoma for subjects with TNFSF15 −358CC genotype (OR 1.42, 95% CI, 1.10 to 2.03, P = 0.04), but not for subjects carrying TNFSF15 TC genotype (OR 1.00, 95% CI, 0.72 to 1.40, P = 0.89), compared with TNFSF15 −358TT genotype carriers. No significant difference between the genotype frequencies of TNFSF15 −638 A>G polymorphism in cases and controls was observed (P = 0.216). In the stratification analysis, no significant differences in distributions of genotype frequencies were found among different stages of tumors.
Table 2. Genotype frequencies of TNFSF15 polymorphisms among gastric cancer patients and controls and their association with the risk of gastric carcinoma.
| Genotype | Patients (n = 470) | Controls (n = 470) | OR (95% CI)* | P-value | ||
| No. | (%) | No. | (%) | |||
| −358 T/C | ||||||
| TT | 107 | (22.8) | 117 | (24.9) | 1.00 (Reference) | |
| TC | 214 | (45.5) | 238 | (50.6) | 1.00 (0.72–1.40) | 0.89 |
| CC | 149 | (31.7) | 115 | (24.5) | 1.42 (1.10–2.03) | 0.04 |
| −638 A/G | ||||||
| AA | 118 | (25.1) | 122 | (26.0) | 1.00 (Reference) | |
| AG | 221 | (47.0) | 240 | (51.0) | 0.94 (0.69–1.30) | 0.729 |
| GG | 131 | (27.9) | 108 | (23.0) | 1.24(0.86–1.78) | 0.256 |
*Adjusted for age, sex and H.pylori infection.
The haplotype frequencies computed using the HaploStats software are presented in Table 3. No significant difference in haplotype frequencies was observed between gastric adenocarcinoma patients and controls (χ2 = 5.65, P = 0.130, df = 3).
Table 3. Risk estimates for extended TNFSF15 haplotypes in gastric cancer patients and controls.
| Haplotype | Patients (n = 470) | Controls (n = 470) | OR (95% CI)* | P-value |
| No. of chromosomes (%) | No. of chromosomes (%) | |||
| A−638–T−358 | 416 (44.3) | 456 (48.5) | 1.00 (Reference) | |
| G−638–T−358 | 471 (50.1) | 441 (46.9) | 1.17 (0.97–1.40) | 0.123 |
| A−638–C−358 | 40 (4.3) | 27 (2.9) | 1.48 (0.94–2.36) | 0.099 |
| G−638–C−358 | 13 (1.3) | 16 (1.7) | 0.83 (0.41–1.71) | 0.564 |
*Adjusted for age, sex, and H. pylori infection.
The interaction of TNFSF15 polymorphisms with H. pylori infection and gastric adenocarcinoma
The percentage of H. pylori infection was significantly higher in cases than in controls (73.2% versus 54.3%, P<0.0001). Logistic regression analysis showed that subjects with H. pylori infection were at a 2-fold higher risk for developing gastric adenocarcinoma compared to those without H. pylori infection (OR 2.31, 95% CI, 1.76 to 3.04, P<0.0001). Consequently, the risk of gastric adenocarcinoma associated with the TNFSF15 genotype was further examined by stratifying gastric cancer patients by H. pylori infection status. Logistic regression analysis (Table 4) showed that the subjects with TNFSF15 −358 CC genotype were at elevated risks for developing gastric adenocarcinoma compared with those with −358 TT genotype in the H. pylori infected group (OR 2.01, 95%CI, 1.65 to 4.25), but not in the H. pylori negative group (OR 0.87, 95% CI, 0.45 to 1.69). In addition, we have examined the potential effect of the −638 A>G variants interacting with H. pylori infection on the susceptibility to develop a gastric adenocarcinoma; however, no significant difference among different genotypes was observed.
