

Abstract
A series of piperidine-based derivatives were identified as novel and potent inhibitors of influenza virus through structural modification of the original compound that was selected from a high-throughput screen. Various analogues were synthesized and confirmed as inhibitors. The structure-activity relationship (SAR) studies suggested that the ether linkage between the quinoline and piperidine is critical to the inhibitory activity. Optimized compoundtert-Butyl 4-(quinolin-4-yloxy)piperidine-1-carboxylate11ehad an excellent inhibitory activity of influenza virus infection against a variety of influenza virus strains with EC50 values as low as 0.05 μM. The selectivity index value (SI = MLD50/EC50) of 11e is over 160,000 based on cytotoxicity measured by MTT assays of three cell lines. We carried out the time-of-addition experiment to delineate the mechanism of inhibition. The result indicates that 11e interferes with an early to middle stage of influenza virus replication.
Introduction
Emergence of influenza A H1N1 virus of swine origin in 2009 serves as a warning once again that new strains of influenza viruses may spread worldwide at an unexpected moment.1-2 In 2011, patients infected by avian influenza virus were again identified in Hong Kong and Shenzhen of China.3 Since March, 2013, a novel avian influenza A H7N9 virus that infects humans has been uncovered in China.4 Therefore, prevention and treatment of influenza virus infection is a constant demand for the public health. The most effective way to control newly emerged influenza virus may be anti-influenza drugs because production and delivery of suitable vaccines could lag behind virus spread. Currently, the popular treatment of influenza virus infection is by neuraminidase inhibitors (NAIs), such as oseltamivir, zanamivir and peramivir.5-9 NAIs bind to the active site of neuraminidase that is a major surface antigen of influenza virus type A and B, and inhibit the release of newly produced influenza virus from infected cells.10-11 However, the efficacy of NAIs may be diminished when mutations occur in the active site of neuraminidase. One of the mutations that occur fairly often in clinical isolates of influenza virus is at residue His274.12-15 When His274 was changed to a tyrosine or other residues, NAIs could no longer bind to neuraminidase tightly so that the mutant influenza virus becomes drug-resistant. If drug-resistant influenza virus begins to circulate in humans, NAI drugs would become obsolete. It is therefore necessary to develop novel inhibitors of influenza virus that target different viral proteins. If novel inhibitors may be developed as anti-influenza drugs, they may be used effectively to treat infection of NAI-resistant influenza virus or be included in combination therapy with NAIs to reduce the chance of emergence of drug-resistant influenza virus.
In the replication cycle of influenza virus in the host cell, there are a number of critical steps that may be blocked by inhibitors to stop virus infection. These steps include virus attachment to cell surface receptors (sialiomolecules on cell surface), membrane fusion during virus entry, virus replication, virus assembly at the plasma membrane, and release. 16Some of the steps occur outside the host cell, such as virus attachment and release, whereas other steps take place inside the host cell. Since NAIs act on neuraminidase outside the host cell, we focus on finding inhibitors that act inside the host cell. It is likely that inhibitors that work inside the host cell would target influenza virus proteins other than neuraminidase. A high throughput screen was developed for identifying such inhibitors among a library of 10,000 compounds.17A novel piperidine-based derivative was found to inhibit influenza virus infection effectively. Virological studies confirmed that this series of compounds inhibit influenza virus replication after virus entry into the host cell. One of the optimized piperidine-based derivatives has a potent anti-influenza activity in nanomolar range. We report here our strategy for inhibitor identification and structure-activity relationship (SAR) studies of this series of piperidine-based derivatives with regard to anti-influenza activities against influenza virus infection.
Results and discussion
HTS leading hit discovery
In order to identify anti-influenza virus agents that can inhibit virus replication inside the host cell, we developed a cell-based assay for influenza virus infection as a high throughput screen (HTS). MDCK cells18 were plated in 96-well trays and allowed to grow for 24 hours in an incubator set at 37°Cunder 5% CO2. When cells became 90% confluent, each well was inoculated with 300 PFU of influenza virus A/Udorn/72 (H3N2). Virus infection was allowed to proceed for 48 hours and the cells were fixed and stained. Without inhibitors, infected cells would be cleared as shown in Fig. 1a. Ribavirin and a fusion inhibitor F13619 that acts outside the host cell were used as controls. To identify inhibitors that act only inside the host cell, compounds were added in the growth media at 100 μg/mL concentrantion after the virus inoculum was incubated with the cells for one hour. Using this HTS strategy, we have discovered a novel piperidine-based derivative, ethyl 4-(quinolin-4-ylthio)piperidine-1-carboxylate (Fig. 1b), (labelled here as P114F5) from a library of 10,000 compounds (MyriaScreen from Sigma-Aldrich). The EC50 value of P114F5 is 13.9 μM based on plaque assay (Fig. 1c). There is no literature report of this compound as an anti-influenza agent based on our literature survey. A time-of-addition experiment was carried out to confirm that P114F5 indeed inhibits influenza virus infection after it enters the host cell. As shown in Fig. 1d, P114F5 was most effective as an inhibitor of influenza virus when it was added to the host cell during virus inoculation and one hour after inoculation. When it was added later, no significant anti-influenza activities were observed, suggesting that P114F5 does not interfere with influenza virus release as NAIs.
Fig. 1.

HTS leading hit P114F5. a) A HTS for inhibitors of influenza A virus infection. Wells A1-D1 was uninfected, whereas wells E1-H1 were infected with influenza virus inoculum incubated with the placebo solution. Ribavirin (50 μM, 1% DMSO) in wells A12-D12 and fusion inhibitor F136 in wells E12-H12 were selected as controls. Well F5 was selected as positive result. b) Structural information of HTS leading hit. c) EC50 measurement. d) Time of addition using P114F5 (30μM, 1% DMSO).
Hit optimization and biological evaluation
In order to gain more insights on the mechanism of action by P114F5, we systematically altered the chemical structure of P114F5 to probe the contribution of P114F5 substructures to the anti-influenza activity. The structure-activity relationship (SAR) studies would help to explore the subunits in P114F5 pertained to its anti-influenza activity. For the convenience of SAR discussion, P114F5 can be readily divided into four subunits, a quinolinyl core (A), a sulfur core (B), a piperidinyl core (C), and a carbamate core (D), leading to a study design where one subunit can be independently varied in the presence of three other fixed subunits (Fig. 2).
Fig. 2.

Diversity-oriented synthesis of derivatives of P114F5 by modifications of four subunits. aquinolinyl core (A), a sulfur core (B), a piperidinyl core (C), and a carbamate core (D).
We initially tested the influence of subunit A (quinolinylgroup) on the anti-influenza activity by replacing quinolinylgroup in P114F5 with substituted quinolinyl (1a and 1b), pyridinyl (1c), substituted phenyl (1dand 1e), naphthyl (1f), substituted 2-oxo-2H-chromen-4-yl (1g and 1h), and 2-oxo-1,2-dihydroquinolin-4-yl (1i) groups, and compounds 1a-i (Table 1) were made by nucleophilic replacement of a halo compound with a thiol compound produced the corresponding compounds1a-i.20Thioether 1a-c and 1g-i were directly obtained from the commercially available 4-chloro compound2a-b and 2g-iinvery good yields by treatment of 321with slightly more than one equivalent of sodiumhydride in DMF at 50 °C. In addition, commercially available benzenethiol 2d-e or naphthalenethiol derivatives 2fwere coupled with bromo compound 422 by the treatment of sodium hydride to generate the desired compound 1d-f (Scheme 1).
Table 1.
Optimization of subunit A. In vitro anti-influenza A virus activities of compounds1a-i produced via Scheme 1a
| Compd. | Structure | EC50 (μM) |
|---|---|---|
| P114F5 |

|
13.9 |
| 1a |

|
>100 |
| 1b |

|
47.0 |
| 1c |
|
>100 |
| 1d |
|
67.5 |
| 1e |
|
52.9 |
| 1f |

|
23.1 |
| 1g |

|
>100 |
| 1h |

|
>100 |
| 1i |

|
>100 |
Using influenza virus A/Udorn/72 strain (H3N2).
Scheme 1.

Reaction conditions. (a) NaH, DMF, 50 °C, 5h, 60-85%.
As shown in Table 1, the anti-influenza activity of these compounds with different modifications of subunit A varied greatly, such as compounds 1a, 1c and 1g-i that did not show any inhibition to influenza virus at a concentration of 100 μM, while other compounds, such as 1b, 1d, 1e and 1f, have moderate inhibitory activities, though they were usually less potent than the original HTS hit P114F5 (EC50 = 13.9 μM). These results indicated that the modification of subunit A didn’t dramatically improve compound’s inhibitory potency.
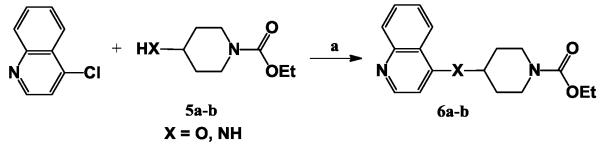
Next, we switched our focus on subunit B, a sulfur subunit in compound P114F5. Compounds 6a and 6b were prepared from 5a and 5b23 (Table 2), in which the sulfur subunit in compound P114F5 was replaced with O and NH subunit, respectively. The synthesis of compounds 6a-b was similar to that of compounds 1a-i (Scheme 2). The nucleophilic displacement of 4-chloroquinoline occurs readily either by an oxygen or amine nucleophile to deliver the corresponding compounds 6a-b.
Table 2.
Optimization of subunit B. In vitro anti-influenza A virus activities of compounds6a-b.a
| Compd. | Structure | EC50 (μM) |
|---|---|---|
| P114F5 |

|
13.9 |
| 6a |

|
2.33 |
| 6b |

|
10.0 |
Using influenza virus A/Udorn/72 strain (H3N2).
Scheme 2.
Reaction conditions. (a) NaH, DMF, 50 °C, 5h, 75-90%.
As we can see from Table 2, the analogue 6a with oxygen as subunit B showed a good inhibitory activity with a 6-fold improvement over the HTS hit P114F5, while analogue 6b with amine as subunit B showed moderate inhibitory activity. The modification of subunit B can clearly affect the anti-influenza activity. Compounds with oxygen as subunit B showed a better inhibitory activity, compared to the HTS hit P114F5. This result implied that using oxygen as subunit B is very important for the anti-influenza activity.
Thirdly an investigation of the substitutions of the piperidinyl ring (subunit C) was carried out, in which a phenyl ring or a cyclohexyl ring was used. The synthesis of 8a-f was also similar to that of compounds 1a-i by use of the nucleophilic displacement reaction from 4-chloroquinoline and 7a-f (Scheme 3).
Scheme 3.
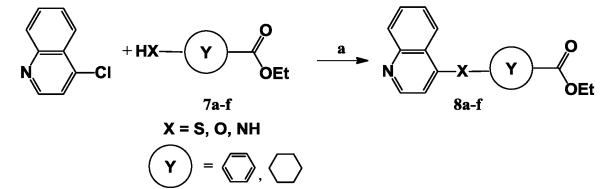
Reaction conditions. (a) NaH, DMF, 50 °C, 5h, 50-85%.
The data for compounds 8a-f identified the role of subunit C on its anti-influenza activity, as summarized in Table 3. The activity of compounds 8a-f varied greatly. Using a phenyl ring as subunit C will result in the loss of inhibitory activity, no matter that sulfur or amine or oxygen was used as subunit B, such as compounds 8a-c. However, compounds with a cyclohexyl ring as subunit C showed a good inhibitory activity with the EC50 value from 10μM to 36μM, such as compounds 8d-f. These results indicated that anon-planar ring used as subunit C can present an important conformation in the inhibitor structure for its interactions with the target, then exhibited an excellent inhibitory activity.
Table 3.
Optimization of subunit C.In vitro anti-influenza A virus activities of compounds8a-f.a
| Compd. | Structure | EC50 (μM) |
|---|---|---|
| P114F5 |
|
13.9 |
| 8a |

