

Abstract
Background
Standardized and well-characterized genetic building blocks are a prerequisite for the convenient and reproducible assembly of novel genetic modules and devices. While numerous standardized parts exist for Escherichia coli, such tools are still missing for the Gram-positive model organism Bacillus subtilis. The goal of this study was to develop and thoroughly evaluate such a genetic toolbox.
Results
We developed five BioBrick-compatible integrative B. subtilis vectors by deleting unnecessary parts and removing forbidden restriction sites to allow cloning in BioBrick (RFC10) standard. Three empty backbone vectors with compatible resistance markers and integration sites were generated, allowing the stable chromosomal integration and combination of up to three different devices in one strain. In addition, two integrative reporter vectors, based on the lacZ and luxABCDE cassettes, were BioBrick-adjusted, to enable β-galactosidase and luciferase reporter assays, respectively. Four constitutive and two inducible promoters were thoroughly characterized by quantitative, time-resolved measurements. Together, these promoters cover a range of more than three orders of magnitude in promoter strength, thereby allowing a fine-tuned adjustment of cellular protein amounts. Finally, the Bacillus BioBrick Box also provides five widely used epitope tags (FLAG, His10, cMyc, HA, StrepII), which can be translationally fused N- or C-terminally to any protein of choice.
Conclusion
Our genetic toolbox contains three compatible empty integration vectors, two reporter vectors and a set of six promoters, two of them inducible. Furthermore, five different epitope tags offer convenient protein handling and detection. All parts adhere to the BioBrick standard and hence enable standardized work with B. subtilis. We believe that our well-documented and carefully evaluated Bacillus BioBrick Box represents a very useful genetic tool kit, not only for the iGEM competition but any other BioBrick-based project in B. subtilis.
Keywords: Bacillus subtilis, BioBrick standard, iGEM, lux, Luminescence, Epitope tag, Integrative vector, Plasmid, Inducible promoter
Introduction
One core aspect of synthetic biology projects that sets them apart from “classical” genetic work is the application of engineering principles such as abstraction, modularity and standardization to assembly strategies and procedures. The characterization and standardization of reusable genetic building blocks is one of the prerequisites for the engineering approach of building complex synthetic biological systems [1]. Towards that end, the Registry of Standard Biological Parts (partsregistry, [2]) was founded by the Massachusetts Institute of Technology in 2003 as a repository for the international Genetically Engineered Machine competition (iGEM) and now maintains and distributes over ten thousand standardized biological parts that adhere to the BioBrick standard as described in the “request for comments 10” (RFC 10) [3]. Such standardized genetic parts – which have for example been successfully used for the construction of novel genetic circuitries, such as a bacterial camera or a push-on-push-off-switch [4,5] – not only significantly simplify devices assembly, but also increase the reproducibility of the resulting constructs [6].
While there are a number of other assembly techniques like Gibson assembly [7], Golden Gate shuffling [8,9] or MoClo [10], the BioBrick standard still plays a key role in the framework of the annual iGEM competition. Moreover, it is also very useful for any other lab, since it is based on standard type II restriction endonucleases used for routine cloning. While the use of such standardized parts and assembly strategies is organism-independent, there is nevertheless a need for specific parts that accommodate organism-specific requirements, such as G+C content, codon preference and different expression and/or regulatory signals.
The classical BioBrick standard allows the free combination of most parts, but does not work for translational fusions, e.g. for addition of gfp or epitope-tags to protein-coding sequences. For this purpose, a number of BioBrick-compatible adaptations were developed, as described in RFC 23 and RFC 25 [11,12]. In each case, the combination of parts is performed via standard restriction digests and subsequent ligations, preferably with vector backbones of different antibiotic resistances to allow the so-called 3A-assembly [13].
Currently, the vast majority of available parts in the Registry of Standard Biological Parts were designed for the Gram-negative model organism Escherichia coli, due to its central role in iGEM and other synthetic biology projects. For other organisms, such as the Gram-positive model organism Bacillus subtilis, the range of available BioBricks is still very limited, especially when looking for well-evaluated and reliable parts. This limitation is unfortunate, given the unique features, powerful genetics and biotechnological relevance of this bacterium.
B. subtilis, together with other Bacillus species, is one of the main producers of industrially relevant enzymes, such as proteases, amylases and lipases. Its excellent fermentation properties, the ability to efficiently secrete proteins and the lack of toxic by-products render it indispensable for the biotechnological industry [14]. B. subtilis is the by far best-characterized Gram-positive bacterium [15], due to its powerful genetics and advantages for industrial use. In addition, B. subtilis has also become a model organism for studying bacterial (multicellular) differentiation, because of its capability to form highly resistant endospores upon nutrient limitation [16-18]. Another transient differentiation strategy is to become naturally competent for genetic transformation by synthesizing the machinery necessary for DNA uptake [17,19,20]. The high efficiency of this process and its tight association with homologous recombination not only enables easy genetic manipulations of the chromosome, but has also led to the development of mostly integrative vectors for use in B. subtilis, even though replicative vectors can also be used [21]. The advantages of integrative vectors are their stable maintenance and hence also defined copy number inside the chromosome.
Based on all those features and differences, the 2012 LMU-Munich iGEM-team decided to develop and provide a set of essential genetic tools for the work with B. subtilis. First of all, empty compatible integrative vectors are needed that can carry any construct of choice. Second, well-evaluated promoters of different strength, both constitutive and inducible, are required to control gene expression levels. For their evaluation and as a tool for measuring the activity of other promoters, reporter vectors are also required. For that purpose, we chose the well-established lacZ reporter, encoding the β-galactosidase, as well as a luciferase reporter (luxABCDE), which we carefully evaluated for the first time. Moreover, a set of epitope tags is provided to enable the convenient detection and purification of proteins. The resulting toolbox presented here, called the Bacillus BioBrick Box, (Table 1) is now completely evaluated and freely available in two DNA repositories, the Registry of Standard Biological Parts [2] and the Bacillus Genetics Stock Center [22]. The vector sources and formatted GeneBank files are given in Table 2 and Additional file 1, respectively.
Table 1.
Overview of the Bacillus BioBrick Box including a summary of the features
| BioBrick | Description and comments a | Reference of source | BGSC # | Registry # |
|---|---|---|---|---|
|
Vectors | ||||
| pBS1C |
Empty vector, integration at amyE, ampr, cmr |
pDG1662-derivative [23] |
ECE257 |
BBa_K823023 |
| pBS2E |
Empty vector, integration at lacA, ampr, mlsr |
pAX01-derivative [24] |
ECE258 |
BBa_K823027 |
| pBS4S |
Empty vector, integration at thrC, ampr, specr |
pDG1731-derivative [23] |
ECE259 |
BBa_K823022 |
| pBS1ClacZ |
lacZ-reporter vector, integration at amyE, ampr, cmr |
pAC6-derivative [25] |
ECE260 |
BBa_K823021 |
| pBS3Clux |
lux-reporter vector, integration at sacA, ampr, cmr |
pAH328-derivative [26] |
ECE261 |
BBa_K823025 |
|
Promoters | ||||
| Pveg |
Very strong constitutive promoter |
[27] |
ECE262 |
BBa_K823003 |
| PliaG |
Constitutive promoter |
[28] |
ECE263 |
BBa_K823000 |
| PlepA |
Strong constitutive promoter |
[29] |
ECE264 |
BBa_K823002 |
| J23101 |
Very weak constitutive promoter |
BBa_J23101 |
ECE266 |
BBa_K823005 |
| PliaI |
Bacitracin-inducible promoter |
[30,31] |
ECE267 |
BBa_K823001 |
| PxylA |
Xylose-inducible promoter |
[32] |
ECE268 |
BBa_K823015 |
|
Epitope-tags | ||||
| His10 |
10xHis-tag |
[33] |
ECE269 |
BBa_K823037 |
| FLAG |
FLAG-tag |
[34] |
ECE270 |
BBa_K823034 |
| StrepII |
Streptactin-tag |
[35,36] |
ECE271 |
BBa_K823038 |
| HA |
HA-tag |
[34,37] |
ECE272 |
BBa_K823035 |
| cMyc | cMyc-tag | [34,38] | ECE273 | BBa_K823036 |
aampr, ampicillin resistance; cmr, chloramphenicol resistance; kanr, kanamycin resistance; spcr, spectinomycin resistance; mlsr, erythromycin-induced resistance to macrolide, lincosamide and streptogramin B-antibiotics (MLS).
Table 2.
Vector sources for the Bacillus BioBrick Box
| Vector | Description a | Used for | Ref. |
|---|---|---|---|
| pAC6 |
Vector for transcriptional promoter fusions to lacZ; integrates at amyE; cmr, ampr |
pBS1ClacZ |
[25] |
| pAH328 |
Vector for transcriptional promoter fusions to luxABCDE (luciferase); integrates at sacA; cmr, ampr |
pBS3Clux |
[26] |
| pDG1662 |
Empty vector, integrates at amyE, cmr, spcr, ampr |
pBS1C |
[23] |
| pDG1731 |
Empty vector; integrates at thrC, spcr, mlsr, ampr |
pBS4S |
[23] |
| pAX01 |
Vector for xylose-dependent gene expression; integrates at lacA, mlsr, ampr |
pBS2E |
[24] |
| pXT |
Vector for xylose-inducible gene expression; integrates in thrC; spcr, ampr |
PxylA |
[39] |
| pSB1C3 |
Replicative E. coli vector, MCS features rfp-cassette; cmr |
MCS |
[40] |
| pGFPamy | Vector for transcriptional promoter fusions to gfpmut1; integrates at amyE; cmr, ampr | gfpmut1 | [41] |
acmr, chloramphenicol resistance; kanr, kanamycin resistance; spcr, spectinomycin resistance; mlsr, erythromycin-induced resistance to macrolid-, lincosamid- and streptogramin B-antibiotics (MLS); MCS, multiple cloning site.