Table 4. Risk of gastric carcinoma associated with TNFSF15 genotypes by H. pylori infection status.
| Genotype | H. pylori infection | Patients (n = 470) | Controls (n = 470) | OR (95% CI )* | P-value |
| No. (%) | No. (%) | ||||
| −358 T/C | |||||
| TT | – | 33 (7.0) | 48 (10.2) | 1.00 (Reference) | |
| TC | – | 61 (13.0) | 110 (23.4) | 0.83 (0.46–1.49) | 0.51 |
| CC | – | 32 (6.8) | 57 (12.1) | 0.87 (0.45–1.69) | 0.65 |
| TT | + | 74 (15.7) | 69 (14.7) | 1.00 (Reference) | |
| TC | + | 153 (32.6) | 128 (27.2) | 1.15 (0.75–1.85) | 0.62 |
| CC | + | 117 (24.9) | 58 (12.3) | 2.01 (1.65–4.25) | 0.006 |
| –638 A/G | |||||
| AA | – | 35 (7.4) | 53 (11.3) | 1.00 (Reference) | |
| AG | – | 61 (13.0) | 110 (23.4) | 0.86 (0.50–1.46) | 0.568 |
| GG | – | 30(6.4) | 52 (11.1) | 0.92 (0.67–1.26) | 0.584 |
| AA | + | 83 (17.7) | 69 (14.7) | 1.00 (Reference) | |
| AG | + | 160 (34.0) | 130 (27.7) | 1.04 (0.70–1.54) | 0.859 |
| GG | + | 101 (21.5) | 56 (11.9) | 1.25 (0.99–1.57) | 0.06 |
*Adjusted for age and sex.
Different transcriptional activity of TNFSF15 −358 T>C, −638 A>G haplotype
Since a linkage disequilibrium between −358 T>C and −638 A>G SNPs was observed, functional evaluation of the haplotype was performed. The transcriptional activity of four haplotypes generated by two TNFSF15 −358 T>C and −638 A>G genetic variants were compared by transiently transfecting the pA−638-C−358, pA−638-T−358, pG−638-C−358, and pG−638-T−358 luciferase reporter constructs into human gastric cell lines MGC-803, AGS and BGC-823. As shown in Figure 1, all constructs containing the −358C allele consistently had lower expression levels of luciferase compared with constructs containing the −358T allele in MGC-803, AGS and BGC-823 cells. Reporter gene expression driven by the constructs with the −638A allele seemed to be higher than the one driven by the constructs with the −638G allele; however, the difference was not statistically significant (P>0.05). Taken together, these data support the influence of TNFSF15 −358 T>C variant on TNFSF15 promoter activity.
Figure 1. Transient reporter gene expression assays with constructs containing full-length TNFSF15 promoter.
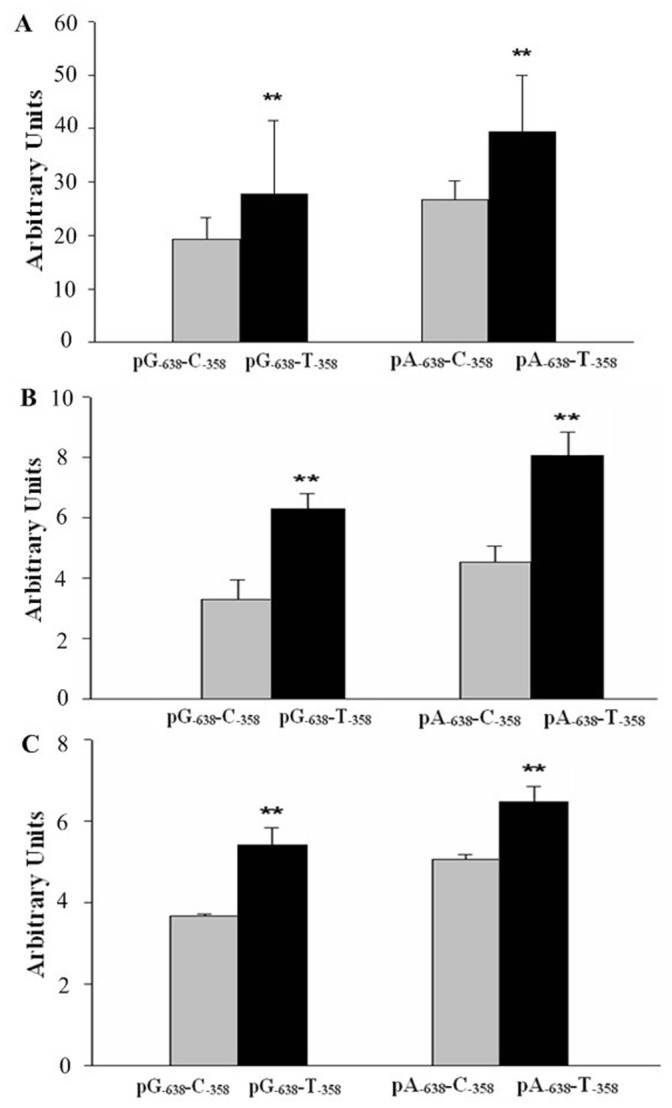
Luciferase expression of constructs in (A) MGC-803 cells, (B) AGS cells, and (C) BGC-823 cells. Luciferase levels were normalized to the results obtained for empty vector and shown as the means and standard deviation (S. D.) of fold increase from 3 independent transfection experiments, each performed in triplicate. **P<0.01 compared with the construct counterparts.