|
>100 |
| 8b |

|
>100 |
| 8c |

|
79.0 |
| 8d |

|
36.2 |
| 8e |

|
19.5 |
| 8f |

|
10.0 |
Using influenza virus A/Udorn/72 strain (H3N2).
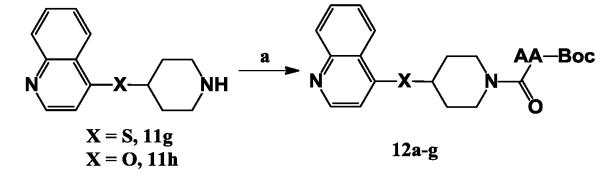
Finally, we explored the role of subunit D (carbamate) of P114F5 by fixing subunit B as S or O and subunit C as piperidine moieties. Ethyl carbamate was firstly converted to other alkyl carbamates, such as methyl carbamate 11a and 11d,tert-butyl carbamate 11b and 11e,n-hexyl carbamate 11c and 11f. These compounds can be prepared following the synthetic route below (Scheme 4): the nucleophilic displacement reaction between 4-chloro-quinoline and tert-butyl 4-mercaptopiperidine-1-carboxylate 9aor tert-butyl 4-hydroxypiperidine-1-carboxylate9b easily offered compound 11b or 11c, following the treatment with TFA to generate compound 11g or 11h, which was treated with N,N’-carbonyldiimidazole(CDI), then R-OH was added to produce the desired compounds 11c and 11f.24-25 The synthesis of 11i-j was prepared viathe nucleophilic displacement reaction according to the published procedure from 4-chloro-quinoline and tetrahydrofuran-3-ol or tetrahydro-2H-pyran-4-ol.
Scheme 4.

Reaction conditions. (a) NaH, DMF, 50 °C, 5h, 70-90%; (b) TFA, DCM, room temperature, 2h, 90-95%; (c) N,N’-carbonyldiimidazole(CDI), then n-C6H13OH, r. t., THF, 12h, 55-70%.
Table 4 revealed that alkyl carbamate analogues with sulfur as subunit B did not show clear enhancement in its inhibitory activity compared to P114F5. However, alkyl carbamate analogues with oxygen as subunit B showed a dramatically improvement in the inhibitory activity, such as compound 11e with tert-butyl carbamate group has a 100-fold better activity compared to HTS hit. Removal of the carbamate group significantly reduced the activity, such as 11h. The in-house synthesized compounds without carbamate motif, such as compounds 11i-j with tetrahydrofuran or tetrahydro-2H-pyran as subunit C and subunit D, led to loss of activity. These results indicated that carbamate group may play an important role on the biological activity by specific interactions with acceptors by forming hydrogen bond. Compound with the bulky alkyl (tert-butyl) in carbamate motif showed an excellent anti-influenza activity. This result can be explained that the bulky alkyl in carbamate motif may form close integration during compound being bound to its target.26
Table 4.
Optimization of subunit D.In vitro anti-influenza A virus activities of compounds11a-j.a
| Compd. | Structure | EC50 (μM) |
|---|---|---|
| P114F5 |

|
13.9 |
| 11a |

|
30.0 |
| 11b |

|
64.5 |
| 11c |

|
>100 |
| 11d |

|
10.0 |
| 11e |

|
0.10 |
| 11f |

|
10.0 |
| 11g |
|
91.0 |
| 11h |

|
>100 |
| 11i |
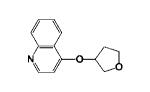
|
>100 |
| 11j |
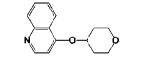
|
>100 |
Using influenza virus A/Udorn/72 strain (H3N2).
A follow up-focused library changing the carbamate group to an amide group by introducing an amino acid moiety was then synthesized. In this library, we kept sulfur or oxygen as subunit B and quinoline as subunit A. The synthesis of compound 12a-g in Table 5 was outlined in Scheme 5. The classic amide bond formation can be easily achieved using 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride(EDC·HCl) as a coupling reagent between amine compound 11g or 11h and Boc-protected amino acid to generate the corresponding compound 12a-g.27
Table 5.
Further optimization of subunit D.In vitro anti-influenza A virus activities of compounds12a-g.a
| Compd. | Structure | EC50 (μM) |
|---|---|---|
| 12a |
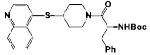
|
15.5 |
| 12b |

|
>100 |
| 12c |

|
>100 |
| 12d |
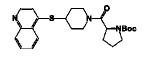
|
38.5 |
| 12e |

|
17.8 |
| 12f |

|
29.1 |
| 12g |

|
>100 |
Using influenza virus A/Udorn/72 strain (H3N2).
Scheme 5.
Reaction conditions. (i) Boc-AA-CO2 H, 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride(EDC·HCl), HOBt, DCM, room temperature, 12h, 65-85%.
The anti-influenza activities of compounds with amino acid as subunit D are summarized in Table 5. The introduction of an amino acid did not clearly improve the inhibitory activity with sulfur or oxygen as subunit B. Usually, these compounds showed a clear inhibitory activity with the EC50 value from 10μM to 100μM. Introducing amino acid motif as subunit D to increase the ability of forming hydrogen bond with acceptors did not clearly improve the anti-influenza activity. We believe that the substantial steric effect of amino acid side chains in 12a-g resulted in their worse anti-influenza activities.
Cytotoxic activity of the most potent compound 11e in different cell lines
To exclude that the observed anti-influenza activity could be due to non-specific cytotoxic activity, MTT assays of the most potent inhibitor 11e were performed by incubating with11e three cell lines, i.e. Madin-Darby canine kidney (MDCK) cells, human embryonic kidney cells (HEK293 cells) and human epithelial carcinoma cell line (HeLa cells).28 As shown in Table 6, 11edid not exhibit cytotoxicity in any of the cell lines up to a concentration of millimolar (mM) range, and its selectivity index value (SI = MLD50/EC50) was over160,000. This result indicated that the excellent anti-influenza activity of the piperidine-based derivatives was not due to non-specific cytotoxic activity.
Table 6.
Cytotoxic study of 11e in Madin-Darby canine kidney (MDCK) cells, human embryonic kidney cells (HEK293 cells) and human epithelial carcinoma cell line (HeLa cells).a
| MDL50 (μM) mean ± SD | |||
|---|---|---|---|
| MDCK cells | HEK 293 cells | Hela cells | |
| 11e | 10061.92 ±235.86 |
8101.97 ±473.16 |
8850.21 ±406.99 |
The cytotoxicity of 11e is expressed by MDL50 in μM, calculated for three cells lines. Data are expressed as the mean ± SD of triplicates for each concentration.
In vitro anti-influenza activity of the most potent compound 11e against various strains
A good anti-influenza drug should have an excellent inhibitory activity against different strains of influenza viruses. After evaluating anti-influenza activities using influenza virus A/Udorn/72 (H3N2), we were interested in investigating the potent inhibitor 11e with other strains of influenza viruses. For this purpose, we selected four strains of influenza A virus (two H3N2 and two H1N1 strains) and one strain of influenza B (Table 7). The results showed that 11e also effectively inhibited all the influenza virus strains tested with varied EC50 values that could be as low as 0.05 μM. These results indicated that 11e is a broad spectrum anti-influenza inhibitor and could potentially provide an effective coverage against infection over different strains of influenza virus.
Table 7.
EC50 values (μM) against various influenza virus strains of compound 11e and HTS hit P114F5.a
| Virus. | 11e | P114F5 |
|---|---|---|
| Influenza A/udorn/72 (H3N2) | 0.10 | 13.9 |
| Influenza A/PR/8/34 (H1N1) | 0.07 | 8.78 |
| Influenza A1/Jingfang 86-1(H1N1) | 0.10 | 9.05 |
| Influenza A3/Lufang 93-1(H3N2) | 0.06 | 11.7 |
| Influenza B/Shenzhen/747 | 0.05 | 10.9 |
EC50 values are the compound concentrations for 50% reduction of plaques.
Time-of-addition studies of the most potent compound 11e
To explore the mechanism of action, time-of-addition studies (Figure 3) were carried out to determine the effectiveness of 11e to reduce the viral yield with regard to the time when the compound was added to the culture medium during the course of the infection.29As shown in Figure 3, significant inhibition of virus replication was observed when compound 11e was added to the cells within 6 h after virus inoculation. However, compound 11e did not show obvious inhibition of influenza virus infection when incubated with the virus inoculum prior to infection, in contrast to the fusion inhibitor that was used as a control. The studies suggested that 11ecould act at early stages of influenza virus replication cycle, including entry and/or an early replication phase.
Fig. 3.