Results and discussion
The vectors of the Bacillus BioBrick Box
To allow the integration of new devices and modules into the chromosome of B. subtilis, we first constructed five vectors (see Table 1 and Figure 1) by modifying established B. subtilis vectors to comply with the BioBrick standard. As described in detail below, we chose the original B. subtilis vectors such that they harbor three different resistance cassettes and also compatible homology regions for integration into the B. subtilis chromosome. This way, all three vectors and derived plasmids can be combined in a single strain. For convenient cloning, they all contain the bla cassette for ampicillin resistance and an E. coli origin of replication (ori).
Figure 1.
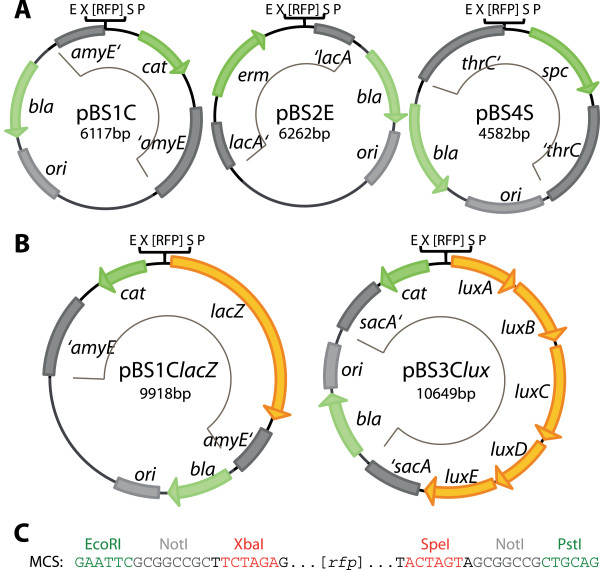
Maps of the modified Bacillus BioBrick Box-vectors in BioBrick (RFC10) standard. Resistance genes are indicated in green, reporter genes in orange and the origin of replication for E. coli as well as the integration loci are shown as gray boxes. The integrative part of the vectors is marked with a thin black line inside each vector map. The vector names are based on the following nomenclature: p = plasmid, BS = B. subtilis; the number refers to the integration locus: 1 = amyE, 2 = lacA, 3 = sacA, 4 = thrC; and the last letter codes for the resistance in B. subtilis: C = chloramphenicol (mediated by cat), E = MLS (mediated by erm: specifies resistance to macrolide, lincosamide and streptogramin B antibiotics if induced by erythromycin), S = spectinomycin (mediated by spc)). The addition in italics refers to the functional part of the reporter vectors: lacZ for β-galactosidase and lux for the luxABCDE-cassette encoding luciferase. (A) The three empty integrative vectors pBS1C, pBS2E and pBS4S contain different resistance genes (cat, erm, spec) and integration loci (amyE, lacA, thrC). This compatibility allows a combined usage in one single strain. They are derived from pDG1662, pAX01 and pDG1731, respectively. (B) Reporter vectors encoding β-galactosidase (lacZ) or luciferase (luxABDCE) as reporter genes. They integrate into amyE or sacA, respectively, and both mediate chloramphenicol resistance. All vectors harbor the RFC10-compatible multiple cloning site with an rfp-cassette, as shown in (C). See text and material and methods for details on construction.
The original B. subtilis vectors pDG1662, pAX01, pDG1731 [23,24] were modified to generate “empty” vectors that lack promoters and reporter genes (Figure 1A). Their integrative part contains the flanking homology regions, a resistance cassette for selection in B. subtilis and the multiple cloning site (MCS), containing an rfp-cassette flanked by the restriction sites EcoRI, NotI, XbaI (upstream) and SpeI, NotI and PstI (downstream) (Figure 1C). They allow cloning in BioBrick standard [3] with selection for white colonies as a result of the removal of the rfp-insert, which – if still present – leads to formation of red colonies in E. coli. All unnecessary parts, such as promoters or additional resistance genes were deleted (see methods for details on construction). In all vectors the restriction sites interfering with the RFC 10 standard were removed. Moreover, for the empty vectors we also removed all AgeI and NgoMIV sites, thus allowing direct cloning in “Freiburg” standard [11] for translational fusions. Alignments with original vectors are provided in Additional file 2.
The three empty vectors, designated pBS1C, pBS2E and pBS4S, integrate into the amyE, lacA and thrC locus and harbor cat, erm and spc resistance cassettes, respectively (Table 1). The reporter vector pBS1ClacZ was derived from pAC6 [25] and contains the β-galactosidase reporter gene lacZ from E. coli with the sacB ribosome binding site (RBS) from B. subtilis downstream of the MCS. The luciferase-reporter vector pBS3Clux, which was derived from pAH328 [26], contains the luxABCDE-operon from Photorabdus luminescence with all RBSs adjusted for use in B. subtilis (Figure 1B). Both reporter vectors allow the measurement of promoter activities based on transcriptional fusions and mediate chloramphenicol resistance. They integrate into the amyE (pBS1ClacZ) and sacA (pBS3Clux) loci, respectively.
For transformation of B. subtilis, the vectors and derived plasmids need to be linearized at a unique ScaI site inside the bla gene. The only exception is pBS2E, which contains a second ScaI site within the integrative part. For this vector, BsaI or PciI can be used for linearization. The functionality of all empty vectors has been verified in a number of different projects (own unpublished results). The reporter vectors were subsequently used to evaluate the different promoters of the Bacillus BioBrick Box (see further below).
Characterization of the thrC integration locus
The four integration sites chosen for the vectors of the Bacillus subtilis BioBrick Box are commonly used for insertion of genetic constructs into the B. subtilis chromosome. The sacA, lacA and amyE genes encode metabolic enzymes required for the degradation of different alternative carbohydrates, i.e. sucrose, galactans and starch, respectively [42-44]. Accordingly, their expression is regulated by sugar availability [42,43,45], which is easy to control by experimental design. The fourth locus, thrC, is required for synthesis of threonine and amino acids derived from it. Expression of the hom-thrC-thrB operon was found to be high under many of the experimental conditions tested in a recent expression profiling study of B. subtilis[46]. While this is not surprising for an amino acid biosynthesis operon, it has major implications for use as an integration site. Regulation of the operon’s promoter, Phom, is not well understood in B. subtilis, and we therefore constructed a transcriptional Phom-luxABCDE fusion to study the promoter activity under different growth conditions.
During growth in LB medium, representing amino acid-replete conditions, expression of Phom-luxABCDE was at an intermediate level, reaching a maximum of 105 RLU/OD (Additional file 3: Figure S1). However, in the defined CSE medium, promoter activities were strongly increased and reached twelve times higher values than in LB medium. Addition of 0.1% casamino acids reduced these activities to two-fold above LB, and in the presence of 1% casamino acids expression was similar to growth in LB (Additional file 3: Figure S1). These data show that some considerations are required when using thrC as an integration site. That is, for applications in minimal or defined media, any construct to be integrated into thrC should contain a strong terminator upstream in order to prevent undesired read-through from Phom.
The promoters of the Bacillus BioBrick Box
Promoters are crucial control elements for developing complex in vivo expression programs. For this purpose, they do not only have to be standardized, but also have to show a predictable and reliable activity under a variety of different conditions. For the optimization of metabolic pathways, the fine-tuning of gene expression is indispensable. Those features can sometimes be provided by inducible promoters or by developing promoter libraries [47-49], as described for E. coli[50,51]. To provide a first set of characterized promoter-parts for B. subtilis, we constructed and evaluated four constitutive promoters of different strength, as well as two inducible promoters that allow inducer-controlled gene expression levels.
Constitutive promoters
For our BioBrick Box, we chose three native σA-dependent B. subtilis promoters and eleven representatives of the Anderson promoter collection [51], which are often used as a gold standard for constitutive gene expression in E. coli. The native B. subtilis promoters comprise PliaG, expressing regulatory components of the cell envelope stress response system [28], PlepA, expressing highly conserved genes in bacteria and mitochondria necessary for exact protein translation [29], and Pveg, which is the strongest constitutive promoter in B. subtilis known to date [27]. The latter controls transcription of the veg gene, which seems to have an important function during the outgrowth of spores [52].
All relevant sequence features of the promoters evaluated in this study are shown in Figure 2; the complete sequences of the cloned fragments can be found in Additional file 1. To determine their activity in B. subtilis, the promoter fragments were cloned into the luciferase reporter vector pBS3Clux and assayed in LB medium as well as in chemically defined MCSE medium (MOPS-based CSE, see Methods for details).
Figure 2.

Nucleotide sequences of the Bacillus BioBrick Box promoter collection. Sequences are aligned according to their -35 and -10 region. All promoter fragments were designed with 3′-end exactly upstream of the RBS. The -10 and -35 regions (if conserved) are shown in bold and underlined. Regulator binding sites for XylR in PxylA and LiaR in PliaI are shown in italics and underlined. The native PxylA also contains a catabolite responsive element (CRE) that suppresses the transcription in the presence of glucose [53]. This binding site is downstream of the promoter and is not present in the PxylA –fragment used here, which is derived from the pXT vector [39].
