

Abstract
In rodents, chronic intermittent ethanol vapor exposure (CIE) produces alcohol dependence, alters the activity of pyramidal neurons and decreases the number of glial progenitors in the medial prefrontal cortex (mPFC). Adult male Wistar rats were exposed to CIE and were injected with mitotic markers to label and phenotype proliferating cells to test the hypothesis that CIE produces concurrent alterations in the structure of pyramidal neurons and the cell cycle kinetics and developmental stages of glial progenitors in the mPFC. Medial prefrontal cortical tissue was processed for Golgi-Cox staining, immunohistochemistry and Western blotting analysis. CIE increased dendritic arborization and spine densities within basal and apical dendrites of pyramidal neurons via aberrant reorganization of actin cytoskeleton-associated molecules. CIE concomitantly increased expression of total NR2B subunits without affecting phosphorylation of NR2B at Tyr-1472 or levels of PSD-95. CIE reduced the length of S phase of the cell cycle of glial progenitors and reduced proliferation and differentiation of progenitors into bHLH transcription factor Olig2-expressing premyelinating oligodendrocyte progenitor cells (OPCs). CIE also produced a corresponding hyperphosphorylation of Olig2, and reduced expression of myelin basic protein. Our findings demonstrate that CIE-induced alterations in OPCs and myelin-related proteins are associated with profound alterations in the structure of pyramidal neurons. In sum, our results not only provide evidence that alcohol dependence leads to pathological changes in the mPFC, which may in part define a cellular basis for cognitive impairments associated with alcoholism, but also show dependence-associated morphological changes in the PFC at the single neuron level.
Keywords: Chronic ethanol, Vapor induced dependence, BrdU, IdU, CldU, Olig2, MBP
Introduction
Alcoholism in humans impairs the structure of, and function dependent on, the frontal cortex (Bechara et al., 2001; George et al., 2001; Tapert et al., 2004a; Tapert et al., 2004b; De Bellis et al., 2005; Medina et al., 2008; Squeglia et al., 2012; Bava et al., 2013; Seo et al., 2013), but the underlying cellular mechanisms contributing to these deleterious effects are unclear. For example, alcoholics show deficits in executive function, declarative memory, and impulsivity, effects that indicate frontal cortex dysfunction (Parsons and Nixon, 1993; Sullivan et al., 2000; Bechara et al., 2001). These deficits are associated with neurodegeneration and loss of myelin (Lewohl et al., 2005; Pfefferbaum and Sullivan, 2005; Ridge et al., 2008; Jacobus et al., 2009; McQueeny et al., 2009; MacKay et al., 2011; Skuja et al., 2012; Bava et al., 2013), suggesting alterations in structure and function of the frontal cortex. Investigating the effects of chronic alcohol exposure on cortical neural systems and myelin in rodent models can help elucidate the neurobiological changes contributing to the pathology of alcoholism in humans (Heilig and Egli, 2006).
Early in vitro studies have reported that chronic intermittent ethanol exposure alters the kinetics and function of N-methyl-D-aspartate-type glutamate receptors (NMDARs) in cortical neurons and these effects were associated with enhanced expression of NMDA receptor subunit 2B (NR2B; (Hu and Ticku, 1995; Hu et al., 1996)). Recent ex vivo slice physiology studies in the medial prefrontal cortex (mPFC) have demonstrated that acute ethanol treatment decreases sustained depolarization that occurs in pyramidal neurons during up-states, indicating that ethanol decreases NMDAR-mediated excitatory postsynaptic currents (Tu et al., 2007; Weitlauf and Woodward, 2008; Woodward and Pava, 2009). Such mechanistic studies have been extended in animal models of chronic ethanol exposure to demonstrate that ethanol alters the functional and structural plasticity of pyramidal neurons in the mPFC. For example, chronic intermittent ethanol vapor exposure (CIE) produces significant, yet opposing effects on pyramidal neuron synaptic activity (persistent increase in NMDAR-mediated excitatory postsynaptic currents), and synaptic plasticity (aberrant increase in NMDAR-mediated spike-timing-dependent plasticity) compared with acute effects on slices, possibly through an NR1 and NR2B-mediated mechanism (Kroener et al., 2012). These adaptive changes in NMDARs during long-term ethanol exposure may be occurring to counterbalance the initial prolonged inhibitory effects of ethanol on NMDAR activity, and may contribute to the aberrant neuronal excitability and neuronal toxicity observed during withdrawal and protracted abstinence (Grant et al., 1990; Chandler, 2003; Kroener et al., 2012). Notably, the altered function of pyramidal neurons is associated with altered structure of pyramidal neurons (increased dendritic arborization and mature spine density), suggesting dysfunctional cortical networking in the mPFC (Holmes et al., 2012; Kroener et al., 2012). Furthermore, CIE alters certain behavioral measures dependent on the PFC, namely, attentional set-shifting and extinction encoding, suggesting maladaptive behavioral flexibility (Holmes et al., 2012; Kroener et al., 2012); these deficits may contribute to the cognitive impairments and loss of behavioral control seen in alcohol-dependent subjects.
Gliogenesis and neurogenesis in the adult brain have been conceptualized to be brain regenerative mechanisms; whether the newly born glia and neurons replace diseased cells or dying cells is a question receiving intense focus. In this context, particularly interesting is the capacity of the mPFC to generate newly born glia, endothelial cells and neurons (Mandyam and Koob, 2012). The number of progenitors in the mPFC that develop into glial fibrillary acidic protein (GFAP)+ astroglia are fewer compared with neuron-glia 2 (NG2)+ glia (also known as oligodendrocyte progenitor cells (OPCs), polydendrocytes or synantocytes) (Mandyam and Koob, 2012); however, the functional significance of NG2 gliogenesis in the adult mPFC is unknown. Emerging studies from several in vitro and in vivo models, show that NG2 glia differentiate into premyelinating oligodendrocytes, mediate nonsynaptic events, and may assist with neuronal transmission (Butt et al., 2002; Watanabe et al., 2002; Belachew et al., 2003; Dawson et al., 2003; Ligon et al., 2006; Rivers et al., 2008; Allen and Barres, 2009; Kang et al., 2010; Sun et al., 2011). These findings have led to proposals that stimulus-related changes in central nervous system myelin could be considered a form of neural plasticity, whereby (presumably active) axons and dendrites undergo myelination (through NG2 gliogenesis) to improve the speed and efficiency of nerve conduction, thus strengthening or synchronizing specific neuronal networks (Fields, 2005, 2010). We have recently demonstrated that CIE reduces the number of newly born progenitors and their capacity to survive in the mPFC (Richardson et al., 2009), suggesting that ethanol’s effect on function and structure of pyramidal neurons in the mPFC could be due to ethanol’s neurotoxicity as revealed by inhibition of mPFC progenitors and gliogenesis. Together, these prior observations lead us to the hypothesis that CIE produces structural reorganization of mPFC pyramidal neurons by reducing the number of myelinating oligodendrocytes and expression of myelin in the mPFC. Golgi-cox staining was used to determine the structure of pyramidal neurons, and Western blot analysis was used to determine changes in proteins associated with maintaining activity of neurons and myelinating glia. Rats were injected with 5-bromo (iodo or chloro)-2′-deoxyuridine (BrdU/IdU or CldU) to label proliferating synthesis (S)-phase progenitors and euthanized 2 h later to measure cell proliferation and cell cycle kinetics and 28 d later to measure cell survival.
Methods
Animals
Adult male Wistar rats (Charles River), weighing 225–275 g and 8 weeks old at the beginning of the experiments, were housed in groups of 2–3 per cage in a temperature-controlled (22°C) vivarium on a 12 h/12 h light/dark cycle (lights on at 8:00 P.M.) with ad libitum access to food and water. All procedures were performed during the dark phase of the light/dark cycle. Thirty-nine rats completed the study (experimental design in Table 1). Experimental procedures were conducted in strict adherence to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publication number 85–23, revised 1996) and approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute.
Table 1.
Experimental design and number of animals in each study.
| Experimental design | |||
|---|---|---|---|
| Number of animals | |||
| Study | Control | CIE | BALs indicated in Figure |
| Western blotting + Golgi Cox | 6 | 6 | 1a |
| Western blotting only | 6 | 1a | |
| IdU/CIdU | 4 | 6 | 1b |
| BrdU | 4 | 7 | 1c |
| Total | 14 | 25 | |
Dependence induction by alcohol vapor chambers
A recent modification of the alcohol dependence model was made to reflect the natural progression of alcohol dependence in which ethanol exposure occurs in a series of extended exposures followed by periods of withdrawal (O’Dell et al., 2004; Gilpin et al., 2008). Chronic exposure to intermittent ethanol vapor exposure elicits even higher alcohol self-administration than continuous vapor (O’Dell et al., 2004), therefore the intermittent procedure was used to induce dependence in alcohol-naïve animals in the present study. Vapors were delivered on a 14 h on/10 h off schedule for 7–10 weeks (Figure 1). This schedule of exposure has been shown to induce physical dependence (Richardson et al., 2008). In the chambers, 95% alcohol flows from a large reservoir to a peristaltic pump (model QG-6, FMI Laboratory, Fluid Metering). Alcohol is delivered from the pump to a sidearm flask at a flow rate that can be regulated. The flask is placed on a heater in which the drops of alcohol hitting the bottom of the flask are vaporized. Air flow controlled by a pressure gauge is delivered to the flask and carries the alcohol vapors to the vapor chamber that contains the animal cages. The flow rate was set to deliver vapors that result in blood alcohol levels between 125 and 250 mg% (Figure 1) or 27.2 and 54.4 mM (Gilpin et al., 2008). In this model, rats exhibit somatic withdrawal signs and negative emotional symptoms reflected by anxiety-like responses, hyperalgesia, and elevated brain reward thresholds (Schulteis et al., 1995; Roberts et al., 2000; Valdez et al., 2002; Rimondini et al., 2003; O’Dell et al., 2004; Zhao et al., 2007; Sommer et al., 2008; Edwards et al., 2012; Vendruscolo et al., 2012). Control rats were not exposed to alcohol vapor.
Figure 1.