Time-of-addition effect of 11e (1.0 μM, 1% DMSO) on influenza A/udorn/72 (H3N2) infection in MDCK cells (Viral yields obtained with respect to different exposure period of 11e to the culture medium during influenza infection).*: P< 0.05.
Conclusions
The threat of a pandemic influenza is a burden that will remain for a long period of time. To date, vaccination is considered as an effective preventive measure to control influenza. However, vaccine development for millions of people, especially with antigenic shift and drift of influenza viruses, is still a multifaceted challenge. To find an alternative, a global trend is to stockpile effective anti-influenza medications. Considering the emergence of resistant strains to current medications it is necessary to develop new therapeutic agents that are effective against emerging drug-resistant influenza viruses. A piperidine-based compound was identified as a novel influenza virus inhibitor from the HTS campaign. Chemistry-driven optimization of the hit compound has resulted in the discovery of the optimized compound 11ewith a high potency in inhibiting influenza virus replication, and a clear SAR has been obtained. Substitutions with different aromatic rings were tolerated for subunit A, but compounds with quinolinyl groups usually offered a better inhibitory activity. Oxygen as subunit B was a game changing replacement for sulfur found in P114F5. Rigid rings as subunit C will lead to a substantial loss of inhibitory activity, most likely due to conformational specificities in interactions with the potential target. The carbamate group as subunit D resulted in compound 11e with an excellent inhibitory potency (EC50 = 0.10 μM). Compound 11e inhibits the activity of both influenza A and B strains in vitro, suggesting that it could act as a starting compound for further anti-influenza drug development. We did not observe cytotoxicity of 11e in any of the cell lines up to a concentration of millimolar (mM) range, and its selectivity index value was over 160,000. The preliminary mechanistic studies of compound 11e suggest that this type of piperidine-based derivatives interferes with an early stage of influenza virus replication. The precise target with which compound 11e interacts remains to be uncovered. The optimized leading compound identified here could open up new opportunities for anti-influenza drug discovery. Work is continued to optimize the selectivity and explore the detailed mechanism of action for this novel inhibitor.
Experimental section
Chemistry
Anhydrous THF and dioxane were distilled from sodium-benzophenone, and dichloromethane was distilled from calcium hydride. All starting materials were purchased from either J&K Chemicals Co. or Sigma Aldrich Chemical Co.. Water was distilled and purified through a Milli-Q water system (Millipore Corp., Bedford, MA). All reactions were carried out under a nitrogen atmosphere with dry solvents under anhydrous conditions, unless otherwise noted. Reactions were monitored by thin-layer chromatography (TLC) carried out on 0.25 mm Tsingdao silica gel plates (60F-254) using UV light as visualizing agent and an ethanolic solution of phosphomolybdic acid and potassiumpermanganate, and heat as developing agents. Tsingdao silica gel (60, particle size 0.040–0.063 mm) was used for flash column chromatography to purified compounds. Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous materials, unless otherwise stated. Melting points were measured in SGW X-4B microscopic melting point apparatus, and were uncorrected. All compounds were characterized by 1H and 13C NMR using Bruker 300 MHz NMR and/or Bruker 400 MHz NMR spectrometers. Chemical shifts are reported in ppm (δ) relative to the residual solvent peak in the corresponding spectra; chloroform δ 7.26 and δ 77.5, DMSO-d6 δ 2.54 and δ 39.5, and coupling constants (J) are reported in Hertz (Hz) (where, s = singlet, b = broad, d = doublet, dd = double doublet, bd = broad doublet, ddd = double doublet of dublet, t = triplet, td = double triplet, q = quartet, m = multiplet) and analyzed using ACD NMR data processing. High resolution mass spectra were taken using Brüker Apex IV RTMSusing electronspray ionisation (ESI) technique. Mass spectra values were reported as m/z.
For the preparation of 1a-c and 1g-i
To a degassed solution of ethyl 4-mercaptopiperidine-1-carboxylate 321 (227mg, 1.20 mmol) in 5 mL of anhydrous DMF was added NaH (60% in mineral oil, 48mg, 1.20 mmol) at 0°C. The suspension mixture was stirred for additional 15 min at room temperature. Then, a solution of the commercial 4-chloro-quinoline derivatives 2a-c or other aromatic halo compounds 2g-i (1.00 mmol) in 5 mL of anhydrous DMF was added to the above suspension mixture, and the mixture was stirred at 50°C until halo compounds disappeared. The reaction mixture was then poured in ice-water and extracted with ethyl acetate, dried over MgSO4, and concentrated. The residue was purified by flash chromatograph (silica, EtOAc/hexane as eluent) to give the title compound 1a-e and 1g-i with the 60-85% yield.For the preparation of 1d-f: To a degassed solution of benzenethiol 2d-e or naphthalene-1-thiol derivatives 2f(1.20 mmol) in 5 mL of degassed DMF was added NaH (60% in mineral oil, 48mg, 1.20 mmol) at 0°C. The suspension mixture was stirred for additional 15 min at room temperature. Then, a solution of the commercial ethyl 4-bromopiperidine-1-carboxylate 422 (236mg, 1.00 mmol) in 5 mL of anhydrous DMF was added to the above suspension mixture, and the mixture was stirred for 5 h at 50°C until 4 disappeared. The reaction mixture was then poured in ice-water and extracted with ethyl acetate, dried over MgSO4, and concentrated. The residue was purified by flash chromatograph (silica, EtOAc/hexane as eluent) to give the title compound 1d-f with the 75-90% yield.
Ethyl 4-((2-phenylquinolin-4-yl)thio)piperidine-1-carboxylate 1a
was prepared from 4-chloro-2-phenylquinoline and ethyl 4-mercaptopiperidine-1-carboxylate in 65% yield as pale yellow solid. Mp: 110-112 °C. 1H NMR (300 MHz, CDCl3): δ 8.22 (dd, J = 8.4, 3.0, 2H, Ar-H), 8.11 (J = 8.4, 1.8, 2H, Ar-H), 7.78 (s, 1H, Ar-H), 7.73 (t, J = 6.9, 1H, Ar-H), 7.56-7.47 (m, 4H, Ar-H), 4.12 (q, J = 6.0, 2H, OCH2), 4.09-4.00 (m, 2H, NCH2), 3.66-3.62 (m, 1H, SCH), 3.20-3.11 (m, 2H, NCH2), 2.13-2.10 (m, 2H, NCH2CH2), 1.78-1.72 (m, 2H, NCH2CH2), 1.26 (t, J = 6.9, 3H, OCH2CH3).13C NMR (75.5 MHz,CDCl3): δ 156.48, 155.33, 147.96, 145.14, 139.46, 130.26, 130.04, 129.44, 128.87, 127.52, 126.53, 126.36, 123.96, 117.74, 61.44, 42.97, 42.30, 31.90, 14.64. HRMS (ESI)calcd for C23H25N2O2S [M+H]+393.1637, found393.1630.
Ethyl 4-((2-chloroquinolin-4-yl)thio)piperidine-1-carboxylate 1b
was prepared from 2,4-dichloroquinoline and ethyl 4-mercaptopiperidine-1-carboxylate in 60% yield as off-white solid. Mp: 98-100 °C. 1H NMR (300 MHz, CDCl3): δ 8.14 (dd, J = 7.5, 3.0, 1H, Ar-H), 7.92 (d, J = 8.1, 1H, Ar-H), 7.73 (dt, J = 6.9, 1.5, 1H, Ar-H), 7.53 (dt, J = 8.1, 1.2, 1H, Ar-H), 7.30 (s, 1H, Ar-H), 4.32-4.26 (m, 1H, SCH), 4.15 (q, J = 7.0, 2H, OCH2), 4.10-3.90 (m, 2H, NCH2), 3.27-3.18 (m, 1H, NCH2), 2.97-2.89 (m, 1H,NCH2CH2), 2.20-2.14 (m, 1H, NCH2CH2), 2.02-1.96 (m, 1H, NCH2CH2), 1.80-1.56 (m, 1H,NCH2CH2), 1.26 (t, J = 7.0, 3H, OCH2CH3). 13C NMR (75.5 MHz,CDCl3): δ 158.92, 155.01, 154.97, 148.61, 141.41, 131.71, 128.43, 127.28, 124.18, 123.73, 120.94, 61.18, 43.47, 36.33, 35.42, 31.75, 21.14, 14.96. HRMS (ESI) calcd for C17H19N2O2NaSCl [M+Na]+, 373.0753, found 373.0746.
Ethyl 4-(pyridin-4-ylthio)piperidine-1-carboxylate 1c