Allelic-specific Binding of Nuclear Proteins to TNFSF15 promoter
Bioinformatics analysis showed that the TNFSF15 −358T>C variant is located within a consensus sequence of the NF-Y binding site, and the T to C transition appears to disrupt NF-Y binding, and this could explain the lower promoter activity. For the −638A>G variant, the TRANSFAC program predicted no change in the binding sites of the transcriptional factors. Furthermore, electrophoretic mobility shift assay was performed to investigate whether the differences in TNFSF15 promoter activity between the −358T and −358C alleles were due to their transcription factor binding activities. Nuclear protein extracts from HeLa cells were incubated with biotin-labeled oligonucleotide probes containing −358T or −358C alleles. As shown in the Figure 2A, a clear DNA-protein complex was detected with −358T probe (lane 2) but not with −358C probe (lane 8). Next, competition experiments were performed in order to determine the sequence specificity of this DNA-protein complex. The band was competed by 100-fold molar excess of unlabeled −358T probe (lane 3) but not by the same concentration of unlabeled −358C probe (lane 4). More so, this DNA-protein complex was completely inhibited by 100-fold molar excess of unlabeled NF-Y recognition element (lane 5) but not by the unlabeled mutated NF-Y recognition element (lane 6). Collectively these results show that −358T allele, but not the −358C allele, is able to bind specifically to the nuclear protein.
Figure 2. EMSA with −358T or −358C containing oligonucleotides allele and nuclear extracts from HeLa cells.

(A) Nuclear protein extracts from HeLa cells were incubated with biotin-labeled oligonucleotide probes containing −358T or −358C. The figure shows mobility of the labeled oligonucleotides without nuclear extracts (lanes 1 and 7), with nuclear extracts in the absence of competitor (lanes 2 and 8), with nuclear extracts in the presence of various unlabeled competitors as indicated at the bottom autoradiograph (lanes 3–6 and 9–12). The arrow indicates a major oligonucleotide-nuclear protein complex. (B) Nuclear protein extracts from HeLa cells were incubated with biotin-labeled oligonucleotide probes containing −358T or consensus NF-Y binding element. The lower arrow indicates major oligonucleotide-nuclear protein complex without antibody to NF-Y, and the upper arrow shows the protein complex with antibody to NF-Y (lane 5 and 10).
To verify that the nuclear protein is indeed NF-Y, the super-EMSA assay was performed using antibodies against NF-Y, in which nuclear protein extracts from HeLa cells were incubated with biotin-labeled oligonucleotide probes containing −358T or a consensus NF-Y binding element. As shown in Figure 2B, when the anti-NF-Y antibody was added, the super shift band was observed (lanes 5 and 10). These findings suggest that NF-Y may possibly bind to the promoter region of TNFSF15 in vivo.
Discussion
Gastric cancer is one of the major health problems worldwide and genetic events underlying its initiation and progression still aren’t completely understood. In the present study, we identified two genetic variants (−358T>C and −638A>G) in the promoter of TNFSF15 gene and examined their influence on TNFSF15 transcriptional activity and the susceptibility to gastric adenocarcinoma in Chinese populations. In our study, subjects with −358CC genotype had a 1.42-fold increased risk for developing gastric cancer compared with −358TT genotype carriers. However, there was no significant difference in TNFSF15 −638A>G genotype frequencies between cases and controls. These data indicated that the TNFSF15 −358T>C polymorphism might be involved in the development of gastric cancer.