Figure 3 shows both cell growth (Figure 3A/D) and the resulting luciferase activities (Figure 3B/E) over an extended growth period, ranging from exponential into the stationary growth phase in the two growth media. The activities, expressed as relative luminescence units (RLU) per OD600, covered about three orders of magnitude (Figure 3B/E), ranging from ~103 RLU/OD for the Anderson promoter J23101 to ~106 RLU/OD for the strong Pveg promoter. For growth in LB, promoter activities varied within a factor of two to three during exponential phase, and declined during stationary phase by more than one order of magnitude (Figure 3B). These variations likely arise from metabolic changes that the cells undergo during growth in this complex medium, as also reflected in the ever-changing growth rate in LB medium (Figure 3A). In contrast, the growth rate in the chemically defined MCSE medium was more constant in exponential phase (Figure 3D) and luciferase activities displayed less dynamic variation than in LB medium (Figure 3E).
Figure 3.
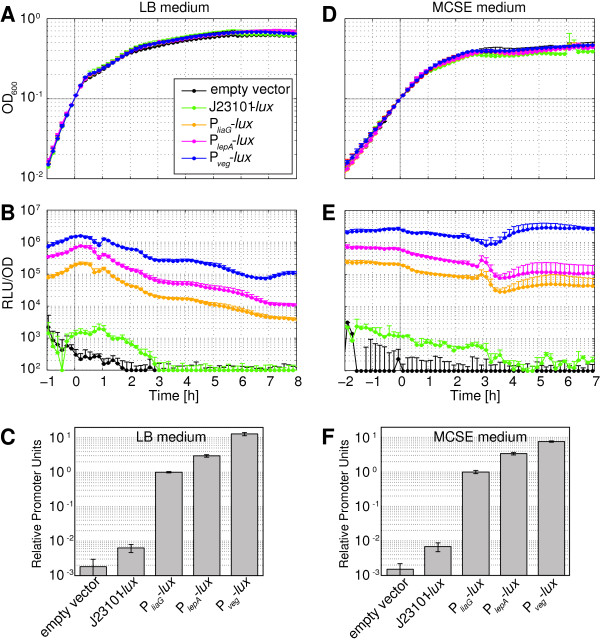
Luciferase activities of the constitutive Bacillus BioBrick Box promoters. Measurement of promoter activity was done based on pBS3Clux-derivatives via luminescence of 100 μl cultures growing in a plate reader (Biotek®, Synergy™2; 96-well plate, 37°C, shaking) over time. The graphs are aligned to an OD600 = 0.1 at time point 0 (grey line). (A)-(C) show data of growth in complex LB medium; (D)-(F) show data of growth in the defined medium MCSE. (A) and (D) Growth curves (OD600) from cultures of the reporter strains TMB1872 (lux without promoter), TMB1862 (J23101-lux), TMB1856 (PliaG-lux), TMB1860 (PlepA-lux) and TMB1930 (Pveg-lux). (B) and (E) Promoter activities of the same strains expressed as luminescence in relative luminescence units (RLU) per OD600 and using the same color code as in panels (A) and (D). (C) and (F) Bar graphs of relative promoter units (RPUs, normalized to PliaG) at time point 0, corresponding to OD600 = 0.1. All graphs show mean values and standard deviations (error bars) of at least four biological replicates.
While the Anderson promoter J23101 is a strong promoter in E. coli[51], deviating from the E. coli consensus promoter sequence by only a single nucleotide exchange in the -35 promoter region (TTGACA to TTTACA), it was the only of eleven different Anderson promoters tested in B. subtilis that generated a measurable luminescence output at all (data not shown). Still, compared to the values measured in E. coli, its activity was surprisingly low and only about ten-fold above the background of the empty vector (Figure 3B/E). This result clearly demonstrates the context-dependence of expression signals and hence stresses the importance of establishing organism-specific parts.
The absolute luciferase activities as documented in Figure 3B/E do not only depend on the promoter strength, but are also dependent on experimental conditions and setups, which can vary significantly between labs and experiments. However, while absolute values might vary, the relative strength of the promoters has proven to be a rather robust and comparable unit between several experiments and even between different research groups [54]. Towards that end, a suitable standard promoter, measured under the same experimental conditions as the promoter under consideration, serves as reference value, and promoter activities are expressed relative to this standard promoter as relative promoter units (RPUs) [54]. The stable expression behavior and intermediate strength of PliaG makes it ideally suited as a reference promoter, and hence we propose to use PliaG for calculating RPUs in B. subtilis. The RPUs for all constitutive promoters, calculated by dividing their activity at an OD600 of 0.1 by the activity of PliaG are shown in Figure 3C/F.
The bacitracin-inducible promoter PliaI
Next, we chose the bacitracin-inducible promoter PliaI for our BioBrick Box, due to its high dynamic range and low basal activity [30,31]. PliaI controls the liaIH operon of B. subtilis, which is the main target of the envelope stress-inducible two-component system LiaRS. Upon induction with the cell wall antibiotic bacitracin, LiaS activates the cognate response regulator LiaR, which in turn strongly induces PliaI[28,55].
For quantitative analysis, PliaI was cloned into the luciferase reporter vector pBS3Clux, grown to exponential phase and induced with different concentrations of bacitracin (see Methods for details). Both in LB as well as in MCSE medium we observed a transient, concentration-dependent increase of luciferase activities for about two to four hours post-induction, which decreased about 100-fold towards stationary phase (Figure 4B/E). The maximal activity of about 300-fold induction was reached 1 hour after bacitracin treatment in LB (30 minutes in MCSE) at a concentration of 30 μg ml-1 bacitracin (Figure 4C/F), as shown previously [31]. Furthermore, induction with 30 μg ml-1 bacitracin leads to minor growth defects, while higher bacitracin concentrations resulted in the lysis of the culture (Figure 4A/D). The respective dose-response curves expressed in RPUs (normalized to PliaG) indicate that PliaI responded more sensitively to low bacitracin concentrations in LB compared to MCSE medium (Figure 4C/F). This might reflect higher stress levels experienced due to faster growth in LB compared to MCSE medium (Figure 4A/D). Notably, the basal activity of PliaI was approximately 0.03 RPU and hence – in the absence of a suitable inducer – perfectly fits into the “activity gap” between the constitutive promoters J23101 (0.006 RPU) and PliaG (1 RPU).
Figure 4.

Luciferase activity of the bacitracin-inducible PliaI promoter. Measurement of PliaI activity in pBS3Clux upon induction with various bacitracin concentrations indicated in the legend. Addition of water was used as non-induced reference. For details of growth and measurement conditions, see legend of Figure 3 and Methods. (A)-(C) Growth in LB medium. (D)-(F) Growth in MCSE medium. (A) and (D) Growth curves (OD600) from cultures of the reporter strain TMB1858 (PliaI-lux). (B) and (E) Promoter activities upon bacitracin induction expressed as luminescence in RLU/OD600.(C) and (F) Dose–response curves one hour post-induction in RPUs (normalized to PliaG). The data on induction with 100 μg/ml bacitracin was omitted in (B) and (E) due to lysis of the culture. All graphs show mean values and standard deviations (error bars) of four biological replicates.
The xylose-inducible promoter PxylA
Natively, PxylA drives the expression of the xylAB operon in B. subtilis, which is required for using xylose as carbon and energy source. This promoter is regulated by XylR and CcpA and hence only induced by xylose if glucose is simultaneously absent, due to carbon catabolite repression [53]. The PxylA derivative used in this study is amplified from the expression vector pXT [39] and lacks the catabolite-responsive element (CRE). As a result, the xylose-inducible promoter of the BioBrick Box is not subject to repression. Nevertheless, the choice of carbon source can still affect the xylose-dependent induction of PxylA as described in more detail below.
The PxylA reporter responded to xylose in a concentration-dependent manner in both LB and MCSE medium and reached its maximum activity about 1-1.5 hours post-induction (Figure 5B/E). Due to a significantly increased basal promoter activity in LB, there was only a ten-fold difference between minimal and maximal activity (Figure 5C), compared to a 100-fold difference in MCSE (Figure 5F). While the maximal activity of PxylA was similar to PliaI, the basal activity of the non-induced promoter, especially in LB medium, was significantly higher for PxylA, resulting in a lower dynamic range. On the other hand, the activity of PxylA was sustained, instead of transient for PliaI, and the sensitivities towards its inducer xylose were comparable in LB and MCSE medium (Figure 5C/F).
Figure 5.

Luciferase activity of the xylose-inducible PxylA promoter. Measurement of Pxyl-activity in pBS3Clux upon induction with various concentrations (see legend) of xylose. Addition of water was used as non-induced reference. For details of growth and measurement conditions, see legend of Figure 3 and methods. (A)-(C) Growth in LB (D)-(F) Growth in MCSE. (A) and (D) Growth curves (OD600) from cultures of the reporter strain TMB1931 (PxylA-lux) in semi-logarithmic scale. (B) and (E) Promoter activities upon xylose-induction expressed as luminescence in RLU/OD600. (C) and (F) Dose–response curves one hour post-induction in RPUs (normalized to PliaG). All graphs show mean values and standard deviations (error bars) of at least four biological replicates.
As mentioned above, removal of the CRE sequence from PxylA removed the repressive effects of glucose via CcpA, but we still observed significantly reduced promoter activities during growth on glucose as carbon source. It was previously shown that glucose-6-phosphate competes with xylose for binding to the xylose repressor protein XylR and prevents its de-activation [56]. We therefore tested xylose-dependent induction of PxylA during growth in CSE medium containing different carbon sources. During growth on glucose, addition of 0.2% xylose led to a 20-fold promoter induction, while up to 70-fold changes were observed during growth on fructose, mannitol or arabinose (Additional file 3: Figure S4). Metabolism of the latter three sugars does not produce glucose-6-phosphate as an intermediate and thus bypasses its repressive effect. The reporter strain showed similar growth rates during growth on glucose, fructose or mannitol (doubling time ca. 50 min), while growth on arabinose was markedly slower at ca. 65 min doubling times (Additional file 3: Figure S4 and data not shown). Based on these results we suggest using fructose as the carbon source for routine experiments in minimal or defined media when working with PxylA.