was prepared from 4-chloropyridine and ethyl 4-mercaptopiperidine- 1-carboxylate in 85% yield as gray solid. Mp: 88-90 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.62 (d, J = 6.6, 2H, Ar-H), 7.90 (d, J = 6.6, 2H, Ar-H), 4.06-3.99 (m, 3H, OCH2 and SCH), 3.92-3.87 (m, 2H, NCH2), 3.15-3.07 (m, 2H, NCH2), 2.05-2.00 (m, 2H, NCH2CH2), 1.56-1.51 (m, 2H, NCH2CH2), 1.17 (t, J = 6.9, 3H, OCH2CH3).13CNMR (75.5 MHz,DMSO-d6): δ 161.39, 154.88, 140.32, 122.97, 61.22, 43.11, 40.73, 31.17, 14.98. HRMS (ESI) calcd for C13H19N2O2S [M+H]+ 267.1167, found 267.1161.
Ethyl 4-(p-tolylthio)piperidine-1-carboxylate 1d
was prepared from 4-methylbenzenethiol and ethyl 4-bromopiperidine- 1-carboxylate in 75% yield as pale yellow solid. Mp: 82-85 °C. 1H NMR (300 MHz,DMSO-d6): δ 7.34 (d, J = 8.1, 2H, Ar-H), 7.12 (d, J = 8.1, 2H, Ar-H), 4.12 (q, J = 7.2, 2H, OCH2), 4.03-3.99 (m, 2H, NCH2), 3.15-3.10 (m, 1H, SCH), 2.99-2.90 (m, 2H, NCH2), 2.34 (s, 3H, Ar-CH3), 1.94-1.88 (m, 2H, NCH2CH2), 1.58-1.49 (m, 2H, NCH2CH2), 1.25 (t, J = 7.2, 3H, OCH2CH3). 13CNMR (75.5 MHz,DMSO-d6): δ 154.89, 137.15, 132.60, 130.07, 61.07, 43.59, 43.20, 32.14, 20.93, 14.85. HRMS (ESI) calcd for C15H22NO2S [M+H]+280.1371, found 280.1367.
Ethyl 4-((4-methoxyphenyl)thio)piperidine-1-carboxylate 1e
was prepared from 4-methoxybenzenethiol and ethyl 4-bromopiperidine-1-carboxylate in 80% yield as pale yellow solid. Mp: 90-92 °C. 1H NMR (300 MHz, CDCl3): δ 7.39 (d, J = 9.0, 2H, Ar-H), 6.84 (d, J = 9.0, 2H, Ar-H), 4.10 (q, J = 7.2, 2H, OCH2), 4.04-3.95 (m, 2H, NCH2), 3.80 (s, 3H, OCH3), 3.05-2.96 (m, 1H, SCH), 2.96-2.86 (m, 2H, NCH2), 1.90-1.84 (m, 2H, NCH2CH2), 1.53-1.41 (m, 2H, NCH2CH2), 1.23 (t, J = 7.0, 3H, OCH2CH3).13CNMR (75.5 MHz,DMSO-d6): δ 159.56, 154.93, 135.67, 123.43, 115.04, 61.10, 55.55, 44.44, 43.25, 32.15, 14.89. HRMS (ESI) calcd for C15H22NO3S [M+H]+ 296.1320 found296.1309.
Ethyl 4-(naphthalen-1-ylthio)piperidine-1-carboxylate 1f
was prepared from naphthalene-1-thiol and ethyl 4-bromopiperidine- 1-carboxylate in 78% yield as pale yellow solid. Mp: 95-97 °C. 1H NMR (300 MHz, CDCl3): δ 8.54 (dd, J = 6.9, 1.2, 1H, Ar-H), 7.89-7.82 (m, 2H, Ar-H), 7.75 (dd, J = 8.0, 1.2, 1H, Ar-H), 7.42 (dd, J = 8.4, 1.2, 1H, Ar-H), 4.12 (q, J = 7.2, 2H, OCH2), 4.03-3.99 (m, 2H, NCH2), 3.28-3.21 (m, 1H, SCH), 2.98-2.89 (m, 2H, NCH2), 1.93-1.88 (m, 2H, NCH2CH2), 1.62-1.58 (m, 2H, NCH2CH2), 1.26 (t, J = 7.2, 3H, OCH2CH3). 13CNMR (75.5 MHz,DMSO-d6): δ154.93, 134.06, 133.76, 131.71, 130.86, 129.02, 128.60, 127.23, 126.78, 126.19, 125.31, 61.12, 43.77, 43.20, 32.24, 14.91. HRMS (ESI) calcd for C18H22NO2S [M+H]+ 316.1371, found 316.1363.
Ethyl 4-((7-methyl-2-oxo-2H-chromen-4-yl)thio)piperidine-1-carboxylate 1g
was prepared from 4-chloro-7-methyl-2H-chromen-2-one and ethyl 4-mercaptopiperidine-1-carboxylate in 85% yield as off-white solid. Mp: 120-122 °C.1H NMR (300 MHz, CDCl3): δ 7.61 (d, J = 8.4, 2H, Ar-H), 7.14-7.08 (m, 2H, Ar-H), 6.15 (s, 1H, Ar-H), 4.16 (q, J = 7.2, 2H, OCH2), 4.11-4.02 (m, 2H, NCH2), 3.59-3.53 (m, 1H, SCH), 3.21-3.13 (m, 2H, NCH2), 2.45 (s, 3H, Ar-CH3), 2.19-2.14 (m, 2H, NCH2CH2), 1.81-1.76 (m, 2H, NCH2CH2), 1.26 (q, J = 7.2, 3H, OCH2CH3). 13CNMR (75.5 MHz, DMSO-d6): δ 158.68, 154.90, 154.25, 152.17, 143.87, 125.87, 123.83, 117.24, 115.49, 106.81, 70.54, 61.19, 31.38, 21.38, 14.96. HRMS (ESI) calcd for C18H22NO4S [M+H]+ 348.1270, found 348.1265.
Ethyl 4-((7-methoxy-2-oxo-2H-chromen-4-yl)thio)piperidine-1-carboxylate 1h
was prepared from 4-chloro-7-methoxy-2H-chromen-2-one and ethyl 4-mercaptopiperidine- 1-carboxylate in 80% yield as off-white solid. Mp: 125-127 °C. 1H NMR (300 MHz, CDCl3): δ 7.63 (d, J = 9.0, 1H, Ar-H), 6.86-6.81 (m, 2H, Ar-H), 6.06 (s, 1H, Ar-H), 4.16 (q, J = 7.2, 2H, OCH2), 4.10-4.03 (m, 2H, NCH2), 3.88 (s, 3H, Ar-OCH3), 3.58-3.55 (m, 1H, SCH), 3.21-3.12 (m, 2H, NCH2), 2.19-2.23 (m, 2H,NCH2CH2), 1.79-1.72 (m,2H,NCH2CH2), 1.28 (t, J = 6.9, 3H, OCH2CH3).13C NMR (75.5 MHz, DMSO-d6): δ 163.10, 158.87, 154.90, 154.40, 154.05, 125.32, 112.74, 111.31, 104.78, 101.36, 61.18, 56.40, 43.16, 31.45, 14.98. HRMS (ESI) calcd for C18H22NO5S [M+H]+ 364.1219, found 364.1209.
Ethyl 4-((2-oxo-1,2-dihydroquinolin-4-yl)thio)piperidine-1-carboxylate 1i
was prepared from 4-chloroquinolin-2(1H)-one and ethyl 4-mercaptopiperidine-1-carboxylate in 72% yield as off-white solid. Mp: 132-135 °C.1H NMR (300 MHz, CDCl3): δ 12.24 (s, 1H, CONH), 7.81 (d, J = 7.2, 1H, Ar-H), 7.47-7.45 (m, 1H, Ar-H), 7.37-7.34 (m, 1H, Ar-H), 7.17-7.15 (m, 1H, Ar-H), 6.49 (s, 1H, Ar-H), 4.10 (q, J = 7.2, 2H, OCH2), 4.03-3.98 (m, 2H, NCH2), 3.56-3.49 (m, 1H, SCH), 2.15-2.09 (m, 2H,NCH2CH2), 1.71-1.67 (m, 2H,NCH2CH2), 1.22 (t, J = 7.2, 3H, OCH2CH3). 13CNMR (100 MHz, CDCl3): δ 163.02, 155.35, 150.73, 137.61, 131.06, 123.95, 122.46, 118.81, 116.77, 114.24, 61.50, 43.15, 40.95, 31.40, 14.65. HRMS (ESI) calcd for C17H21N2O3S [M+H]+ 333.1273, found 333.1272.
For the preparation of 6a-b
To a degassed solution of ethyl 4-hydroxypiperidine-1-carboxylate 5a or ethyl 4-aminopiperidine-1-carboxylate 5b24 (1.20 mmol) in 5 mL of anhydrous DMF was added NaH (60% in mineral oil, 48mg, 1.20 mmol) at 0°C. The suspension mixture was stirred for additional 15 min at room temperature. Then, a solution of the commercial 4-chloro-quinoline (163mg, 1.00 mmol) in 5 mL of anhydrous DMF was added to the above suspension mixture, and the mixture was stirred at 90°C until 4-chloro-quinoline disappeared. The reaction mixture was then poured in ice-water and extracted with ethyl acetate, dried over MgSO4, and concentrated. The residue was purified by flash chromatograph (silica, EtOAc/hexane as eluent) to give the title compound 6a-b with the 75-90% yield
Ethyl 4-(quinolin-4-yloxy)piperidine-1-carboxylate 6a
was prepared from 4-chloroquinoline and ethyl 4-hydroxypiperidine-1-carboxylate in 90% yield as pale yellow solid. Mp: 95-97°C.1H NMR (300 MHz, CDCl3): δ 8.73 (d, J = 5.0, 1H, Ar-H), 8.23 (dd, J = 8.4, 0.9, 1H, Ar-H), 8.03 (d, J = 8.7, 1H, Ar-H), 7.70 (dd, J = 9.9, 1.5, 1H, Ar-H), 7.51 (dd, J = 8.4, 0.9, 1H, Ar-H), 6.74 (d, J = 5.4, 1H, Ar-H), 4.82-4.68 (m, 1H, OCH), 4.17 (q, J = 7.2, 2H, OCH2), 3.78-3.70 (m, 2H, NCH2), 3.64-3.56 (m, 2H, NCH2), 2.04-1.93 (m, 4H,NCH2CH2), 1.26 (m, J = 7.2, 3H, OCH2CH3). 13C NMR (100 MHz, CDCl3): δ 159.77, 155.47, 151.07, 149.45, 129.83, 128.88, 125.63, 121.78, 101.39, 72.12, 61.42, 40.40, 29.97, 14.66. HRMS (ESI) calcd for C17H21N2O3 [M+H]+ 301.1552, found 301.1544.
Ethyl 4-(quinolin-4-ylamino)piperidine-1-carboxylate 6b
was prepared from 4-chloroquinoline and ethyl 4-aminopiperidine-1-carboxylate in 75% yield as white solid. Mp: 88-90 °C.1H NMR (300 MHz, DMSO-d6): δ 8.16 (d, J = 7.8, 1H, Ar-H), 7.73 (dd, J = 8.1, 1.8, 1H, Ar-H), 7.42-7.35 (m, 2H, Ar-H), 7.26(t, J = 8.1, 1H, Ar-H), 7.07 (d, J = 8.1, 1H, Ar-H), 6.61 (d, J = 7.8, 1H, Ar-H), 5.79 (d, J = 7.8, 1H, Ar-NH-CH), 4.13-3.94 (m, 4H, NCH2 and OCH2CH3), 3.75-3.68 (m, 1H, Ar-NH-CH), 3.03-2.92 (m, 2H,NCH2), 2.01-1.97 (m, 2H,NCH2CH2), 1.50-1.38 (m, 2H,NCH2CH2), 1.18(t, J = 6.9, 3H, OCH2CH3). 13C NMR (100 MHz, DMSO-d6): δ 155.17, 143.07, 134.59, 128.33, 127.16, 126.00, 124.30, 123.53, 122.10, 115.88, 104.15, 61.11, 49.45, 43.06, 31.58, 15.01. HRMS (ESI) calcd for C17H21N3O2 [M]+ 299.1634, found 299.1750.
The synthetic procedure of 8a-f is similar to that of synthetic procedure of 6a-b, respectively.
Ethyl 4-(quinolin-4-ylthio)benzoate 8a
was prepared from 4-chloroquinoline and ethyl 4-mercaptobenzoate in 85% yield as white solid. Mp: 115-117 °C. 1H NMR (300 MHz, CDCl3): δ 8.68 (d, J = 9.4, 1H, Ar-H), 8.23 (d, J = 0.9, 1H, Ar-H), 8.20 (d, J = 0.9, 1H, Ar-H), 8.10 (d, J = 8.4, 2H, Ar-H), 7.77 (dt, J = 5.7, 1.5, 1H, Ar-H), 7.60 (dt, J = 6.2, 1.2, 1H, Ar-H) 7.53 (dd, J = 6.6, 1.2, 1H, Ar-H) 7.03 (d, J = 4.5, 1H, Ar-H), 4.40 (q, J = 7.2, 2H, OCH2), 1.40 (t, J = 7.2, 3H, OCH2CH3). 13C NMR (75.5 MHz, DMSO-d6): δ 165.46, 150.50, 148.02, 144.15, 137.37, 132.91, 130.92, 130.71, 130.29, 130.26, 127.85, 126.38, 124.10, 121.70, 61.41, 14.50. HRMS (ESI) calcd for C18H16NO2S [M+H]+ 310.0902, found 310.0891.
