

Abstract
Hemophagocytic lymphohistiocytosis (HLH) is an inborn disorder of immune regulation caused by mutations affecting perforin-dependent cytotoxicity. Defects of this pathway impair negative feedback between cytotoxic lymphocytes and APCs, leading to prolonged and pathologic activation of T cells. Etoposide, a widely used chemotherapeutic drug which inhibits topoisomerase II, is the mainstay of treatment for HLH, though its therapeutic mechanism remains unknown. We utilized a murine model of HLH, involving lymphocytic choriomeningitis virus infection of perforin deficient mice to study the activity and mechanism of etoposide for treating HLH and found that it substantially alleviated all symptoms of murine HLH and allowed prolonged survival. This therapeutic effect was relatively unique among chemotherapeutic agents tested, suggesting distinctive effects on the immune response. We found that the therapeutic mechanism of etoposide in this model system involved potent deletion of activated T cells and efficient suppression of inflammatory cytokine production. This effect was remarkably selective; etoposide did not exert a direct anti-inflammatory effect on macrophages or dendritic cells and it did not cause deletion of quiescent naive or memory T cells. Finally, etoposide’s immunomodulatory effects were similar in wild type and perforin deficient animals. Thus, etoposide treats HLH by selectively eliminating pathologic, activated T cells and may have utility as a novel immune modulator in a broad array of immunopathologic disorders.
Introduction
While immunity against pathogens is necessary for survival, the specificity and magnitude of immune responses must be tightly regulated to avoid dangerous immunopathology. Hemophagocytic lymphohistiocytosis (HLH) is a unique inborn disorder of immune regulation in which immune responses are appropriately directed (not autoimmune), but may be rapidly fatal due to inefficient control of T cell activation.(1) Mutations in perforin and related genes have been found to be causal in most affected families. Studies in animal models have established a chain of pathogenesis in which defective perforin-mediated cytotoxic feedback allows prolonged/excessive antigen presentation by dendritic cells (DC), which drives abnormal CD8+ T cell activation, IFN-γ production, and subsequent macrophage-mediated immunopathology.(2) (3) (4) (5) Though this pathologic immune activation may have varied manifestations, a constellation of features typifies patients with HLH: fever, splenomegaly, severe cytopenias, elevated ferritin (often extreme), depressed levels of fibrinogen (paradoxical, in the context of inflammation), elevated markers of T cell and macrophage activation (e.g., soluble CD25 and CD163), and hemophagocytosis (the appearance of macrophages engulfing blood and marrow cells) in various tissues.(6, 7)
Though HLH is rapidly fatal without specific therapy, treatment with the chemotherapeutic agent etoposide was empirically discovered to be life-saving over 30 years ago.(8) Follow-up international studies have established etoposide-based regimens as the standard of care.(9) (10) Despite this extensive clinical study, it remains unclear how etoposide treats HLH or why it has succeeded while other chemotherapeutic or immunosuppressive agents tried sporadically in the 1970’s and 1980’s were largely unsuccessful.(11) The only study to examine this question analyzed etoposide-driven apoptosis of lymphocytes from patients with HLH and found that they responded in a similar fashion as those from normal individuals.(12) The question of how a chemotherapeutic agent treats an intrinsic immune regulatory disorder is also broadly relevant to the fields of autoimmunity and transplantation, where DNA-damaging chemotherapeutic agents are used to treat rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, and to prevent graft-versus-host disease.
In the current report, we utilized a well-described murine model of HLH, involving lymphocytic choriomeningitis virus (LCMV) infection of perforin deficient (prf-/-) mice, to study the in vivo therapeutic mechanism of action for etoposide. We found that etoposide has clear therapeutic activity in this model, alleviating all features of HLH. When compared to a broad array of chemotherapeutic agents, we found that it was relatively unique. Because etoposide is often observed to have rapid and dramatic effects in clinical HLH, it has been commonly speculated that it may be acting directly on inflamed macrophages. Contrary to this expectation, we found no direct beneficial effects on either macrophage activation or antigen presentation by DC’s. Instead, we found that etoposide strongly suppressed IFN-γ levels. This, in turn, was due to near complete ablation of pathologically activated, virus-specific T cells. Notably, this depletion was also seen in wild type (WT) animals and was extremely specific in vivo- quiescent naïve and memory cells were largely spared. Because of its potency and selectivity, we conclude that etoposide has unique immunomodulating qualities in HLH and may have broader utility as an immunosuppressive agent.
Methods
Mice, viruses, and in vivo treatments
C57BL/6, prf-/-, and IFN-γ-/- mice were obtained from The Jackson Laboratory. TCR-transgenic P14, SMARTA, and OT1 mice were a gift from P. Marrack. Mice were housed in a specific pathogen-free facility and animal experiments were approved by the CCHMC IACUC. LCMV-WE viral stocks were generated and tittered as described.(13) Mice were infected via intraperitoneal (IP) injection of 200 plaque-forming units. Vaccinia-ova was obtained from P. Marrack and 105 pfu were injected IV. All chemotherapeutics were obtained from the CCHMC clinical pharmacy [Etoposide(14) (ETOP, 50 mg/kg, cyclophosphamide(15) (CPM, 100 mg/kg), methotrexate(16) (MTX, 70 mg/kg), cisplatin(17) (CDDP, 10 mg/kg), clofarabine(18, 19) (CLO, 8 mg/kg), dexamethasone(20) (DEX, 3 mg/kg), doxorubicin(21) (DOXO, 1 mg/kg), fludarabine(22) (FLU, 300 mg/kg), 5-fluorouracil(23) (5FU, 20 mg/kg), or vinblastine(24) (VBL, 0.5 mg/kg)], and were administered as single doses via IP injection after dilution or reconstitution in saline or 5% dextrose. Etoposide carrier (65% peg300, 30% ethanol, 5% tween80; 50mcl/mouse) was used as indicated. Mice were examined longitudinally, typically 3 times per week, for development of signs of HLH-like disease using a clinical scoring system to gauge disease severity (see supplemental table 1 for details). For IFN-γ infusion, osmotic pumps (Durect) were loaded with IFN-γ and implanted subcutaneously as previously described.(4)
ELISA and complete blood counts
ELISAs were performed on either serum (IFN-γ) or plasma (soluble CD25, ferritin, fibrinogen). Commercially available ELISA kits were used to assess fibrinogen and ferritin levels, and assays were performed according to manufacturer’s instructions (Innovative Research). For other ELISA’s, capture and detecting antibodies were purified from hybridoma supernantants as previously described.(4) (3) Complete blood counts were obtained using a dedicated veterinary CBC machine (Hemavet950FS; Drew Scientific).