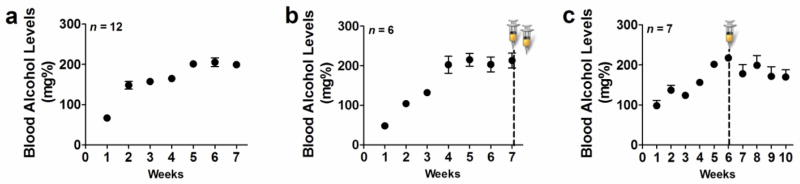
Steady blood alcohol levels (BALs) can be maintained via chronic intermittent ethanol vapor exposure paradigm. (a–c) BALs expressed in mg% over several weeks of CIE. (a) BALs from animals used for Golgi-Cox and Western blotting analysis; (b) BALs from animals used for CldU/IdU cell cycle kinetics analysis; (c) BALs from animals used for BrdU/Olig2 analysis. Syringes in (b, c) indicate the time point when IdU/CldU (b) or BrdU (c) were injected.
Measurement of blood alcohol levels
Blood sampling (tail bleedings) was performed immediately after daily bouts of alcohol vapor exposure in dependent animals twice during the first week of vapor exposure and once during each subsequent week of vapor exposure. Plasma (5 μL) was used for measurement of blood alcohol levels using an Analox AM 1 analyzer (Analox Instruments). The reaction is based on the oxidation of alcohol by alcohol oxidase in the presence of molecular oxygen (alcohol+O2→acetaldehyde+H2O2). The rate of oxygen consumption is directly proportional to the alcohol concentration. Single-point calibrations were done for each set of samples with reagents provided by Analox Instruments (25–400 mg% or 5.4–87.0 mM). When blood samples were outside the target range (125–250 mg%), vapor levels were adjusted.
Golgi-Cox staining and analysis to determine the structure of mPFC pyramidal neurons
For determination of the effect of alcohol dependence on cortical pyramidal neuronal architecture, a group of vapor exposed rats (n = 6) and controls (n = 6), were killed by rapid decapitation under light isofluorane anesthesia 3 h after the last vapor exposure and whole brain was quickly removed and cut mid sagitally. One half of the brain (the left hemisphere for each animal in control and CIE groups) was processed for Golgi-Cox staining. The other half (right hemisphere) was used for Western blotting analysis. For Golgi-Cox staining, the brain was submerged in Golgi-Cox solution A+B (FD Neurotechnologies Inc.) for 8 d at room temperature, followed by solution C for 4 d at room temperature and stored at −80°C until processed for staining. Frozen brain tissue was coronally cut on a cryostat at 100um-thick sections and stained with solution D+E and dehydrated according to manufacturer’s instructions. Brains were coded before sectioning to ensure that experimenters were blind to treatments.
To evaluate pyramidal neuron morphology, a Zeiss Axiophot microscope and a computer-based system (Neurolucida; Micro-BrightField) was used to generate three-dimensional neuron tracings that were subsequently visualized and analyzed using NeuroExplorer (MicroBrightField). In order for a neuron to be selected the following four criteria were met: (1) the pyramidal neuron was in the layer 2/3 of the medial prefrontal cortex, (2) the pyramidal neuron was distinct from other pyramidal and interneurons to allow for identification of dendrites, (3) the pyramidal neuron was not truncated, and (4) the pyramidal neuron exhibited dark, well filled staining throughout (Gould et al., 1990).
For each animal, 4 pyramidal neurons in layer 2/3 of the medial prefrontal cortex (3.7 to 2.0 mm from bregma; infralimbic and prelimbic cortices were combined) were traced at 40x magnification with an oil immersion lens (equipped with a 10x eye piece). Both the apical and basal trees were traced, and morphological measurements were analyzed separately. For each reconstructed neuron, an estimate of dendritic complexity was obtained using the Sholl ring method. A 3D Sholl analysis was performed in which concentric spheres of increasing radius (starting sphere 10 um and increasing in 20 um increments) were layered around the cell body until dendrites were completely encompassed (Figure 2c). The number of dendritic intersections at each increment was counted, and results were expressed as total intersections and the number of intersections per radial distance from the soma.
Figure 2.

CIE alters the structure of pyramidal neurons in the mPFC. (a–d) 3D reconstruction and Sholl ring analysis at 20 μm ring distance with a starting ring at 10 μm. A representative example of Golgi-Cox stained pyramidal neuron in the mPFC (a) along with neuron tracings in Neurolucida (b) and corresponding 3D reconstruction and Sholl ring (c) from one CIE animal. xyz orientation of the cell in (a) and the corresponding depth of the cell shown in micrometers are indicated in (d). xyz analysis indicated that the entire cell in its xyz axis is within 60 microns thickness. (e–f) Neuron tracings from one control (e) and one CIE (f) animal. (g) Total number of intersections of basal dendrites (filled circles, control in black and CIE in red) and apical dendrites (open circles, control in black and CIE in red) of mPFC pyramidal neurons. (h) Length of basal and apical dendrites shown as soma to tip distance of the dendrites or total length of all dendritic branches; controls are in black and CIE is in red. n= 6 animals in each group and neuron data is from 24 neurons in each group. ***p < 0.001, **p < 0.01, *p < 0.05 compared with controls.
For all neurons, spine density was measured at 63x–100x magnification with an oil immersion lens (equipped with a 10x eye piece) on the same dendritic branches used for Sholl analysis. Dendritic spine density was measured on four dendritic segments from both the basal and apical tree. For the basal tree, spines were counted along 10–12 um segments of 2–5° order branches located at least 50 um away from the cell body. For the apical tree, spines were counted along 10 um segments of primary apical branches located at least 80 um away from the cell body. Dendritic spines were classified into 2 categories (mushroom or nonmushroom) based on their length and neck and head width (Figure 3a–d). Stringent criteria were used for the categorization of spine heads into mushroom and nonmushroom type spines and were based on several seminal reports (Peters and Kaiserman-Abramof, 1970; Harris et al., 1992; Markham et al., 2005; Jurgens et al., 2012). All measurements of spine density were taken from 1–2-um-thick dendritic segments to minimize the number of spines hidden by the dendritic shaft and ensure that the number of hidden spines was proportional across all segments counted and among treatment groups. The total number of dendritic spines visible along both sides of the segment were counted and expressed as number of spines per 10 microns of dendrite.
Figure 3.

CIE increases the spine density of pyramidal neurons in the mPFC. (a–d) Representative images of 10 μm segments of apical (a, b) and basal (c, d) dendrites from distal portions of the dendrite from one control (a, c) and one CIE (b, d) animal. Arrowhead in (b), thin arrow in (c) and thick arrow in (d) point to nonmushroom type spines. Arrowhead in (d) points to mushroom type spine. Scale bar in (d) is 2 um, applies (a–d). (e–f) Quantitative analysis of total number of spines (nonmushroom + mushroom type; e) and mushroom type spines (f) from control (open bars) and CIE (red hatched bars) animals from apical and basal dendrites. n= 6 animals in each group and spine data is from 24 neurons in each group. ***p < 0.001 compared with controls, #p < 0.05 compared with apical dendrites.
Western blotting
Procedures optimized for measuring neuronal levels of both phosphoproteins and total proteins were employed (Edwards et al., 2013). Tissue punches from alcohol-naive rats (n = 6) and alcohol-dependent rats (n = 12) from 500 um thick PFC sections were homogenized on ice by sonication in buffer (320 mM sucrose, 5 mM HEPES, 1 mM EGTA, 1 mM EDTA, 1% SDS, with Protease Inhibitor Cocktail and Phosphatase Inhibitor Cocktails II and III diluted 1:100; Sigma), heated at 100 degrees C for five minutes, and stored at −80 degrees C until determination of protein concentration by a detergent-compatible Lowry method (Bio-Rad, Hercules, CA). Samples were mixed (1:1) with a Laemmli sample buffer containing β-mercaptoethanol. Each sample containing protein from one animal was run (10–30 μg per lane) on 8–12% SDS-PAGE gels (Bio-Rad) and transferred to polyvinylidene fluoride membranes (PVDF pore size 0.2 μm). Blots were blocked with 5% milk (w/v) in TBST (25 mM Tris-HCl (pH 7.4), 150 mM NaCl and 0.1% Tween 20 (v/v)) for 16–20 h at 4 °C and were incubated with the primary antibody for 16–20 h at 4 °C: antibody to NR2B (1:200, Santa Cruz cat. no. sc-9057, predicted molecular weight 178 kDa, observed band ~180 kDa), antibody to pNR2B Tyr-1472 (1:200, Cell Signaling cat. no. 4208S, predicted molecular weight 190 kDa, observed band ~180 kDa), antibody to Arp2/3 (1:500, Millipore, cat. no. MABT95, predicted molecular weight 20 kDa, observed band ~23 kDa), antibody to neuronal Wiskott–Aldrich Syndrome protein (nWASP; 1:500, Cell Signaling, cat. no. 4848S, predicted molecular weight ~60 kDa, observed band ~60 kDa), antibody to PSD-95 (1:500, Millipore, cat. no. 04-1066, predicted band size 95 kDa, observed band ~95 kDa), antibody to oligodendrocyte lineage transcription factor 2 (Olig2; 1:10000, predicted molecular weight 37 kDa (Ligon et al., 2006), observed band ~37 kDa), antibody to phosphorylated Olig2 (pOlig2) Ser-10, 13,14 (1:500, predicted molecular weight 35 kDa, observed band ~35 kDa (Sun et al., 2011)), antibody to MBP (1:500, Abcam, cat. no. ab40390, predicted band size 18–23 kDa, observed band ~20 kDa). β-tubulin (1:8,000, Santa cruz cat. no. sc-5274, predicted band size 50 kDa, observed band ~50 kDa) was used as a loading control. Blots were then washed three times for 15 min in TBST, and then incubated for 1 h at room temperature (24 °C), appropriately with either horseradish peroxide–conjugated goat antibody to rabbit (1:10,000, BioRad) or horseradish peroxide–conjugated goat antibody to mouse IgG1 (1:10,000, BioRad) in TBST. After another three washes for 15 min with TBST, immunoreactivity was detected using SuperSignal West Dura chemiluminescence detection reagent (Thermo Scientific) and collected using HyBlot CL Autoradiography film (Denville Scientific) and a Kodak film processor. Net intensity values were determined using the ImageJ software (version 1.45S, NIH). Following chemiluminescence detection, blots were stripped for 20 minutes at room temperature (Restore, Thermo Scientific) and reprobed for total protein levels of β-Tubulin for normalization purposes. Unpaired Student’s t tests were used to compare values between groups.