Ethyl 4-(quinolin-4-yloxy)benzoate 8b
was prepared from 4-chloroquinoline and ethyl 4-hydroxybenzoate in 80% yield as pale white solid. Mp: 106-108 °C. 1H NMR (300 MHz, CDCl3): δ 8.74 (d, J = 4.2, 1H, Ar-H), 8.32 (d, J = 1.0, 1H), 8.17 (m, 3H, Ar-H), 7.80 (dt, J = 5.1, 0.9, 1H, Ar-H), 7.60 (dt, J = 6.3, 0.9, 1H, Ar-H), 7.23 (d, J = 3.6, 2H, Ar-H), 6.67 (d, J = 3.9, 1H, Ar-H), 4.41 (d, J = 5.1, 2H, OCH2), 1.42 (t, J = 5.4, 3H, OCH2CH3). 13CNMR (75.5 MHz, CDCl3): δ 165.39, 159.99, 158.73, 151.89, 149.81, 132.16, 130.83, 129.32, 127.12, 121.78, 121.28, 120.67, 106.71, 61.24, 40.65, 38.99, 14.54. HRMS (ESI) calcd for C18H16NO3 [M+H]+ 294.1130, found 294.1126.
Ethyl 4-(quinolin-4-ylamino)benzoate 8c
was prepared from 4-chloroquinoline and ethyl 4-aminobenzoate in 50% yield as pale white solid. Mp: 98-100 °C.1H NMR (300 MHz, DMSO-d6): δ 9.32 (s, 1H, Ar-H), 8.59 (d, J = 4.0, 1H, Ar-H), 8.33 (d, J = 6.0, 1H, Ar-NH-Ar), 7.93 (m, 3H, Ar-H), 7.73 (t, J = 1.0, 1H, Ar-H), 7.59 (t, J = 1.0, 1H, Ar-H), 7.44 (d, J = 6.0, 2H, Ar-H), 7.26 (d, J = 4.0, 1H, Ar-H), 4.29 (q, J = 6.0, 2H, OCH2), 1.31 (t, J = 6.0, 3H, OCH2CH3). 13C NMR (75.5 MHz,DMSO-d6): δ 165.78, 151.08, 149.41, 146.50, 146.17, 131.15, 129.96, 129.63, 125.59, 123.33, 122.77, 121.10, 119.35, 105.18, 60.74, 14.64. HRMS (ESI) calcd for C18H17N2O2 [M+H]+ 293.1290, found 293.1287.
Ethyl 4-(quinolin-4-yloxy)cyclohexanecarboxylate 8d
as cis/trans mixture was prepared from 4-chloroquinoline and ethyl 4-hydroxycyclohexanecarboxylate (cis/trans isomers) in 65% yield as pale white solid. Mp: 91-96°C. 1H NMR (300MHz, CDCl3): δ 8.73 (d, J = 5.4, 1H, Ar-H), 8.20 (d, J = 7.5, 0.9, 1H, Ar-H), 8.03 (d, J = 8.4, 1H, Ar-H), 7.70 (dt, J = 8.1, 1.5, 1H, Ar-H), 7.50 (dd, J = 8.1, 0.9, 1H, Ar-H), 6.74 (d, J = 5.4, 1H, Ar-H), 4.56-4.52 (m, 1H, Ar-OCH), 4.18 (q, J = 6.9, 2H, OCH2), 2.51-249 (m, 1H, CHCO2Et),2.32-2.28 (m, 2H, CH2CH2), 2.20-2.14 (m, 2H, CH2CH2), 1.74-1.66 (m, 4H, CH2CH2), 1.29 (t, J = 6.9, 3H, OCH2CH3). 13CNMR (75.5 MHz,DMSO-d6): δ 174.91, 159.83, 151.84, 149.29, 130.04, 128.90, 125.90, 122.03, 121.62, 102.56, 75.27, 60.25, 55.23, 41.37, 30.04, 26.46, 14.46. HRMS (ESI) calcd for C18H23NO3 [M+H]+ 300.1600, found 300.1591.
Ethyl 4-(quinolin-4-ylthio)cyclohexanecarboxylate 8e
as cis/trans mixture was prepared from 4-chloroquinoline and ethyl 4-mercaptocyclohexanecarboxylate (cis/trans isomers) in 80% yield as pale white solid. Mp: 94-99°C. 1H NMR (300 MHz, CDCl3): δ 8.72 (d, J = 4.8, 1H, Ar-H), 8.22 (d, J = 8.4, 1H, Ar-H), 8.09 (d, J = 8.4, 1H, Ar-H), 7.73 (dt, J = 8.1, 0.9, 1H, Ar-H), 7.59 (dt, J = 8.1, 0.9, 1H, Ar-H), 7.26 (s, 1H, Ar-H), 4.17 (q, J = 7.2, 2H, OCH2), 3.82-3.6 (m, 1H, Ar-SCH), 2.53-2.48 (m, 1H, CHCO2Et), 2.14-2.05 (m, 2H, CH2CH2), 2.01-1.96 (m, 4H, CH2CH2), 1.86-1.79 (m, 2H, CH2CH), 1.28 (t, J = 6.8, 3H2, OCH2CH3). 13CNMR (75.5 MHz,DMSO-d6): δ 174.66, 149.95, 147.50, 145.83, 130.23, 130.05, 127.02, 126.49, 123.83, 117.99, 70.54, 60.24, 41.53, 29.40, 25.50, 14.50. HRMS (ESI) calcd for C18H22NO2S [M+H]+ 316.1371, found 316.1365.
Ethyl 4-(quinolin-4-ylamino)cyclohexanecarboxylate 8f
as cis/trans mixture was prepared from 4-chloroquinoline and ethyl 4-aminocyclohexanecarboxylate (cis/trans isomers) in 55% yield as pale white solid. Mp: 87-92°C. 1H-NMR (400 MHz, CDCl3): δ 8.54 (t, J = 5.2,1H, Ar-H), 7.98 (d, J = 8.4, 1H, Ar-H), 7.71 (t, J = 7.6, 1H Ar-H), 7.63 (dt, J = 6.8, 0.9, 1H, Ar-H), 7.43 (t, J = 7.2, 1H, Ar-H), 6.44 (t, J = 5.6, 1H, Ar-H), 5.01 (d, J = 6.8, 0.6H, Ar-NH-CH), 4.90-4.80 (m, 0.4H, Ar-NH-CH), 4.20-4.11 (m, 2H, OCH2), 3.72 (b, 0.6H Ar-NH-CH), 3.54-3.50 (m, 0.4H, Ar-NH-CH), 2.60-2.56 (m, 0.6H, CHCO2Et), 2.39-2.31 (m, 0.4H, CHCO2Et), 2.16-2.08 (m, 1H, CH2CH2), 2.03-1.92 (m, 2H, CH2CH2), 1.84-1.79 (m, 2H, CH2CH2), 1.72-1.62 (m, 1H, CH2CH2), 1.39-1.32 (m, 1H, CH2CH2), 1.28 (t, J = 6.8, 3H, OCH2CH3). 13C NMR (75.5 MHz, CDCl3): δ 175.23, 174.57, 151.00, 149.36, 149.31, 148.86, 129.33, 129.29, 129.05, 123.94, 122.46, 122.35, 119.30, 119.25, 99.00, 70.55, 60.15, 50.60, 49.84, 42.34, 31.09, 28.65, 28.00, 25.66, 14.58, 14.52. HRMS (ESI) calcd for C18H23N2O2 [M+H]+ 299.1741.
For the preparation of 11a-b and 11d-e
To a degassed solution of alkyl 4-mercaptopiperidine-1-carboxylate 9a-b or alkyl 4-hydroxypiperidine-1-carboxylate 9d-e(1.20 mmol) in 5 mL of anhydrous DMF was added NaH (60% in mineral oil, 48mg, 299.1760, found 1.20 mmol) at 0°C. The suspension mixture was stirred for additional 15 min at room temperature. Then, a solution of 4-chloro-quinoline (1.00 mmol) in 5 mL of anhydrous DMF was added to the above suspension mixture, and the mixture was stirred for 5 h at 50°C until 4-chloro-quinoline disappeared. The reaction mixture was then poured in ice-water and extracted with ethyl acetate, dried over MgSO4, and concentrated. The residue was purified by flash chromatograph (silica, EtOAc/hexane as eluent) to give the title compound 11a-b and 11d-e with the 70-90% yield.
For the preparation of 11g-h
To a solution of compound 11b or 11e (1.00 mmol) in 5 mL of CH2Cl2 was added trifluoroacetic acid (1.0 mL, 1.00 mmol) at 0°C. Then, the mixture was stirred at room temperature until 11b or 11e disappeared. The reaction mixture was concentrated under reduced pressure. Then the residue was resolved in EtOAc, washed with saturated NaHCO3 solution, dried over MgSO4, and concentrated. The residue was purified by flash chromatograph (silica, EtOAc/hexane as eluent) to give the title compound 11g-h with the 90-95% yield.
For the preparation of 11c and 11f
To a degassed solution of n-hexyl alcohol (1.00 mmol) in 5 mL of degassed THF was added N,N’-carbonyldiimidazole (162mg, 1.00 mmol) at 0°C. The mixture was stirred for additional 15 min at room temperature. Then, a solution of the 11g or 11h(1.00 mmol) in 5 mL of anhydrous THF was added to the above mixture, and the mixture was stirred at room temperature for 12h. The reaction mixture was concentrated under reduced pressure, and the residue was dissolved in ethyl acetate, washed with 10% HCl and saturated NaCl solution, dried over MgSO4, and concentrated. The residue was purified by flash chromatograph (silica, EtOAc/hexane as eluent) to give the title compound 11c and 11f with the 55-70% yield.
For the preparation of 11i-j
To a degassed solution of tetrahydrofuran-3-ol or tetrahydro-2H-pyran-4-ol (1.20 mmol) in 5 mL of anhydrous DMF was added NaH (60% in mineral oil, 48mg, 1.20 mmol) at 0°C. The suspension mixture was stirred for additional 15 min at room temperature. Then, a solution of 4-chloroquinoline (1.00 mmol) in 5 mL of anhydrous DMF was added to the above suspension mixture, and the mixture was stirred for 5 h at 50°C until 4-chloro-quinoline disappeared. The reaction mixture was then poured in ice-water and extracted with ethyl acetate, dried over MgSO4, and concentrated. The residue was purified by flash chromatograph (silica, EtOAc/hexane as eluent) to give the title compound 11i-j with the 90-95% yield.
Methyl 4-(quinolin-4-ylthio)piperidine-1-carboxylate 11a
was prepared from 4-chloro-quinoline and methyl 4-mercaptopiperidine-1-carboxylate in 90% yield as yellow solid. Mp:105-107 °C. 1H NMR (400 MHz, CDCl3): δ 8.74 (d, J = 4.8, 1H, Ar-H), 8.19 (d, J = 8.4, 1H, Ar-H), 8.09 (d, J = 8.4, 1H, Ar-H), 7.73 (dt, J = 8.4, 4.8, 1H, Ar-H), 7.56 (dt, J = 8.4, 4.8, 1H, Ar-H), 7.29 (d, J = 4.8, 1H, Ar-H), 4.10-3.95 (b, 2H, NCH2), 3.70 (s, 3H, CO2CH3), 3.64-3.59 (m, 1H, Ar-SCH), 3.19-3.12 (m, 2H, NCH2), 2.16-2.05 (m, 2H,NCH2CH2), 1.80-1.65 (m, 2H,NCH2CH2). 13CNMR (100 MHz, CDCl3): δ 155.93, 149.16, 147.76, 145.69, 130.16, 130.03, 127.46, 126.83, 124.15, 118.62, 52.89, 43.29, 41.95, 31.77. HRMS (ESI) calcd for C16H19N2O2S [M+H]+ 303.1167, found 303.1160.