Cell isolation, adoptive transfers, and cell culture
For in vivo generation of quiescent memory T cells, 3000 CD45.1+ OT1 T cells were transferred into naïve prf-/- mice one day prior to vaccinia-ova infection. In vitro etoposide cultures were as follows: spleens cells from uninfected or LCMV-infected mice were ficoll purified, then cultured overnight in DMEM with either IL-7 (0.5ng/ml, peprotech) or IL-2 (100 U/ml, human clinical grade) for cells from naïve or LCMV infected mice, respectively. After overnight culture, cells were once again ficolled, then cultured with etoposide for 4 hours. Drug was then washed out and cells were cultured overnight in cytokine, then assessed for apoptosis 12 hours after etoposide pulse. For ex vivo antigen presentation assays, dendritic cells were isolated using magnetic beads (70-80% CD11c+/MHC II+; mostly CD11b+ after LCMV) and cultured with LCMV-specific T cells (P14 or SMARTA) as described.(25) Supernatants were harvested after 24 hours for analysis of IFN-γ production by the T cells in response to antigen presentation.
Flow cytometry, microscopy
All Abs were obtained from either Biolegend or eBioscience. Tetrameric major histocompatibility complex (MHC) reagents were produced in SF9 insect cells as described.(26) For quantitation of T cells producing IFN-γ in vivo, mice were injected with brefeldin A prior to sacrifice and spleens were crushed in PBS/1% paraformaldehyde, as described.(3) For measuring phosphatidylserine exposure, cells were stained with recombinant, Alexa-647 labeled MFG-E8-D89E, a high affinity PS-binding protein.(27) For microscopy, tissues were fixed in 10% buffered formalin and paraffin embedded, followed by sectioning and staining with hematoxylin/eosin. Images were obtained with an Evos XL core microscope using a 10x or 20x objective.
Statistics
All statistical tests were performed using NCSS2007 statistical analysis software. Where appropriate, results are given as the mean ± standard error with statistical significance determined by two-tailed t-test.
Results
Etoposide treatment rescues LCMV-infected prf-/- mice from HLH-like disease
LCMV-infected perforin-deficient (prf-/-) mice were previously characterized by our group and validated as a robust murine model for human HLH.(2) These mice develop a fatal HLH-like syndrome within weeks following LCMV infection, displaying all of the diagnostic and typical features seen in patients with HLH. We tested the effects of etoposide on HLH-like disease development in this model by giving a single IP drug injection of 50mg/kg, 5 days post-LCMV infection. We chose this time point because it is around the time that immune responses in wild type (WT) and prf-/- mice begin to diverge and this dose because it is within the range of xenograft studies and is the murine equivalent of what is used clinically for treating HLH.(1, 28, 29) When treated with etoposide, we found that prf-/- mice survived long-term after LCMV infection, while those treated with drug vehicle died within three weeks of infection (Figure 1A). A dose titration revealed that etoposide’s effects on survival and immune activation (see below) were titratable (Fig. S1). In order to more accurately measure global disease severity in LCMV-infected mice, we developed a clinical scoring system, ranging from asymptomatic (0) to dead (12), based on weight loss, stance, coordination, skin tenting, conjunctivitis, ascites, and mortality (see supplemental table 1 for further details). As expected, LCMV-infected prf-/- mice displayed much higher clinical scores, whereas LCMV-infected WT mice experienced mild clinical symptoms and generally recovered by day 20 of illness (Figure 1B). Etoposide treated prf-/- mice had higher initial scores than WT mice, but recovered and displayed prolonged survival (Figure 1A). Etoposide treated WT mice displayed no clear benefit or worsening on this scale, compared to WT controls (data not shown).
Figure 1. Etoposide treatment rescues LCMV-infected prf-/- mice from HLH-like disease.
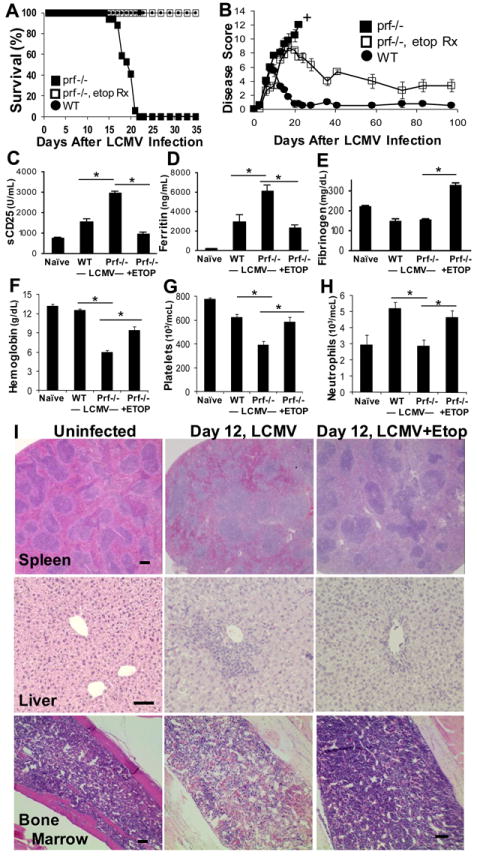
LCMV-infected prf-/- mice were treated with etoposide (ETOP), or carrier by ip injection 5 days post-infection. LCMV-infected wild type mice treated with carrier 5 days post-infection are included for comparison. Mice were monitored longitudinally for survival (A) and disease severity using a clinical scoring system described in the methods section (B). Plasma was assessed by ELISA at day 8 after infection for soluble CD25 (C) and at day 12 for ferritin (D), and fibrinogen (E). Blood was drawn on day 15 day after LCMV infection for assessment of blood counts, including hemoglobin (F), platelet (G), and neutrophil (H) levels. Tissues were obtained 12 days after infection and stained with hematoxylin/ eosin, revealing preservation of normal splenic architecture, decreased liver infiltrates, and improved marrow cellularity with etoposide treatment (markers = 100 microns) (I). Data are presented as mean ± SE and are compiled from 8-23 mice per group assayed across 3 or more experiments. † = all mice in group deceased. *p<0.005,
We also examined HLH disease-related biomarkers and cytopenias in these mice. In human HLH, elevations in plasma soluble-CD25 (IL2-receptor, α-chain) and ferritin levels are thought to correlate with systemic T cell activation and acute-phase inflammation, respectively, while variable decreases in plasma fibrinogen levels are less well understood.(7) These biomarker profiles are recapitulated in LCMV-infected prf-/- mice, relative to infected WT controls, but were efficiently reversed with etoposide administration (Figure 1C-E). Furthermore, etoposide treatment rescued LCMV-infected prf-/- mice from the development of cytopenias (Figure 1F-H). Finally, we examined multiple tissues for evidence of histologic improvement and found that etoposide preserved normal splenic architecture (which is largely effaced in prf-/- mice after LCMV infection), decreased inflammatory liver infiltrates, and, ironically, increased marrow cellularity (Figure 1I). Thus, etoposide treatment leads to improvements in disease severity, mortality, disease-related biomarkers, cytopenias, and histology in LCMV-infected prf-/- mice.