Injections of mitotic markers
Cell cycle kinetics group
Alcohol-naïve (control, n = 4) and alcohol dependent rats (n = 6) 2h after the last vapor exposure received one injection of 50 mg/kg 5-Iodo-2′-deoxyuridine (IdU; MP Biomedicals, cat# 100357; dissolved in 0.9% saline, 0.2N NaOH at 2.5 mg/ml, normalized to pH 7 with 10N HCl) followed by 50 mg/kg CldU 2 h later (Yuan et al., 2011). These rats survived for 30 min after the CldU injection and were killed by transcardial perfusions.
Cell survival group
A separate group of alcohol-naive rats (n = 4) and alcohol dependent rats (n = 7) received one injection of 150 mg/kg BrdU 2h after the vapor exposure. The alcohol dependent rats continued to receive ethanol vapors for additional 4 weeks and were killed by transcardial perfusions 3–4h after the last vapor exposure.
Perfusions and brain tissue collection
After the last vapor exposure, rats were fully anaesthetized using chloral hydrate (240 mg/kg, i.p.). Rats were then transcardially perfused with phosphate-buffered saline (over 2 minutes at 15ml/min and 4% paraformaldehyde (over 20 minutes at 15ml/min). The brains were dissected out and postfixed in 4% paraformaldehyde at 4°C for 16–20h and sectioned in the coronal plane at a thickness of 40μm on a freezing microtome. The sections through the brain were collected in nine vials (containing 0.1% NaN3 in 1X phosphate-buffered saline (PBS)) and stored at 4°C. One ninth of the brain region was used for immunohistochemical analysis.
Antibodies, immunohistochemistry, microscopic analysis, and quantification
The following primary antibodies were used for immunohistochemistry (IHC): mouse monoclonal anti-BrdU (1:10, catalog # ab8152, Abcam), rat monoclonal anti-BrdU (1:400; catalog # MCA2060, Serotec) and rabbit polyclonal anti-Olig2 (1:10000). The left and right hemispheres of every ninth section through the rat medial prefrontal cortex (Figure 5a) were slide-mounted, coded, and dried overnight prior to IHC. The sections were pretreated (Mandyam et al., 2004), blocked, and incubated with the primary antibodies followed by biotin-tagged (Olig2) or fluorescent-tagged (CldU, IdU, BrdU and Olig2) secondary antibodies. Absolute cell counting (as demonstrated in Noori and Fornal, (Noori and Fornal, 2011)) rather than unbiased stereological estimates in sections through the mPFC (Figure 5a; mPFC; 3.7 to 2.2 mm from bregma; (Paxinos and Watson, 1997)) were quantified with a Zeiss Axiophot Microscope equipped with MicroBrightField Stereo Investigator software, a three-axis Mac 5000 motorized stage, a Zeiss digital charge-coupled device ZVS video camera, PCI color frame grabber, and computer workstation. Live video images were used to draw contours delineating the mPFC and were traced with 2.5x objective (equipped with 10x eyepiece). Cell counting was performed visually by the observer blind to the study with a 40x objective (equipped with a 10x eyepiece). A 150 × 150 μm frame was placed over the regions of interest using the Stereo Investigator stereology platform. The frame was moved systematically over the tissue to cover the entire contoured area and labeled cells in each region falling entirely within the borders of the contour were marked and analyzed. Immunoreactive cells were quantified bilaterally from sections representing 3.7, 3.2, 2.7, and 2.2 mm from bregma and were summed up for each brain region. Cells in the mPFC were summed and multiplied by 9 to give the total number of cells (Mandyam et al., 2007).
Figure 5.

CIE reduces the length of the S-phase of the cell cycle of mPFC progenitors. (a) Schematic of bregma regions and the area of prefrontal cortex included for quantification. Area shaded in gray included the cingulate cortex (Cg), prelimbic cortex (PrL), and infralimbic cortex (IL). (b–c) Representative epifluorescent images of IdU (b) and CldU (c) labeling in the subventricular zone (SVZ) and mPFC from one control animal. Thin arrow in (b, c) points to cells in the SVZ and arrowhead in (b, c) points to a IdU/CldU double labeled cell in the mPFC. (d–e) Orthogonal view of IdU/CldU labeled cells in the SVZ (d) and mPFC (e) showing equal penetration of antibodies in the xz and yz planes. (f) Schematic representation of the cell cycle of proliferating progenitors in the mPFC. S, synthesis phase; G1, gap1 phase; G2, gap2 phase and M, mitosis. Ns is number of cells in S phase, NEX is number of cells exiting the S phase, NEN is number of cells entering the S phase of the cell cycle. Injections of IdU were given at T, time = 0h and Cldu were given at T = 2h. (g–i) Representative confocal images of IdU (red, g) and CldU (green, h) labeled cells in the mPFC from one control animal. (j) Quantitative analysis of number of IdU or CldU or IdU/CldU cells in the mPFC per animal from 4 pairs of bilateral mPFC sections. (k) Length of S phase of proliferating progenitors in the mPFC expressed in hours. (l) Total number of CldU only cells in the mPFC represented as number of cells per mm2 of the mPFC. n = 6 animals in each group. *p < 0.05 compared with controls.
Length of S-phase
Fluorescent double-labeling of CldU/IdU was performed for S-phase analysis as described previously (Yuan et al., 2011). To determine the S-phase of the cell cycle, proliferating cells in the S-phase were pulse-chased with low doses of exogenous markers IdU and CldU (50 mg/kg each, i.p).; (Maslov et al., 2004; Burns and Kuan, 2005; Vega and Peterson, 2005; Breunig et al., 2007), and the length of the S-phase was calculated using the parameters according to Cameron and McKay (Cameron and McKay, 2001) and Hayes and Nowakowski (Hayes and Nowakowski, 2002). ldU and CIdU (Figure 5b–h) were used sequentially to label S-phase cells at two time-points (T1 = 0 h, T2 = 2 h), a time-point predicted to be less than the G2/M-phase based on cell cycle studies in the rat hippocampus; (Cameron and McKay, 2001). Control and CIE rats were given one injection of IdU followed by CIdU 2 h (T2) later after 7 weeks of chronic CIE. Every ninth section through the mPFC was used for analysis. We used a mouse monoclonal anti-BrdU antibody from Abcam (1:10; cat# ab8152) to detect IdU cells and a rat monoclonal anti-BrdU antibody from Serotec (1:400; cat# MCA2060) to detect CldU cells (Yuan et al., 2011). The lack of nonspecific staining was confirmed in a separate set of animals that received either IdU injections only or CldU injections only (data not shown). In addition, both the antibodies for IdU and CldU showed specific staining in brain tissue from BrdU-injected rats (data not shown).
Fluorescently labeled brain tissue from animals sequentially injected with IdU and CldU (CY3 for IdU and CY2 for CldU, respectively; Figure 5b–h) revealed three distinct populations of proliferating cells in the S-phase, some cells labeled with rat antibody only, some labeled with mouse antibody only, and most colabeled with both antibodies (Figure 5b–e). The labeling pattern was quantified as the following: single-labeled IdU (number of cells that exit the S-phase during T2 = 2 h, NEX), single-labeled CldU (number of cells that enter the S-phase during TCldU = 30 min, NEN), and double-labeled IdU/CldU cells. Based on previous reports, the expected value (E) of the proportion of dividing cells in the S-phase at any given time point was equal to the proportion of cell cycle time of the S-phase labeled with a single S-phase marker (Nowakowski et al., 1989; Hayes and Nowakowski, 2000; Cameron and McKay, 2001; Yuan et al., 2011). The calculation of the length of the S-phase (TS) was performed as the following:
TEX = T2 = interinterval time (2 h; Figure 5f)
Total number of cells labeled with mouse BrdU antibody (IdU) = NIdU
Total number of cells labeled with rat BrdU antibody = CldU cells = cells in S-phase NS = NCldU
Cells that exited the S-phase and entered the G2/M-phase at T2 = (NIdU single) = NEX
Time-line of S-phase TS = T2/(NEX/NC)
TS values were calculated individually for each rat.
Number of S-phase cells
The number of CldU-immunoreactive (IR) cells was quantified from each control and CIE rat in every ninth section through the mPFC using a stereology platform. The number of cells was multiplied by 9 to give an estimate for the total number of cells in the S-phase of the cell cycle per mm2 of the mPFC.
Cell survival study
Every ninth section through the mPFC from control and CIE rats that received BrdU was mounted and stained for BrdU (CY3) and Olig2 (FITC). All BrdU labeled cells (15–22 cells) from each rat were processed for confocal microscopy to determine the ratio of BrdU/Olig2 colabeling.
Confocal microscopy
Confocal analysis was performed on individual CldU-immunoreactive (IR), IdU-IR and BrdU-IR cells using a 60× oil immersion objective (equipped with a 10x eyepiece). Optical sectioning in the z-plane was performed using multitrack scanning with a section thickness of 0.45 μm. Colocalization of antibodies was assessed with the confocal system by analysis of adjacent z-sections (gallery function and orthogonal function for equal penetration of the antibodies). For the S-phase study, the number of cells that were IdU alone, CldU alone, and IdU/CldU labeled were quantified and are shown as raw data for each animal. For cell survival study the number of BrdU cells colabeled with Olig2 were estimated and the percent of BrdU cells colabeled with Olig2 in each rat was used for analysis.
Statistical analysis
The effects of group (control vs. CIE) and distance from the soma on dendritic length were analyzed using two-factor ANOVA, with repeated measures for distance from the soma, followed by Fisher’s LSD post hoc tests. Unpaired Student’s t tests were used to compare overall apical length between groups. Differences in cell cycle kinetics, number of progenitors and density of proteins were also analyzed by Student’s t tests. Data are expressed as mean ± SEM. Values of p < 0.05 were considered statistically significant. Graphs were generated using GraphPad Prism 5.0 software. Images presented here were collected on a confocal microscope (LaserSharp 2000, version 5.2, emission wavelengths 488, 568, and 647 nm; Bio-Rad Laboratories) and imported into Photoshop (version CS2). Only the gamma adjustment in the Levels function was used.