tert-Butyl 4-(quinolin-4-ylthio)piperidine-1-carboxylate 11b
was prepared from 4-chloro-quinoline and tert-butyl 4-mercapto-piperidine-1-carboxylate 9a in 80% yield as yellow solid. Mp: 100-102 °C. 1H NMR (300 MHz, CDCl3): δ 8.69 (d, J = 4.8, 1H, Ar-H), 8.15 (dd, J = 8.7, 0.9, 1H, Ar-H),8.03 (d, J = 8.4, 1H, Ar-H),7.67 (dt, J = 6.9, 1.5, 1H, Ar-H),7.51 (dt, J = 5.7, 1.5, 1, Ar-H H),7.24 (d, J = 4.8, 1H, Ar-H),3.95-3.88 (m, 2H, NCH2), 3.58-3.52 (m, 1H, SCH), 3.08-2.99 (m, 2H, NCH2), 2.03-1.99 (m,2H,NCH2CH2), 1.68-1.58 (m, 2H,NCH2CH2), 1.41 (s, 9H, OC4H9). 13CNMR (75.5 MHz, CDCl3): δ 154.52, 149.10, 147.69, 145.34, 129.91, 129.80, 127.19, 126.48, 123.91, 118.21, 79.77, 41.79, 31.60, 28.28. HRMS (ESI) calcd for C19H25N2O2S [M+H]+ 345.1637, found 345.1626.
n-Hexyl 4-(quinolin-4-ylthio)piperidine-1-carboxylate 11c
was prepared from 11g and n-hexyl alcohol in 55% yield as pale yellow solid. Mp: 96-99°C. 1H-NMR (400 MHz, CDCl3): δ 8.75 (d, J = 4.8, 1H, Ar-H), 8.20 (d, J = 8.4, 1H, Ar-H),8.13 (d, J = 8.4, 1H, Ar-H),7.75 (dt, J = 8.4, 1.2, 1H, Ar-H),7.60 (d, J = 7.2, 1H, Ar-H),7.31 (d, J = 4.8, 1H, Ar-H), 4.10-3.90 (m, 4H, OCH2C5H11 and NCH2), 3.65-3.63 (m, 1H, SCH), 3.20-3.13 (m, 2H, NCH2), 2.21-2.11 (m, 2H,NCH2CH2), 1.80-1.70 (m, 2H,NCH2CH2), 1.65-1.55 (m, 2H,OC2H4CH2C3H7), 1.37-1.25 (m, 6H,OC2H4C3H6CH3), 0.91 (t, J = 3.6, 3H, OC5H10CH3). 13C NMR (100 MHz, CDCl3): δ 155.61, 150.02, 146.83, 145.42, 132.93, 129.95, 128.85, 128.34, 117.63, 68.07, 45.43, 40.12, 31.56, 31.27, 30.45, 28.86, 25.52, 22.40, 14.16, 149.17, 147.77, 145.69, 130.12, 130.03, 127.45, 126.80, 124.14, 118.60, 65.90, 43.22, 42.00, 31.79, 31.59, 29.08, 25.75, 22.68, 14.16.HRMS (ESI) calcd for C21H29N2O2S [M+H]+ 373.1950 found 373.1945.
Methyl 4-(quinolin-4-yloxy)piperidine-1-carboxylate 11d
was prepared from 4-chloroquinoline and methyl 4-hydroxypiperidine-1-carboxylate in 82% yield as orange colour solid. Mp: 102-104 °C. 1H-NMR (300 MHz, CDCl3): δ 8.73 (d, J = 5.1, 1H, Ar-H), 8.22 (dd, J = 8.4, 1H, Ar-H), 8.04 (d, J = 8.4, 1H, Ar-H), 7.70 (dt, J = 8.1, 0.9, 1H, Ar-H), 7.50 (dt, J = 8.4, 0.9, 1H, Ar-H), 6.74 (d, J = 5.1, 1H, Ar-H), 4.87-4.75 (m, 1H, OCH), 3.79-3.68 (m, 5H, CO2CH3 and, NCH2), 3.64-3.56 (m, 2H, NCH2), 2.08-1.95 (m, 4H,NCH2CH2). 13C NMR (75.5 MHz,DMSO-d6): δ 159.61, 155.52, 151.76, 149.13, 130.23, 128.78, 126.12, 122.12, 121.61, 102.74, 72.51, 52.78, 40.82, 30.05. HRMS (ESI) calcd for C16H19N2O3 [M+H]+ 287.1396, found 287.1400.
tert-Butyl 4-(quinolin-4-yloxy)piperidine-1-carboxylate 11e
was prepared from 4-chloroquinoline and tert-butyl 4-hydroxypiperidine-1-carboxylate 9b in 75% yield as pale yellow solid. Mp: 95-97°C.1H NMR (400 MHz, CDCl3): δ 8.74 (d, J = 5.1, 1H, Ar-H), 8.23 (dd, J = 6.3, 0.9, 1H, Ar-H), 8.03 (d, J = 8.4, 1H, Ar-H), 7.71 (t, J = 6.6, 1H, Ar-H), 7.51 (t, J = 6.6, 1H, Ar-H), 6.74 (d, J = 4.2, 1H, Ar-H), 4.84-4.81 (m, 1H, OCH), 3.74-3.68 (m, 2H,NCH2), 3.56-3.50 (m, 2H,NCH2), 2.07-1.94 (m, 4H,NCH2CH2), 1.29 (s, 9H, OC4H9). 13C NMR (75.5 MHz,DMSO-d6): δ 159.48, 154.32, 151.84, 149.37, 130.10, 128.97, 126.01, 122.11, 121.64, 102.69, 79.20, 72.62, 30.20, 28.68.HRMS (ESI) calcd for C19H26N2O3 [M+H]+ 329.1865, found 329.1855.
n-Hexyl 4-(quinolin-4-yloxy)piperidine-1-carboxylate 11f
was prepared from 11h and n-hexyl alcohol in 65% yield as pale yellowsolid. Mp: 88-90 °C. 1H-NMR (400 MHz, CDCl3): δ 8.73 (d, J = 5.4, 1H, Ar-H), 8.21 (dd, J = 8.4, 1.2, 1H, Ar-H), 8.06 (d, J = 8.4, 1H, Ar-H), 7.70 (dt, J = 5.4, 1.2, 1H, Ar-H), 7.52 (dd, J = 8.0, 0.8, 1H, Ar-H), 6.74 (d, J = 5.6, 1H, Ar-H), 4.90-4.80 (m, 1H, OCH), 4.09 (d, J = 6.7, 2HOCH2C5H11), 3.80-3.70 (m, 2H), 3.65-3.51 (m, 2H,NCH2), 2.10-1.90 (m, 4H,NCH2CH2), 1.70-1.60 (m, 2H,OC2H4CH2C3H7), 1.40-1.30 (m, 6H,OC2H4C3H6CH3), 1.28 (t, J = 6.3, 3H, OC5H10CH3). 13C NMR (100 MHz, CDCl3): δ 160.22, 155.76, 151.01, 149.25, 130.23, 128.82, 125.97, 122.02, 121.98, 101.53, 72.42, 65.92, 40.58, 31.61, 30.16, 29.11, 25.77, 22.69, 14.16. HRMS (ESI) calcd for C21H29N2O2S [M+H]+373.1950 373.1948.
4-(Piperidin-4-ylthio)quinoline hydrochloride 11g
was prepared from 11b by the treatment of TFA in 90% yield, which was converted its hydrochloride salt as yellow solid. Mp: found 99-103 °C. 1H NMR (300 MHz, DMSO-d6): δ 9.20 (b, 2H, (CH2)NH·HCl), 8.99 (d, J = 6.0, 1H, Ar-H), 8.27 (d, J = 9.0, 2H, Ar-H), 8.10 (t, J = 6.9, 1H, Ar-H), 7.96 (d, J = 6.6, 1H, Ar-H), 7.85 (t, J = 7.2, 1H, Ar-H), 4.40-4.15 (m, 1H, SCH), 3.40-3.31 (m, 2H,NCH2), 3.15-3.10 (m, 2H,NCH2), 2.31-2.25 (m, 2H,NCH2CH2), 1.96-1.93 (m, 2H,NCH2CH2). 13C NMR (75.5 MHz,DMSO-d6): δ 157.75, 143.67, 137.86, 134.47, 129.69, 125.65, 124.30, 122.99, 116.96, 42.87, 40.66, 28.15. HRMS (ESI) calcd for C14H17N2S [M+H-HCl]+ 245.1112, found 245.1111.
4-(Piperidin-4-yloxy)quinolone 11h
was prepared from 11e by the treatment of TFA in 95% yield as off-yellow solid. Mp: 93-95°C. 1H NMR (400 MHz, CDCl3): δ 8.72 (d, J = 5.2, 1H, Ar-H), 8.26 (d, J = 8.4, 1H, Ar-H), 8.02 (d, J = 8.4, 1H, Ar-H), 7.70 (dt, J = 6.0, 1.2, 1H, Ar-H), 7.51 (t, J = 7.6, 1H, Ar-H), 6.74 (d, J = 5.2, 1H, Ar-H), 5.69-5.31 (b, 1H, (CH2)NH), 4.47-4.71 (m, 1H, OCH), 3.25-3.19 (m, 2H,NCH2), 2.88-2.81 (m, 2H,NCH2), 2.15-2.10 (m, 2H,NCH2CH2), 1.91-1.75 (m, 2H,NCH2CH2). 13C NMR (75.5 MHz,DMSO-d6): δ 159.63, 151.87, 149.38, 130.04, 129.00, 125.93, 122.07, 121.74, 102.70, 74.37, 43.62, 40.69, 32.00. HRMS (ESI) calcd for C14H17N2O [M+H]+ 229.1341, found 229.1338.
4-((Tetrahydrofuran-3-yl)oxy)quinolone 11i
was prepared from 4-chloroquinoline and tetrahydrofuran-3-ol in 90% yield as pale yellow solid. Mp: 87-90 °C. 1H NMR (300 MHz, CDCl3): δ 9.07 (b, 1H, Ar-H), 8.63 (d, J = 6.3 1H, Ar-H), 8.34 (d, J = 6.3, 1H, Ar-H), 7.99 (t, J = 6.0 1H, Ar-H), 7.78 (t, J = 6.3 1H, Ar-H), 7.34 (b, 1H, Ar-H), 5.52 (s, 1H, OCH), 4.26-4.11 (m, 3H, OCH2CHCH2 and CH2OCH2CH2), 4.05-3.95 (m, 1H, CH2OCH2CH2), 2.33 (b, 1H, CH2OCH2CH2), 2.20 (b, 1H, CH2OCH2CH2). 13C NMR (75.5 MHz, CDCl3): δ 166.91, 146.81, 139.27, 134.84, 129.12, 123.46, 120.86, 103.80, 82.05, 72.35, 70.54, 66.98, 32.69. HRMS (ESI) calcd for C13H14NO2 [M+H]+ 216.1025, found 216.1022.
4-((Tetrahydro-2H-pyran-4-yl)oxy)quinolone 11j
was prepared from 4-chloroquinoline and tetrahydro-2H-pyran-4-ol in 95% yield as yellow solid. Mp: 87-91 °C. 1H NMR (400 MHz, CDCl3): δ 8.97 (b, 1H, Ar-H), 8.69 (d, J = 6.3, 1H, Ar-H), 8.38 (d, J = 6.3, 1H, Ar-H), 8.01 (t, J = 6.0, 1H, Ar-H), 7.80 (t, J = 5.7, 1H, Ar-H), 7.26-7.16 (m, 1H, Ar-H), 5.12 (b, 1H, OCH), 4.10-4.07 (m, 2H, CH2OCH2CH2), 3.77-3.72 (m, 2H, CH2OCH2CH2), 2.35-2.25 (m, 2H, CH2OCH2CH), 2.06-2.04 (m, 2H2,CH2OCH2CH2). 13C NMR (75.5 MHz, CDCl3): δ 166.80, 146.72, 139.30, 134.81, 129.06, 123.47, 120.94, 120.78, 103.63, 75.82, 64.48, 31.21. HRMS (ESI) calcd for C14H16NO2 [M+H]+ 230.1181, found 230.1180.
For the preparation of 12a-g
To a degassed solution of 11g-h(1.00 mmol) and Boc-AA-CO2H (1.10 mmol), and HOBt (1.10 mmol) in 10 mL of anhydrous DCM was added EDC*HCl (230 mg, 1.20 mmol) at 0°C. The mixture was stirred over night at room temperature. Then, the reaction was quenched with H2O (15.00 mL). The mixture was extracted with DCM (20.0 mL), and organic phase was sequently washed with 10% HCl, saturated NaCl solution, saturated NaHCO3 solution, and saturated NaCl solution, dried over MgSO4, and concentrated. The residue was purified by flash chromatograph (silica, EtOAc/hexane as eluent) to give the title compound 12a-g with the 65-85% yield.
(S)-tert-Bbutyl (1-oxo-3-phenyl-1-(4-(quinolin-4-ylthio)piperidin-1-yl)propan-2-yl)carbamate 12a