Only select chemotherapeutic agents have efficacy for treating murine HLH
We next sought to determine whether etoposide is unique in its ability to treat murine HLH or whether this is a general attribute of cytotoxic chemotherapy. Figure 2 shows clinical score (A,B) and blood hemoglobin (C) levels for LCMV-infected prf-/- mice given a single IP injection of various chemotherapy drugs 5 days post-LCMV infection. For the sake of clarity, drugs that exhibited therapeutic activity are shown in Figure 2A, while those that failed to improve the disease course are shown in Figure 2B. As was seen with etoposide, mice treated with either cyclophosphamide or methotrexate survived (Figure 2A). The kinetics of their response after treatment with CPM or MTX and reversal of HLH-related anemia (Figure 2C) was similar to that seen with etoposide. In contrast, no improvement in the disease course was demonstrated in LCMV-infected prf-/- mice treated with other cytolytic or anti-inflammatory agents using published murine doses (Figure 2B, C). Attempts at dose escalation (CDDP, CLO, DOXO, FLU) resulted in early mortality and attempts at serial dosing on a daily schedule (DEX, FLU) did not alter the disease course (data not shown). Thus, etoposide appears relatively unique among a range of agents tested for its ability to alleviate murine HLH. The two exceptions to this finding, cyclophosphamide and methotrexate are notable for their uses as immune suppressive agents in a variety of clinical contexts or for intrathecal treatment of HLH-associated inflammation of the central nervous system.
Figure 2. Only select chemotherapeutic agents have efficacy in treating murine HLH.

LCMV-infected prf-/- mice were treated with cyclophosphamide (CPM), etoposide (ETOP), methotrexate (MTX), cisplatin (CDDP), clofarabine (CLO), dexamethasone (DEX), doxorubicin (DOXO), fludarabine (FLU), 5-fluorouracil (5FU), vinblastine (VBL), or carrier control (Prf-/- CON) by IP injection 5 days post-infection. LCMV-infected wild type mice treated with carrier control (WT CON) 5 days post-infection are included for comparison. Mice were monitored serially for disease severity using a clinical scoring system to measure therapeutic effect (A) or lack thereof (B). Blood was drawn on day 15 post-LCMV infection to assess hemoglobin levels (C). Data are the mean ± SE of 3-4 mice per group, representative of 3 or more independent experiments. † = all mice in treatment group deceased, *p<0.004.
Etoposide treatment does not directly decrease macrophage activation or antigen presentation by dendritic cells in LCMV-infected prf-/- mice
Due to clinical observations and more recent experimental data(4), it is widely believed that systemic activation of macrophages plays a prominent role in the development of HLH. This fact combined with the prompt clinical response observed in some patients after starting etoposide has suggested to many that etoposide exerts its therapeutic effect by suppressing macrophage activation and/or antigen presentation.(30) In order to assess this potential mechanism of action, we examined macrophage phenotypes after etoposide treatment of LCMV-infected prf-/-mice. When we examined tissues 1-2 weeks after etoposide treatment, we observed decreased macrophage infiltration by histology and flow cytometry (F4/80+ cells decreasing from approx. 13% to 9%) (Figure 1 and data not shown). However, because disease development was completely reversed in these animals, it was not clear whether this was simply a secondary effect. In contrast, when we examined tissues on day 7, after treating with etoposide on day 5, we found that the highly activated phenotype of splenic macrophages (assessing CD40, CD80, CD86, and MHC class II on F4/80+ cells in spleen, marrow, and liver) was unchanged (Figure 3A and B and data not shown). Furthermore, the numbers of macrophages and DC’s, as a percentage of splenocytes, were not decreased after etoposide (approx. 13% F4/80+ and 2% CD11c+/MHC II+, with or without treatment). In addition to surface phenotype and numbers, we sought to assess the function of antigen presenting cells after etoposide. We have recently reported that abnormalities of DC function, rather than DC numbers, are a critical determinant of immune dysregulation in prf-/- mice.(5) To investigate this, we isolated DC’s from etoposide or carrier-treated, LCMV-infected prf-/- mice (treatment on day 5, DC isolation on day 7) and cultured them with LCMV-specific CD8+ and CD4+ T cells. As expected, we found that DC’s from prf-/- mice presented endogenously-derived viral antigen much more potently than those from WT mice (Figure 3C and D). Notably, DC’s from etoposide-treated prf-/- mice revealed no decrease in antigen presentation potency. In fact, in some experiments, antigen presentation was actually increased by etoposide treatment (Figure 3C and data not shown). This finding was similar to prior observations that in vivo T cell depletion leads to increases of antigen presentation by DC’s.(5) Thus, etoposide does not appear to have a direct suppressive effect on macrophage activation or antigen presentation by DC’s.
Figure 3. Etoposide treatment does not directly decrease macrophage activation or the quality of antigen presentation by dendritic cells in LCMV-infected prf-/- mice.