The reporters of the Bacillus BioBrick Box
Reporter genes are important genetic tools for the measurement of promoter activities. The BioBrick Box therefore provides the established lacZ[57] and the luxABCDE reporter in the aforementioned reporter vectors pBS1ClacZ and pBS3Clux. To evaluate both vectors, we used the bacitracin-inducible liaI promoter to compare the characteristics of both reporters. The Miller units obtained for the lacZ reporter were compared to the RLU/OD600 values of the luxABCDE reporter one hour post-induction at identical inducer concentrations.
Both reporters showed a high dynamic range over about four orders of magnitude between the empty vector as background and the maximal PliaI activity (Additional file 3: Figure S2). Importantly, we observed a linear correlation between PliaI-lux and PliaI-lacZ activity, demonstrating that each of the reporters provides an accurate measurement of promoter activity. Nevertheless, the lux-reporter has several advantages over lacZ: luminescence can be monitored online in growing cultures and neither requires the addition of a substrate nor performing a separate assay. Moreover, we have determined an average half-life of only 4.2 ± 0.3 min for the luciferase signal (Additional file 3: Figure S3), compared to a lifetime in the order of hours for β-galactosidase (data not shown). Hence, the lux reporter is particularly very well suited to resolve transcriptional responses occurring at timescales shorter than the cell-cycle. In fact, given this short lifetime one might expect that the luminescence activity follows the promoter activity almost instantly.
In order to test how closely the luminescence may follow the underlying promoter activity, we devised a simplified mathematical model that includes the processes of lux mRNA transcription and degradation as well as Lux protein translation and decay (Figure 6A); see Methods for all assumptions and details. This model then enabled us to relate the time-dependent luminescence signal back to the time-dependent changes in the apparent promoter activity, by making the simplifying assumption that all changes in the luminescence signal are caused by changes in the transcription rate alone. As a proof-of-principle, we fitted the experimental luminescence activity of the PliaI promoter induced with 30 μg/ml bacitracin in MCSE (Figure 6B; symbols) by our model (Figure 6B; colored lines), and extracted the underlying time-dependent apparent promoter activity (Figure 6C). Here it is clearly visible that the fit of the experimental data is best for an mRNA half-life of 5 min or shorter, since otherwise rapid changes in the luminescence signal cannot be reproduced within this model (Figure 6B; arrow). Strikingly, the model shows that the time course of the apparent promoter activity is almost identical to the luminescence activity, with only a ~10 min delay between promoter activity and the luminescence signal (Figure 6C), further underlining the power of the lux reporter in resolving highly dynamic transcriptional responses. It should be noted that the large fluctuations in apparent promoter activities at values below 10-2 arbitrary units are likely due to experimental noise and do not necessarily reflect true changes in transcription rate.
Figure 6.
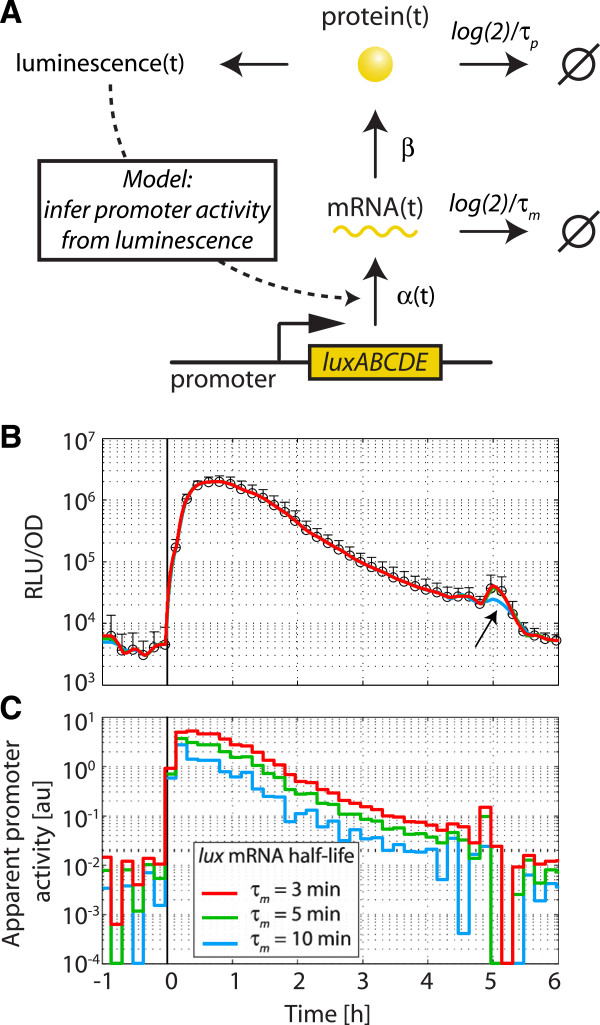
Inference of promoter activity from luminescence measurements. (A) Quantitative mathematical model used to infer the underlying promoter activity from luminescence time-series. Here, the promoter activity is reflected in a time-dependent transcription rate, α(t). luxABCDE mRNA is degraded at rate log(2)/τm , translated at rate β into Lux proteins, which are in turn degraded at rate log(2)/τp. For simplicity, the model assumes a single Lux protein species, which is rate-limiting for luciferase activity and considered to be directly proportional to luminescence output. (B) As a test case for our method, the luciferase activity of pBS3Clux-PliaI (TMB1858) induced with 30 μg/ml bacitracin (symbols) was fitted by our quantitative mathematical model (solid lines). Since the luxABCDE mRNA half-life is unknown in B. subtilis, we fitted the data using a physiological spectrum of half-lives (τm = 3, 5 and 10 min) and found that the model can reproduce the highly dynamic luciferase activity (arrow) only for mRNA half-lives of 5 min and shorter. (C) Underlying apparent promoter activity in arbitrary units (au) for the fits in (B). For all details see main text.
The epitope tags of the Bacillus BioBrick Box
Epitope tags are useful tools for both protein detection by Western blot analysis using tag-specific antibodies and for purification purposes or protein interaction studies. In our Bacillus BioBrick Box, we provide five widely used tags – FLAG-, His10-, StrepII-, HA-, and cMyc-tag – in a modified Freiburg standard that was developed to allow the generation of both N- and C-terminal translational fusions (Figure 7A). The DNA and protein sequences of all tags are shown in Figure 7B. To evaluate our tags, fusions to gfpmut1 under control of a constitutive promoter were generated as described in Methods. Western blot analyses with GFP-specific antibodies showed that all proteins could be detected in the soluble protein fraction and were therefore correctly produced (Figure 7C). Analyses with tag-specific antibodies showed that addition of the different tags led to a slight shifting of apparent sizes between the N- and C-terminal fusions in the range of 25 to 30 kDa on the SDS-PAGE (Figure 7C).
Figure 7.
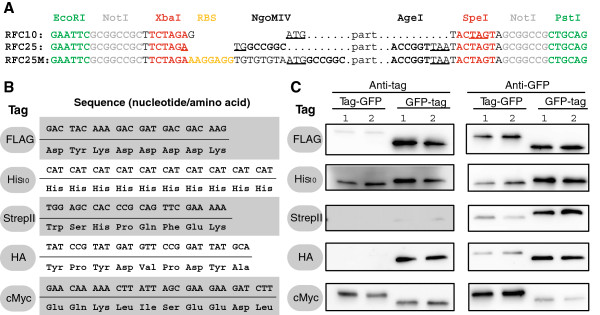
Epitope tags of the Bacillus BioBrick Box. (A) Pre- and suffixes of different cloning standards. The start- and the first stop-codon are underlined. RFC10 [3] describes the commonly used BioBrick standard. To combine two parts, the upstream part is cut with SpeI, the downstream one with XbaI (red sites). The compatible overhangs of the two isocaudomers can be ligated, and result in an eight basepair scar that cannot be recut but is optimal for RBS spacing. This causes a frameshift that is not suitable for translational fusions. RFC25 [11] describes the “Freiburg standard” which was developed by the 2007 iGEM-Team from Freiburg to allow translational fusions. Two parts can be combined similar to RFC10, but using NgoMIV and AgeI as restriction enzymes. The resulting scar ACCGGC codes for Threonine and Glycine. RFC25M refers to our modification of RFC25 by adding an optimized RBS (yellow). While this convenient modification simplifies the cloning procedure, it should be noted that as a consequence it also prevents downward compatibility, since the resulting BioBrick directly adds two functional DNA parts in a single step. The tag-BioBricks and gfpmut1 are designed in that standard. (B) Nucleotide and amino acid sequences of the epitope tags, without the RFC25M pre- and suffixes shown in (A). (C) Western blots of N- and C-terminal fusions of each tag to GFP, using the strains TMB1920 (Flag-gfp), TMB1921 (gfp-Flag), TMB1922 (HA-gfp), TMB1923 (gfp-HA), TMB1924 (cMyc-gfp), TMB1925 (gfp-cMyc), TMB1926 (His-gfp), TMB1927 (gfp-His), TMB1928 (StrepII-gfp) and TMB1929 (gfp-StrepII). For each construct, two independent clones were tested with epitope tag- and GFP-specific antibodies as a positive control.