was prepared from 11g and Boc-Phe-OH in 85% yield as pale yellow solid. Mp: 180-182 °C. 1H NMR (400 MHz, CDCl3): δ 8.71(d, J = 4.8, 1H, Ar-H), 8.15 (d, J = 6.0, 1H, Ar-H), 8.09-8.06 (m, 1H, Ar-H), 7.73-7.70 (m, 1H, Ar-H), 7.60-7.54 (m, 1H, Ar-H), 7.35-7.12 (m, 6H, Ar-H), 5.50-5.44 (m, 1H, NHBoc), 4.87-4.84 (m, 1H, CHNHBoc), 4.32-4.10 (m, 1H, SCH), 3.63-3.57 (m, 1H, NCH2), 3.51-3.45 (m, 1H, NCH2), 3.20-3.01 (m, 1HNCH2), 2.99-2.90 (m, 2.5H, PhCH2CH and NCH2),2.75-2.60 (m, 0.5H, PhCH2CH), 2.10-1.92 (m, 2H,NCH2CH2), 1.80-1.53 (m, 2H,NCH2CH2), 1.40 (s, 9H, NHCO2C4H9). 13C NMR (100 MHz, CDCl3): δ 170.35, 155.22, 149.26, 147.89, 145.32, 136.64, 130.12, 129.73, 128.86, 127.49, 127.34, 127.11, 126.86, 124.16, 118.92, 79.94, 51.12, 45.13, 44.49, 41.49, 40.62, 31.84, 28.50. HRMS (ESI) calcd for C28H34N3O3S [M+H]+ 492.2321, found 492.2316.
(S)-tert-Butyl (4-amino-1,4-dioxo-1-(4-(quinolin-4-ylthio)piperi-din-1-yl)butan-2-yl)carbamate 12b
was prepared from 11g and Boc-Asn-OH in 73% yield as pale yellow solid. Mp: 118-120 °C.1H NMR (300 MHz, CDCl3): δ 8.75 (d, J = 4.0, 1H, Ar-H), 8.18 (d, J = 8.0, 1H, Ar-H), 8.10 (d, J = 8.4, 1H, Ar-H), 7.73 (t, J = 7.2, 1H, Ar-H), 7.56 (d, J = 7.2, 1H, Ar-H), 7.29 (d, J = 3.6, 1H, Ar-H), 6.20-6.18 (m, 1H, CONH2), 5.66-5.63 (m, 1H, NHBoc), 5.42-5.39 (m, 1H, CONH2), 4.94-4.89 (m, 1H, CHNHBoc), 4.26-3.96 (m, 1H,NCH2), 3.66-3.60 (m, 1H,NCH2), 3.35-3.08 (m, 2H,NCH2CH2), 2.65-2.47 (m, 2H, CH2CONH2), 2.15-2.06 (m, 2H,NCH2CH2), 1.37 (s, 9H, NHCO2C4H9). 13C NMR (100 MHz, CDCl3): 167.30, 154.81, 149.35, 147.95, 144.99, 144.86, 130.18, 127.47, 126.94, 124.15, 119.04, 116.88, 81.12, 47.04, 45.32, 44.69, 42.07, 41.69, 41.57, 41.47, 32.35, 32.08, 31.57, 31.27, 28.40. HRMS (ESI) calcd for C23H31N4O4S [M+H]+ 459.2066, found 459.2068.
(S)-tert-Butyl (2-oxo-1-phenyl-2-(4-(quinolin-4-ylthio)piperidin-1-yl)ethyl)carbamate 12c
was prepared from 11g and L-2-((tert-butoxycarbonyl)amino)-2-phenylacetic acid 65% yield as pale yellow solid. Mp: 90-92 °C. 1H NMR (400 MHz, CDCl3): δ 8.70 (t, J = 2.8, 1H, Ar-H), 8.20 (d, J = 5.2, 1H, Ar-H), 8.10-8.05 (m, 1H, Ar-H), 7.73-7.70 (m, 1H, Ar-H),7.58-7.50 (m, 1H, Ar-H),7.35-7.26 (m, 5H, Ar-H),7.20 (d, J = 4.8, 1H, Ar-H), 6.08-6.03 (m, 1H, NHBoc), 5.57 (t, J = 6.8, 1H, PhCHNHBoc), 4.48-4.30 (m, 0.5H, SCH),4.20-4.10 (m, 0.5H, SCH),3.77-3.71 (m, 1H,NCH2), 3.60-3.54 (m, 1H,NCH2),3.48-3.46 (m, 0.5H,NCH2),3.40-3.08 (m, 1.5H,NCH2), 2.10-2.00 (m, 2H,NCH2CH2),1.90-1.50 (m, 2H,NCH2CH2),1.42 (s, 9H, NHCO2C4H9). 13C NMR (100 MHz, CDCl3): δ 168.48, 155.21, 149.23, 147.90, 144.92, 138.26, 130.14, 129.34, 128.54, 127.76, 126.90, 124.13, 118.94, 79.93, 55.44, 44.78, 41.74, 31.43, 28.51. HRMS (ESI) calcd for C27H32N3O3S [M+H]+ 478.2164, found 478.2145.
(S)-tert-Butyl 2-(4-(quinolin-4-ylthio)piperidine-1-carbonyl)-pyrrolidine-1-carboxylate12das tautomer was prepared from 11g
and Boc-Pro-OH in 72% yield as white solid. Mp: 168-171 °C. 1H NMR (400 MHz, CDCl3): δ 8.69 (s, 1H, Ar-H), 8.12 (s, 1H, Ar-H), 8.03 (s, 1H, Ar-H), 7.67 (s, 1H, Ar-H), 7.51 (s, 1H, Ar-H), 7.24 (s, 1H, Ar-H), 4.62-4.49 (m, 1H, COCHNBocCH2), 4.40-4.04 (m, 1H, SCH), 3.96-3.00 (m, 6H,NCH2 and CH2CH2NBoc), 2.40-1.50 (m, 8H,NCH2CH2, CH2CH2NBoc and CHCH2CH2CH2NBoc), 1.41 (s, 9H, NHCO2C4H9). 13C NMR (100 MHz, CDCl3): δ 170.82, 170.73, 154.60, 154.03, 149.20, 147.81, 147.72, 147.62, 145.43, 145.13, 130.15, 130.06, 129.87, 127.38, 126.86, 124.08, 118.93, 118.63, 79.64, 57.06, 56.84, 56.46, 46.84, 46.61, 44.68, 44.46, 44.09, 42.03, 41.88, 41.77, 41.59, 41.43, 41.10, 32.58, 32.31, 32.12, 32.02, 31.49, 30.76, 30.57, 30.01, 29.77, 28.64, 28.54, 24.32, 23.68, 23.45.HRMS (ESI) calcd for C24H32N3O3S [M+H]+ 442.2164, found 442.2158.
(S)-tert-Butyl (1-oxo-3-phenyl-1-(4-(quinolin-4-yloxy)piperidin-1-yl)propan-2-yl)carbamate 12e
was prepared from 11h and Boc-Phe-OH in 85% yield as white solid. Mp: 132-135 °C. 1H NMR (400 MHz, CDCl3): δ 8.67 (s, 1H, Ar-H), 8.10 (d, J = 5.4, 1H, Ar-H), 8.01 (t, J = 9.2, 1H, Ar-H), 7.70-7.60 (m, 1H, Ar-H), 7.51 (td, J = 28.2, 6.9, 1H, Ar-H), 7.26-7.10 (m, 5H, Ar-H), 6.60 (dd, J = 10.5, 5.2, 1H, Ar-H), 5.59 (d, J = 6.0, 1H, NHBoc), 4.87 (d, J = 6.8, 1H, CHNHBoc), 4.65 (d, J = 40.0, 1H, OCH), 3.80 (dd, J = 50.2,13.2, 1H, PhCH2CH), 3.65-3.40 (m, 2H, PhCH2CH and NCH2), 3.35-3.10 (m, 1H, NCH2), 3.05-2.90 (m, 2H, PhCH2CH and NCH2), 2.00-1.60 (m, 2H,NCH2CH2), 1.39 (s, 9H, NHCO2C4H9). 13C NMR (100 MHz, CDCl3): δ 170.3 159.82, 155.28, 151.13, 149.34, 136.84, 130.15, 129.80, 129.71, 128.98, 128.90, 128.70, 127.11, 125.94, 121.85, 101.53, 79.86, 71.62, 51.12, 42.13, 40.64, 38.59, 30.21, 28.50. HRMS (ESI) calcd for C28H34N3O4 [M+H]+ 476.2549, found 476.2540.
(S)-tert-Butyl 2-(4-(quinolin-4-yloxy)piperidine-1-carbonyl)-pyrrolidine-1-carboxylate 12f
as tautomer was prepared from 11h and Boc-Pro-OH in 77% yield as white solid. Mp: 106-108 °C. 1H NMR (400 MHz, CDCl3): δ 8.72 (d, J = 5.2, 1H, Ar-H), 8.18 (t, J = 8.0, 1Hv), 8.03 (d, J = 8.4, 1H, Ar-H), 7.69 (t, J = 7.6, 1H, Ar-H), 7.50 (d, J = 6.8, 1H, Ar-H),6.73 (d, J = 4.4, 1H, Ar-H), 4.90-4.80 (m, 1H, COCHNBocCH2), 4.60 (dd, J = 54.5, 6.8, 1H, OCH), 4.01-3.40 (m, 6H,NCH2 and CH2CH2NBoc), 2.47-1.75 (m, 8H,NCH2CH2, CH2CH2NBoc and CHCH2CH2CH2NBoc), 1.44 (s, 4.5H, NHCO2C4H9), 1.41 (s, 4.5H, NHCO2C4H9). 13C NMR (100 MHz, CDCl3): δ 170.82, 170.70, 159.82, 159.65, 154.46, 153.83, 150.94, 149.24, 149.12, 129.99, 128.81, 128.72, 128.56, 125.71, 121.68, 121.59, 101.32, 79.48, 79.43, 71.89, 56.89, 56.73, 56.31, 56.21, 46.68, 41.93, 41.69, 41.44, 38.93, 38.76, 38.53, 38.43, 30.78, 30.47, 30.37, 30.06, 29.83, 29.68, 28.44, 28.40, 24.13, 23.44, 23.33. HRMS (ESI) calcd for C24H32N3O4 [M+H]+ 426.2393, found 426.2386.
(S)-tert-Butyl (4-amino-1,4-dioxo-1-(4-(quinolin-4-yloxy)-piperidin-1-yl)butan-2-yl)carbamate 12g was prepared from 11h
and Boc-Asn-OH in 85% yield as white solid. Mp: 108-110 °C. 1H NMR (400 MHz, CDCl3): δ 8.75 (d, J = 5.6, 1H, Ar-H), 8.22 (dd, J = 8.4, 2.9, 1H, Ar-H), 8.06 (d, J = 8.4, 1H, Ar-H), 7.72 (dt, J = 6.9, 1.6, 1H, Ar-H), 7.52 (d, J = 7.5, 1H, Ar-H), 6.75 (dd, J = 4.8, 3.2, 1H, Ar-H), 6.36 (b, 1H, CONH2), 5.76 (dd, J = 38.0, 9.0, 1H, OCH),5.58 (b, 1H, CONH2), 5.03 (q, J = 8.0, 1H,CHNHBoc),4.92 (d, J = 3.6, 1H, NHBoc),4.08-3.61 (m, 4H,NCH2), 2.75-2.68 (m, 1H, CH2CONH2), 2.65-2.55 (m, 1H, CH2CONH2), 2.30-2.00 (m, 4H,NCH2CH2), 1.48 (s, 9H, NHCO2C4H9). 13C NMR (100 MHz, CDCl3): δ 172.56, 170.05, 160.68, 155.48, 150.52, 148.17, 130.73, 128.16, 126.34, 122.01, 121.87, 101.67, 80.47, 72.49, 72.07, 39.23, 30.69, 29.92, 28.47. HRMS (ESI) calcd for C23H31N4O5 [M+H]+ 443.2294, found 443.2290.
Cells and viruses
Madin-Darby canine kidney cell (MDCK), HEK 293 cells and Hela cells were obtained from China Center for Type Culture Collection. Influenza A/udorn/72 (H3N2), A/PR/8/34 (H1N1), A1/Jingfang 86-1(H1N1), and A3/Lufang93-1(H3N2)China Center for Type Culture Collection. B/Shenzhen/747 were obtained from Shenzhen Centers for Disease Control and Prevention (China).
MDCK cell-based anti-influenza assay for the high-throughput screening protocol in 96-well plate
The flat-bottomed 96-well plate (plate A) was added 110 μL of DMEM with 0.1% TCPK-trypsin and 1% antibiotics in each well. Then, 1 μL of 5mM of the positive control solution (Ribavirin) was added to wells 12-A, B, C and D, and 1 μL of 1μM of negative control solution (F136) was added to 12-E, F, G and H in plate A. The remaining wells was added 1 μL of compound solution (each well contains a compound at 10 μg/100 μL in 1% DMSO, from MyriaScreen from Sigma-Aldrich). Another 96-well plates, named as plate B, with MDCK cells growing in monolayer was washed with PBS, and infected with 100 PFU A/Udorn/72 influenza viruses in each well except for well 1-A, B, C and D as blank. After infection for one hour,plate B was washed with PBS. Then, 100 μL of compound-medium in plate A was added to plate B. The resulting plate was incubated for 48h in the incubator at 37°C under 5% CO2. The plate were fixed and stained with 0.5% crystal violet solution. Wells as blank control and Ribavirin control will be colour as deep purple. Wells in which cytopathic effects occurred will be clear or partially clear. Positive inhibition of virus infection by the screening compound is identified when the well is stained the same as the ribavirin control.