Results
Chronic inhalation of ethanol vapors over an intermittent schedule produces and maintains robust blood alcohol levels
Measurements of experimenter-adjusted BALs indicated an increase over the 6–7 weeks compared with the first week of vapor exposure in the groups of animals assigned for Western blotting analysis, the IdU/CldU study and the BrdU study (F(6, 114) = 63.9, p < 0.001 (Figure 1a–c) by repeated measures two-way ANOVA). Although the flow rate was set to deliver ethanol vapors that result in BALs between 125 and 250 mg%, this level was not reached in the first week of vapor exposure in certain animals. However, BALs were not significantly different between the groups (F(2, 114) = 0.16, n.s. (Figure 1a–c)) and was maintained between 150 and 250 mg% (target BAL) during weeks 4–7 until week 10.
CIE produces significant alterations in the structure of pyramidal neurons in the mPFC
Dendritic arborization
Both the apical and basal trees of mPFC pyramidal neurons were traced, and morphological measurements were analyzed separately (Figure 2a–f). Morphological analysis demonstrated that the total number of dendritic intersections in the basal dendrites were significantly higher when compared with apical dendrites. A two-way ANOVA revealed a main effect of type of dendrite (Figure 2g; F(1, 736) = 95.5, p < 0.001), and a significant interaction (type of dendrite x distance from soma; F(15, 736) = 30.4, p = 0.001)), indicating that the number of intersections varied over the distance from soma. CIE increased the number of dendritic intersections in the basal dendrites. A two-way ANOVA revealed a significant effect of ethanol (Figure 2g; F(1, 460) = 4.2, p = 0.04), and a significant interaction (ethanol x distance from soma; F(9, 460) = 5.9, p < 0.001). CIE also increased the number of dendritic intersections in the apical dendrites. A two-way ANOVA revealed a significant effect of ethanol (Figure 2g; F(1, 736) = 4.3, p = 0.03), and a significant interaction (ethanol x distance from soma; F(15, 736) = 3.3, p < 0.001). Post-hoc analyses indicated higher number of intersections at distances 20 – 60 μm from the soma in both basal and apical dendrites in CIE animals compared with controls (Figure 2g, p < 0.01), indicating an increase in dendritic extent near the soma.
Dendritic Length
CIE did not alter the total combined length of the apical and basal dendritic trees, but significantly reduced the soma-tip distance of the apical and basal dendritic trees (Figure 2h; p < 0.05), indicating dendritic retraction.
Dendritic Spine Density
Spine density was calculated and analyzed separately for apical and basal dendritic trees. A two-way ANOVA revealed a main effect of the number of spines on type of dendrite (Figure 3e; F(1, 92) = 3.7, p = 0.05), indicating more number of spines on the basal dendrites compared with apical dendrites. CIE increased the spine density of mPFC apical and basal dendrites (Figure 3a–d). A two-way ANOVA revealed a significant effect of ethanol (Figure 3e; F(1, 92) = 83.45, p = 0.0001), and a strong trend for a significant interaction (ethanol x type of dendrite; F(1, 92) = 3.5, p = 0.06)). There were no differences in the number of mushroom-like spines in apical dendrites when compared with basal dendrites and CIE did not alter the number of mushroom-like spines (Figure 3f).
CIE alters the levels of NR2B NMDA receptor subunits in the mPFC
To determine the effects of CIE on the total levels and the phosphorylation state of NR2B subunits, Western blot analyses were carried out on mPFC-enriched tissue punches. Consistent with a previous report (Kroener et al., 2012), CIE significantly increased the levels of total NR2B subunits (Figure 4a–b; p = 0.04 by Student’s t-test). Increases in total NR2B levels were not associated with enhanced phosphorylation of NR2B at Tyr-1472 (Figure 4a–b).
Figure 4.

(a–b) CIE increases levels of total NR2B without effecting phosphorylation of NR2B (pNR2B) at Tyr-1472 in the mPFC. (a) Representative immunoblots of total NR2B vs. pNR2B compared with b-tubulin. (b) Densitometric analysis of proteins in CIE animals expressed as percent change compared with controls. (c–d) CIE elevates levels of Arp2/3 without effecting levels of PSD-95 and nWASP in the mPFC. (c) Representative immunoblots of Arp2/3 compared with b-tubulin. (d) Densitometric analysis of proteins in CIE animals expressed as percent change compared with controls. n = 11–12 in CIE group and 6 in controls; *p ≤ 0.05 compared with controls.
CIE alters the levels of actin nucleator complex Arp2/3 in the mPFC
To determine the effects of CIE on protein complexes that regulate actin cytoskeleton and microtubule formation that is necessary for the formation of filament protrusions or spines on dendrites, Western blot analyses were carried out to determine changes in the levels of PSD-95, Arp2/3 and nWASP, an activator of Arp2/3. Consistent with a recent report (Holmes et al., 2012), CIE did not alter levels of PSD-95 (Figure d). CIE significantly increased levels of Arp2/3 (Figure 4c, d; p = 0.04 by Student’s t-test) when compared with controls, although these changes were not associated with altered levels of nWASP (Figure 4d).
CIE reduces the number of newly born glial progenitors in the mPFC by altering their cell cycle kinetics
Previous work from our laboratory demonstrated that IdU and CldU (analogs of BrdU) produce equivalent labeling of hippocampal progenitors compared with BrdU at lower doses (Yuan et al., 2011). We also demonstrated that in the hippocampus, IdU cells can be labeled with mouse monoclonal BrdU antibody, and CldU cells can be distinctly labeled with rat monoclonal BrdU antibody. In the mPFC, we demonstrate that sequential labeling with IdU followed by CldU display three distinct populations of proliferating cells (Figure 5b–e; g–i). We used this approach to determine the length of the S-phase of the cell cycle of mPFC progenitors from the control and CIE groups. Animals were sequentially injected with IdU followed by CldU 2 h later (see Methods). According to our recently published mathematical calculations (Yuan et al., 2011), that is based on several seminal reports (Cameron and McKay, 2001; Hayes and Nowakowski, 2002), IdU+ cells in the S-phase at T = 0 h would have moved into the G2/M phases of the cell cycle at T = 2 h when CldU was injected (Figure 5f). At T = 30 min after CldU injection, CldU cells will mostly be in the S-phase (Figure 5f). According to previously published reports, pulse-chasing S-phase cells at any given time-point with two S-phase markers, such as [3H]thymidine and BrdU (Cameron and McKay, 2001; Hayes and Nowakowski, 2002) or IdU and CldU (Vega and Peterson, 2005), produces three distinct populations of cells. For example, the set of S-phase cells labeled at T1 = 0 h with IdU will be single- (NEX) and double-labeled (NS) at T2 = 2 h (Figure 5f). The set of S-phase cells labeled at T2 = 2 h with CldU will be single- (NEN) and double-labeled (NS) when euthanized (TCldU = 30 min; Figure 5f). All three groups of S-phase cells were visible in all of the rats analyzed, with most of the S-phase cells double-labeled with CldU and IdU (Figure 5j–k). The calculation of the S-phase length in naive controls revealed a timeframe of greater than 30 h (Figure 5k), demonstrating that the S-phase of the cell cycle of mPFC progenitors is longer than the S-phase of hippocampal subgranular zone progenitors in adult rats (Olariu et al., 2007; Yuan et al., 2011). CIE did not alter the number of cells in NEN and NS, but showed a trend towards higher number of cells in NEX (p = 0.07; Figure 5j). S-phase analysis indicated that CIE decreased the duration of S-phase compared with controls (Figure 5k; p = 0.04 by Student’s t-test). Separate stereological analysis of CldU+ cells showed a significant reduction in the number of S-phase cells in CIE animals compared with controls (Figure 5l; p = 0.04 by Student’s t-test).
CIE reduces the number of newly born and pre-existing premyelinating oligodendrocytes and levels of myelin basic protein in the mPFC by inducing hyperphosphorylation of the bHLH transcription factor Olig2
In controls and CIE animals, twenty eight-day-old BrdU cells were co-labeled with Olig2 (Figure 6a–d). As expected, in the controls, approximately 60% of BrdU cells were colabeled with Olig2, indicating that most of the progenitors in the mPFC differentiated into premyelinating oligodendrocytes (Figure 6e). CIE reduced the proportion of BrdU-labeled Olig2 cells (Figure 6e; p = 0.03 by Student’s t-test). Immunohistochemistry of myelin basic protein showed no significant change in the location and morphology of myelin (Figure 6f–g). Western blot analyses were performed to determine whether CIE reduced the total levels of Olig2 and whether CIE altered the phosphorylation of Olig2 (Figure 6h). CIE reduced total Olig2 levels in the mPFC (Figure 6i; p = 0.02 by Student’s t-test) and this reduction was associated with enhanced phosphorylation of Olig2 at Ser-10, 13, 14 (Figure 6i; p = 0.01 by Student’s t-test). The reduction in premyelinating oligodendrocytes was also associated with lower levels of myelin basic protein (Figure 6i; p = 0.03 by Student’s t-test).
Figure 6.

CIE reduces the proportion of newly born Olig2 labeled cells in the mPFC and reduces myelin expression in the mPFC. (a–c) Epifluorescent images of mPFC cortex section stained for Olig2 (FITC, green, a), BrdU (CY3, red, b) and DAPI (blue, c). Thin arrow in (a–b) shows a BrdU labeled cell that is not Olig2+. Arrowhead in (a–b) shows two BrdU labeled cells that are Olig2+. (d) Orthogonal section of a BrdU+/Olig2+ cell in the mPFC (cell shown in (b)) showing equal penetration of the antibodies in xz and yz planes. (e) Ratio of 28-day-old BrdU+ cells colabeled with Olig2. (f–g) immunohistochemistry of myelin basic protein in the mPFC from one control (f) and one CIE (g) animal. (h) Representative immunoblots of phosphorylated Olig2 (pOlig2), total Olig2, MBP and corresponding blots for b Tubulin. (i) Densitometric analysis of total Olig2, pOlig2 and MBP in the mPFC represented as percent change from controls. n = 7 animals in CIE group and n = 5 animals in control group for Olig2/BrdU IHC analysis, and n = 12 animals in CIE group, n = 6 animals in control group for Western blotting analysis. **p < 0.01 and *p < 0.05 when compared with controls.