Recent studies point to a possible anti-tumor role of TNFSF15. For instance, it has been shown that recombinant TNFSF15 exhibits anti-neoplastic effects by inhibiting angiogenesis and TNFSF15 overexpression was associated with improved cancer prognosis [20], [22], [27]. Moreover, TNFSF15 can suppress endothelial cell proliferation and induce apoptosis in endothelial cells via different kinase pathways [11], [28], [29].
To the best of our knowledge, this is the first study to evaluate the association of functional TNFSF15 polymorphisms with susceptibility to cancer. Nevertheless, several studies have examined the relationship between TNFSF15 polymorphisms and susceptibility to inflammatory bowel disease and Crohn’s disease in Japanese and European populations [23], [24], [30]. In our study, we have sequenced the full-length of TNFSF15 promoter in a subset of 40 healthy Han Chinese subjects, and we have identified two polymorphisms (−358T>C and −638A>G). The −358C allele frequency in our control subjects was 0.498, which differs from that reported via GWAS in Japanese population (−358C: 0.593). The −638G allele frequency in our control population (−638G: 0.485) differed from that observed in Japanese (−638G: 0.593) and European populations (−638G: 0.655) as well [23], [24]. These differences might be due to relatively small sample size in our study and different ethnic backgrounds of the study participants. In addition, the allele frequencies of the 2 SNPs in this cohort were different to those listed in 1000 Genome project (−358C: 0.278 and −638G: 0.279). Additional studies based on larger sample sizes are needed to further verify these frequencies in different populations. Since genetic variants may have different influences on TNFSF15 expression, that ethnic differences in the distribution of TNFSF15 genetic variants might partially correlate with their different effects on cancer phenotypes within these populations.
The function of promoter motifs and transcriptional factors in the regulation of TNFSF15 expression are still not fully elucidated. In our study, the in silico analysis of the −638A>G change in the TNFSF15 promoter conferred neither gain nor loss of binding activities for any transcription factor. However, the change of nuclear transcription factor NF-Y binding activity was observed for the TNFSF15 −358T>C promoter polymorphism with EMSA experiments, indicating NF-Y might play important roles in TNFSF15 expression. NF-Y is the CCAAT box activator which is one of the most frequent promoter elements [31], [32]. NF-Y has widespread activity, and is specifically required for genes regulated during the cell-cycle which is important for many critical cellular and developmental events including cancer progression [31]. Studies showed that inactivation of NF-Y gene in mouse model is lethal at early stage of development [33]. Furthermore, our reporter constructs encompassing the TNFSF15 −358T allele showed stronger transcription activity than the constructs encompassing the −358C allele. These findings are in line with the results of our gastric cancer association study in which the −358CC genotype was associated with higher susceptibility to gastric cancers, indicating that the −358C allele might be a cancer-risk allele.
Taken together, these results point to the possible influence of TNFSF15 −358T>C promoter polymorphism in the regulation of TNFSF15 expression in gastric tissue. In this case, it would be expected that TNFSF15 genetic variants might contribute to the development of gastric cancer by inhibiting the TNFSF15 expression. However, Kakuta et al. reported that the reporter construct with TNFSF15 −358C allele showed higher transcription activity than that with −358T allele in stimulated Jurkat cells. However, in the aforementioned study, no difference was observed in the luciferase activity in U937 cells between pGL4 constructs with −358C and −358T alleles. These conflicting findings pointed to a possible different regulation of TNFSF15 by various regulators in specific cell types [34].
Besides genetic variants, environmental factors also play an important role in the etiology of gastric cancer. Many epidemiological studies have shown that increased risk of gastric cancer was associated with H. pylori infection in gastric tissues [4], [35], [36]. In animal experiments, H. pylori infection has been demonstrated to result in gastric inflammation and eventually lead to gastric cancer in mice [37], [38]. It is known that tumor necrosis factors play important roles in the progression and severity of inflammation, thereby contributing to the development of H. pylori-induced gastric cancer [39]–[41]. For instance, levels of TNF-α, a pro-inflammatory cytokine, are elevated within H. pylori infected gastric mucosa [42]. More so, functional genetic variants of the TNF-α gene have been associated with the susceptibility to gastric cancer [43]. TNFSF15, as a member of the tumor necrosis factor superfamily, modulates cell growth and inflammation in many different cell types [13], [20], [28], [44]. In this study we have evaluated the influence of TNFSF15 and H. pylori infection on gastric cancer susceptibility and found there was a joint effect of −358C allele and H. pylori infection in raising the risk of gastric cancer. The subjects with TNFSF15 −358CC genotype were at elevated risks for developing gastric adenocarcinoma compared with those with −358TT genotype in the H. pylori infected group, but not in the H. pylori negative group. These results implicate a synergistic interaction between H. pylori-induced inflammation and host genetic factors in the development of gastric cancer.