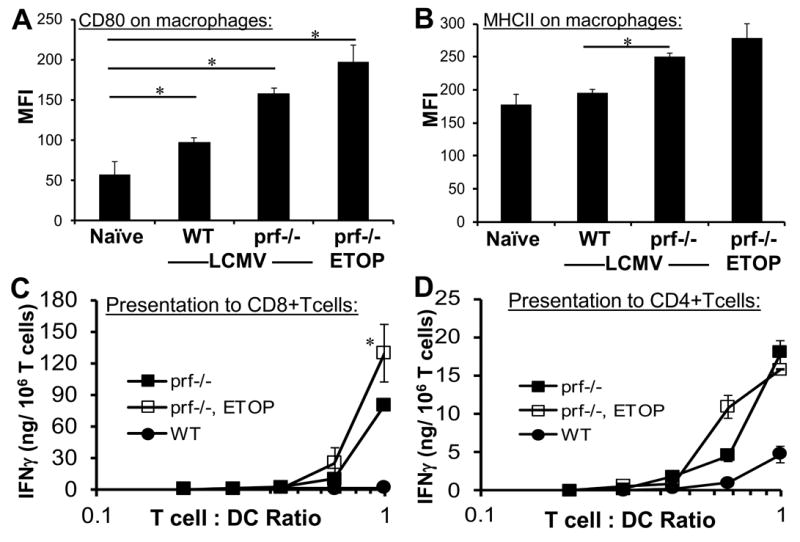
LCMV-infected prf-/- mice were treated with etoposide or carrier 5 days post-infection. LCMV-infected wild type mice treated with carrier are included for comparison. Surface phenotype of splenic macrophages (F4/80+) was assessed two days later by flow cytometry, including CD80 and MHC class II levels (A and B). Dendritic cells (CD11c+/ MHC II+) were magnetically sorted from collagenase treated spleens (pooled, 4 animals/group) seven days after infection and plated with LCMV-specific transgenic CD8+ or CD4+ T cells (P14 or SMARTA) to assess MHC class I and class II restricted presentation of endogenously acquired viral antigens. Antigen presentation to T cells was quantitated by IFN-γ production after overnight culture. No IFN-γ was detected when T cells of irrelevant specificity were cultured with DC’s (not shown). Data are the mean ± SE of 3-4 animals per group, representative of 3 independent experiments. *P<0.05
Etoposide alleviates murine HLH by suppressing IFN-γ levels in LCMV-infected prf-/- mice
Prior studies have indicated that hyperproduction of IFN- γ is necessary for development of HLH in LCMV-infected prf-/- mice.(2) Therefore we hypothesized that the therapeutic effect of etoposide in these mice is due to a reduction of IFN- γ levels. Accordingly, we observed that etoposide treatment on day 5 led to a dramatic suppression of serum IFN-γ levels in prf-/- mice (Figure 4A). Furthermore, we predicted that if suppression of IFN-γ levels is a component of etopside’s therapeutic mechanism, then drug treatment after the in vivo peak of cytokine production would have little efficacy, because macrophages have already been exposed to high levels of IFN-γ and etoposide does not appear to act directly on macrophages.(2),(4) On the contrary, if etoposide directly affected macrophage activation, then treatment would still be therapeutic at later time points. To test our prediction, we treated LCMV-infected prf-/- mice with etoposide at day 10, a timepoint after peak cytokines levels, but before significant anemia has developed.(4) In this case, neither the disease course (Figure 4B) nor the disease-related anemia (Figure 4C) were ameliorated by late etoposide therapy. In contrast, mice deficient for both perforin and IFN-γ (DKO) were protected from anemia and fatal disease (Figure 4B and C).
Figure 4. Etoposide alleviates murine HLH by suppressing IFN-γ levels in LCMV-infected prf-/- mice.
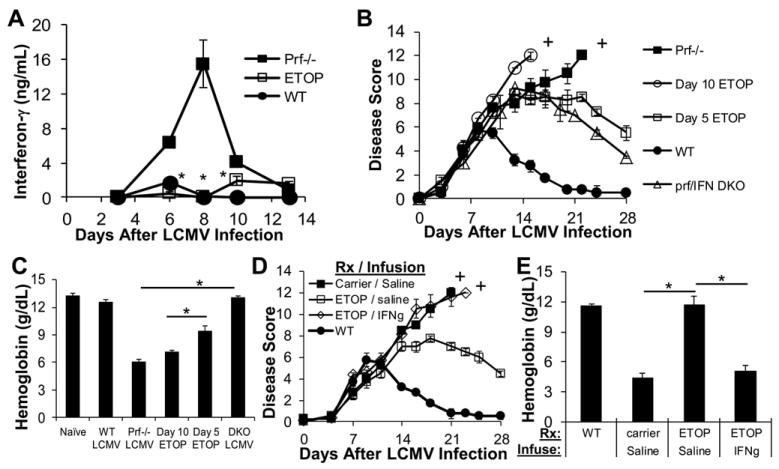
LCMV-infected prf-/- mice were treated with carrier or etoposide 5 days post-infection and serum was obtained on days 3, 6, 8, 10, and 13 for assessment of IFN-γ levels by ELISA (A). LCMV-infected prf-/- mice were treated with etoposide either before or after peak IFN-γ production (day 5 or 10) and monitored longitudinally for disease severity using a clinical scoring system (B) or bled on day 15 to assess blood hemoglobin level (C). LCMV-infected wild type mice and prf/ IFN-γ (DKO) deficient mice are included for comparison. LCMV-infected prf-/- mice were treated on day 5 post-infection with carrier or etoposide. On day 5 these mice were also implanted with osmotic pumps (containing either IFN-γ or saline) to ‘add back’ IFN-γ that is suppressed by etoposide treatment. Pumps were calibrated to maintain serum IFN-γ levels between 3 and 6 ng/ml for up to 7 days. Mice were monitored longitudinally using a clinical scoring system (D) and blood was drawn for assessment of hemoglobin 15 days post-LCMV infection (E). Data are the mean ± SE of 3-4 mice per group, representative of 3 or more experiments. †=all mice in treatment group deceased, *p<0.01 when comparing etoposide vs. carrier treated mice, DKO vs. prf-/- mice, treatment at day 5 vs. day 10, or IFN-γ vs. saline infused animals.
If decreasing inflammatory cytokine production is an important aspect of etoposide’s mechanism in HLH, then ‘adding back’ IFN-γ to etoposide treated prf-/- mice, would abolish its therapeutic benefit. To test this prediction, we utilized subcutaneous osmotic pumps to deliver IFN-γ or saline to LCMV-infected prf-/- mice treated with etoposide on day 5. We implanted pumps shortly after administering etoposide or carrier, titrated to maintain serum IFN-γ levels of 3-6 ng/ml for up to 7 days. This level is somewhat lower than that seen in control LCMV-infected prf-/- mice, but much higher than seen in etoposide treated or WT mice. Consistent with previous results, mice treated with etoposide and infused with saline showed improvement in disease symptoms and survival (Figure 4D and E). However, when mice were given exogenous IFN-γ, the benefit of etoposide therapy was completely abrogated (Figure 4D and E). Thus, suppression of excessive IFN-γ production is a key aspect of etoposide’s mechanism of action in murine HLH.