The original FLAG-tag was developed as hydrophilic hexapeptide marker sequence (Lys-Asp-Asp-Asp-Asp-Lys) that is located at the outside of protein surfaces. Adaptation to B. subtilis was performed by Kaltwasser et al.[34]. In Western blots, the C-terminal fusion to GFP can be detected very well, while the signal for the N-terminal fusion was weak. The His10-tag was developed and is used for affinity purification on Ni2+-NTA-columns and Western blotting [33]. Both the N- and C-terminal fusions to GFP were easily detectable in Western blots (Figure 7C). The Strep-tag II, which was optimized from the original Strep-tag to allow both N- and C-terminal fusion, can be used together with the streptavidin-derivative Strep-Tactin as its specific binding partner [35,36]. The expression of the N-terminal fusion seemed to be rather low (anti-GFP blot) and detection with anti-strep antibodies was weak. Nevertheless, Strep-tagII is the epitope of choice for co-immunoprecipitation in protein-protein interaction studies in B. subtilis due to the highly specific interaction with Strep-Tactin-covered magnetic beads or resins [58]. The HA-tag was named after the human influenza hemagglutinin, a viral surface protein that is important for host cell infection [37]. The corresponding HA-tag was codon-optimized for B. subtilis[34] and worked very well for detection of C-terminally tagged GFP. The cMyc-tag is derived from the human c-myc proto-oncogene [38], which was also optimized for B. subtilis[34]. C-terminal fusion to gfp seemed to negatively influence the GFP-expression levels. However, the detection with anti-cMyc-antibodies worked well for both N- and C-terminal fusions.
In summary, all five epitope tags were shown to be functional. The ideal terminus for creating protein-tag fusions as well as the most suitable tag will have to be determined experimentally for each target protein and application. For such studies, the parts from the BioBrick Box offer a convenient and fast way for optimization of conditions and constructs.
Conclusion
In this study, we described the construction and evaluation of a toolkit containing essential vectors, promoters and epitope-tags for standardized genetic assembly in B. subtilis, a biotechnological workhorse and Gram-positive model organism.
We modified and adapted five integrative B. subtilis vectors to the BioBrick standard. Three are empty vectors that are fully compatible to each other and allow their combination in one single strain due to different integration loci and resistance cassettes. Two additional reporter vectors harbor lacZ and luxABCDE cassettes for evaluating transcriptional promoter fusions. While β-galactosidase is already long established as a reporter and needs no further introduction, we conducted a detailed study of the lux-reporter system. We determined the half-life of the luciferase signal as 4.2 ± 0.3 min, making the reporter instable enough to directly monitor both induction and repression of promoters almost in real time, as revealed by our mathematical model for the lux-reporter system. This feature, together with the very low background activity and the possibility to determine promoter activities directly in growing cultures, makes this an ideal reporter system, especially since the lux cassette from P. luminescence present in pBS3Clux does not require the addition of any luciferase substrate.
Four constitutive promoters and the uninduced PliaI offer an activity range of three orders of magnitude with stable expression levels in defined MCSE medium and during exponential growth phase in LB medium. We adopted the concept of RPUs for B. subtilis and propose the PliaG promoter to be used as a reference standard for RPU determination in this organism. The Bacillus BioBick Box also contains two inducible promoters, PliaI and PxylA, which were thoroughly evaluated with the inducers bacitracin and xylose, respectively. Using inducer concentrations over about three orders of magnitude, the two promoters revealed a dynamic range of 300- or 100-fold activity over background, respectively. PliaI has a transient activity in complex and defined medium, whereas PxylA displays sustained induction.
Finally, we evaluated five widely used epitope tags as N- and C-terminal fusions to GFP via Western blotting. All are functional, but results for the test case GFP indicate a context-dependence in immuno-detection: The FLAG- and HA-tag were only suitable for C-terminal fusions to GFP, whereas His10 and cMyc worked both for N- and C-terminal fusions.
Taken together, we provide a set of well-evaluated standardized tools for the work in B. subtilis. We hope that this toolkit will inspire not only future iGEM-teams to choose B. subtilis as their chassis organism, but also greatly facilitate the engineering of this versatile model organism for synthetic biology applications in the future.
Methods
Bacterial strains and growth conditions
B. subtilis and E. coli were routinely grown in Luria-Bertani (LB) medium (1% (w/v) tryptone, 0.5% (w/v) yeast extract, 1% (w/v) NaCl) at 37°C with agitation (200 rpm). All strains used in this study are listed in Additional file 3: Table S2. Selective media for B. subtilis contained kanamycin (10 μg ml-1), spectinomycin (100 μg ml-1), chloramphenicol (5 μg ml-1), or erythromycin in combination with lincomycin (1 μg ml-1; 25 μg ml-1 for mlsr). Selective media for E. coli contained ampicillin (100 μg ml-1) or chloramphenicol (35 μg ml-1). Solid media additionally contained 1.5% (w/v) agar.
Transformation
E. coli (XL1 blue, Stratagene) competent cells were prepared and transformed according to OpenWetWare [59]. Transformations of B. subtilis were carried out as described previously [21]. The integration of plasmids into the B. subtilis genome was checked on starch plates (amyE), with minimal medium (thrC) or colony PCR (sacA, lacA). Detailed protocols can be found in the Additional file 3 and [60].
MOPS-based chemically defined medium with succinate and glutamate (MCSE)
The chemically defined CSE medium (3.3 g l-1 (NH4)2SO4, 29 mM KH2PO4, 70 mM K2HPO4, 1 × III’-salts (100 × III’-salts: 0.232 g l-1 MnSO4 × 4 H2O, 12.3 g l-1 MgSO4 × 7 H2O), 50 mg l-1 Tryptophan, 22 mg l-1ammonium ferric citrate, 0.8% (w/v) K-glutamate, 0.6% (w/v) Na-succinate, up to 2.5% (w/v) C-source) is used for experiments with stable growth conditions and a defined C-source [61]. In long-term experiments (18 hours), we noticed a white precipitate disturbing the OD600-measurements in the microplate reader. We tested the use of a phosphate-reduced 3-(N-Morpholino)-Propanesulfonic acid (MOPS) buffer (10 × MOPS buffer: 83.72 g l-1 MOPS, 33 g l-1 (NH4)2SO4, 3.85 mM KH2PO4, 6.15 mM K2HPO4; adjusted to pH 7 with KOH), which did not cause precipitation. This is the composition of the newly developed MOPS-based MCSE medium: 1 × MOPS, 50 mg l-1 Tryptophan, 22 mg l-1ammonium ferric citrate, 1 × III’-salts, 0.8% (w/v) K-glutamate, 0.6% (w/v) Na-succinate, 0.2% (w/v) fructose. The C-source for MCSE can be varied according to the experimental requirements.
DNA manipulation and plasmid construction
General cloning procedure, such as endonuclease restriction digest, ligation and PCR, was performed with enzymes and buffers from New England Biolabs (NEB; Ipswich, MA, USA) according to the respective protocols. For subsequent cloning, Phusion® polymerase was used for PCR amplifications, otherwise OneTaq® was the polymerase of choice. PCR-purification was performed with the HiYield PCR Gel Extraction/PCR Clean-up Kit (Süd-Laborbedarf GmbH (SLG), Gauting, Germany). Plasmid preparation for mutagenesis-PCR templates was performed with the HiYield Plasmid Mini-Kit (SLG) according to the manufacturer’s protocol, otherwise by alkaline lysis plasmid preparation. All plasmids created during this study are listed in Additional file 3: Table S1, all primer sequences are given in Additional file 3: Table S3.
Vectors of the Bacillus BioBrick Box
In all vectors, the PstI site in the bla gene was removed by site-directed mutagenesis using the oligonucleotides TM2206 and TM2207. The resulting vector is marked with the auxiliary “blamut”. Primer design and mutagenesis were performed according to the manufacturer’s instructions for the QuikChange® II Site-Directed Mutagenesis Kit (Agilent Technologies). All plasmids were test-digested to confirm removal of the restriction site.
To create the empty vector pBS1C, pDG1662blamut was cut with PstI to remove one XbaI site and the spcr outside of the integrative part. The 6 kb fragment was religated. The remaining PstI site was mutated via site-directed mutagenesis with the primers TM2845 and TM2846. To insert the MCS, the vector was cut with EcoRI. The MCS was amplified by PCR from pSB1C3 with the Primers TM2843 and TM2844, cut with EcoRI and BsaI (EcoRI-compatible overhang) and ligated into the vector. The remaining NgoMIV sites were removed by subsequent site-directed mutagenesis with TM3005 + TM3006, TM3011 + TM3012 and TM3013 + TM3014, respectively, resulting in pBS1C.
To create the empty vector pBS2E, pAX01 was cut with SacI. The 6.3 kb fragment was religated to remove the xylR-PxylA-fragment. The PstI site in bla was removed. The vector was cut with XbaI and the 6 kb fragment religated to reduce the amount of forbidden restriction sites. The lost terminator of erm was replaced by the PCR-amplified terminator with BsaI-overhangs (primers: TM2975 and TM2976). The vector was opened with XbaI and ligated with the PCR product cut with BsaI (XbaI-compatible overhang). The correct direction was checked by sequencing and removal of the XbaI-site was confirmed. Finally, the MCS was amplified from pSB1C3 (TM2608 and TM2609) with PstI and NsiI overhangs, cut with PstI and NsiI and ligated into the PstI-cut vector. Correct orientation of the insert was confirmed by restriction digest and sequencing. The remaining NgoMIV sites were removed by subsequent site-directed mutagenesis with TM3011 + TM3012 and TM3028 + TM3029, respectively, resulting in pBS2E.
To create the empty vector pBS4S, the erm-resistance outside of the integrative part of pDG1731 was removed via cut-ligation with MluI and BssHI, resulting in a 4.7 kb vector. The PstI site in bla was removed as described above. The PstI site in thrB was removed performing site-directed mutagenesis with the primers TM2835 and TM2836. The spc-promoter with upstream PstI-overhang was amplified with the primers TM2837 and TM2838 and cut with PstI and PciI. The MCS was cut from pSB1C3 with EcoRI and PstI. The vector was cut with EcoRI and PciI and the 4.3 kb-fragment ligated with both cut DNA-fragments. The remaining NgoMIV sites were removed by subsequent site-directed mutagenesis with TM3005 + TM3006 and TM3011 + TM3012, respectively, resulting in pBS4S.