Acknowledgments
We thank Dr. Dongxin Lin from Chinese Academy of Medical Sciences and Peking Union Medical College for providing three cell lines.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.
Funding Statement
This work was supported by Program for New Century Excellent Talents in University (NCET-11-0933), the National Natural Science Foundation of China (NSFC-81101483), Beijing Science and Technology Project (Z131101002813044) and the Science Fund for Distinguished Young Scholars of Hebei Scientific Committee (H2012401022). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Ferlay JSI, Ervik M, Dikshit R, Eser S, Mathers C, et al. (2012) GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet]. Lyon, France: International Agency for Research on Cancer; 2013. Globocan 2012 website. Available: http://globocan.iarc.fr/Default.aspx. Accessed 2014 Sep 1.
- 2. Eid R, Moss SF (2002) Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 346: 65–67. [PubMed] [Google Scholar]
- 3. Normark S, Nilsson C, Normark BH, Hornef MW (2003) Persistent infection with Helicobacter pylori and the development of gastric cancer. Adv Cancer Res 90: 63–89. [DOI] [PubMed] [Google Scholar]
- 4. Sugiyama T (2004) Development of gastric cancer associated with Helicobacter pylori infection. Cancer Chemother Pharmacol 54 Suppl 1S12–20. [DOI] [PubMed] [Google Scholar]
- 5. Tracey KJ, Wei H, Manogue KR, Fong Y, Hesse DG, et al. (1988) Cachectin/tumor necrosis factor induces cachexia, anemia, and inflammation. J Exp Med 167: 1211–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schaffler A, Scholmerich J, Salzberger B (2007) Adipose tissue as an immunological organ: Toll-like receptors, C1q/TNFs and CTRPs. Trends Immunol 28: 393–399. [DOI] [PubMed] [Google Scholar]
- 7. Kohchi C, Mizuno D, Soma G (1991) Expression of tumor necrosis factor-alpha and -beta transcripts in embryonal carcinoma and trophoblast cell lines: inflammation-like state as possible regulatory mechanism for ontogenesis. Eur Cytokine Netw 2: 245–255. [PubMed] [Google Scholar]
- 8. de Silva PS, Nguyen DD, Sauk J, Korzenik J, Yajnik V, et al. (2012) Long-term outcome of a third anti-TNF monoclonal antibody after the failure of two prior anti-TNFs in inflammatory bowel disease. Aliment Pharmacol Ther 36: 459–466. [DOI] [PubMed] [Google Scholar]
- 9. Zhang N, Sanders AJ, Ye L, Jiang WG (2009) Vascular endothelial growth inhibitor in human cancer (Review). International journal of molecular medicine 24: 3–8. [DOI] [PubMed] [Google Scholar]
- 10. Metheny-Barlow LJ, Li LY (2006) Vascular endothelial growth inhibitor (VEGI), an endogenous negative regulator of angiogenesis. Seminars in ophthalmology 21: 49–58. [DOI] [PubMed] [Google Scholar]
- 11. Grimaldo S, Tian F, Li LY (2009) Sensitization of endothelial cells to VEGI-induced apoptosis by inhibiting the NF-kappaB pathway. Apoptosis : an international journal on programmed cell death 14: 788–795. [DOI] [PubMed] [Google Scholar]
- 12. Zhang Z, Li LY (2012) TNFSF15 Modulates Neovascularization and Inflammation. Cancer Microenviron 5: 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhai Y, Yu J, Iruela-Arispe L, Huang WQ, Wang Z, et al. (1999) Inhibition of angiogenesis and breast cancer xenograft tumor growth by VEGI, a novel cytokine of the TNF superfamily. International journal of cancer Journal international du cancer 82: 131–136. [DOI] [PubMed] [Google Scholar]
- 14. Jin T, Guo F, Kim S, Howard A, Zhang YZ (2007) X-ray crystal structure of TNF ligand family member TL1A at 2.1A. Biochem Biophys Res Commun 364: 1–6. [DOI] [PubMed] [Google Scholar]