Etoposide acts via selective destruction of activated T cells in LCMV-infected prf-/- mice
Because we have previously demonstrated that most detectable IFN-γ in LCMV-infected prf-/- mice is derived from highly activated CD8+ T cells,(2) we hypothesized that etoposide was suppressing excessive IFN-γ production by depleting or potentially ‘de-activating’ these cells. To test this hypothesis directly, we measured the number of CD8+ T cells producing IFN-γ in vivo using a previously described technique.(3) We found that etoposide decreased the number of IFN-γ producing T cells to levels that were similar to (or lower than) those seen in WT mice (Figure 5A). Next, we directly assessed depletion of virus-specific T cells by staining with a peptide-MHC tetrameric staining reagent (H-2Db-Gp33) incorporating an immunodominant LCMV epitope (Gp33-41). We found that etoposide treatment lead to near complete ablation of LCMV-specific CD8+ T cells in all animals (Figure 5B). The absolute number of Db-Gp33+ CD8+ T cells dropped from approximately 2-4 million cells/ spleen to less than 50,000 in most animals, with almost 1/3 of etoposide-treated animals displaying no Db-Gp33-specific T cells above staining background. In order to account for the practical limits of detection using MHC-tetrameric reagents, we (conservatively) scored animals in which no antigen-specific T cells could be detected as ‘100 fold’ depleted. When we compiled data from multiple experiments involving a large number of animals (>20/ group), we found that in both prf-/- and WT animals, etoposide treatment caused a nearly 100 fold depletion (compared to carrier-treated mice in the same experiment) of virus-specific CD8+ T cells (Figure 5C). Thus, etoposide was suppressing IFN-γ production in prf-/- mice by potently ablating (not ‘deactivating’) pathologically activated, virus-specific T cells. In contrast to this profound depletion of LCMV-specific T cells, naïve T cells (defined as CD44lo) and pre-existing (quiescent) memory T cells (see methods) were largely spared. A similar depletional effect of etoposide was seen on CD4+ T cells, where virus-specific cells (those staining with IAb-GP61 tetramer) were largely ablated, while naïve CD4+ T cells were spared (Figure 5D). Though the role of CD4+ T cells in driving HLH pathogenesis is not as clear as CD8+ T cells, etoposide’s ability to selectively affect activated CD8+ and CD4+ T cells in both WT and prf-/- animals suggests that it could be useful for immunomodulation in a broad range of contexts.
Figure 5. Etoposide acts via selective destruction of activated effector T cells in LCMV-infected prf-/- mice.
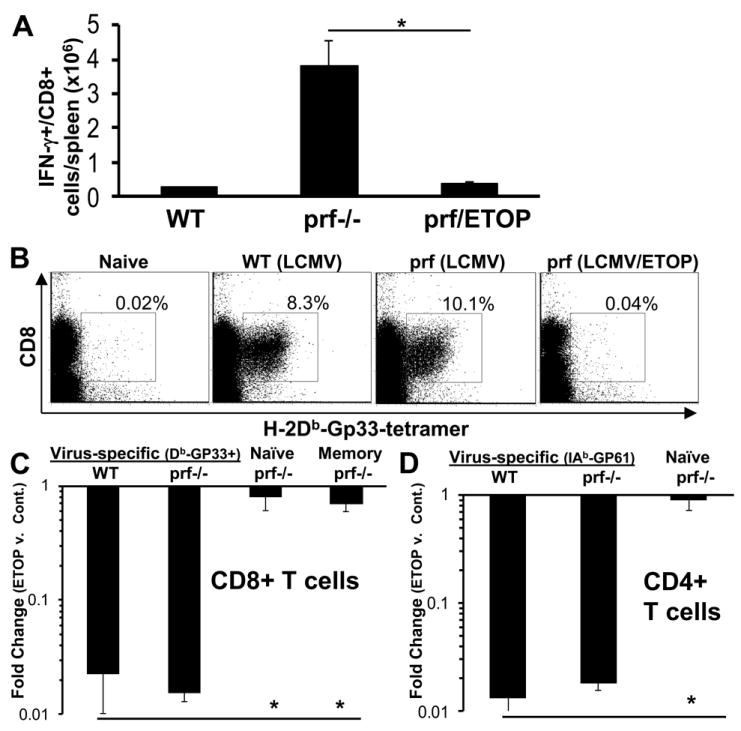
LCMV-infected WT or prf-/- mice were treated with etoposide or carrier 5 days after LCMV infection. Spleens were harvested 8 days post-infection after in vivo brefeldin administration for quantitation of T cells which were producing IFN-γ in vivo (A). In parallel experiments, day 8 spleen cells were stained using MHC-peptide tetramers (Gp33 in the context of Db) to delineate virus-specific CD8+ T cells. Representative plots of live gated-spleen cells from each group are shown. Percentage shown reflect % of live-gated/CD8+ cells. (B). Absolute numbers of virus-specific (Db-GP33 tetramer+), naïve (CD44lo), and quiescent memory CD8+ T cells were quantitated in LCMV-infected, etoposide (or carrier) treated animals (C). Fold change with etoposide treatment was calculated by comparing the total number of each cell population in spleens of carrier and drug treated animals. For assessment of quiescent memory T cells, ovalbumin-specific T cells primed by vaccinia-ova infection >1 month prior to LCMV infection were tracked. Virus-specific CD4+ T cells were enumerated with IAb-GP61-80 tetramer and compared to naïve CD4+ T cells in LCMV-infected, etoposide (or carrier) treated animals (D). *P<0.01 Data are ± SE, with >8 mice per group, from 3 or more experiments.
Etoposide directly and preferentially induces apoptosis of activated T cells
To determine whether etoposide was acting directly on activated T cells, we cultured spleen cells from LCMV-infected or uninfected prf-/- mice with a titration of drug and measured apoptosis by assessing cell permeability and phosphatidylserine exposure. We found that etoposide induced apoptosis of activated T cells at physiologically relevant concentrations after overnight culture (Figure 6A and B).(31) We observed similar results with highly purified, in vitro activated T cells, further confirming a direct effect (data not shown). When we assessed the kinetics of in vivo therapy, we found that etoposide-induced T cell ablation was evident within 24 hours of treatment, suggesting a similar mechanism (Fig. S2). Finally, we observed that activated CD8+ T cells from LCMV-infected mice (CD44hi, day 6) were significantly more sensitive to the apoptosis-inducing effects of etoposide than naïve T cells (CD44lo) from uninfected animals (Figure 6B). Thus, etoposide acts directly on CD8+ T cells to induce apoptosis and activation potentiates this effect.
Figure 6. Etoposide directly and preferentially induces apoptosis of activated T cells.