To create the reporter vector pBS1ClacZ, pAC6blamut was cut with EcoRI and PstI. The primers TM2301 and TM2302 (1 pM) were mixed, heated (95°C, 10 min), re-annealed (50°C, 10 min) to become double stranded with 4 bp overhangs and ligated into the vector. The vector was cut with EcoRI and PstI and ligated with the MCS from pSB1C3 cut with EcoRI and PstI. A PstI site in the E. coli ori was removed by cutting with BglII and religation of the 11 kb fragment.
To create the reporter vector pBS3Clux, the PstI site in the sacA locus of pAH328blamut was removed by site-directed mutagenesis with the primers TM2885 and TM2886. Then, the XbaI site of luxD was removed similarly with the Primers TM2887 and TM2888. The MCS was amplified by PCR from pSB1C3 with the primers TM2843 and TM2884, cut with EcoRI and BsaI (SpeI-overhang) and ligated into the EcoRI/SpeI-cut vector.
Promoters of the Bacillus BioBrick Box
The promoter J23101 was used from the parts registry [2]. The promoters PliaG, PlepA, Pveg and PliaI were amplified from B. subtilis W168 genomic DNA using the primer pairs TM2891/TM2892, TM2899/TM2900, TM2903/TM2904 and TM2895/TM2896, respectively. PxylA was amplified from pXT [39] with the primers TM2968 + TM2969. The PCR products were cut with EcoRI and SpeI, ligated into pSB1C3, pBS3Clux and pBS1ClacZ (PliaI only) and verified by sequencing.
Epitope-tags of the Bacillus BioBrick Box
All eptitope tags were synthesized with pre- and suffixes (Figure 7A) together as a single part in a plasmid from GeneArt (Life Technologies Corporation, USA). The plasmid was cut with EcoRI and PstI and, without purification steps, ligated into pSB1C3 cut with EcoRI and PstI. The products were checked by sequencing for their correct insert. gfpmut1 was adapted to modified Freiburg standard by PCR with the primers TM2934/TM2935 and pGFPamy as template and cloned into pSB1C3 (both cut with EcoRI and SpeI). For translational fusion, the pSB1C3-tag-constructs were cut open with AgeI + PstI (tag is N-terminal) or EcoRI + NgoMIV (tag is C-terminal) and purified. gfp was cut with the corresponding restriction enzymes of the Freiburg standard, purified from an agarose gel and ligated into the vector. The tag-gfp or gfp-tag fusions as well as pBS0KPspac* were cut with EcoRI and PstI, purified from agarose gels, ligated and verified by sequencing.
Luciferase assay
Luciferase activities of strains harbouring pBS3Clux-derivates were assayed using a Synergy™2 multi-mode microplate reader from BioTek® (Winooski, VT, USA). The reader was controlled using the software Gen5™. Culture volumes were 100 μl per well in 96-well plates (black walls, clear bottom; Greiner Bio-One, Frickenhausen, Germany), and incubation occurred at 37°C with agitation (intensity: medium). Cell growth was monitored by optical density at 600 nm wavelength (OD600). Raw luminescence output (relative luminescence units, RLU) was normalized to cell density by dividing each data-point by its corresponding corrected OD600 value (RLU/OD).
For constitutive promoters, LB or MCSE medium was inoculated 1:500 from overnight cultures of each strain. Cultures were incubated at 37°C with agitation and OD600 as well as luminescence were monitored every 10 min for at least 13 hours. For inducible promoters, 10 ml of LB or MCSE medium were inoculated 1:500 from overnight cultures and grown to OD600 = 0.2-0.5. Those pre-cultures were diluted to OD600 = 0.05 (MCSE) or 0.01 (LB), respectively, and transferred to eight (PliaI) or ten (PxylA) wells of a 96-well plate. The OD600 as well as luminescence were monitored every 10 min for one hour. At an OD600 ~ 0.1 (corresponding to OD600 ~ 0.4 in cuvettes of 1 cm light path length) 5 μl of the inducer was added to final concentrations of 0.1, 0.3, 1, 3, 10, 30 or 100 μg ml-1 Zn2+-bacitracin or 0.001, 0.003, 0.005, 0.01, 0.02, 0.03, 0.07, 0.2, 0.5% (w/v) xylose. To one culture-well, 5 μl of water was added as uninduced control. Cultures were incubated at 37°C with agitation and the OD600 as well as luminescence were monitored every 10 min for at least 13 hours.
β-Galactosidase assay
LB medium (30 ml) was inoculated 1:100 from fresh overnight culture of strains harboring pBS1ClacZ-PliaI or pBS1ClacZ without promoter and grown at 37°C, shaking at 200 rpm. At OD600 = 0.4-0.6, the culture was split into 3 ml samples and induced with 30 μl of Zn2+-bacitracin to the final concentrations of 0.1, 0.3, 1, 3, 10, 30 or 100 μg ml-1. To one sample, 30 μl of water was added as a negative control. After incubation for another 30 min, 2 ml of the samples were harvested by centrifugation (13000 × g, 1-3 min) and the pellets were stored at -20°C. For the assay, the pellets were resuspended in 1 ml of working buffer and assayed for β-galactosidase activity as described elsewhere, with normalization to cell density [62].
Western blot analysis
To verify the functionality of the epitope tags, Western blot analyses of the strains TMB1920-TMB1929 were performed. LB medium (15 ml) was inoculated 1:100 from overnight culture and grown at 37°C and 200 rpm to OD600 ~ 0.5. Of this, 10 ml were harvested by centrifugation (8000 × g, 5 min) and the pellets stored at -20°C. Pellets were resuspended in 1 ml disruption buffer (50 mM Tris–HCl pH 7.5, 100 mM NaCl) and lysed by sonication. Samples (12 μl of lysate) were loaded per lane on two 12.5% SDS-polyacrylamide gels and SDS-PAGE was performed according standard procedure [60]. One gel was stained with colloidal coomassie, the other one was used for protein transfer to a PVDF membrane (Merck Millipore, Billerica, MA, USA) by submerged blotting procedure (Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad, Hercules, CA, USA)). After protein transfer, the membranes were treated with the following antibodies and conditions. Detailed protocols can be found in the Additional file 3.
GFP
Probing with primary antibodies takes place with rabbit anti-GFP antibodies (1:3000, Epitomics, No. 1533). Horseradish-peroxidase (HRP)-conjugated anti-rabbit antibodies (1:2000, Promega, W401B) were used as secondary antibody. Hybridization of both antibodies was carried out in Blotto-buffer (2.5% (w/v) skim milk powder, 1 × TBS (50 mM Tris–HCl pH 7.6, 0.15 M NaCl)).
FLAG
Rabbit anti-FLAG (1:2000, Sigma, Anti-Flag polyclonal, F7425) and anti-rabbit-HRP (1:2000, Promega, W401B) were used in Blotto-buffer.
His10
Mouse-anti-Penta-His (1:2000, Qiagen, Penta-His, No. 34660) in 1 × TBS, 5% bovine serum albumin (BSA, Fraction V; Carl ROTH) and anti-mouse-HRP (1:2000, Promega, W402B1) in 1 × TBS, 10 % (w/v) skim milk powder were used.
StrepII
Strep-Tactin-HRP conjugate (IBA, Strep-Tactin-HRP conjugate, No. 2-1502-001) 1:100 in 1 × PBS (4 mM KH2PO4; 16 mM Na2HPO4; 115 mM NaCl) with 0.1% (w/v) Tween20 was used.
HA
Rabbit anti-HA (1:500, Sigma, H6908) in TBS, 0.05% (w/v) Tween20, 5% (w/v) skim milk powder and anti-rabbit-HRP (1:2000, Promega, W401B) in Blotto-buffer were used.
cMyc
Rabbit anti-Myc (1:2000, Abcan, ab9106) in TBS, 0.05% (w/v) Tween20, 5% (w/v) skim milk powder and anti-rabbit-HRP (1:2000, Promega, W401B) in Blotto-buffer were used.
Chemiluminescence signals were detected after addition of the HRP-substrate Ace Glow (Peqlab, Erlangen, Germany) using a Fusion™ imaging system (Peqlab).
Determination of luciferase half-life
To determine the half-life of the output of the luciferase reporter system, B. subtilis harboring the Pxyl-luxABCDE reporter construct was grown in CSE medium in the presence of 0.15% (w/v) xylose under the conditions described for luciferase assays with constitutive promoters. When luciferase activities reached approximately 105 RLU/OD600 (early exponential phase), further protein synthesis was stopped by the addition of 500 μg ml-1 tetracycline, and luminescence and OD600 were monitored every 5 min for 1 h. The half-life of the luminescence output was determined from a fit of the data from eight replicate assays with an exponential decay function (Additional file 3: Figure S3).
Data analysis of luciferase assays
For each individual sample, the optical density (OD600) and luminescence values (RLU) were background-corrected by subtracting the respective values measured for wells containing 100 μl of the corresponding medium. Only for the measurements of the constitutive promoters, in which wells were inoculated with cells right from the start of the experiment, the OD600 of pure medium was initially higher than the OD600 of culture-filled wells, such that the average values of culture-filled wells at t = 0 h were used as blank for OD600. In order to compensate for sample-to-sample variations in the growth lag of individual cultures, we aligned all individual measurements of the constitutive promoters on the time-axis, such that OD600 = 0.1 was shifted to t = 0 h. Subsequently RLU/OD600 values were calculated for individual measurements and for each condition both mean and standard deviation of RLU/OD600 values were determined from at least three biological replicates.