Discussion
Exposure to chronic alcohol via the CIE paradigm provides several advantages for studying the neurobiological consequences of alcohol dependence (Macey et al., 1996; Liu and Weiss, 2002, 2003; Moore et al., 2004; Budygin et al., 2007; Miki et al., 2008; Gilpin et al., 2009; Richardson et al., 2009; Zahr et al., 2009). However, little is known about the impact of CIE on neuron structure and function, and regenerative events in the prefrontal cortex (Weitlauf and Woodward, 2008; Richardson et al., 2009; Holmes et al., 2012; Kroener et al., 2012), a brain region injured by alcohol abuse (Parsons and Nixon, 1993; Sullivan et al., 2000; Bechara et al., 2001). Such investigations of chronic alcohol on mPFC neuron structure and development of glial progenitors could help clarify the relationship between neuron structure and gliogenesis in alcohol dependence. The present results highlight how CIE alters the structure of pyramidal neurons in the mPFC via disrupting glial plasticity by reducing myelinating oligodendrocytes and myelin-associated proteins in the mPFC. Specifically, we demonstrate that CIE increases dendritic arborization and spine density within the basal and apical dendrites of pyramidal neurons with corresponding increases in the levels of the actin nucleator complex Arp2/3. CIE also increased the levels of NR2B subunits without significant changes in the phosphorylation state of NR2B at Tyr-1472 or levels of PSD-95. This suggests an aberrant relocation of NMDARs to extrasynaptic sites in the mPFC neurons that may contribute to synaptic activity and excitotoxic cell death in the mPFC. We also demonstrate that newly born progenitors in the mPFC differentiate into neurogenic bHLH transcription factor Olig2-expressing myelinating oligodendrocytes, and that CIE reduces the process of myelinating oligodendrogenesis by altering cell cycle kinetics of progenitors and inducing aberrant hyperphosphorylation of Olig2. The reduction in myelinating oligodendrogenesis is also associated with reduction of myelin basic protein in the mPFC in CIE animals. These effects of chronic alcohol exposure produced by the CIE paradigm on pyramidal neurons and myelin indicate one possible neurobiological alteration contributing to the pathology of alcohol dependence.
NMDA receptors in the mPFC are subject to dynamic regulation during ethanol exposure and during withdrawal from ethanol (Hu and Ticku, 1997; Tsai and Coyle, 1998; Chandler, 2003; Prendergast and Mulholland, 2012). For example, acute ethanol exposure inhibits NMDA receptor function (Tu et al., 2007; Weitlauf and Woodward, 2008; Woodward and Pava, 2009), and chronic ethanol exposure (via CIE) increases NMDA function and/or expression of NMDA receptors (Hu and Ticku, 1995; Holmes et al., 2012; Kroener et al., 2012). CIE-induced NMDA receptor potentiation is hypothesized to be associated with the excitotoxic processes associated with CNS hyperexcitability during ethanol withdrawal episodes (Crews et al., 1996; Chandler et al., 1998). The mechanism(s) underlying CIE-induced adaptive changes in NMDARs are not clear, and could be associated with increases in NMDA receptor density, particularly functional changes in NMDARs (Hu and Ticku, 1995; Chandler et al., 1998). Notably, functional changes in NMDARs are significantly linked with phosphorylation and dephosphorylation of NR2B subunits (Ali and Salter, 2001; Nakazawa et al., 2001; Nakazawa et al., 2006). For example, activity-dependent phosphorylation of Tyr-1472 by the Fyn family of tyrosine kinases is essential for proper localization of NR2B-containing receptors at synapses (Yaka et al., 2002; Lavezzari et al., 2003; Prybylowski et al., 2005; Nakazawa et al., 2006) and plays a mechanistic role in the behavioral and physiological tolerance produced by ethanol (Miyakawa et al., 1997). Furthermore, phosphorylation of NR2B at Tyr-1472 and interactions of NR2B with PSD-95 regulate internalization of NMDARs via clathrin-mediated endocytosis, such that phosphorylation suppresses NMDAR endocytosis and dephosphorylation enhances NMDAR endocytosis (Lavezzari et al., 2003; Prybylowski et al., 2005). In this context, the acute effects of ethanol on reduced NMDA receptor activity in the hippocampus are associated with reduced phosphorylation of NR2B subunits at Tyr-1472 without significant changes in total NR2B levels (Alvestad et al., 2003), suggesting mislocalization of NR2B subunits and NMDARs by ethanol. Chronic ethanol exposure (via CIE) in mice increases NMDA receptor activity (Holmes et al., 2012; Kroener et al., 2012) and increases the expression of total NR2B (Kroener et al., 2012). The present findings in rats are consistent with the previous studies in mice and demonstrate that chronic CIE enhances expression of NR2B subunits in the mPFC, and significantly extend the findings to demonstrate that the increase in total NR2B receptor density is not associated with enhanced phosphorylation of NR2B at Tyr-1472 or enhanced levels of PSD-95. Altered NR2B levels, together with a lack of regulation from Fyn phosphorylation and PSD-95, suggests that during CIE, NR2B-containing NMDARs may relocate to extrasynaptic sites, contributing to the dysregulation of NMDAR activity associated with withdrawal and protracted abstinence (Clapp et al., 2010).
Dendritic arborization and spine density are forms of structural plasticity governed by intrinsic genetic programs and extrinsic signals, and are subject to dynamic regulation during CNS excitotoxicity (Smart and Halpain, 2000; Cline, 2001). For example, CIE in mice produces dendritic hypertrophy of mPFC neurons, suggesting significant dendritic remodeling in the mPFC (Holmes et al., 2012). While it has been demonstrated that alterations in neuron structure are not a prerequisite for neuron function (Lang et al., 2004), the abnormal structure of mPFC neurons in CIE animals correlated with deficits in mPFC neuronal function (Holmes et al., 2012; Kroener et al., 2012). The present findings in rats are consistent with previous studies in mice and demonstrate that chronic CIE in rats increases dendritic arborization of pyramidal neurons within the basal and apical dendrites, and significantly extend the findings to demonstrate that the increase in arborization is associated with an enhanced density of spines within the basal and apical dendrites of pyramidal neurons. Spine plasticity (generation and elimination) is regulated by NMDARs, but the precise consequences of NMDAR activation on spine pruning is variable and is under intense investigation (Smart and Halpain, 2000; Cline, 2001). In this context, aberrant NMDAR synaptic activity increases the number of spines (Kirov and Harris, 1999) and such a neuroadaptation could contribute to CIE-induced enhanced spine density within the apical and basal dendrites in mPFC pyramidal neurons. We next wanted to determine whether the enhanced spine density was associated with increased levels of specialized cytoskeleton molecules that are dedicated to maintaining structure, stability and activity of newly formed spines. For example, actin filament dynamics via actin-related proteins (Arp2 and Arp3) are known to be a key player in the formation of filaments on dendrites that elongate into protrusions and eventually form spine heads (Cooper and Schafer, 2000). Notably, the interaction of Arp2/3 with other key players such as nWASP and PSD-95 is critical to functional mature spine formation (Wegner et al., 2008). Our findings of enhanced Arp2/3 levels without associated increases in nWASP and PSD-95 in CIE animals suggests immature spine formation following CIE. These biochemical findings are supported by detailed morphological characteristics of spines in CIE animals, which indicate a CIE enhancement of nonmushroom-type spines (Harris et al., 1992). Taken together, enhanced expression of NR2B subunits and aberrant arborization within basal and apical dendrites associated with enhanced formation of nonmushroom type spines in the pyramidal neurons in the mPFC demonstrates a dysfunctional reorganization of prefrontal neuronal networks by CIE.
We have previously shown that CIE reduces the number of proliferating and surviving progenitors in the mPFC (Richardson et al., 2009). The present findings significantly extend our previous studies to demonstrate that CIE reduces the number of progenitors actively dividing in the S phase of the cell cycle. CIE also reduces the length of the S phase of the cell cycle, indicating a mechanism contributing to CIE-induced reduction in proliferation of glial progenitors in the mPFC. For example, it has been hypothesized that during development, regulation of cell cycle kinetics (such as length of time a cell spends in the cell cycle) controls the expansion versus differentiation of cortical neural progenitors (Bally-Cuif and Hammerschmidt, 2003; Ohnuma and Harris, 2003; Dehay and Kennedy, 2007); such that reducing the length of the cell cycle assists with maintenance of the undifferentiated state (Lange et al., 2009; Salomoni and Calegari, 2010). Furthermore, alterations in the length of G1 phase of the cell cycle have been causally linked to the switch from proliferation to neurogenesis of cortical progenitors during development (Lange et al., 2009). In vitro models have demonstrated that increases in the length of S phase of the cell cycle of human neuroblastoma cells corresponded to enhanced expression of neuronal differentiation markers (Guarnieri et al., 2009), suggesting that length of S phase also regulates the fate of proliferating cells. These studies in developmental models and in vitro models have led to the hypothesis that shortening of the length of S phase of hippocampal neural progenitors by binge ethanol treatment in adolescent rats may result in reduced differentiation of hippocampal neural progenitors (McClain et al., 2011). Based on these studies, it appears that CIE-mediated reduction in the length of S phase of mPFC progenitors may promote an expansion of the glial progenitor pool and maintain glial progenitors in an undifferentiated state.
In the postnatal brain, Olig2 (a bHLH transcription factor and a proneural factor with significant roles in gliogenesis) is expressed in premyelinating oligodendrocyte progenitors and in mature and terminally differentiating myelinating oligodendrocytes, where it appears to have ongoing biological functions (Rivers et al., 2008). During development, the function of Olig2 is dynamically modulated by regulated phosphorylation of a conserved triple serine motif (S10, 13 and 14) within the amino-terminal domain (Sun et al., 2011). For example, phosphorylation status of the triple serine motif of Olig2 undergoes a dramatic decrease as proliferating progenitors in the embryo mature into terminally differentiated, myelinating oligodendrocytes (Sun et al., 2011). These findings suggest a positive correlation between hyperphosphorylation of Olig2 and diminished differentiation of Olig2 progenitors into terminally differentiated myelinating oligodendrocytes and myelin. In this context, CIE produced hyperphosphorylation of Olig2 and concomitantly reduced the number of differentiating premyelinating oligodendrocytes (BrdU+/Olig2+) and the levels of myelin in the mPFC. These results, combined with altered cell cycle kinetics of proliferating progenitors provide a relationship between CIE-induced reduction in myelin and altered structure of pyramidal neurons.