- 15. Jin T, Kim S, Guo F, Howard A, Zhang YZ (2007) Purification and crystallization of recombinant human TNF-like ligand TL1A. Cytokine 40: 115–122. [DOI] [PubMed] [Google Scholar]
- 16. Zhang E, Zhu X, Han S, Peng Z, Wang W, et al. (2013) Increased expression of TNF ligand-related molecule 1A and death receptor 3 in bladder tissues of patients with painful bladder syndrome/interstitial cystitis. Exp Ther Med 5: 282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Das SK, Vasudevan DM (2007) Essential factors associated with hepatic angiogenesis. Life Sci 81: 1555–1564. [DOI] [PubMed] [Google Scholar]
- 18. Zhang N, Sanders AJ, Ye L, Kynaston HG, Jiang WG (2010) Expression of vascular endothelial growth inhibitor (VEGI) in human urothelial cancer of the bladder and its effects on the adhesion and migration of bladder cancer cells in vitro. Anticancer Res 30: 87–95. [PubMed] [Google Scholar]
- 19. Chew LJ, Pan H, Yu J, Tian S, Huang WQ, et al. (2002) A novel secreted splice variant of vascular endothelial cell growth inhibitor. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 16: 742–744. [DOI] [PubMed] [Google Scholar]
- 20. Hou W, Medynski D, Wu S, Lin X, Li LY (2005) VEGI-192, a new isoform of TNFSF15, specifically eliminates tumor vascular endothelial cells and suppresses tumor growth. Clinical cancer research : an official journal of the American Association for Cancer Research 11: 5595–5602. [DOI] [PubMed] [Google Scholar]
- 21. Wu J, Jiang Y, Yang W, He Z, Meng S, et al. (2012) Dual function of RGD-modified VEGI-192 for breast cancer treatment. Bioconjug Chem 23: 796–804. [DOI] [PubMed] [Google Scholar]
- 22. Parr C, Gan CH, Watkins G, Jiang WG (2006) Reduced vascular endothelial growth inhibitor (VEGI) expression is associated with poor prognosis in breast cancer patients. Angiogenesis 9: 73–81. [DOI] [PubMed] [Google Scholar]
- 23. Yamazaki K, McGovern D, Ragoussis J, Paolucci M, Butler H, et al. (2005) Single nucleotide polymorphisms in TNFSF15 confer susceptibility to Crohn’s disease. Hum Mol Genet 14: 3499–3506. [DOI] [PubMed] [Google Scholar]
- 24. Tremelling M, Berzuini C, Massey D, Bredin F, Price C, et al. (2008) Contribution of TNFSF15 gene variants to Crohn’s disease susceptibility confirmed in UK population. Inflamm Bowel Dis 14: 733–737. [DOI] [PubMed] [Google Scholar]
- 25. Yang M, Guo Y, Zhang X, Miao X, Tan W, et al. (2007) Interaction of P53 Arg72Pro and MDM2 T309G polymorphisms and their associations with risk of gastric cardia cancer. Carcinogenesis 28: 1996–2001. [DOI] [PubMed] [Google Scholar]
- 26. Zhao D, Sun T, Zhang X, Guo Y, Yu D, et al. (2007) Role of CD14 promoter polymorphisms in Helicobacter pylori infection–related gastric carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research 13: 2362–2368. [DOI] [PubMed] [Google Scholar]
- 27. Sethi G, Sung B, Aggarwal BB (2009) Therapeutic Potential of VEGI/TL1A in Autoimmunity and Cancer. Adv Exp Med Biol 647: 207–215. [DOI] [PubMed] [Google Scholar]
- 28. Yue TL, Ni J, Romanic AM, Gu JL, Keller P, et al. (1999) TL1, a novel tumor necrosis factor-like cytokine, induces apoptosis in endothelial cells. Involvement of activation of stress protein kinases (stress-activated protein kinase and p38 mitogen-activated protein kinase) and caspase-3-like protease. J Biol Chem 274: 1479–1486. [DOI] [PubMed] [Google Scholar]
- 29. Haridas V, Shrivastava A, Su J, Yu GL, Ni J, et al. (1999) VEGI, a new member of the TNF family activates nuclear factor-kappa B and c-Jun N-terminal kinase and modulates cell growth. Oncogene 18: 6496–6504. [DOI] [PubMed] [Google Scholar]