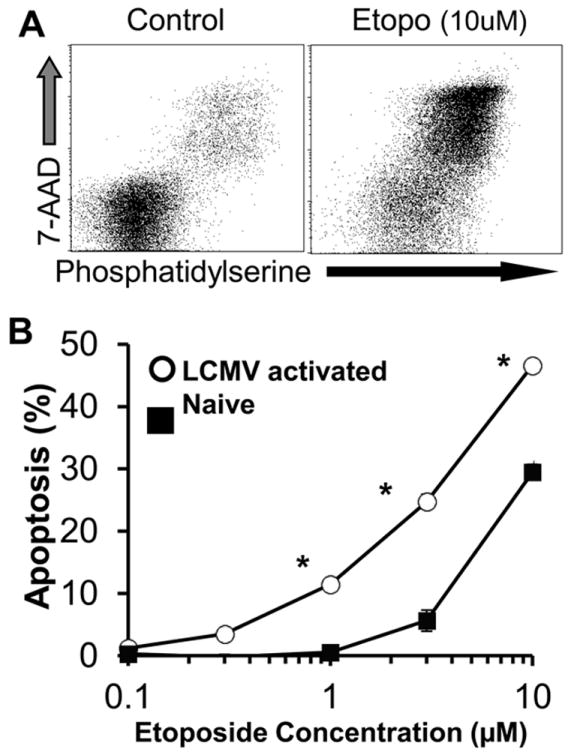
Spleen cells from LCMV-infected (day 6) prf-/- mice were cultured in the presence of etoposide or carrier for 4 hours, then washed and cultured overnight. Cells were then assessed for permeability (with 7-AAD) and phosphatidylserine exposure (using a recombinant PS-binding protein, see methods). CD8+ cells are shown (A). LCMV-activated (day 6, CD44hi cells) and naïve (CD44lo from uninfected mice) CD8+ cells were exposed to a titration of etoposide (or carrier) as in A, and apoptosis induction (%phosphatidylserine+) was measured (B). *P<0.01
Discussion
Though etoposide is the foundation of modern HLH therapy, there has been little understanding of how it reverses life-threatening inflammation in patients with HLH, or why it has succeeded over other chemotherapeutic or anti-inflammatory agents. In the current report we have found that etoposide’s therapeutic activity can be robustly reproduced in a well-established murine model of HLH. This observation has given us a unique window to dissect its mechanism of action in a disease relevant context. Consistent with very sparse clinical data, we found that etoposide does indeed have a relatively unique ability to suppress HLH-like immunopathology. Furthermore, we have found that etoposide’s effects on this pathologic immune response are highly specific for activated T cells and largely spare quiescent T cells and innate immune cells. This high degree of selectivity suggests that etoposide may have unique immunomodulatory qualities in a wider array of immunopathologic conditions. Our findings also underscore the potential utility of studying HLH as a prototype for T cell-driven disorders.
Our findings have several implications related to the pathophysiology and treatment of HLH. First, it is notable that dexamethasone, a broadly immunosuppressive agent, had minimal therapeutic effects, compared with etoposide (Figure 1). In additional studies (not shown), we have found that the combination of etoposide with dexamethasone does have additive effects, but these benefits are modest. Because HLH appears to entail abnormal regulation of antigen presentation,(5) we interpret this finding to suggest that pharmacologic suppression of T cell activation (such as with corticosteroids or calcineurin inhibitors) may be largely futile in the presence of ongoing and/or strong antigenic stimuli. Instead, deletion of the offending T cells appears to be the most effective approach. Indeed, the main clinical alternative to etoposide for treating HLH is the use of T cell depleting antibodies (anti-thymocyte globulin or alemtuzumab).(1) Second, though an indirect consequence of etoposide-mediated T cell deletion, the key role of IFN-γ suppression underscores the importance of this inflammatory mediator in HLH and provides additional impetus for translational efforts testing IFN-γ blockade. One caveat: the relatively narrow time window we observed for effective etoposide treatment suggests that kinetics or other aspects (perhaps IFN-γ related) of this model diverge from clinical HLH, where etoposide may be effective even after prolonged illness. Third, our findings underscore the importance of T cells as therapeutic targets in HLH, reinforcing existing clinical and experimental data.
First synthesized in 1966, etoposide exhibits anti-neoplastic activity against AML, lymphomas, and a variety of solid-tumor cancers.(32) Etoposide inhibits topoisomerase II, leading to double stranded DNA breaks (32), though precisely how this specific genotoxic lesion exerts a selective effect on activated T cells is not understood. Ongoing studies on this topic may reveal new aspects of T cell biology and new therapeutic targets for immunologic diseases.
Of the agents tested, three chemotherapeutic drugs were capable of attenuating murine HLH disease pathology: etoposide, cyclophosphamide, and methotrexate. Though these agents are biochemically diverse (a topoisomerase inhibitor, DNA alkylator, and an antimetabolite, respectively) they share clinical utility as immunosuppressive drugs. Although etoposide is the basis of modern HLH therapy, methotrexate is used as intrathecal therapy for CNS inflammation in HLH(6, 33) while cyclophosphamide has been rarely reported as monotherapy(34) or as part of combination chemotherapy(35) of HLH. Though etoposide has not been studied in the context of auto- or allo-immunity, both methotrexate and cyclophosphamide are used in a variety of severe autoimmune conditions and for the prevention/treatment of graft versus host disease.(36, 37) Surprisingly, fludarabine, a purine analog with strong anti-lymphocyte qualities (38), was not therapeutic in our system, despite multiple dosing strategies. Therefore, it appears that mechanistic differences between these agents are relevant to their immunomodulatory qualities and will require further study.
One of the most notable findings of the current study is the remarkable selectivity we observed for etoposide as a T cell depleting and immunomodulating agent. It ablated activated, anti-viral T cells nearly 100 fold and (indirectly) suppressed inflammatory cytokine levels, while essentially sparing quiescent naïve and memory T cells. Our findings are consistent with prior reports noting that etoposide causes apoptosis of activated, but not resting lymphocytes in vitro. (39) (40) (41) Notably, we observed similar potency and selectivity in prf-/- and WT animals (Figure 5 and data not shown). Together, these findings suggest that etoposide could be used to efficiently eliminate unwanted or pathologic T cells in a wider variety of contexts, based on their activation status. Sparing of quiescent T cells would preserve protective memory responses and would not impair responses to newly encountered pathogens. Additional studies are needed, however, as other investigators have observed temporary suppressive effects on T cells surviving etoposide exposure. (42) Despite potential limits, a selective deletional approach could compare very favorably to non-selective T cell depleting agents (such as alemtuzumab) or broadly immunosuppressive drugs (such as corticosteroids). However, additional preclinical studies will be needed to assess the potential efficacy of etoposide in immune disorders beyond HLH.
The principle adverse effects of etoposide and other chemotherapeutic agents relate to off-target genotoxicity, including dose-dependent suppression of hematopoiesis and risk of secondary leukemia. In general, therapy-related leukemia has been reported after treatment with prolonged or high intensity combination regimens incorporating etoposide and other genotoxic agents, though patients with HLH receiving unusually prolonged courses of etoposide monotherapy have developed leukemia. (43, 44) Paradoxically, we found that etoposide prevented the development of cytopenias and marrow hypocellularity in treated animals, presumably because its desirable effects on T cells were more potent that its off target effects on marrow cells. Though such adverse effects may be managed clinically, further studies into the mechanistic basis of immune modulation, genotoxicity, and marrow suppression may help to further improve the therapeutic index of etoposide and related drugs for the treatment of HLH and other immunologic disorders.
Supplementary Material
Acknowledgments
We would also like to thank Erin Zoller, PhD for technical assistance in osmotic pump placement and Robert Thacker, PhD for assistance with the ex vivo antigen stimulation assays. In addition, we appreciate the gracious support of the Cincinnati Children’s Hospital Medical Center clinical pharmacy.