Collectively, these data propose that CIE produces an abnormal structure of pyramidal neurons in the mPFC, with accompanying abnormalities in gliogenesis, myelinating oligodendrogenesis and myelin. The finding that the structure of pyramidal neurons is vulnerable to CIE via alterations in myelin plasticity implies that a chronic history of alcohol abuse may increase the risk of maladaptive cognitive and emotional behaviors dependent on the PFC via myelin loss.
Acknowledgments
The study was supported by funds from the National Institute on Drug Abuse (DA022473 to CDM), National institute on Alcoholism and Alcohol Abuse (AA020098 and AA06420 to CDM, AA020839 to SE) and Alcohol Beverage Medical Research Foundation to CDM. The authors thank Drs. Charles Stiles and John Alberta, Harvard Medical School, for generously providing Olig2 and phosphorylated Olig2 antibodies. We thank Dr. Heidi Jurgens, University of Illinois, for her assistance with the dendritic arborization analysis and spine density analysis. We appreciate the technical support of Elena Crawford for immunohistochemical analyses, StereoInvestigator and Neurolucida, and Maury Cole for assistance with alcohol vapor chambers. We thank Dr. Miranda Staples for critical reading of the manuscript and McKenzie Fannon for editorial assistance. This is publication number 25057 from The Scripps Research Institute.
References
- Ali DW, Salter MW. NMDA receptor regulation by Src kinase signalling in excitatory synaptic transmission and plasticity. Curr Opin Neurobiol. 2001;11:336–342. doi: 10.1016/s0959-4388(00)00216-6. [DOI] [PubMed] [Google Scholar]
- Allen NJ, Barres BA. Neuroscience: Glia - more than just brain glue. Nature. 2009;457:675–677. doi: 10.1038/457675a. [DOI] [PubMed] [Google Scholar]
- Alvestad RM, Grosshans DR, Coultrap SJ, Nakazawa T, Yamamoto T, Browning MD. Tyrosine dephosphorylation and ethanol inhibition of N-Methyl-D-aspartate receptor function. J Biol Chem. 2003;278:11020–11025. doi: 10.1074/jbc.M210167200. [DOI] [PubMed] [Google Scholar]
- Bally-Cuif L, Hammerschmidt M. Induction and patterning of neuronal development, and its connection to cell cycle control. Curr Opin Neurobiol. 2003;13:16–25. doi: 10.1016/s0959-4388(03)00015-1. [DOI] [PubMed] [Google Scholar]
- Bava S, Jacobus J, Thayer RE, Tapert SF. Longitudinal changes in white matter integrity among adolescent substance users. Alcohol Clin Exp Res. 2013;37(Suppl 1):E181–189. doi: 10.1111/j.1530-0277.2012.01920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara A, Dolan S, Denburg N, Hindes A, Anderson SW, Nathan PE. Decision-making deficits, linked to a dysfunctional ventromedial prefrontal cortex, revealed in alcohol and stimulant abusers. Neuropsychologia. 2001;39:376–389. doi: 10.1016/s0028-3932(00)00136-6. [DOI] [PubMed] [Google Scholar]
- Belachew S, Chittajallu R, Aguirre AA, Yuan X, Kirby M, Anderson S, Gallo V. Postnatal NG2 proteoglycan-expressing progenitor cells are intrinsically multipotent and generate functional neurons. J Cell Biol. 2003;161:169–186. doi: 10.1083/jcb.200210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breunig JJ, Silbereis J, Vaccarino FM, Sestan N, Rakic P. Notch regulates cell fate and dendrite morphology of newborn neurons in the postnatal dentate gyrus. Proc Natl Acad Sci U S A. 2007;104:20558–20563. doi: 10.1073/pnas.0710156104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budygin EA, Oleson EB, Mathews TA, Lack AK, Diaz MR, McCool BA, Jones SR. Effects of chronic alcohol exposure on dopamine uptake in rat nucleus accumbens and caudate putamen. Psychopharmacology (Berl) 2007;193:495–501. doi: 10.1007/s00213-007-0812-1. [DOI] [PubMed] [Google Scholar]
- Burns KA, Kuan CY. Low doses of bromo- and iododeoxyuridine produce near-saturation labeling of adult proliferative populations in the dentate gyrus. Eur J Neurosci. 2005;21:803–807. doi: 10.1111/j.1460-9568.2005.03907.x. [DOI] [PubMed] [Google Scholar]
- Butt AM, Kiff J, Hubbard P, Berry M. Synantocytes: new functions for novel NG2 expressing glia. J Neurocytol. 2002;31:551–565. doi: 10.1023/a:1025751900356. [DOI] [PubMed] [Google Scholar]
- Cameron HA, McKay RD. Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. Journal of Comparative Neurology. 2001;435:406–417. doi: 10.1002/cne.1040. [DOI] [PubMed] [Google Scholar]
- Chandler LJ. Ethanol and brain plasticity: receptors and molecular networks of the postsynaptic density as targets of ethanol. Pharmacol Ther. 2003;99:311–326. doi: 10.1016/s0163-7258(03)00096-2. [DOI] [PubMed] [Google Scholar]
- Chandler LJ, Harris RA, Crews FT. Ethanol tolerance and synaptic plasticity. Trends Pharmacol Sci. 1998;19:491–495. doi: 10.1016/s0165-6147(98)01268-1. [DOI] [PubMed] [Google Scholar]
- Clapp P, Gibson ES, Dell’acqua ML, Hoffman PL. Phosphorylation regulates removal of synaptic N-methyl-D-aspartate receptors after withdrawal from chronic ethanol exposure. J Pharmacol Exp Ther. 2010;332:720–729. doi: 10.1124/jpet.109.158741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline HT. Dendritic arbor development and synaptogenesis. Curr Opin Neurobiol. 2001;11:118–126. doi: 10.1016/s0959-4388(00)00182-3. [DOI] [PubMed] [Google Scholar]
- Cooper JA, Schafer DA. Control of actin assembly and disassembly at filament ends. Curr Opin Cell Biol. 2000;12:97–103. doi: 10.1016/s0955-0674(99)00062-9. [DOI] [PubMed] [Google Scholar]
- Crews FT, Morrow AL, Criswell H, Breese G. Effects of ethanol on ion channels. Int Rev Neurobiol. 1996;39:283–367. doi: 10.1016/s0074-7742(08)60670-4. [DOI] [PubMed] [Google Scholar]
- Dawson MR, Polito A, Levine JM, Reynolds R. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci. 2003;24:476–488. doi: 10.1016/s1044-7431(03)00210-0. [DOI] [PubMed] [Google Scholar]
- De Bellis MD, Narasimhan A, Thatcher DL, Keshavan MS, Soloff P, Clark DB. Prefrontal cortex, thalamus, and cerebellar volumes in adolescents and young adults with adolescent-onset alcohol use disorders and comorbid mental disorders. Alcohol Clin Exp Res. 2005;29:1590–1600. doi: 10.1097/01.alc.0000179368.87886.76. [DOI] [PubMed] [Google Scholar]
- Dehay C, Kennedy H. Cell-cycle control and cortical development. Nat Rev Neurosci. 2007;8:438–450. doi: 10.1038/nrn2097. [DOI] [PubMed] [Google Scholar]
- Edwards S, Baynes BB, Carmichael CY, Zamora-Martinez ER, Barrus M, Koob GF, Gilpin NW. Traumatic stress reactivity promotes excessive alcohol drinking and alters the balance of prefrontal cortex-amygdala activity. Transl Psychiatry. 2013;3:e296. doi: 10.1038/tp.2013.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S, Vendruscolo LF, Schlosburg JE, Misra KK, Wee S, Park PE, Schulteis G, Koob GF. Development of mechanical hypersensitivity in rats during heroin and ethanol dependence: alleviation by CRF(1) receptor antagonism. Neuropharmacology. 2012;62:1142–1151. doi: 10.1016/j.neuropharm.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD. Myelination: an overlooked mechanism of synaptic plasticity? Neuroscientist. 2005;11:528–531. doi: 10.1177/1073858405282304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD. Neuroscience. Change in the brain’s white matter. Science. 2010;330:768–769. doi: 10.1126/science.1199139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George MS, Anton RF, Bloomer C, Teneback C, Drobes DJ, Lorberbaum JP, Nahas Z, Vincent DJ. Activation of prefrontal cortex and anterior thalamus in alcoholic subjects on exposure to alcohol-specific cues. Arch Gen Psychiatry. 2001;58:345–352. doi: 10.1001/archpsyc.58.4.345. [DOI] [PubMed] [Google Scholar]
- Gilpin NW, Richardson HN, Cole M, Koob GF. Vapor inhalation of alcohol in rats. Curr Protoc Neurosci. 2008;Chapter 9(Unit 9):29. doi: 10.1002/0471142301.ns0929s44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Smith AD, Cole M, Weiss F, Koob GF, Richardson HN. Operant behavior and alcohol levels in blood and brain of alcohol-dependent rats. Alcohol Clin Exp Res. 2009;33:2113–2123. doi: 10.1111/j.1530-0277.2009.01051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E, Woolley CS, Frankfurt M, McEwen BS. Gonadal steroids regulate dendritic spine density in hippocampal pyramidal cells in adulthood. J Neurosci. 1990;10:1286–1291. doi: 10.1523/JNEUROSCI.10-04-01286.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant KA, Valverius P, Hudspith M, Tabakoff B. Ethanol withdrawal seizures and the NMDA receptor complex. Eur J Pharmacol. 1990;176:289–296. doi: 10.1016/0014-2999(90)90022-x. [DOI] [PubMed] [Google Scholar]
- Guarnieri S, Pilla R, Morabito C, Sacchetti S, Mancinelli R, Fano G, Mariggio MA. Extracellular guanosine and GTP promote expression of differentiation markers and induce S-phase cell-cycle arrest in human SH-SY5Y neuroblastoma cells. Int J Dev Neurosci. 2009;27:135–147. doi: 10.1016/j.ijdevneu.2008.11.007. [DOI] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci. 1992;12:2685–2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes NL, Nowakowski RS. Exploiting the dynamics of S-phase tracers in developing brain: interkinetic nuclear migration for cells entering versus leaving the S- phase. Dev Neurosci. 2000;22:44–55. doi: 10.1159/000017426. [DOI] [PubMed] [Google Scholar]
- Hayes NL, Nowakowski RS. Dynamics of cell proliferation in the adult dentate gyrus of two inbred strains of mice. Brain Res Dev Brain Res. 2002;134:77–85. doi: 10.1016/s0165-3806(01)00324-8. [DOI] [PubMed] [Google Scholar]