- 30. Thiebaut R, Kotti S, Jung C, Merlin F, Colombel JF, et al. (2009) TNFSF15 polymorphisms are associated with susceptibility to inflammatory bowel disease in a new European cohort. Am J Gastroenterol 104: 384–391. [DOI] [PubMed] [Google Scholar]
- 31. Donati G, Gatta R, Dolfini D, Fossati A, Ceribelli M, et al. (2008) An NF-Y-dependent switch of positive and negative histone methyl marks on CCAAT promoters. PLoS One 3: e2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ceribelli M, Dolfini D, Merico D, Gatta R, Vigano AM, et al. (2008) The histone-like NF-Y is a bifunctional transcription factor. Mol Cell Biol 28: 2047–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhattacharya A, Deng JM, Zhang Z, Behringer R, de Crombrugghe B, et al. (2003) The B subunit of the CCAAT box binding transcription factor complex (CBF/NF-Y) is essential for early mouse development and cell proliferation. Cancer Res 63: 8167–8172. [PubMed] [Google Scholar]
- 34. Kakuta Y, Ueki N, Kinouchi Y, Negoro K, Endo K, et al. (2009) TNFSF15 transcripts from risk haplotype for Crohn’s disease are overexpressed in stimulated T cells. Hum Mol Genet 18: 1089–1098. [DOI] [PubMed] [Google Scholar]
- 35. Lamb A, Chen LF (2013) Role of the Helicobacter pylori-induced inflammatory response in the development of gastric cancer. J Cell Biochem 114: 491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Epplein M, Nomura AM, Hankin JH, Blaser MJ, Perez-Perez G, et al. (2008) Association of Helicobacter pylori infection and diet on the risk of gastric cancer: a case-control study in Hawaii. Cancer Causes Control 19: 869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang TC, Dangler CA, Chen D, Goldenring JR, Koh T, et al. (2000) Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology 118: 36–47. [DOI] [PubMed] [Google Scholar]
- 38. Oshima H, Matsunaga A, Fujimura T, Tsukamoto T, Taketo MM, et al. (2006) Carcinogenesis in mouse stomach by simultaneous activation of the Wnt signaling and prostaglandin E2 pathway. Gastroenterology 131: 1086–1095. [DOI] [PubMed] [Google Scholar]
- 39. Zhao C, Lu X, Bu X, Zhang N, Wang W (2010) Involvement of tumor necrosis factor-alpha in the upregulation of CXCR4 expression in gastric cancer induced by Helicobacter pylori. BMC Cancer 10: 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Suganuma M, Kurusu M, Suzuki K, Nishizono A, Murakami K, et al. (2005) New tumor necrosis factor-alpha-inducing protein released from Helicobacter pylori for gastric cancer progression. J Cancer Res Clin Oncol 131: 305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kirikoshi H, Sekihara H, Katoh M (2001) Up-regulation of WNT10A by tumor necrosis factor alpha and Helicobacter pylori in gastric cancer. International journal of oncology 19: 533–536. [PubMed] [Google Scholar]
- 42. Crabtree JE, Shallcross TM, Heatley RV, Wyatt JI (1991) Mucosal tumour necrosis factor alpha and interleukin-6 in patients with Helicobacter pylori associated gastritis. Gut 32: 1473–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Melo Barbosa HP, Martins LC, Dos Santos SE, Demachki S, Assumpcao MB, et al. (2009) Interleukin-1 and TNF-alpha polymorphisms and Helicobacter pylori in a Brazilian Amazon population. World J Gastroenterol 15: 1465–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yu J, Tian S, Metheny-Barlow L, Chew LJ, Hayes AJ, et al. (2001) Modulation of endothelial cell growth arrest and apoptosis by vascular endothelial growth inhibitor. Circ Res 89: 1161–1167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.