This work was supported by NIH grant R01HL091769 and the Primary Immune Deficiency Treatment Consortium as a sub-award from NIH grant U54 AI082973.
Abreviations used in this article
- HLH
hemophagocytic lymphohistiocytosis
- DC
dendritic cell
- LCMV
lymphocytic choriomeningitis virus
- WT
wild type
- DKO
double knockout
- ETOP
etoposide
- CPM
cyclophosphamide
- MTX
methotrexate
- CDDP
cisplatin
- CLO
clofarabine
- DEX
dexamethasone
- DOXO
doxorubicin
- FLU
fludarabine
- 5FU
5-fluorouracil
- VBL
vinblastine
Footnotes
Disclosures
The authors have no financial conflicts of interest to report.
References
- 1.Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118:4041–4052. doi: 10.1182/blood-2011-03-278127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104:735–743. doi: 10.1182/blood-2003-10-3413. [DOI] [PubMed] [Google Scholar]
- 3.Lykens JE, Terrell CE, Zoller EE, Risma K, Jordan MB. Perforin is a critical physiologic regulator of T-cell activation. Blood. 2011;118:618–626. doi: 10.1182/blood-2010-12-324533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zoller EE, Lykens JE, Terrell CE, Aliberti J, Filipovich AH, Henson PM, Jordan MB. Hemophagocytosis causes a consumptive anemia of inflammation. The Journal of experimental medicine. 2011;208:1203–1214. doi: 10.1084/jem.20102538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Terrell CE, Jordan MB. Perforin deficiency impairs a critical immunoregulatory loop involving murine CD8+ T cells and dendritic cells. Blood. 2013 doi: 10.1182/blood-2013-04-495309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatric blood & cancer. 2007;48:124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 7.Johnson TS, Villanueva J, Filipovich AH, Marsh RA, Bleesing JJ. Contemporary diagnostic methods for hemophagocytic lymphohistiocytic disorders. J Immunol Methods. 2011;364:1–13. doi: 10.1016/j.jim.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 8.Ambruso DR, Hays T, Zwartjes WJ, Tubergen DG, Favara BE. Successful treatment of lymphohistiocytic reticulosis with phagocytosis with epipodophyllotoxin VP 16-213. Cancer. 1980;45:2516–2520. doi: 10.1002/1097-0142(19800515)45:10<2516::aid-cncr2820451008>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 9.Henter JI, Samuelsson-Horne A, Arico M, Egeler RM, Elinder G, Filipovich AH, Gadner H, Imashuku S, Komp D, Ladisch S, Webb D, Janka G. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100:2367–2373. doi: 10.1182/blood-2002-01-0172. [DOI] [PubMed] [Google Scholar]
- 10.Trottestam H, Horne A, Arico M, Egeler RM, Filipovich AH, Gadner H, Imashuku S, Ladisch S, Webb D, Janka G, Henter JI, Histiocyte S. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011;118:4577–4584. doi: 10.1182/blood-2011-06-356261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janka GE. Familial hemophagocytic lymphohistiocytosis. European journal of pediatrics. 1983;140:221–230. doi: 10.1007/BF00443367. [DOI] [PubMed] [Google Scholar]
- 12.Fadeel B, Orrenius S, Henter JI. Induction of apoptosis and caspase activation in cells obtained from familial haemophagocytic lymphohistiocytosis patients. British journal of haematology. 1999;106:406–415. doi: 10.1046/j.1365-2141.1999.01538.x. [DOI] [PubMed] [Google Scholar]
- 13.Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. The Journal of experimental medicine. 1984;160:521–540. doi: 10.1084/jem.160.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wood LJ, Nail LM, Perrin NA, Elsea CR, Fischer A, Druker BJ. The cancer chemotherapy drug etoposide (VP-16) induces proinflammatory cytokine production and sickness behavior-like symptoms in a mouse model of cancer chemotherapy-related symptoms. Biol Res Nurs. 2006;8:157–169. doi: 10.1177/1099800406290932. [DOI] [PubMed] [Google Scholar]
- 15.Barbon CM, Yang M, Wands GD, Ramesh R, Slusher BS, Hedley ML, Luby TM. Consecutive low doses of cyclophosphamide preferentially target Tregs and potentiate T cell responses induced by DNA PLG microparticle immunization. Cellular immunology. 2010;262:150–161. doi: 10.1016/j.cellimm.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 16.Dicker AP, Waltham MC, Volkenandt M, Schweitzer BI, Otter GM, Schmid FA, Sirotnak FM, Bertino JR. Methotrexate resistance in an in vivo mouse tumor due to a non-active-site dihydrofolate reductase mutation. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:11797–11801. doi: 10.1073/pnas.90.24.11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shin JN, Seo YW, Kim M, Park SY, Lee MJ, Lee BR, Oh JW, Seol DW, Kim TH. Cisplatin inactivation of caspases inhibits death ligand-induced cell death in vitro and fulminant liver damage in mice. The Journal of biological chemistry. 2005;280:10509–10515. doi: 10.1074/jbc.M413865200. [DOI] [PubMed] [Google Scholar]
- 18.Bonate PL, Arthaud L, Stuhler J, Yerino P, Press RJ, Rose JQ. The distribution, metabolism, and elimination of clofarabine in rats. Drug Metab Dispos. 2005;33:739–748. doi: 10.1124/dmd.104.002592. [DOI] [PubMed] [Google Scholar]
- 19.Parker WB, Shaddix SC, Gilbert KS, Shepherd RV, Waud WR. Enhancement of the in vivo antitumor activity of clofarabine by 1-beta-D-[4-thio-arabinofuranosyl]-cytosine. Cancer chemotherapy and pharmacology. 2009;64:253–261. doi: 10.1007/s00280-008-0862-z. [DOI] [PubMed] [Google Scholar]
- 20.Ben S, Li X, Xu F, Xu W, Li W, Wu Z, Huang H, Shi H, Shen H. Treatment with anti-CC chemokine receptor 3 monoclonal antibody or dexamethasone inhibits the migration and differentiation of bone marrow CD34 progenitor cells in an allergic mouse model. Allergy. 2008;63:1164–1176. doi: 10.1111/j.1398-9995.2008.01747.x. [DOI] [PubMed] [Google Scholar]
- 21.Pajic M, Iyer JK, Kersbergen A, van der Burg E, Nygren AO, Jonkers J, Borst P, Rottenberg S. Moderate increase in Mdr1a/1b expression causes in vivo resistance to doxorubicin in a mouse model for hereditary breast cancer. Cancer research. 2009;69:6396–6404. doi: 10.1158/0008-5472.CAN-09-0041. [DOI] [PubMed] [Google Scholar]