- Heilig M, Egli M. Pharmacological treatment of alcohol dependence: target symptoms and target mechanisms. Pharmacol Ther. 2006;111:855–876. doi: 10.1016/j.pharmthera.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Holmes A, Fitzgerald PJ, MacPherson KP, DeBrouse L, Colacicco G, Flynn SM, Masneuf S, Pleil KE, Li C, Marcinkiewcz CA, Kash TL, Gunduz-Cinar O, Camp M. Chronic alcohol remodels prefrontal neurons and disrupts NMDAR-mediated fear extinction encoding. Nat Neurosci. 2012;15:1359–1361. doi: 10.1038/nn.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XJ, Ticku MK. Chronic ethanol treatment upregulates the NMDA receptor function and binding in mammalian cortical neurons. Brain Res Mol Brain Res. 1995;30:347–356. doi: 10.1016/0169-328x(95)00019-o. [DOI] [PubMed] [Google Scholar]
- Hu XJ, Ticku MK. Functional characterization of a kindling-like model of ethanol withdrawal in cortical cultured neurons after chronic intermittent ethanol exposure. Brain Res. 1997;767:228–234. doi: 10.1016/s0006-8993(97)00581-7. [DOI] [PubMed] [Google Scholar]
- Hu XJ, Follesa P, Ticku MK. Chronic ethanol treatment produces a selective upregulation of the NMDA receptor subunit gene expression in mammalian cultured cortical neurons. Brain Res Mol Brain Res. 1996;36:211–218. doi: 10.1016/0169-328x(95)00223-f. [DOI] [PubMed] [Google Scholar]
- Jacobus J, McQueeny T, Bava S, Schweinsburg BC, Frank LR, Yang TT, Tapert SF. White matter integrity in adolescents with histories of marijuana use and binge drinking. Neurotoxicol Teratol. 2009;31:349–355. doi: 10.1016/j.ntt.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgens HA, Amancherla K, Johnson RW. Influenza infection induces neuroinflammation, alters hippocampal neuron morphology, and impairs cognition in adult mice. J Neurosci. 2012;32:3958–3968. doi: 10.1523/JNEUROSCI.6389-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Fukaya M, Yang JK, Rothstein JD, Bergles DE. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron. 2010;68:668–681. doi: 10.1016/j.neuron.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirov SA, Harris KM. Dendrites are more spiny on mature hippocampal neurons when synapses are inactivated. Nat Neurosci. 1999;2:878–883. doi: 10.1038/13178. [DOI] [PubMed] [Google Scholar]
- Kroener S, Mulholland PJ, New NN, Gass JT, Becker HC, Chandler LJ. Chronic alcohol exposure alters behavioral and synaptic plasticity of the rodent prefrontal cortex. PLoS ONE. 2012;7:e37541. doi: 10.1371/journal.pone.0037541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang C, Barco A, Zablow L, Kandel ER, Siegelbaum SA, Zakharenko SS. Transient expansion of synaptically connected dendritic spines upon induction of hippocampal long-term potentiation. Proc Natl Acad Sci U S A. 2004;101:16665–16670. doi: 10.1073/pnas.0407581101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange C, Huttner WB, Calegari F. Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell. 2009;5:320–331. doi: 10.1016/j.stem.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Lavezzari G, McCallum J, Lee R, Roche KW. Differential binding of the AP-2 adaptor complex and PSD-95 to the C-terminus of the NMDA receptor subunit NR2B regulates surface expression. Neuropharmacology. 2003;45:729–737. doi: 10.1016/s0028-3908(03)00308-3. [DOI] [PubMed] [Google Scholar]
- Lewohl JM, Wixey J, Harper CG, Dodd PR. Expression of MBP, PLP, MAG, CNP, and GFAP in the Human Alcoholic Brain. Alcohol Clin Exp Res. 2005;29:1698–1705. doi: 10.1097/01.alc.0000179406.98868.59. [DOI] [PubMed] [Google Scholar]
- Ligon KL, Kesari S, Kitada M, Sun T, Arnett HA, Alberta JA, Anderson DJ, Stiles CD, Rowitch DH. Development of NG2 neural progenitor cells requires Olig gene function. Proc Natl Acad Sci U S A. 2006;103:7853–7858. doi: 10.1073/pnas.0511001103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Weiss F. Additive effect of stress and drug cues on reinstatement of ethanol seeking: exacerbation by history of dependence and role of concurrent activation of corticotropin-releasing factor and opioid mechanisms. J Neurosci. 2002;22:7856–7861. doi: 10.1523/JNEUROSCI.22-18-07856.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Weiss F. Stimulus conditioned to foot-shock stress reinstates alcohol-seeking behavior in an animal model of relapse. Psychopharmacology (Berl) 2003;168:184–191. doi: 10.1007/s00213-002-1267-z. [DOI] [PubMed] [Google Scholar]
- Macey DJ, Schulteis G, Heinrichs SC, Koob GF. Time-dependent quantifiable withdrawal from ethanol in the rat: effect of method of dependence induction. Alcohol. 1996;13:163–170. doi: 10.1016/0741-8329(95)02030-6. [DOI] [PubMed] [Google Scholar]
- MacKay RK, Colson NJ, Dodd PR, Lewohl JM. Differential expression of 14-3-3 isoforms in human alcoholic brain. Alcohol Clin Exp Res. 2011;35:1041–1049. doi: 10.1111/j.1530-0277.2011.01436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandyam CD, Koob GF. The addicted brain craves new neurons: putative role for adult-born progenitors in promoting recovery. Trends Neurosci. 2012;35:250–260. doi: 10.1016/j.tins.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandyam CD, Norris RD, Eisch AJ. Chronic morphine induces premature mitosis of proliferating cells in the adult mouse subgranular zone. Journal of Neuroscience Research. 2004;76:783–794. doi: 10.1002/jnr.20090. [DOI] [PubMed] [Google Scholar]
- Mandyam CD, Wee S, Eisch AJ, Richardson HN, Koob GF. Methamphetamine self-administration and voluntary exercise have opposing effects on medial prefrontal cortex gliogenesis. J Neurosci. 2007;27:11442–11450. doi: 10.1523/JNEUROSCI.2505-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham JA, McKian KP, Stroup TS, Juraska JM. Sexually dimorphic aging of dendritic morphology in CA1 of hippocampus. Hippocampus. 2005;15:97–103. doi: 10.1002/hipo.20034. [DOI] [PubMed] [Google Scholar]
- Maslov AY, Barone TA, Plunkett RJ, Pruitt SC. Neural stem cell detection, characterization, and age-related changes in the subventricular zone of mice. J Neurosci. 2004;24:1726–1733. doi: 10.1523/JNEUROSCI.4608-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain JA, Hayes DM, Morris SA, Nixon K. Adolescent binge alcohol exposure alters hippocampal progenitor cell proliferation in rats: effects on cell cycle kinetics. J Comp Neurol. 2011;519:2697–2710. doi: 10.1002/cne.22647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQueeny T, Schweinsburg BC, Schweinsburg AD, Jacobus J, Bava S, Frank LR, Tapert SF. Altered white matter integrity in adolescent binge drinkers. Alcohol Clin Exp Res. 2009;33:1278–1285. doi: 10.1111/j.1530-0277.2009.00953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina KL, McQueeny T, Nagel BJ, Hanson KL, Schweinsburg AD, Tapert SF. Prefrontal cortex volumes in adolescents with alcohol use disorders: unique gender effects. Alcohol Clin Exp Res. 2008;32:386–394. doi: 10.1111/j.1530-0277.2007.00602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Kuma H, Yokoyama T, Sumitani K, Matsumoto Y, Kusaka T, Warita K, Wang ZY, Hosomi N, Imagawa T, KSB, Itoh S, Nakamura Y, Takeuchi Y. Early postnatal ethanol exposure induces fluctuation in the expression of BDNF mRNA in the developing rat hippocampus. Acta Neurobiol Exp (Wars) 2008;68:484–493. doi: 10.55782/ane-2008-1714. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Yagi T, Kitazawa H, Yasuda M, Kawai N, Tsuboi K, Niki H. Fyn-kinase as a determinant of ethanol sensitivity: relation to NMDA-receptor function. Science. 1997;278:698–701. doi: 10.1126/science.278.5338.698. [DOI] [PubMed] [Google Scholar]
- Moore DB, Madorsky I, Paiva M, Barrow Heaton M. Ethanol exposure alters neurotrophin receptor expression in the rat central nervous system: Effects of neonatal exposure. J Neurobiol. 2004;60:114–126. doi: 10.1002/neu.20010. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Komai S, Tezuka T, Hisatsune C, Umemori H, Semba K, Mishina M, Manabe T, Yamamoto T. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2001;276:693–699. doi: 10.1074/jbc.M008085200. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Komai S, Watabe AM, Kiyama Y, Fukaya M, Arima-Yoshida F, Horai R, Sudo K, Ebine K, Delawary M, Goto J, Umemori H, Tezuka T, Iwakura Y, Watanabe M, Yamamoto T, Manabe T. NR2B tyrosine phosphorylation modulates fear learning as well as amygdaloid synaptic plasticity. Embo J. 2006;25:2867–2877. doi: 10.1038/sj.emboj.7601156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noori HR, Fornal CA. The appropriateness of unbiased optical fractionators to assess cell proliferation in the adult hippocampus. Front Neurosci. 2011;5:140. doi: 10.3389/fnins.2011.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski RS, Lewin SB, Miller MW. Bromodeoxyuridine immunohistochemical determination of the lengths of the cell cycle and the DNA-synthetic phase for an anatomically defined population. J Neurocytol. 1989;18:311–318. doi: 10.1007/BF01190834. [DOI] [PubMed] [Google Scholar]
- O’Dell LE, Roberts AJ, Smith RT, Koob GF. Enhanced alcohol self-administration after intermittent versus continuous alcohol vapor exposure. Alcohol Clin Exp Res. 2004;28:1676–1682. doi: 10.1097/01.alc.0000145781.11923.4e. [DOI] [PubMed] [Google Scholar]