Mathematical model for promoter activity
To get an estimate for the time-dependence of the promoter activity that results in a given luciferase activity, we set up a simple mathematical model for the expression of the Lux-system (Figure 6). For simplicity, the model assumes that one of the Lux proteins is rate-limiting for light production, such that the luminescence is proportional to this protein species. The corresponding differential equations for the concentration of luxABCDE mRNA, m(t), and the rate-limiting Lux protein, p(t), read
| (1) |
and
| (2) |
where α(t) is the time-dependent apparent transcription rate, β the translation rate and τm and τp the mRNA and protein half-lives, respectively. Although mRNA and protein stability might also depend on cell physiology, time depended changes in these parameters were ignored to avoid further unknown parameters. Likewise, we did not model the kinetics of inducer uptake/signal transduction explicitly, but subsumed these processes in the kinetics of the time-dependent apparent transcription rate. For simplicity, the translation rate was directly expressed in units of RLU/mRNA/min, such that p(t) has units of RLU. The model in Eqs. (1) and (2) was solved analytically for p(t), and then fitted to the experimental luminescence trajectories. To this end, all parameters except α(t) were fixed to the following values: τp = 4.2 min (experimentally determined in Additional file 3: Figure S3), β = 104 RLU/mRNA/min (arbitrary, but adjusted to meet typical transcription rates), τm = 3, 5 and 10 min (typical values for bacterial mRNA half-lives [63]). Then α(t) was discretized into N intervals of constant transcription rate αi(i = 1,…N) (the length of the intervals was adjusted to ∆t = 10 min as in the luciferase assay). Finally, the αi were estimated by fitting p(t) to the luminescence trajectories using a trust-region reflective Newton method for least-squares minimization (MATLAB, The MathWorks, Inc.).
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
TM, JR, KK, JB, TC, FD, JE and SK conceptualized the contents of the Bacillus BioBrick Box and designed strategies for the modification of the vectors. JR constructed all vectors. JR and KK cloned and evaluated the promoters. KK cloned the tag-gfp-fusions and performed all Western blots. SG and CS performed the analyses of Phom activities and analyzed the influence of C-sources on Pxyl activation. GF performed the quantitative data analysis and mathematical modeling. JR, GF, SG and TM wrote the manuscript. TM supervised the 2012 iGEM team LMU-Munich and coordinated the experimental work. All authors read and approved the final manuscript.
Authors’ information
Jara Radeck, Korinna Kraft, Julia Bartels, Tamara Cikovic, Franziska Dürr, Jennifer Emenegger and Simon Kelterborn were members of the 2012 iGEM-team LMU-Munich.
Supplementary Material
GeneBank files of Bacillus BioBricks.
Alignment of original and final vectors.
Supplemental Figures, Tables and Text.
Contributor Information
Jara Radeck, Email: jara.radeck@campus.lmu.de.
Korinna Kraft, Email: korinna.kraft@gmx.de.
Julia Bartels, Email: julia_bartels222@yahoo.de.
Tamara Cikovic, Email: tamara.cikovic@gmail.com.
Franziska Dürr, Email: franzis.post@gmx.de.
Jennifer Emenegger, Email: jenn@emenegger.com.
Simon Kelterborn, Email: simon.kelterborn@campus.lmu.de.
Christopher Sauer, Email: c.sauer@ncl.ac.uk.
Georg Fritz, Email: georg.fritz@lmu.de.
Susanne Gebhard, Email: susanne.gebhard@bio.lmu.de.
Thorsten Mascher, Email: mascher@bio.lmu.de.
Acknowledgements
The authors would like to thank Tina Wecke for supervising the iGEM-Team 2012 and all initiators, organizers and volunteers of iGEM, because this work would never have been initiated without this competition. We thank Richard Losick for providing the pAH328, Angelika Böttger for providing antibodies against the cMyc- and HA-tag, and Ainhoa Revilla-Guarinos, Tobias Bauer and Nicole Treichelt for initial cloning and preliminary data.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG MA2837/3-1 to TM and GE2164/3-1 to SG) and from the Fonds der Chemischen Industrie (to SG). Finally, the LMU-Munich 2012 iGEM team would like to acknowledge the generous support from all of their sponsors: BioTek, DSM, ERA Synbio, GeneArt (Life Technologies), LMU Faculty of Biology, the LMU genomics service unit, Lehre@LMU, Münchner Universitätsgesellschaft, Sarstedt, and Semadeni (gold sponsors); AppliChem and Bio-Rad (silver sponsors); the Bacillus Genetic Stock Center BGSC and Süd-Laborbedarf GmbHSLG (bronze sponsors).
References
- Endy D. Foundations for engineering biology. Nature. 2005;438:449–453. doi: 10.1038/nature04342. [DOI] [PubMed] [Google Scholar]
- Registry of standard biological parts. http://partsregistry.org.
- Draft standard for BioBrick biological parts. http://hdl.handle.net/1721.1/45138.
- Levskaya A, Chevalier AA, Tabor JJ, Simpson ZB, Lavery LA, Levy M, Davidson EA, Scouras A, Ellington AD, Marcotte EM. et al. Synthetic biology: engineering Escherichia coli to see light. Nature. 2005;438:441–442. doi: 10.1038/nature04405. [DOI] [PubMed] [Google Scholar]
- Lou C, Liu X, Ni M, Huang Y, Huang Q, Huang L, Jiang L, Lu D, Wang M, Liu C. et al. Synthesizing a novel genetic sequential logic circuit: a push-on push-off switch. Mol Sys Biol. 2010;6:350. doi: 10.1038/msb.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller KM, Arndt KM. Standardization in synthetic biology. Methods Mol Biol. 2012;813:23–43. doi: 10.1007/978-1-61779-412-4_2. [DOI] [PubMed] [Google Scholar]
- Gibson DG. Synthesis of DNA fragments in yeast by one-step assembly of overlapping oligonucleotides. Nucleic Acids Res. 2009;37:6984–6990. doi: 10.1093/nar/gkp687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler C, Gruetzner R, Kandzia R, Marillonnet S. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS ONE. 2009;4:e5553. doi: 10.1371/journal.pone.0005553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler C, Kandzia R, Marillonnet S. A one pot, one step, precision cloning method with high throughput capability. PLoS ONE. 2008;3:e3647. doi: 10.1371/journal.pone.0003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner S, Engler C, Weber E, Gruetzner R, Marillonnet S. Fast track assembly of multigene constructs using golden gate cloning and the MoClo system. Bioeng Bugs. 2012;3:38–43. doi: 10.4161/bbug.3.1.18223. [DOI] [PubMed] [Google Scholar]
- Fusion Protein (Freiburg) BioBrick assembly standard. [ http://hdl.handle.net/1721.1/45140]
- A New BioBrick Assembly Strategy Designed for Facile Protein Engineering. [ http://hdl.handle.net/1721.1/32535]
- Shetty R, Lizarazo M, Rettberg R, Knight TF. Assembly of BioBrick standard biological parts using three antibiotic assembly. Methods Enzymol. 2011;498:311–326. doi: 10.1016/B978-0-12-385120-8.00013-9. [DOI] [PubMed] [Google Scholar]
- van Dijl JM, Hecker M. Bacillus subtilis: from soil bacterium to super-secreting cell factory. Microb Cell Fact. 2013;12:3. doi: 10.1186/1475-2859-12-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl AM, Losick R, Kolter R. Ecology and genomics of Bacillus subtilis. Trends Microbiol. 2008;16:269–275. doi: 10.1016/j.tim.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington J. Regulation of endospore formation in Bacillus subtilis. Nat Rev Microbiol. 2003;1:117–126. doi: 10.1038/nrmicro750. [DOI] [PubMed] [Google Scholar]
- Lopez D, Vlamakis H, Kolter R. Generation of multiple cell types in Bacillus subtilis. FEMS Microbiol Rev. 2009;33:152–163. doi: 10.1111/j.1574-6976.2008.00148.x. [DOI] [PubMed] [Google Scholar]
- McKenney PT, Driks A, Eichenberger P. The Bacillus subtilis endospore: assembly and functions of the multilayered coat. Nat Rev Microbiol. 2013;11:33–44. doi: 10.1038/nrmicro2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I, Christie PJ, Dubnau D. The ins and outs of DNA transfer in bacteria. Science. 2005;310:1456–1460. doi: 10.1126/science.1114021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I, Dubnau D. DNA uptake during bacterial transformation. Nat Rev Microbiol. 2004;2:241–249. doi: 10.1038/nrmicro844. [DOI] [PubMed] [Google Scholar]
- Harwood CR, Cutting SM. Molecular Biological Methods for Bacillus. Chichester: John Wiley & Sons; 1990. [Google Scholar]
- Bacillus Genetic Stock Center. http://www.bgsc.org/
- Guerout-Fleury AM, Frandsen N, Stragier P. Plasmids for ectopic integration in Bacillus subtilis. Gene. 1996;180:57–61. doi: 10.1016/S0378-1119(96)00404-0. [DOI] [PubMed] [Google Scholar]
- Hartl B, Wehrl W, Wiegert T, Homuth G, Schumann W. Development of a new integration site within the Bacillus subtilis chromosome and construction of compatible expression cassettes. J Bacteriol. 2001;183:2696–2699. doi: 10.1128/JB.183.8.2696-2699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stülke J, Martin-Verstraete I, Zagorec M, Rose M, Klier A, Rapoport G. Induction of the Bacillus subtilis ptsGHI operon by glucose is controlled by a novel antiterminator, GlcT. Mol Microbiol. 1997;25:65–78. doi: 10.1046/j.1365-2958.1997.4351797.x. [DOI] [PubMed] [Google Scholar]