- 22.Plunkett W, Saunders PP. Metabolism and action of purine nucleoside analogs. Pharmacol Ther. 1991;49:239–268. doi: 10.1016/0163-7258(91)90057-s. [DOI] [PubMed] [Google Scholar]
- 23.Saif MW, von Borstel R. 5-Fluorouracil dose escalation enabled with PN401 (triacetyluridine): toxicity reduction and increased antitumor activity in mice. Cancer chemotherapy and pharmacology. 2006;58:136–142. doi: 10.1007/s00280-005-0129-x. [DOI] [PubMed] [Google Scholar]
- 24.Cervi D, Klement G, Stempak D, Baruchel S, Koki A, Ben-David Y. Targeting cyclooxygenase-2 reduces overt toxicity toward low-dose vinblastine and extends survival of juvenile mice with Friend disease. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11:712–719. [PubMed] [Google Scholar]
- 25.Weninger W, Manjunath N, von Andrian UH. Migration and differentiation of CD8+ T cells. Immunological reviews. 2002;186:221–233. doi: 10.1034/j.1600-065x.2002.18618.x. [DOI] [PubMed] [Google Scholar]
- 26.Crawford F, Kozono H, White J, Marrack P, Kappler J. Detection of antigen-specific T cells with multivalent soluble class II MHC covalent peptide complexes. Immunity. 1998;8:675–682. doi: 10.1016/s1074-7613(00)80572-5. [DOI] [PubMed] [Google Scholar]
- 27.Bu HF, Zuo XL, Wang X, Ensslin MA, Koti V, Hsueh W, Raymond AS, Shur BD, Tan XD. Milk fat globule-EGF factor 8/lactadherin plays a crucial role in maintenance and repair of murine intestinal epithelium. The Journal of clinical investigation. 2007;117:3673–3683. doi: 10.1172/JCI31841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Senter PD, Saulnier MG, Schreiber GJ, Hirschberg DL, Brown JP, Hellstrom I, Hellstrom KE. Anti-tumor effects of antibody-alkaline phosphatase conjugates in combination with etoposide phosphate. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:4842–4846. doi: 10.1073/pnas.85.13.4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 30.Janka GE. Hemophagocytic syndromes. Blood reviews. 2007;21:245–253. doi: 10.1016/j.blre.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 31.Colombo T, Broggini M, Torti L, Erba E, D’Incalci M. Pharmacokinetics of VP16-213 in Lewis lung carcinoma bearing mice. Cancer chemotherapy and pharmacology. 1982;7:127–131. doi: 10.1007/BF00254534. [DOI] [PubMed] [Google Scholar]
- 32.Hande KR. Etoposide: four decades of development of a topoisomerase II inhibitor. European journal of cancer. 1998;34:1514–1521. doi: 10.1016/s0959-8049(98)00228-7. [DOI] [PubMed] [Google Scholar]
- 33.Fischer A, Virelizier JL, Arenzana-Seisdedos F, Perez N, Nezelof C, Griscelli C. Treatment of four patients with erythrophagocytic lymphohistiocytosis by a combination of epipodophyllotoxin, steroids, intrathecal methotrexate, and cranial irradiation. Pediatrics. 1985;76:263–268. [PubMed] [Google Scholar]
- 34.Bell RJ, Brafield AJ, Barnes ND, France NE. Familial haemophagocytic reticulosis. Arch Dis Child. 1968;43:601–606. doi: 10.1136/adc.43.231.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shin HJ, Chung JS, Lee JJ, Sohn SK, Choi YJ, Kim YK, Yang DH, Kim HJ, Kim JG, Joo YD, Lee WS, Sohn CH, Lee EY, Cho GJ. Treatment outcomes with CHOP chemotherapy in adult patients with hemophagocytic lymphohistiocytosis. Journal of Korean medical science. 2008;23:439–444. doi: 10.3346/jkms.2008.23.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zandman-Goddard G, Shoenfeld Y. Novel approaches to therapy for systemic lupus erythematosus. Eur J Intern Med. 2000;11:130–134. doi: 10.1016/s0953-6205(00)00074-1. [DOI] [PubMed] [Google Scholar]
- 37.Luznik L, Bolanos-Meade J, Zahurak M, Chen AR, Smith BD, Brodsky R, Huff CA, Borrello I, Matsui W, Powell JD, Kasamon Y, Goodman SN, Hess A, Levitsky HI, Ambinder RF, Jones RJ, Fuchs EJ. High-dose cyclophosphamide as single-agent, short-course prophylaxis of graft-versus-host disease. Blood. 2010;115:3224–3230. doi: 10.1182/blood-2009-11-251595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davis JC, Jr, Fessler BJ, Tassiulas IO, McInnes IB, Yarboro CH, Pillemer S, Wilder R, Fleisher TA, Klippel JH, Boumpas DT. High dose versus low dose fludarabine in the treatment of patients with severe refractory rheumatoid arthritis. The Journal of rheumatology. 1998;25:1694–1704. [PubMed] [Google Scholar]
- 39.Ferraro C, Quemeneur L, Fournel S, Prigent AF, Revillard JP, Bonnefoy-Berard N. The topoisomerase inhibitors camptothecin and etoposide induce a CD95-independent apoptosis of activated peripheral lymphocytes. Cell death and differentiation. 2000;7:197–206. doi: 10.1038/sj.cdd.4400595. [DOI] [PubMed] [Google Scholar]
- 40.Stahnke K, Fulda S, Friesen C, Strauss G, Debatin KM. Activation of apoptosis pathways in peripheral blood lymphocytes by in vivo chemotherapy. Blood. 2001;98:3066–3073. doi: 10.1182/blood.v98.10.3066. [DOI] [PubMed] [Google Scholar]
- 41.Newton K, Strasser A. Ionizing radiation and chemotherapeutic drugs induce apoptosis in lymphocytes in the absence of Fas or FADD/MORT1 signaling. Implications for cancer therapy. The Journal of experimental medicine. 2000;191:195–200. doi: 10.1084/jem.191.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Slater LM, Stupecky M, Sweet P, Osann KE. Enhancement of leukemia rejection by mice successfully treated for L1210 leukemia due to low dose compared to high dose VP-16. Leukemia research. 2002;26:203–206. doi: 10.1016/s0145-2126(01)00105-9. [DOI] [PubMed] [Google Scholar]
- 43.Seiter K. Toxicity of the topoisomerase II inhibitors. Expert opinion on drug safety. 2005;4:219–234. doi: 10.1517/14740338.4.2.219. [DOI] [PubMed] [Google Scholar]
- 44.Imashuku S. Etoposide-related secondary acute myeloid leukemia (t-AML) in hemophagocytic lymphohistiocytosis. Pediatric blood & cancer. 2007;48:121–123. doi: 10.1002/pbc.21082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.