- Ohnuma S, Harris WA. Neurogenesis and the cell cycle. Neuron. 2003;40:199–208. doi: 10.1016/s0896-6273(03)00632-9. [DOI] [PubMed] [Google Scholar]
- Olariu A, Cleaver KM, Cameron HA. Decreased neurogenesis in aged rats results from loss of granule cell precursors without lengthening of the cell cycle. J Comp Neurol. 2007;501:659–667. doi: 10.1002/cne.21268. [DOI] [PubMed] [Google Scholar]
- Parsons OA, Nixon SJ. Neurobehavioral sequelae of alcoholism. Neurol Clin. 1993;11:205–218. [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 3. San Diego: Academic Press; 1997. [DOI] [PubMed] [Google Scholar]
- Peters A, Kaiserman-Abramof IR. The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. Am J Anat. 1970;127:321–355. doi: 10.1002/aja.1001270402. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV. Disruption of brain white matter microstructure by excessive intracellular and extracellular fluid in alcoholism: evidence from diffusion tensor imaging. Neuropsychopharmacology. 2005;30:423–432. doi: 10.1038/sj.npp.1300623. [DOI] [PubMed] [Google Scholar]
- Prendergast MA, Mulholland PJ. Glucocorticoid and polyamine interactions in the plasticity of glutamatergic synapses that contribute to ethanol-associated dependence and neuronal injury. Addict Biol. 2012;17:209–223. doi: 10.1111/j.1369-1600.2011.00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47:845–857. doi: 10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson HN, Lee SY, O’Dell LE, Koob GF, Rivier CL. Alcohol self-administration acutely stimulates the hypothalamic-pituitary-adrenal axis, but alcohol dependence leads to a dampened neuroendocrine state. Eur J Neurosci. 2008;28:1641–1653. doi: 10.1111/j.1460-9568.2008.06455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson HN, Chan SH, Crawford EF, Lee YK, Funk CK, Koob GF, Mandyam CD. Permanent impairment of birth and survival of cortical and hippocampal proliferating cells following excessive drinking during alcohol dependence. Neurobiol Dis. 2009;36:1–10. doi: 10.1016/j.nbd.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridge JP, Ho AM, Innes DJ, Dodd PR. The expression of NMDA receptor subunit mRNA in human chronic alcoholics. Ann N Y Acad Sci. 2008;1139:10–19. doi: 10.1196/annals.1432.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimondini R, Sommer W, Heilig M. A temporal threshold for induction of persistent alcohol preference: behavioral evidence in a rat model of intermittent intoxication. J Stud Alcohol. 2003;64:445–449. doi: 10.15288/jsa.2003.64.445. [DOI] [PubMed] [Google Scholar]
- Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson WD. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–1401. doi: 10.1038/nn.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AJ, Heyser CJ, Cole M, Griffin P, Koob GF. Excessive ethanol drinking following a history of dependence: animal model of allostasis. Neuropsychopharmacology. 2000;22:581–594. doi: 10.1016/S0893-133X(99)00167-0. [DOI] [PubMed] [Google Scholar]
- Salomoni P, Calegari F. Cell cycle control of mammalian neural stem cells: putting a speed limit on G1. Trends Cell Biol. 2010;20:233–243. doi: 10.1016/j.tcb.2010.01.006. [DOI] [PubMed] [Google Scholar]
- Schulteis G, Markou A, Cole M, Koob GF. Decreased brain reward produced by ethanol withdrawal. Proc Natl Acad Sci U S A. 1995;92:5880–5884. doi: 10.1073/pnas.92.13.5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo D, Lacadie CM, Tuit K, Hong KI, Constable RT, Sinha R. Disrupted ventromedial prefrontal function, alcohol craving, and subsequent relapse risk. JAMA Psychiatry. 2013;70:727–739. doi: 10.1001/jamapsychiatry.2013.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skuja S, Groma V, Smane L. Alcoholism and cellular vulnerability in different brain regions. Ultrastruct Pathol. 2012;36:40–47. doi: 10.3109/01913123.2011.629770. [DOI] [PubMed] [Google Scholar]
- Smart FM, Halpain S. Regulation of dendritic spine stability. Hippocampus. 2000;10:542–554. doi: 10.1002/1098-1063(2000)10:5<542::AID-HIPO4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Sommer WH, Rimondini R, Hansson AC, Hipskind PA, Gehlert DR, Barr CS, Heilig MA. Upregulation of voluntary alcohol intake, behavioral sensitivity to stress, and amygdala crhr1 expression following a history of dependence. Biol Psychiatry. 2008;63:139–145. doi: 10.1016/j.biopsych.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Squeglia LM, Pulido C, Wetherill RR, Jacobus J, Brown GG, Tapert SF. Brain response to working memory over three years of adolescence: influence of initiating heavy drinking. J Stud Alcohol Drugs. 2012;73:749–760. doi: 10.15288/jsad.2012.73.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan EV, Rosenbloom MJ, Pfefferbaum A. Pattern of motor and cognitive deficits in detoxified alcoholic men. Alcohol Clin Exp Res. 2000;24:611–621. [PubMed] [Google Scholar]
- Sun Y, Meijer DH, Alberta JA, Mehta S, Kane MF, Tien AC, Fu H, Petryniak MA, Potter GB, Liu Z, Powers JF, Runquist IS, Rowitch DH, Stiles CD. Phosphorylation state of Olig2 regulates proliferation of neural progenitors. Neuron. 2011;69:906–917. doi: 10.1016/j.neuron.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapert SF, Brown GG, Baratta MV, Brown SA. fMRI BOLD response to alcohol stimuli in alcohol dependent young women. Addict Behav. 2004a;29:33–50. doi: 10.1016/j.addbeh.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Tapert SF, Pulido C, Paulus MP, Schuckit MA, Burke C. Level of response to alcohol and brain response during visual working memory. J Stud Alcohol. 2004b;65:692–700. doi: 10.15288/jsa.2004.65.692. [DOI] [PubMed] [Google Scholar]
- Tsai G, Coyle JT. The role of glutamatergic neurotransmission in the pathophysiology of alcoholism. Annu Rev Med. 1998;49:173–184. doi: 10.1146/annurev.med.49.1.173. [DOI] [PubMed] [Google Scholar]
- Tu Y, Kroener S, Abernathy K, Lapish C, Seamans J, Chandler LJ, Woodward JJ. Ethanol inhibits persistent activity in prefrontal cortical neurons. J Neurosci. 2007;27:4765–4775. doi: 10.1523/JNEUROSCI.5378-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez GR, Roberts AJ, Chan K, Davis H, Brennan M, Zorrilla EP, Koob GF. Increased ethanol self-administration and anxiety-like behavior during acute ethanol withdrawal and protracted abstinence: regulation by corticotropin-releasing factor. Alcohol Clin Exp Res. 2002;26:1494–1501. doi: 10.1097/01.ALC.0000033120.51856.F0. [DOI] [PubMed] [Google Scholar]
- Vega CJ, Peterson DA. Stem cell proliferative history in tissue revealed by temporal halogenated thymidine analog discrimination. Nat Methods. 2005;2:167–169. doi: 10.1038/nmeth741. [DOI] [PubMed] [Google Scholar]
- Vendruscolo LF, Barbier E, Schlosburg JE, Misra KK, Whitfield TW, Jr, Logrip ML, Rivier C, Repunte-Canonigo V, Zorrilla EP, Sanna PP, Heilig M, Koob GF. Corticosteroid-dependent plasticity mediates compulsive alcohol drinking in rats. J Neurosci. 2012;32:7563–7571. doi: 10.1523/JNEUROSCI.0069-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Toyama Y, Nishiyama A. Differentiation of proliferated NG2-positive glial progenitor cells in a remyelinating lesion. J Neurosci Res. 2002;69:826–836. doi: 10.1002/jnr.10338. [DOI] [PubMed] [Google Scholar]
- Wegner AM, Nebhan CA, Hu L, Majumdar D, Meier KM, Weaver AM, Webb DJ. N-wasp and the arp2/3 complex are critical regulators of actin in the development of dendritic spines and synapses. J Biol Chem. 2008;283:15912–15920. doi: 10.1074/jbc.M801555200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitlauf C, Woodward JJ. Ethanol selectively attenuates NMDAR-mediated synaptic transmission in the prefrontal cortex. Alcohol Clin Exp Res. 2008;32:690–698. doi: 10.1111/j.1530-0277.2008.00625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward JJ, Pava MJ. Effects of ethanol on persistent activity and up-States in excitatory and inhibitory neurons in prefrontal cortex. Alcohol Clin Exp Res. 2009;33:2134–2140. doi: 10.1111/j.1530-0277.2009.01059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaka R, Thornton C, Vagts AJ, Phamluong K, Bonci A, Ron D. NMDA receptor function is regulated by the inhibitory scaffolding protein, RACK1. Proc Natl Acad Sci U S A. 2002;99:5710–5715. doi: 10.1073/pnas.062046299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan CJ, Quiocho JM, Kim A, Wee S, Mandyam CD. Extended access methamphetamine decreases immature neurons in the hippocampus which results from loss and altered development of neural progenitors without altered dynamics of the S-phase of the cell cycle. Pharmacol Biochem Behav. 2011;100:98–108. doi: 10.1016/j.pbb.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahr NM, Mayer D, Vinco S, Orduna J, Luong R, Sullivan EV, Pfefferbaum A. In vivo evidence for alcohol-induced neurochemical changes in rat brain without protracted withdrawal, pronounced thiamine deficiency, or severe liver damage. Neuropsychopharmacology. 2009;34:1427–1442. doi: 10.1038/npp.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Weiss F, Zorrilla EP. Remission and resurgence of anxiety-like behavior across protracted withdrawal stages in ethanol-dependent rats. Alcohol Clin Exp Res. 2007;31:1505–1515. doi: 10.1111/j.1530-0277.2007.00456.x. [DOI] [PubMed] [Google Scholar]