- Schmalisch M, Maiques E, Nikolov L, Camp AH, Chevreux B, Muffler A, Rodriguez S, Perkins J, Losick R. Small genes under sporulation control in the Bacillus subtilis genome. J Bacteriol. 2010;192:5402–5412. doi: 10.1128/JB.00534-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam KH, Chow KC, Wong WK. Construction of an efficient Bacillus subtilis system for extracellular production of heterologous proteins. J Biotechnol. 1998;63:167–177. doi: 10.1016/S0168-1656(98)00041-8. [DOI] [PubMed] [Google Scholar]
- Jordan S, Junker A, Helmann JD, Mascher T. Regulation of LiaRS-dependent gene expression in Bacillus subtilis: Identification of inhibitor proteins, regulator binding sites and target genes of a conserved cell envelope stress-sensing two-component system. J Bacteriol. 2006;188:5153–5166. doi: 10.1128/JB.00310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Polacek N, Vesper O, Staub E, Einfeldt E, Wilson DN, Nierhaus KH. The highly conserved LepA is a ribosomal elongation factor that back-translocates the ribosome. Cell. 2006;127:721–733. doi: 10.1016/j.cell.2006.09.037. [DOI] [PubMed] [Google Scholar]
- Mascher T, Zimmer SL, Smith TA, Helmann JD. Antibiotic-inducible promoter regulated by the cell envelope stress-sensing two-component system LiaRS of Bacillus subtilis. Antimicrob Agents Chemother. 2004;48:2888–2896. doi: 10.1128/AAC.48.8.2888-2896.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toymentseva AA, Schrecke K, Sharipova MR, Mascher T. The LIKE system, a novel protein expression toolbox for Bacillus subtilis based on the liaI promoter. Microb Cell Fact. 2012;11:143. doi: 10.1186/1475-2859-11-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzer P, Gartner D, Allmansberger R, Hillen W. Identification and sequence analysis of the Bacillus subtilis W23 xylR gene and xyl operator. J Bacteriol. 1989;171:3840–3845. doi: 10.1128/jb.171.7.3840-3845.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochuli E, Bannwarth W, Dobeli H, Gentz R, Stuber D. Genetic approach to facilitate purification of recombinant proteins with a novel metal chelate adsorbent. Nat Biotech. 1988;6:1321–1325. doi: 10.1038/nbt1188-1321. [DOI] [Google Scholar]
- Kaltwasser M, Wiegert T, Schumann W. Construction and application of epitope- and green fluorescent protein-tagging integration vectors for Bacillus subtilis. Appl Env Microbiol. 2002;68:2624–2628. doi: 10.1128/AEM.68.5.2624-2628.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerra A, Schmidt TG. Applications of a peptide ligand for streptavidin: the strep-tag. Biomol Eng. 1999;16:79–86. doi: 10.1016/S1050-3862(99)00033-9. [DOI] [PubMed] [Google Scholar]
- Voss S, Skerra A. Mutagenesis of a flexible loop in streptavidin leads to higher affinity for the strep-tag II peptide and improved performance in recombinant protein purification. Protein Eng. 1997;10:975–982. doi: 10.1093/protein/10.8.975. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Niman HL, Houghten RA, Cherenson AR, Connolly ML, Lerner RA. The structure of an antigenic determinant in a protein. Cell. 1984;37:767–778. doi: 10.1016/0092-8674(84)90412-4. [DOI] [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5(12):3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derre I, Rapoport G, Msadek T. The CtsR regulator of stress response is active as a dimer and specifically degraded in vivo at 37°C. Mol Microbiol. 2000;38:335–347. doi: 10.1046/j.1365-2958.2000.02124.x. [DOI] [PubMed] [Google Scholar]
- Part: pSB1C3. [ http://parts.igem.org/wiki/index.php/Part:pSB1C3]
- Bisicchia P, Botella E, Devine KM. Suite of novel vectors for ectopic insertion of GFP, CFP and IYFP transcriptional fusions in single copy at the amyE and bglS loci in Bacillus subtilis. Plasmid. 2010;64(3):143–149. doi: 10.1016/j.plasmid.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Arnaud M, Vary P, Zagorec M, Klier A, Debarbouille M, Postma P, Rapoport G. Regulation of the sacPA operon of Bacillus subtilis: identification of phosphotransferase system components involved in SacT activity. J Bacteriol. 1992;174:3161–3170. doi: 10.1128/jb.174.10.3161-3170.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel RA, Haiech J, Denizot F, Errington J. Isolation and characterization of the lacA gene encoding beta-galactosidase in Bacillus subtilis and a regulator gene, lacR. J Bacteriol. 1997;179:5636–5638. doi: 10.1128/jb.179.17.5636-5638.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi K, Nagata Y, Maruo B. Genetic control of the rate of alpha-amylase synthesis in Bacillus subtilis. J Bacteriol. 1974;119:410–415. doi: 10.1128/jb.119.2.410-415.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Guvener ZT, Cho JY, Chung KC, Chambliss GH. Specificity of DNA binding activity of the Bacillus subtilis catabolite control protein CcpA. J Bacteriol. 1995;177:5129–5134. doi: 10.1128/jb.177.17.5129-5134.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas P, Mäder U, Dervyn E, Rochat T, Leduc A, Pigeonneau N, Bidnenko E, Marchadier E, Hoebeke M, Aymerich S. et al. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science. 2012;335:1103–1106. doi: 10.1126/science.1206848. [DOI] [PubMed] [Google Scholar]
- Dehli T, Solem C, Jensen PR. Tunable promoters in synthetic and systems biology. Subcell Biochem. 2012;64:181–201. doi: 10.1007/978-94-007-5055-5_9. [DOI] [PubMed] [Google Scholar]
- Hammer K, Mijakovic I, Jensen PR. Synthetic promoter libraries–tuning of gene expression. Trends Biotechnol. 2006;24:53–55. doi: 10.1016/j.tibtech.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Mutalik VK, Guimaraes JC, Cambray G, Lam C, Christoffersen MJ, Mai QA, Tran AB, Paull M, Keasling JD, Arkin AP. et al. Precise and reliable gene expression via standard transcription and translation initiation elements. Nat Methods. 2013;10:354–360. doi: 10.1038/nmeth.2404. [DOI] [PubMed] [Google Scholar]
- Braatsch S, Helmark S, Kranz H, Koebmann B, Jensen PR. Escherichia coli strains with promoter libraries constructed by Red/ET recombination pave the way for transcriptional fine-tuning. Biotechniques. 2008;45:335–337. doi: 10.2144/000112907. [DOI] [PubMed] [Google Scholar]
- Anderson promoter collection. [ http://parts.igem.org/Promoters/Catalog/Anderson]
- Fukushima T, Ishikawa S, Yamamoto H, Ogasawara N, Sekiguchi J. Transcriptional, functional and cytochemical analyses of the veg gene in Bacillus subtilis. J Biochem. 2003;133:475–483. doi: 10.1093/jb/mvg062. [DOI] [PubMed] [Google Scholar]
- Jacob S, Allmansberger R, Gartner D, Hillen W. Catabolite repression of the operon for xylose utilization from Bacillus subtilis W23 is mediated at the level of transcription and depends on a cis site in the xylA reading frame. Mol Gen Genet. 1991;229:189–196. doi: 10.1007/BF00272155. [DOI] [PubMed] [Google Scholar]
- Kelly JR, Rubin AJ, Davis JH, Ajo-Franklin CM, Cumbers J, Czar MJ, de Mora K, Glieberman AL, Monie DD, Endy D. Measuring the activity of BioBrick promoters using an in vivo reference standard. J Biol Eng. 2009;3:4. doi: 10.1186/1754-1611-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrecke K, Jordan S, Mascher T. Stoichiometry and perturbation studies of the LiaFSR system of Bacillus subtilis. Mol Microbiol. 2013;87:769–788. doi: 10.1111/mmi.12130. [DOI] [PubMed] [Google Scholar]
- Dahl MK, Schmiedel D, Hillen W. Glucose and glucose-6-phosphate interaction with Xyl repressor proteins from Bacillus spp. may contribute to regulation of xylose utilization. J Bacteriol. 1995;177:5467–5472. doi: 10.1128/jb.177.19.5467-5472.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH. Experiments in molecular genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1972. [Google Scholar]
- Herzberg C, Weidinger LA, Dörrbecker B, Hübner S, Stülke J, Commichau FM. SPINE: a method for the rapid detection and analysis of protein-protein interactions in vivo. Proteomics. 2007;7:4032–4035. doi: 10.1002/pmic.200700491. [DOI] [PubMed] [Google Scholar]
- Competent E. coli cells. http://openwetware.org/wiki/TOP10_chemically_competent_cells.
- Sambrook J, Russell DW. Molecular Cloning - A Laboratory Manual. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Stülke J, Hanschke R, Hecker M. Temporal activation of β-glucanase synthesis in Bacillus subtilis is mediated by the GTP pool. J Gen Microbiol. 1993;139:2041–2045. doi: 10.1099/00221287-139-9-2041. [DOI] [PubMed] [Google Scholar]
- Miller JH. Experiments in molecular genetics. Cold Spring Harbor Laboratory: Cold Spring Harbor, New York; 1972. [Google Scholar]
- Bernstein JA, Khodursky AB, Lin P-H, Lin-Chao S, Cohen SN. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proc Natl Acad Sci U S A. 2002;99:9697–9702. doi: 10.1073/pnas.112318199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
GeneBank files of Bacillus BioBricks.
Alignment of original and final vectors.
Supplemental Figures, Tables and Text.