Determination of EC50 of piperidine-based derivatives by MDCK cell-based anti-influenza assay
The anti-influenza virus activities of piperidine-based derivatives were measured by the EC50 values. MDCK cells were seeded (1.2 × 105cells/well in DMEM with 10% FBS) in 12-well tissue culture plates and incubated for 48 hours at 37 °C in a humidified 5% CO2 incubator. The cells were infected with virus (100PFU, Udorn-4) in the incubator for 1 h. After adsorption of the virus, the virus suspension was removed and the cells were washed with DMEM, The cell monolayer were then overlaid with DMEM medium containing 1% agar and serial compounds dilutions (2-fold serial dilutions for 6 gradients, each concentration measured 2 wells) in the presence of 1%thouTCPK-trypsinand 1% antibiotics. After infection for 48 hours, the monolayer were fixed and stained with 0.5% crystal violet solution. The number of virus-induced plaques was counted.
in vitro Cytotoxic studies of 11e
Cytotoxicity of 11e was assessed by MTT assays. 5×104 cells/well were seeded on a flat-bottomed 96-well tissue culture plates, and incubated for 18 h at 37 °C for cell adherence. Then the medium was removed and replenished with 100μl of DMEM containing different concentrations of 11e ranging from 32μM to 10000μM for 24 h in an atmosphere of 5% CO2 at 37 °C. For the MTT assay, which evaluates mitochondrial viability, 20 μL of MTT solution (5 mg/mL) was added, and the plates were incubated for an additional 4 h. After incubation, the supernatant was carefully removed from the wells followed by the addition of 150 μL DMSO with thorough mixing. The optical density at 570 and 630 nm (background) was determined on an ELISA reader. The cell viability was expressed as the percentage of control absorbance obtained in untreated cells after subtracting the absorbance from an appropriate background. Last, the minimum lethal dose for 50% of the cells (MLD50) was determined.
Time-of-addition assay
MDCK cells (1.2 × 105 cells/well) were seeded in 12-well tissue culture plates and incubated for 48 hours. The cells were infected with virus (100PFU) in the incubator for 1 h. After adsorption of the virus, the virus suspension was removed and the cells were washed with DMEM, then replenished with fresh virus medium (DMEM with 1% antibiotics and 1%thou TPCK-trypsin). The test medium containing the test compound solution was added during the periods −2 to 0 h (adsorption), 0-2, 1-3, 2-4, 4-6, 6-8, and 18-20 h. After each incubation period, the monolayer was washed with DMEM and incubated with fresh virus medium. After 24 hours, the supernatant was collected and the viral yield was measured. Statistical analysis was analysed by ANOVA followed by Tukey’smultiple comparison test using Prism 5.0b.
Supplementary Material
Acknowledgements
The work was supported financially by the National Natural Science Foundation of China (Grants 81161120401) to Guoxin Wang; in part by a NIH grant (AI080669) to Ming Luo. We also thank the Shenzhen Science and Technology Plan Project (Grant No. ZDSY20130331145112855) for the funding support.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
References
- 1.Smith GJD, Vijaykrishn D, Bahl J, Lycett SJ, Worobey M, Pybus OG, Ma SK, Cheung CL, Raghwani J, Bhatt S, Peiris JSM, Guan Y, Rambaut A. Nature. 2009;459:1122. doi: 10.1038/nature08182. [DOI] [PubMed] [Google Scholar]
- 2.Nalluswami K, Nambia A, Perrianne L, Moll M, Lute J, Simwale O. MMWR, Morbidity Mortality WklyRep. 2011;60:1213. [Google Scholar]
- 3.Global animal epidemic situation review in 2011. China Journal of Animal Quarantine. 2011;28(12):82. [Google Scholar]
- 4.Gao R, Cao B, Hu Y. N. Engl. J. Med. 2013;368:1888. doi: 10.1056/NEJMoa1304459. [DOI] [PubMed] [Google Scholar]
- 5.Hayden F. Clin Infect Dis. 2009;48(Suppl 1):S3. doi: 10.1086/591851. [DOI] [PubMed] [Google Scholar]
- 6.LAgoja IM, De Clercq E. Med. Res. Rev. 2008;28:1. doi: 10.1002/med.20096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Itzstein M. Nat. Rev. Drug Discovery. 2007;6:967. doi: 10.1038/nrd2400. [DOI] [PubMed] [Google Scholar]
- 8.Hsieh HP, Hsu JT. Curr. Pharm. Des. 2007;13:3531. doi: 10.2174/138161207782794248. [DOI] [PubMed] [Google Scholar]
- 9.De Clercq E E. Nat. Rev. Drug Discovery. 2006;5:1015. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palese P, Compans RW. The Journal of General Virology. 1976;33:159. doi: 10.1099/0022-1317-33-1-159. [DOI] [PubMed] [Google Scholar]
- 11.Liu C, Eichelberger MC, Compans RW, Air GM. Journal of Virology. 1995;69:1099. doi: 10.1128/jvi.69.2.1099-1106.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gubareva LV, Kaiser L, Matrosovich MN, Soo-Hoo Y, Hayden FG. J. Infect. Dis. 2001;183:523. doi: 10.1086/318537. [DOI] [PubMed] [Google Scholar]
- 13.Moscona A. N. Engl. J. Med. 2009;360:953. doi: 10.1056/NEJMp0900648. [DOI] [PubMed] [Google Scholar]
- 14.Carrat F, Flahault A. Vaccine. 2007;25:6852. doi: 10.1016/j.vaccine.2007.07.027. [DOI] [PubMed] [Google Scholar]
- 15.Treanor J. N. Engl. J. Med. 2004;350:218. doi: 10.1056/NEJMp038238. [DOI] [PubMed] [Google Scholar]
- 16.Teissier E, Penin F, Pécheur E-I. Molecules. 2011;16:221. doi: 10.3390/molecules16010221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MyriaScreen library was obtained from Sigma-Aldrich.
- 18.MDCK was obtained from China Center for Type Culture Collection.
- 19.The fusion inhibitor F136 was prepared according to the reference: Wang GX, Luo M M, Yang Z. 2011 WO2011160024.
- 20.Savini L, Gaeta A, Fattorusso C, Catalanotti B, Campiani G, Chiasserini L, Pellerano C, Novellino E, McKissic D, Saxena A. J. Med. Chem. 2003;46:1. doi: 10.1021/jm0255668. [DOI] [PubMed] [Google Scholar]
- 21.Ethyl 4-mercaptopiperidine-1-carboxylate 3 was prepared following to the literature: Hornberger K, Cheung M, Pobanz MA, Emmitte KA, Kuntz KY, Badiang JB. JB. 2007 WO2007030366.
- 22.Ethyl 4-bromopiperidine-1-carboxylate 4 was prepared following to the literature: Astles PC, Baker SR, Bonnefous C, Vernier JM, Keenan M, Sanderson AJ. 2003 WO2003062224.
- 23.Ethyl 4-aminopiperidine-1-carboxylate 5b was prepared following to the literature: Miriyala B, Bhattacharyya S, Williamson JS. Tetrahedron. 2004;60:1463.
- 24.Kong J, Chen CY, Balsells-Padros J, Cao Y, Dunn RF, Dolman SJ, Janey J, Li H, Zacuto MJ. J. Org.Chem. 2012;77:3820. doi: 10.1021/jo3001595. [DOI] [PubMed] [Google Scholar]
- 25.Jung ME, Deng G. J. Org.Chem. 2012;77:11002. doi: 10.1021/jo302308q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyer E, Botos I, Scapozza L, Zhang D. Perspectives in Drug Discovery and Design. 1995;3:168. [Google Scholar]
- 27.Mazaleyrat JP, Rage I, Xie J, Savrda J, Wakselman M. Tetrahedron Letters. 1992;33:4453. [Google Scholar]
- 28.An J, Lee DCW, Law AHY, Yang CLH, Poon LLM, Lau ASY, Jones SJM. J. Med. Chem. 2009;52:2667. doi: 10.1021/jm800455g. [DOI] [PubMed] [Google Scholar]
- 29.Yeh JY, Coumar MS, Horng JT, Shiao HY, Kuo FM, Lee HL, Chen IC, Chang CW, Tang WF, Tseng SN, Chen CJ, Shih SR, Hsu JTA, Liao CC, Chao YS, Hsieh HP. J. Med. Chem. 2010;53:1519. doi: 10.1021/jm901570x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.